

Abstract
An increasing number of transcription factors have been shown to activate DNA replication. However, the underlying mechanism remains to be elucidated. Here it is shown that when tethered to a cellular replication origin, the acidic transcriptional activation domain of the breast cancer protein BRCA1 alters the local chromatin structure and stimulates chromosomal DNA replication. Cancer-predisposing mutations in BRCA1 that abolish transcriptional activation also prevent chromatin remodeling and activation of replication. Chromatin remodeling occurs even in the absence of a functional replication origin. Thus, increasing chromatin accessibility may be an important mechanism used by transcription factors to facilitate multiple nuclear processes.
Keywords: Chromatin remodeling, replication, transcription factors, BRCA1, yeast, ARS1
Initiation of eukaryotic DNA replication is enhanced by transcription factors that bind to the vicinity of an origin of replication. This phenomenon is well documented by numerous studies of viral DNA replication in mammalian cells (Van der Vliet 1996). The fact that one common regulatory factor can stimulate both transcription and replication poses an intriguing mechanistic problem. In vitro biochemical studies with reconstituted chromatin templates have shown that transcription factors activate viral DNA replication by counteracting the inhibitory effect of the nucleosomes (Cheng and Kelly 1989; Li and Botchan 1994). This, in turn, may facilitate assembly of the replication initiation complex. Furthermore, specific interactions between transcriptional activation domains and components of the basal replication machinery may provide additional mechanisms by which transcription factors activate viral DNA replication (Dutta et al. 1993; He et al. 1993; Li and Botchan 1993). The proposed role of transcription factors in activation of viral DNA replication is reminiscent of their function in transcriptional activation.
Compared with the viral studies, the role of transcription factors in cellular DNA replication is less well understood. One of the best defined origins of replication in Saccharomyces cerevisiae, ARS1, contains four genetically defined cis-acting elements: A, B1, B2, and B3. The A element is the binding site for an initiator protein complex called origin recognition complex (ORC) (Bell and Stillman 1992) and is absolutely required for ARS function. The B1 element provides an additional ORC binding site and other functions in initiation of replication (Rao and Stillman 1995; Rowley et al. 1995). The function of the B2 element is not clear. The B3 element contains a binding site for the yeast transcription factor Abf1p. B3, in conjunction with B1 and B2, is required for efficient origin function at ARS1 (Marahrens and Stillman 1992). Similar to the transcription factors involved in activation of viral DNA replication, Abf1p function in activation of ARS1 replication can be replaced by other transcription factors from a variety of organisms, including viruses, yeast, and mammals (Marahrens and Stillman 1992; Li et al. 1998). Although these findings suggest functional conservation between activation of viral and chromosomal DNA replication, the exact mechanism by which transcription factors activate cellular replication in vivo remains to be determined.
In this report we provide in vivo evidence that chromatin remodeling is an important pathway utilized by transcription factors to activate yeast chromosomal replication. In addition, mutations that abolish chromatin remodeling also severely impair activation of replication and transcription. Therefore, alteration of chromatin structure induced by transcription factors may be a common step in activation of multiple nuclear processes in eukaryotes.
Results
The BRCA1 transcriptional activation domain can activate yeast DNA replication in both plasmid and chromosomal contexts
The carboxy-terminal region of breast cancer protein BRCA1 contains an acidic transcriptional activation domain. When fused to yeast GAL4 DNA-binding domain, this BRCA1 domain can activate transcription in both mammalian and yeast cells (Chapman and Verma 1996; Monteiro et al. 1996). Interestingly, the transcriptional activation is abolished by cancer-predisposing mutations that occur in this region of BRCA1. We and others have shown previously that several acidic transcriptional activation domains can activate initiation of DNA replication in yeast (Marahrens and Stillman 1992; Li et al. 1998). To determine whether the BRCA1 activation domain is also capable of activating replication, we tethered it to a replication origin (ARS1) in Saccharomyces cerevisiae via the GAL4 DNA-binding domain. First, a plasmid stability assay was used to determine the effect of GAL4–BRCA1 on a plasmid-borne ARS1 origin (Li et al. 1998). The test plasmids used in this assay contained a functional centromere (CEN4), a selectable marker gene (URA3), and a modified ARS1 with one GAL4 binding site. Replication efficiency of the plasmid was reflected by the percentage of yeast cells that still retained the plasmid after 14 generations of nonselective growth.
As shown in Figure 1A (lane 1), very few cells (0.5 ± 0.1%) expressing GAL4 DNA-binding domain alone (GAL4–DBD) were able to retain the ARS1-containing plasmid. In contrast, expression of GAL4–BRCA1(1560–1863) resulted in a significant increase in plasmid stability (26 ± 3%; lane 2). This effect was due to the tethering of the activation domain to the replication origin, as no enhancement was detected when the GAL4 binding site at the origin was mutated (0.1 ± 0.1%; lane 3). The BRCA1 activation domain contains two repeats of the so-called BRCT motif, which is present in a variety of proteins involved in repair, replication, and checkpoint control (Koonin et al. 1996). Deletion of the first BRCT repeat of BRCA1 (amino acids 1560–1759) led to a moderate decrease in activation of origin function (lane 9), whereas removal of the second BRCT repeat (amino acids 1760–1863) completely abolished the ability of GAL4–BRCA1 to stimulate plasmid stability (lane 8).
Figure 1.
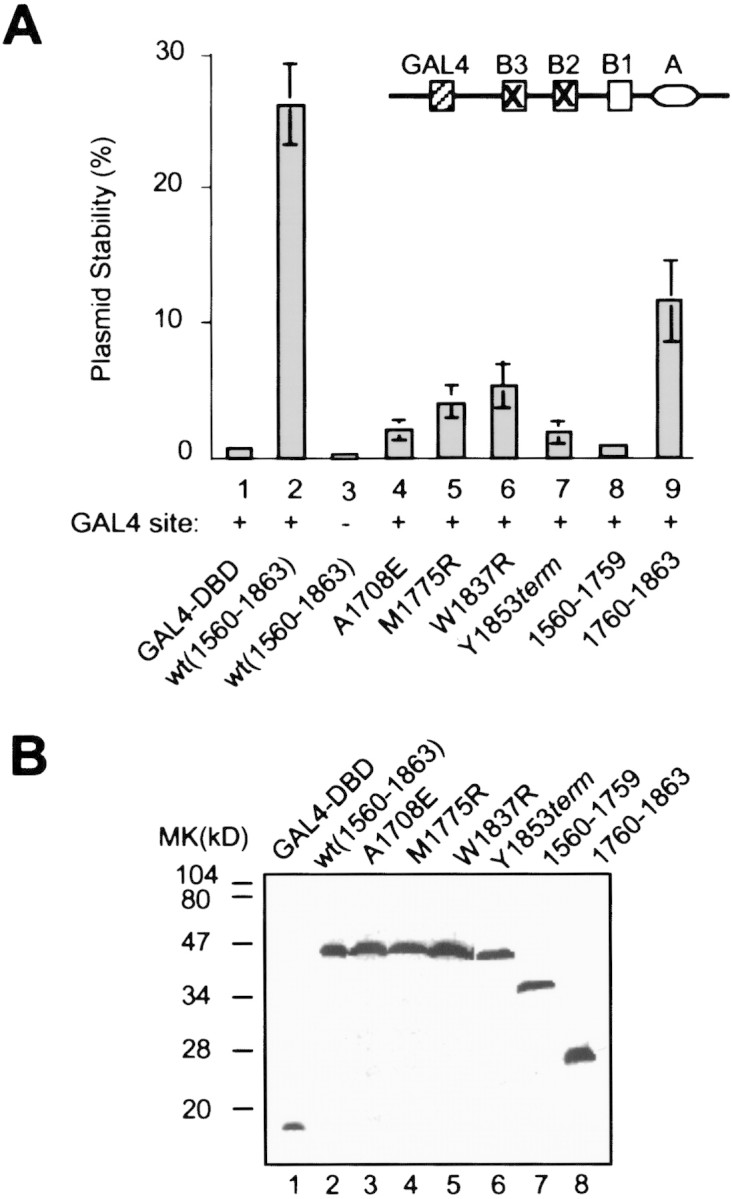
Activation of plasmid replication by GAL4–BRCA1. (A) Yeast cells expressing GAL4 derivatives were transformed with a test plasmid containing the modified ARS1 sequence (top). (Lane 3) Cells were transformed with a test plasmid bearing a mutant GAL4 binding site instead (Li et al. 1998). The values are averages of at least four independent experiments. (B) Anti-HA immunoblot of an equal amount of whole-cell extracts from the BP1ΔH strain expressing various GAL4 derivatives.
To determine the effect of tumor-associated BRCA1 mutations on activation of the replication origin, we introduced four cancer-predisposing mutations into GAL4–BRCA1. Three of the missense mutations lead to a single amino acid change (A1708E, M1775R and W1837R), whereas Y1853term results in deletion of the last 10-amino-acid residues of BRCA1. These mutations abolish the transcriptional activation by BRCA1 (Chapman and Verma 1996; Monteiro et al. 1996; Somasundaram et al. 1997; Y. Hu and R. Li, unpubl.). Western analysis showed that the hemagglutinin (HA)-tagged wild-type and mutant GAL4–BRCA1 fusion proteins were expressed at a similar level in yeast cells (Fig. 1B). Strikingly, all mutants were defective in stimulating origin function (Fig. 1A, lanes 4–7). Thus, the cancer-predisposing mutations had the same effect on the BRCA1-dependent activation of transcription and DNA replication.
To confirm the results from the plasmid-based replication assay, we replaced the native ARS1 in chromosome IV with a modified ARS1 that contained five GAL4 binding sites (diagram in Fig. 2A). First, a chromatin immunoprecipitation assay (Aparicio et al. 1997) was used to ascertain binding of the HA-tagged GAL4 derivatives to the modified chromosomal ARS1. As shown in Figure 2A (bottom), all GAL4 derivatives, including GAL4–DBD alone, the wild-type and mutant GAL4–BRCA1, coprecipitated with the modified ARS1 region. In contrast, none of the proteins was associated with the control replication origin ARS305. Thus, all GAL4 derivatives were capable of binding to the GAL4 binding sites on chromosome.
Figure 2.


GAL4–BRCA1 stimulates initiation of replication from a chromosomal locus. (A) Binding of the GAL4 derivatives to the GAL4 sites engineered at the chromosomal ARS1 in vivo. Chromatin immunoprecipitation assay was performed in RL1 strain with a high-affinity anti-HA antibody. Both input (top) and immunoprecipitated DNA (bottom) were used in the PCR reactions to amplify the sequences corresponding to ARS1 and ARS305, respectively. The strain expressed either no HA-tagged protein (lane 1) or various HA-tagged GAL4 derivatives (lanes 2–5). (B) Two-dimensional gel analysis of chromosomal ARS1 replication in RL1 strain expressing various GAL4 derivatives. The position of the bubble arc is indicated by an arrow in panel B. The GAL4 derivatives expressed in each sample are indicated.
Next, a two-dimensional gel electrophoretic technique was used to directly monitor the effect of the GAL4 derivatives on chromosomal DNA replication. Southern blotting analysis was used to distinguish the following two types of replication intermediates that encompassed the ARS1 origin: those that initiated from ARS1 and therefore traced a characteristic bubble arc, and those that were replicated passively by a replication fork from origins outside the ARS1-containing fragment and therefore traced a Y arc. In the presence of GAL4–DBD alone, very little initiation in the modified ARS1 could be detected, as indicated by a very weak bubble arc (Fig. 2B, panel A). In contrast, expression of GAL4–BRCA1 yielded a much stronger bubble arc (panel B). The same fusion protein did not affect replication efficiency of a control chromosomal origin that lacks GAL4 binding sites (data not shown). Two cancer-predisposing mutants of BRCA1 (M1775R and Y1853term), although capable of binding to the modified chromosomal ARS1 (Fig. 2A), failed to activate initiation of replication (Fig. 2B, panels C and D). Taken together, these data establish that the acidic activation domain of BRCA1 can stimulate a eukaryotic replication origin in both plasmid and chromosomal contexts.
Chromatin remodeling is an important step in activation of replication by acidic transcriptional activators
To determine the mechanism by which transcription factors activate cellular DNA replication, we examined the impact of the GAL4-derived activator on chromatin structure at the modified chromosomal ARS1 origin in vivo. The micrococcal nuclease (MNase) digestion pattern around the ARS1 region was analyzed by an indirect end-labeling method (Kadonaga 1998). In contrast to GAL4–DBD alone (Fig. 3, lanes 2,3), GAL4–BRCA1 caused prominent changes in the nuclease digestion pattern at the replication origin (lanes 4,5). In particular, a hypersensitive site (band A) was partially protected in the presence of GAL4–BRCA1, whereas the sensitivity of a neighboring site (band B) was greatly enhanced. Notably, cancer-predisposing mutations in the BRCA1 activation domain that abolished activation of replication also abrogated the chromatin remodeling at the replication origin (lanes 6–13).
Figure 3.

GAL4–BRCA1 induces alteration of the chromatin structure at the replication origin. Indirect end-labeling assay was used for analyzing the MNase digestion pattern around the ARS1 region. The radioactive probe is indicated by a vertical bar (left). The arrows (A,B) indicate the two bands, the intensity of which was most significantly affected by GAL4–BRCA1. The approximate positions of the GAL4 binding sites and the four cis elements of ARS1 are indicated at left. As a control, genomic DNA from the RL1 strain was isolated and the naked DNA was digested with a limited amount of MNase (lane 1).
To determine the causal relationship between chromatin remodeling and activation of replication, we introduced a point mutation into the ARS consensus sequence (ACS) that serves as the binding site for the ORC. This mutation completely abolishes formation of the prereplicative complex (pre-RC) and thereby initiation of replication (Aparicio et al. 1997; Marahrens and Stillman 1994; R. Li et al. unpubl.). As shown in Figure 4A, alteration of chromatin structure by the GAL4-derived activator was not affected at all by the absence of a functional replication origin (cf. lanes 3 and 4 with 5 and 6). Thus, chromatin remodeling is likely to be a cause, rather than an effect, of active replication.
Figure 4.


Chromatin remodeling by GAL4–BRCA1 is independent of a functional origin of replication. (A) Indirect end-labeling assay in RL1(G5/-B) and RL5(G5/-A/-B) strains. (B) Nucleosome array assay. The same MNase-digested genomic DNA as used in A was resolved by agarose gel electrophoresis without prior restriction digestion. The blot was first probed with a 200-bp fragment encompassing the ARS1 sequence (top) and subsequently stripped and reprobed with a SalI–HindIII fragment located 6 kb upstream of the chromosomal ARS1 locus (bottom). The positions of the two radioactive probes relative to the ARS1 locus are illustrated at right (thick bars).
To confirm that the GAL4-derived activator induces chromatin remodeling, we also carried out a nucleosome array assay (Kadonaga 1998). As shown in Figure 4B (top), GAL4–DBD alone yielded a DNA ladder indicative of a regular nucleosomal array at the origin (lanes 1–3). In contrast, the presence of GAL4–BRCA1 led to an apparent DNA smear (lanes 4–6), suggesting disruption of the regularity of the nucleosome array. Furthermore, the BRCA1-triggered loss of a canonical nucleosome array was unaffected by inactivation of the ORC binding site (Fig. 4B, top; cf. lanes 4–6 with 7–9). When the ARS1-specific hybridization blot was stripped and reprobed with a radioactive fragment corresponding to a genomic region 6 kb upstream of ARS1, regular DNA ladders were observed in all cases (Fig. 4B, bottom). Therefore, chromatin remodeling by GAL4–BRCA1 was confined to the vicinity of the GAL4 binding sites.
To ascertain that chromatin remodeling is the mechanism that is actually used by endogenous yeast transcription factors to activate a native replication origin, we also examined the nuclease digestion pattern around the wild-type and mutant ARS1 origins that harbor either point or linker substitution mutations in each of the four cis elements (Fig. 5; WT, -A, -B1, -B2, and -B3). These mutations are known to abolish function of the corresponding cis elements in initiation of replication (Marahrens and Stillman 1992, 1994). As shown in Figure 5, limited MNase digestion of the wild-type origin resulted in three prominent hypersensitive sites around ARS1 (lanes 1,2). Mutations in A (lanes 3,4) or B1 (lanes 5,6) did not change the overall digestion pattern, although the intensity of the bottom band was somewhat reduced. This may be due to lack of ORC binding in the A and B1 mutants (Bell and Stillman 1992; Rao and Stillman 1995). Likewise, the B2 mutation (lanes 7,8) slightly reduced the intensity of the middle band but did not affect the global digestion pattern. In contrast, abolishing the Abf1 binding site (B3) caused the most salient changes in the digestion pattern at ARS1 (lanes 9,10). Specifically, the top hypersensitive site disappeared completely, whereas a novel band appeared right above (indicated by asterisks at right). These results suggest that chromatin remodeling may be the mechanism used by the endogenous Abf1p to activate initiation of replication from the native ARS1.
Figure 5.

Chromatin remodeling by Abf1p at the native chromosomal ARS1. The parental and derivative SP1 strains carrying various mutations at the chromosomal ARS1 locus were used in the indirect end labeling assay. The two B3-dependent changes in nuclease sensitivity are indicated by asterisks right.
Discussion
The results presented here provide in vivo molecular evidence that chromatin remodeling is an important mechanism of transcription factors to activate cellular DNA replication. This extends and substantiates previous in vitro studies of the transcription factor’s role in activation of viral DNA replication (Cheng and Kelly 1989; Li and Botchan 1994). Taken together, these data strongly suggest that chromatin reconfiguration is a highly conserved feature of transcription factors in activation of eukaryotic DNA replication. It is not yet clear what other factors are required for mediating chromatin remodeling at the cellular replication origins. Nevertheless, in view of the multiple chromatin remodeling systems identified in recent years and their roles in mediating transcriptional activation (Kadonaga 1998), it seems plausible that such complexes may also play similar functions in activation of DNA replication by transcription factors. In keeping with this notion, it has been shown that structural changes at the SV40 origin of DNA replication in vitro can be mediated by a chromatin accessibility complex (CHRAC) from Drosophila (Alexiadis et al. 1998).
Mutations in the BRCA1 activation domain that affect chromatin remodeling abolish its ability to activate transcription as well as DNA replication. Moreover, chromatin remodeling persists even in the absence of a functional replication origin. These findings support the notion that chromatin remodeling is a common theme for many, if not all, transcription factors to activate various chromosomal events. In addition to activation of replication and transcription, transcription factors are also known to confer hot spot activity in meiotic recombination in Schizosaccharomyces pombe and S. cerevisiae by virtue of their ability to modify local chromatin structure (Nicolas 1998). Context may have an important role in determining which nuclear machinery a transcription factor can stimulate at a particular time and location. This may include the chromosomal region with which the transcription factor is associated (e.g., replication origins vs. transcriptional promoters), the physiological state of the cell in which it functions (e.g., proliferating vs. nonproliferating), and the specific interactions that it may have with components of each basal machinery. In this regard, it is noteworthy that BRCA1 has been implicated in transcription (Chapman and Verma 1996; Monteiro et al. 1996; Somasundaram et al. 1997), repair (Gowen et al. 1998), replication, and recombination (Scully et al. 1997b,c). BRCA1 is associated, directly or indirectly, with chromatin-modifying proteins such as p300, hBRG1 (Neish et al. 1998), and BRCA2 (Chen et al. 1998), which itself is associated with histone acetyltransferase activity (Fuks et al. 1998; Siddique et al. 1998). In addition, BRCA1 also interacts with components of transcription and repair machineries, such as the mammalian RNA polymerase II holoenzyme (Scully et al. 1997a) and RAD51 (Scully et al. 1997b). Therefore, it is conceivable that the chromatin remodeling activity associated with the BRCA1 activation domain may be an important feature of the multifunctional protein in regulation of different chromosomal events.
Although the yeast replication assays show that the BRCA1 activation domain is capable of activating a cellular replication origin, direct involvement of BRCA1 in mammalian DNA replication remains to be established. Nevertheless, a role of BRCA1 in replication, at least under certain physiological conditions, would be consistent with several S-phase-specific behaviors of BRCA1. Scully et al. (1997b) report that BRCA1 forms discrete nuclear foci in S-phase cells. Following DNA damage, BRCA1 foci in S-phase nuclei are dispersed, and BRCA1, together with the repair protein RAD51, colocalizes with proliferating cell nuclear antigen (PCNA), an essential replication protein (Scully et al. 1997c). In addition, when quiescent cells are induced to proliferate, both the total protein and phosphorylation levels of BRCA1 increase in late G1 phase and reach a maximum in S phase (Chen et al. 1996; Jin et al. 1997; Scully et al. 1997c). Finally, the homozygous Brca1 knockout mice are embryonic lethal at an early stage right before the first proliferative burst required for the development (Gowen et al. 1996; Hakem et al. 1996; Liu et al. 1996). These observations suggest a role for BRCA1 in S-phase progression, repair, and checkpoint control. It has been well documented that proteins with dual functions in replication and damage response provide important links between the replication machinery and the checkpoint pathways. For example, two other members of the BRCT domain family, Cut5/Rad4 in fission yeast and Dpb11 in budding yeast, are essential for promoting chromosomal DNA replication as well as S-phase checkpoint (Saka and Yanagida 1993; Araki et al. 1995). In a similar manner, the connection of BRCA1 with replication, repair, and checkpoint may ensure high fidelity of DNA replication and orderly S-phase progression in mammalian cells.
Materials and methods
Plasmids and yeast strains
The test plasmid for the plasmid stability assay (pARS1/-B23/G24) was described previously (Li et al. 1998). To construct the vectors for expression of the GAL4 fusion proteins in yeast, the wild-type and mutant BRCA1 sequences were amplified by a standard PCR method and cloned into the XbaI and BamHI sites immediately downstream of the GAL4(1–94) sequence in the CUP1 vector (Li et al. 1998), which was then integrated into the LEU2 locus on chromosome.
The yeast strain used for the plasmid stability assay BP1ΔH was described previously (Li et al. 1998). To construct the yeast strains that contain the modified chromosomal ARS1 sequences (5xG/-B and 5xG/-A/-B), an XbaI–HindIII fragment bearing five GAL4 binding sites from G5BCAT (Lillie and Green 1989) was used to replace a 119-bp genomic sequence (616–735; coordinates according to Marahrens and Stillman 1992) immediately upstream of the B3 element of ARS1. The mutations at the corresponding four cis elements of ARS1 are 860T-G (A), XhoI linker substitutions at 835–842 (B1), 798–805 (B2), and double point mutations at 756 and 758 (B3) (Marahrens and Stillman 1992). A pop-in and pop-out gene replacement method was used to replace the wild-type ARS1 sequence in the BP1ΔH strain with the modified ARS1. The resulting strains were named RL1 (5xG/-B) and RL5 (5xG/-A/-B), respectively.
The yeast strains that were used in the nuclease digestion experiment shown in Figure 5 were described previously (Marahrens and Stillman 1994). The parental strain is SP1 (MATa, ura3-52, his3, trp1-289, leu2-2,113, ade8, can1). The mutations in the cis elements of ARS1 are the same as described above for the RL1 and RL5 strains.
Immunoblotting, plasmid stability assay, and two-dimensional gel electrophoresis
These assays were performed as described previously (Li et al. 1998), except that all liquid media contained 100 μm copper sulfate for induction of the GAL4 derivatives. An anti-HA antibody (12CA5) was used in the immunoblotting.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation assay was performed as described previously (Aparicio et al. 1997). The sequences for the PCR primers will be provided on request. A high-affinity anti-HA antibody from Boehringer Mannheim was used for immunoprecipitation.
MNase digestion assays
The assays were adapted from a published protocol for in vivo footprint analysis (Huibregtse and Engelke 1991), with the following modifications. The lysed spheroplasts were treated with MNase (Worthington) at a final concentration of 50 U/ml for 2 and 5 min in Figures 3, 4A, and 5, and 2, 5, and 10 min in Figure 4B. For the indirect end-labeling assay, genomic DNA was digested with EcoRI, resolved by agarose gel electrophoresis, and probed with a 358-bp RsaI–EcoRI fragment 3′ to the ARS1 sequence. For the nucleosome array experiment, genomic DNA was resolved by electrophoresis without prior restriction digestion and probed with an EcoRI–HindIII ARS1-containing fragment from pARS1 (Marahrens and Stillman 1992) or a SalI–HindIII fragment 6 kb upstream of the ARS1 region.
Acknowledgments
We thank B. Stillman, I. Verma, and H. Hanafusa for providing the reagents, T. Miyake for helpful discussions, and M. Alexandrow, J. Hamlin, A. Pemov, and M. Smith for critical reading of the manuscript. This work was supported in part by a research grant (5-FY97-0667) from the March of Dimes Birth Defects Foundation and a grant from the Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rl2t@virginia.edu; FAX (804) 924-5069.
References
- Alexiadis V, Varga-Weisz PD, Bonte E, Becker PB, Gruss C. In vitro chromatin remodeling by chromatin accessibility complex (CHRAC) at the SV40 origin of DNA replication. EMBO J. 1998;17:3428–3438. doi: 10.1093/emboj/17.12.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Araki H, Leem S-H, Phongdara A, Sugino A. Dpb11, which interacts with DNA polymerase II(e) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- Chen Y, Farmer AA, Chen C-F, Jones DC, Chen P-L, Lee W-H. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res. 1996;56:3168–3172. [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber B, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Cheng L, Kelly TJ. Transcriptional activator nuclear factor I stimulates the replication of SV40 minichromosomes in vivo and in vitro. Cell. 1989;59:541–551. doi: 10.1016/0092-8674(89)90037-8. [DOI] [PubMed] [Google Scholar]
- Dutta A, Ruppert JM, Aster JC, Winchester E. Inhibition of DNA replication factor RPA by p53. Nature. 1993;365:79–82. doi: 10.1038/365079a0. [DOI] [PubMed] [Google Scholar]
- Fuks F, Milner J, Kouzarides T. BRCA2 associates with acetyltransferase activity when bound to P/CAF. Oncogene. 1998;17:2531–2534. doi: 10.1038/sj.onc.1202475. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Avrutskaya AV, Latour AM, Koller BH, Leadon SA. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- Hakem R, Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- He Z, Brinton BT, Greenblatt J, Hassell JA, Ingles CJ. The transcription protein VP16 and GAL4 bind replication factor A. Cell. 1993;73:1223–1232. doi: 10.1016/0092-8674(93)90650-f. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Engelke DR. Direct sequence and footprint analysis of yeast DNA by primer extension. Methods Enzymol. 1991;194:550–562. doi: 10.1016/0076-6879(91)94042-b. [DOI] [PubMed] [Google Scholar]
- Jin Y, Xu XL, Yang M-C, Wei F, Ayi T-C, Bowcock AM, Baer R. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc Natl Acad Sci. 1997;94:12075–12080. doi: 10.1073/pnas.94.22.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell. 1998;92:307–313. doi: 10.1016/s0092-8674(00)80924-1. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Altschul SF, Bork P. BRCA1 protein products: Functional motifs. Nat Genet. 1996;13:266–268. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- Li R, Botchan MR. The acidic transcription activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell. 1993;73:1207–1221. doi: 10.1016/0092-8674(93)90649-b. [DOI] [PubMed] [Google Scholar]
- ————— Acidic transcription factors alleviate nucleosome-mediated repression of DNA replication of bovine pappilomavirus type 1. Proc Natl Acad Sci. 1994;91:7051–7055. doi: 10.1073/pnas.91.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu DS, Tanaka M, Zheng L, Berger SL, Stillman B. Activation of chromosomal DNA replication in Saccharomyces cerevisiae by acidic transcriptional activation domains. Mol Cell Biol. 1998;18:1296–1302. doi: 10.1128/mcb.18.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie JW, Green MR. Transcription activation by the adenovirus E1A protein. Nature. 1989;338:39–44. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee W-H. Inactivation of the mouse Brca1gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes & Dev. 1996;10:1835–1843. doi: 10.1101/gad.10.14.1835. [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science. 1992;255:817–823. doi: 10.1126/science.1536007. [DOI] [PubMed] [Google Scholar]
- ————— Replicator dominance in a eukaryotic chromosome. EMBO J. 1994;13:3395–3400. doi: 10.1002/j.1460-2075.1994.tb06642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neish AS, Anderson SF, Schlegel BP, Wei W, Parvin JD. Factors associated with the mammalian RNA polymerase II holoenzyme. Nucleic Acids Res. 1998;26:847–853. doi: 10.1093/nar/26.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A. Relationship between transcription and initiation of meiotic recombination: Toward chromatin accessibility. Proc Natl Acad Sci. 1998;95:87–89. doi: 10.1073/pnas.95.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao H, Stillman B. The origin recognition complex interacts with a bipartitie DNA binding site within yeast replicators. Proc Natl Acad Sci. 1995;92:2224–2228. doi: 10.1073/pnas.92.6.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley A, Cocker JH, Harwood J, Diffley JFX. Initiation complex assembly at budding yeast replication origins begins with the recognition of a bipartitie sequence by limiting amounts of the initiator, ORC. EMBO J. 1995;14:2631–2641. doi: 10.1002/j.1460-2075.1995.tb07261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y, Yanagida M. Fission yeast cut5+, required for S phase onset and M phase restraint, is identical to the radiation-damage repair gene rad4+ Cell. 1993;74:383–393. doi: 10.1016/0092-8674(93)90428-s. [DOI] [PubMed] [Google Scholar]
- Scully R, Anderson SF, Chao D, Wei W, Ye L, Young RA, Livingston DM, Parvin JD. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci. 1997a;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997b;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs R, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997c;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Siddique H, Zou J-P, Rao VN, Reddy ESP. The BRCA2 is a histone acetyltransferase. Oncogene. 1998;16:2283–2285. doi: 10.1038/sj.onc.1202003. [DOI] [PubMed] [Google Scholar]
- Somasundaram K, Zhang H, Zeng Y-X, Houvras Y, Peng Y, Zhang H, Wu GS, Licht JD, Weber BL, Ei-Deiry WS. Arrest of the cell cycle by the tumor-suppressor BRCA1 requires the CDK-inhibitor p21(WAF1/CIP1) Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- Van der Vliet PC. DNA replication in eukaryotic cells. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. Roles of transcription factors in DNA replication; pp. 87–118. [Google Scholar]