

Abstract
Listeria monocytogenes is a Gram-positive intracellular pathogen that is naturally resistant to lysozyme. Recently, it was shown that peptidoglycan modification by N-deacetylation or O-acetylation confers resistance to lysozyme in various Gram-positive bacteria, including L. monocytogenes. L. monocytogenes peptidoglycan is deacetylated by the action of N-acetylglucosamine deacetylase (Pgd) and acetylated by O-acetylmuramic acid transferase (Oat). We characterized Pgd−, Oat−, and double mutants to determine the specific role of L. monocytogenes peptidoglycan acetylation in conferring lysozyme sensitivity during infection of macrophages and mice. Pgd− and Pgd− Oat− double mutants were attenuated approximately 2 and 3.5 logs, respectively, in vivo. In bone-marrow derived macrophages, the mutants demonstrated intracellular growth defects and increased induction of cytokine transcriptional responses that emanated from a phagosome and the cytosol. Lysozyme-sensitive mutants underwent bacteriolysis in the macrophage cytosol, resulting in AIM2-dependent pyroptosis. Each of the in vitro phenotypes was rescued upon infection of LysM− macrophages. The addition of extracellular lysozyme to LysM− macrophages restored cytokine induction, host cell death, and L. monocytogenes growth inhibition. This surprising observation suggests that extracellular lysozyme can access the macrophage cytosol and act on intracellular lysozyme-sensitive bacteria.
INTRODUCTION
Lysozyme is a potent antimicrobial peptide identified in all major taxa of living organisms that represents an ancient host defense mechanism against invading microbes (4, 37). Lysozyme is typically found in host fluids, including tears, saliva, airway fluid, serum, and mucus, and in lysosomal granules of neutrophils and macrophages (4). Lysozyme activity results in bacteriolysis and the potential release of microbial ligands that may be detected by host innate immune pathways (18). The ubiquitous presence of lysozyme has undoubtedly selected pathogens that avoid its effects.
Lysozyme displays muramidase activity that hydrolyzes the β1-4 glycosidic linkage between N-acetyl muramic acid and N-acetylglucosamine residues, which make up the glycan backbone of peptidoglycan (2). Lysozyme also displays cationic antimicrobial activity independent of muramidase action (42). Two lysozyme genes, those encoding lysozymes M and P, are expressed in mice (10), while only a single lysozyme is expressed in humans. Lysozyme M (LysM) is the predicted ortholog of human lysozyme and is the predominant form expressed in most cells, including bone marrow-derived macrophages (BMM) (10). Previous studies have demonstrated that disruption of LysM in mice resulted in increased susceptibility to some bacterial infections. For instance, LysM− mice had increased bacterial burden when infected with Klebsiella pneumoniae or Pseudomonas aeruginosa (8, 35). Similarly, Shimada et al. showed increased susceptibility to middle ear infection in LysM− mice infected with Streptococcus pneumoniae (53).
Many pathogens modify their peptidoglycan to resist host lysozyme (12). Common modifications of peptidoglycan include N-deacetylation, N-glycolylation, and O-acetylation (63). Bacillus anthracis, S. pneumoniae, and L. monocytogenes are Gram-positive pathogens that N-deacetylate the N-acetylglucosamine residues of their peptidoglycan and are consequently resistant to lysozyme (3, 64, 68). For example, Vollmer and Tomasz identified pgdA in S. pneumoniae, which encodes an N-acetylglucosamine deacetylase and contributes to lysozyme resistance (64). Boneca et al. identified pgdA in L. monocytogenes and showed that it provides resistance to lysozyme in broth culture and contributes to virulence (3). Staphylococcus aureus is resistant to lysozyme due to modification of its peptidoglycan by O-acetylation. Bera et al. identified oatA in S. aureus and showed that its action mediated resistance to lysozyme and that its presence in some Staphylococcus species correlated with lysozyme resistance (1, 2). S. pneumoniae also carries an O-acetyl transferase, Adr, and Davis et al. showed that both adr and pgdA contribute to lysozyme sensitivity and pathogenesis (9, 11). Interestingly, the double mutants were hypersensitive to lysozyme and were attenuated in vivo but were able to outcompete wild-type (wt) S. pneumoniae in LysM− mice, demonstrating a fitness cost for the expression of pgdA and adr (11).
L. monocytogenes is a Gram-positive facultative intracellular pathogen that has been used for decades as a model system to examine basic aspects of infection and immunity both in tissue culture and in murine models of listeriosis (60). Following internalization by host cells, L. monocytogenes is subjected to antimicrobial conditions of a maturing phagosome (40). Bacteria subsequently either are killed in the phagosome or escape into the host cell cytosol, where they grow rapidly. Escape from the phagosome is mediated largely by a secreted pore-forming cholesterol-dependent cytolysin, listeriolysin O (LLO) (52). Cytosolic L. monocytogenes exploits a host system of actin-based motility to move from cell to cell, thereby avoiding extracellular defense mechanisms (57).
Three primary cellular pathways of innate immunity are triggered during infection by L. monocytogenes. The first represents a generic, MyD88-dependent response that emanates from cell surface and phagosomal recognition and leads to the transcription of inflammatory cytokines, including interleukin-12 (IL-12) and IL-1β (31, 43). Presumably, Toll-like receptor 2 (TLR2), which recognizes components of the bacterial cell wall, is the primary receptor that contributes to this response (58). However, the precise ligand(s) that stimulates this pathway has yet to be identified. A second pathway is triggered by listerial cyclic di-AMP and stimulates an interferon regulatory factor 3/Sting-dependent response, leading to the expression of beta interferon (IFN-β) and coregulated genes (26, 43, 50, 51, 66). L. monocytogenes DNA and muramyl dipeptide (MDP) were shown to reconstitute the IFN-β response in vitro, and this pathway was amplified by NOD2 (31). A third pathway triggered by L. monocytogenes infection, but only to a small extent, is activation of the DNA-dependent AIM2 inflammasome and the consequent activation of caspase-1 and cell death by pyroptosis (28). Pyroptosis is a programmed cell death pathway dependent on caspase-1 and associated with inflammation, in contrast with other cell death pathways, such as apoptosis and necrosis (36). AIM2-dependent induction of pyroptosis is caused by the infrequent lysis of intracellular L. monocytogenes (51). AIM2-dependent pyroptosis is dependent on the downstream adaptor ASC and is associated with secretion of IL-1β (15, 33, 45).
In this study, we addressed the role of L. monocytogenes N-acetylglucosamine deacetylase (Pgd; lmo0415) and O-acetylmuramic acid transferase (Oat; lmo1291) during infection. Pgd− and Pgd− Oat− mutants were lysozyme sensitive and had intracellular growth defects, increased induction of cytokine transcriptional responses, and increased bacteriolysis and subsequent pyroptosis.
MATERIALS AND METHODS
Bacterial strains.
All L. monocytogenes strains used and generated in this study were in the 10403S background. In-frame deletion mutants were constructed by splice overlap extension and introduced by allelic exchange (5). Strains were grown to stationary phase in brain heart infusion (BHI) medium at 30°C overnight without shaking for macrophage infections.
Broth growth curves.
Stationary-phase cultures were backdiluted 1:40 in BHI medium and grown to mid-log phase at 37°C with shaking. Titrations of hen egg white lysozyme in phosphate-buffered saline (PBS) were added in constant volumes at mid-log phase (optical density at 600 nm [OD600] of 0.3 at 240 min), and growth was measured at OD600 at 15-min intervals over the course of 8 h. CFU were measured by plating culture dilutions on LB agar plates. Plates were incubated overnight at 37°C, and colonies were counted the following day.
Disc diffusion assay.
A total of 3 × 108 stationary-phase L. monocytogenes cells were plated in 250 μl BHI medium on LB agar plates. One milligram of hen egg white lysozyme was added to Whatman paper absorbent discs on the plate in a 10-μl volume. Plates were incubated overnight at 37°C, and the diameter of the zone of growth inhibition around each disc was measured the following day.
Macrophages.
Bone marrow-derived macrophages were prepared from 6- to 8-week-old female mice as previously described (27). All mice were in the C57BL/6 genetic background. LysM− mice were a gift from Tomas Ganz. AIM2 short hairpin RNA (shRNA) knockdown vectors were a gift from Katherine Fitzgerald, and immortalized C57BL/6 macrophages were a gift from Russell Vance.
Intracellular growth curves.
A total of 2 × 106 BMM were plated overnight and infected with LLO-expressing strains at a multiplicity of infection (MOI) of 0.1 and LLO− L. monocytogenes strains at an MOI of 1. Thirty minutes after infection, macrophage monolayers were washed with PBS and fresh medium was added. At 1 h postinfection, 50 μg/ml gentamicin was added to kill extracellular bacteria. Replication was quantified as previously described (47). Briefly, three coverslips at each time point were washed with water to lyse macrophages. Bacteria recovered from each coverslip were plated on LB agar plates, and CFU were determined.
Macrophage gene expression by quantitative RT-PCR.
A total of 2 × 106 BMM were infected at an MOI of 1 for 4 h. Thirty minutes after infection, macrophage monolayers were washed with PBS and fresh medium was added. At 1 h postinfection, 50 μg/ml gentamicin was added to kill extracellular bacteria. RNA was purified using an RNAqueous kit according to the manufacturer's instructions (Ambion, Austin, TX). RNA was DNase treated, processed, and analyzed by real-time quantitative reverse transcription (RT)-PCR as previously described (31).
Luciferase reporter delivery system.
A total of 5 × 105 IFN-α/β receptor (IFN-α/βR)-negative BMM were infected at an MOI of 5 with L. monocytogenes strains bearing a plasmid reporter, pBHE573, that encodes luciferase under a cytomegalovirus (CMV) promoter as previously described (51). Extracellular hen egg white lysozyme (1 mg/ml) was added at 1 h postinfection where indicated. IFN-α/βR− BMM were used to prevent L. monocytogenes-induced IFN signaling and host protein synthesis inhibition.
LDH release and IL-1β ELISA.
A total of 5 × 105 BMM were pretreated 12 to 16 h with 100 ng/ml Pam3CSK4 (Invivogen, San Diego, CA) and infected at an MOI of 5 as previously described (51). Extracellular hen egg white lysozyme (Sigma) was added at 1 mg/ml at 1 h postinfection where indicated. Supernatants were collected at 6 h postinfection and analyzed for lactate dehydrogenase (LDH) release and IL-1β secretion. To measure LDH release, 60 μl supernatant was added to 60 μl LDH detection reagent as previously described in duplicate in 96-well plates (51). IL-1β secretion was determined using the mouse IL-1β enzyme-linked immunosorbent assay (ELISA) Ready-SET-Go! according to the manufacturer's instructions (eBioscience, San Diego, CA).
Immunofluorescence microscopy.
Infected BMM were fixed with 4% paraformaldehyde. Samples were stained using a rabbit polyclonal anti-L. monocytogenes primary antibody (Difco) and an Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen) secondary antibody. Cells were stained with rhodamine-phalloidin for actin and DAPI (4′,6-diamidino-2-phenylindole) for nucleic acid. Images were acquired with an Olympus IX81 epifluorescence microscope using a 60× objective. Images were digitally overlaid using Metamorph software (Universal Imaging). BMM were scored positive for bacterial degradation based on the presence of multiple fluorescent specks smaller than L. monocytogenes.
In vivo infections.
C57BL/6 mice (The Jackson Laboratory) or LysM− mice were injected intravenously with 1 50% lethal dose (1 × 105 CFU) of wild-type, Pgd−, or Pgd− Oat− L. monocytogenes bacteria, and spleen and liver were harvested 48 h later as previously described (50).
Lysozyme detection assay.
Serum was collected from C57BL/6 or LysM− mice using BD Microtainer serum separator tubes (Becton, Dickinson and Company, Franklin Lakes, NJ) and was analyzed for the presence of lysozyme activity using an EnzChek lysozyme assay kit according to the manufacturer's instructions (Invitrogen).
RESULTS
Pgd− L. monocytogenes bacteria are sensitive to lysozyme.
To determine the relative lysozyme sensitivity of Pgd−, Oat−, and Pgd− Oat− L. monocytogenes bacteria, we assayed growth in broth culture and on LB agar in the presence or absence of hen egg white lysozyme. Bacteria were grown for 4 h to mid-log phase, followed by the addition of titrated concentrations of lysozyme within the physiological range (6, 7, 19–21, 38, 48, 61), and growth was monitored for an additional 4 h (Fig. 1A to D). Wild-type and Oat− strains grew similarly in the presence or absence of lysozyme at all concentrations tested. In contrast, the growth of the Pgd− strain was inhibited by 100 μg/ml lysozyme, while growth of the Pgd− Oat− strain was inhibited by a concentration of 50 μg/ml. In addition, the optical density of the cultures decreased in the presence of 100 μg/ml lysozyme, indicating bacteriolysis. Similarly, bacterial CFU of Pgd− and Pgd− Oat− strains decreased more than 2 and 3 logs, respectively, in the presence of lysozyme, while the wild-type strain was unaffected (data not shown). Pgd− and Pgd− Oat− stationary-phase cultures were also sensitive to lysozyme (data not shown). Similar results were seen using a disc diffusion assay. Bacterial lawns, grown on LB plates with Whatman paper discs containing 1 mg lysozyme, were measured for zones of clearance of each strain of L. monocytogenes (Fig. 1E to H). As expected, no clearance was observed for wild-type or Oat− L. monocytogenes, while the average zone of clearance was 16.8 mm in diameter for Pgd− L. monocytogenes and 18.75 mm for the Pgd− Oat− strain. These data indicate that wild-type and Oat− L. monocytogenes strains were lysozyme resistant whereas the Pgd− Oat− double mutant was more sensitive to lysozyme than the Pgd− mutant alone.
Fig. 1.

Sensitivity of L. monocytogenes strains to lysozyme. Growth of wt (A), Pgd− (B), Oat− (C), and Pgd− Oat− (D) L. monocytogenes strains in BHI medium in the presence of titrated lysozyme (LZ). Lysozyme was added to cultures in equal volumes at 240 min after bacterial inoculation, and OD600 was monitored at 15-min intervals. Growth of wt (E), Pgd− (F), Oat− (G), and Pgd− Oat− (H) L. monocytogenes strains on LB plates containing 1 mg lysozyme on a Whatman paper disc. Bacterial lawns were grown overnight, and the zone of clearance was measured the next day. Data are representative of more than 3 independent experiments with similar results.
Lysozyme-sensitive L. monocytogenes bacteria demonstrate intracellular growth defects that are rescued in the absence of LysM.
To determine the role of Pgd and Oat during the intracellular life cycle, bone marrow-derived macrophages (BMM) from C57BL/6 (wild-type) mice were infected with either wild-type, Pgd−, Oat−, or Pgd− Oat− L. monocytogenes. Oat− L. monocytogenes had no detectable growth defect (data not shown), the Pgd− strain had a small defect, and Pgd− Oat− L. monocytogenes demonstrated a more significant defect at early time points (Fig. 2A). The number of CFU between 0.5 h and 2 h increased 1.5-fold during wt infection and 1.1-fold during infection with Pgd−. In contrast, the number of Pgd− Oat− CFU decreased 2.4-fold during this time, and the difference between wt and Pgd− Oat− CFU was statistically significant at 2 h by Student's t test (P = 0.0163). An LLO− mutation was introduced into each of these strains to directly examine the fate of bacteria trapped in phagosomes. LLO− Pgd− and LLO− Pgd− Oat− had approximately a half-log and 2-log reduction in CFU, respectively, at 8 h in wild-type BMM (Fig. 2C). These data suggest that the primary observed defect occurred within phagosomes, while cytosolic bacteria grew nearly as well as the wild type. The growth defect of the Pgd− and Pgd− Oat− strains was rescued in LysM− BMM (Fig. 2B). Similarly, the loss of CFU in LLO− Pgd− and LLO− Pgd− Oat− infections was rescued in LysM− BMM (Fig. 2D). These data suggest that the lysozyme sensitivity of Pgd− and Pgd− Oat− L. monocytogenes strains accounted for the defects in intracellular growth in BMM.
Fig. 2.

Growth of L. monocytogenes strains in BMM. A total of 2 × 106 wt (A) or LysM− (B) BMM were infected with wt, Pgd−, or Pgd− Oat− L. monocytogenes at an MOI of 0.1. A total of 2 × 106 wt (C) or LysM− (D) BMM were infected with LLO− L. monocytogenes strains at an MOI of 1. BMM were lysed, and bacterial CFU were quantified. Error bars represent standard deviations of the means of technical triplicates, and data are representative of more than 3 independent experiments with similar results. Student's t test was used to analyze statistical significance compared to wt L. monocytogenes infection at each time point. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Lysozyme-sensitive L. monocytogenes bacteria induce increased vacuolar and cytosolic cytokine signaling.
To determine whether lysozyme susceptibility results in a differential host innate immune response, we analyzed vacuolar and cytosolic cytokine expression levels in C57BL/6 (wild-type) or LysM− BMM infected with lysozyme-sensitive L. monocytogenes. L. monocytogenes bacteria that secrete LLO, a pore-forming hemolysin, access the cytosol and induce strong MyD88 and TRIF-independent IFN-β expression (43). L. monocytogenes bacteria that lack LLO are unable to escape from the phagosome and fail to induce IFN-β but instead stimulate a specific MyD88-dependent cytokine expression program that emanates from the vacuole that includes IL-12 and IL-1β transcription (43).
The expression of IL-1β (Fig. 3A) and IL-12 (Fig. 3B) increased during infection with LLO− Pgd− and LLO− Pgd− Oat− L. monocytogenes strains compared to that during wild-type infection. Increased IL-1β and IL-12 expression by lysozyme-sensitive strains was not observed in LysM− BMM (Fig. 3A and B). Surprisingly, Pgd− and Pgd− Oat− L. monocytogenes strains, which expressed LLO, also stimulated increased IL-1β and IL-12 expression in a lysozyme-dependent manner (Fig. 3A and B).
Fig. 3.

Pgd− and Pgd− Oat− L. monocytogenes strains induce increased cytosolic and vacuolar cytokine responses dependent on the presence of host lysozyme. A total of 106 wt (black bars) or LysM− (gray bars) BMM were infected with wt or lysozyme-sensitive L. monocytogenes at an MOI of 1 for 4 h. LLO− infections were also performed at an MOI of 1. RNA was harvested, and IL-1β (A), IL-12 (B), and IFN-β (C) transcripts were measured relative to those of β-actin by quantitative PCR. Error bars represent standard deviations of the means determined in triplicate. Data are representative of at least 3 independent experiments with similar results. One-way analysis of variance (ANOVA) and Tukey's post hoc test were used to analyze the significance of infections in either wt or LysM− BMM (dashed lines). Student's t test was used to analyze significance for each bacterial strain. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Pgd− and Pgd− Oat− L. monocytogenes strains stimulated increased IFN-β transcription in wild-type BMM but not LysM− BMM (Fig. 3C). Pgd− and Pgd− Oat− infection induced 2.75- and 1.70-fold more IFN-β transcription, respectively, than wt infection. Increased IFN-β expression was not observed in LysM− BMM infected with lysozyme-sensitive strains. As expected, LLO− Pgd− and LLO− Pgd− Oat− L. monocytogenes strains did not induce IFN-β (data not shown). These data suggest that lysozyme-sensitive L. monocytogenes bacteria that have escaped the phagosome are more readily sensed by a cytosolic surveillance pathway, leading to increased IFN-β expression. Similarly, lysozyme-sensitive L. monocytogenes bacteria are more readily sensed by TLRs, leading to enhanced transcription of IL-1β and IL-12.
Lysozyme-sensitive L. monocytogenes strains undergo increased bacteriolysis in the macrophage cytosol.
We sought to determine whether lysozyme-sensitive strains lyse during infection. Wild-type or LysM− BMM were infected with wild-type, Pgd−, or Pgd− Oat− L. monocytogenes strains and were visualized by fluorescence microscopy for evidence of bacteriolysis. After 2 h, degradation of Pgd− Oat− L. monocytogenes was clearly observed in wild-type BMM (Fig. 4A) but not in LysM− BMM (Fig. 4B). Evidence of bacterial degradation was observed in 20.7% of wild-type BMM infected with Pgd− Oat− L. monocytogenes and 4.0% of wild-type BMM infected with Pgd− L. monocytogenes. Less than 1% of LysM− BMM infected with Pgd− or Pgd− Oat− L. monocytogenes showed evidence of bacterial degradation. None of the BMM infected with wild-type L. monocytogenes showed evidence of degradation.
Fig. 4.

Intracellular bacteriolysis of lysozyme-sensitive L. monocytogenes. Immunofluorescence microscopy of wt (A) or LysM− (B) BMM infected with Pgd− Oat− L. monocytogenes (green) imaged at 90 min postinfection (actin and nuclei were stained red and blue, respectively). Insets depict bacterial degradation. Size bars represent 10 μm. Cytosolic DNA delivery is increased in Pgd− and Pgd− Oat− L. monocytogenes strains (C). IFN-α/βR− BMM were infected with L. monocytogenes bearing a plasmid-containing luciferase under a CMV promoter at an MOI of 5. Luciferase expression was detected at 6 h postinfection. Error bars represent standard deviations of the means determined in triplicate. Statistical significance was evaluated using Student's t test. Data are representative of at least 3 separate experiments with similar results. *, P < 0.01; **, P < 0.0001.
To indirectly measure cytosolic bacteriolysis, we used a reporter plasmid bearing a luciferase gene under the control of a CMV promoter (51). Lysis of cytosolic bacteria bearing the reporter plasmid results in delivery of the plasmid to the cytosol and host expression of luciferase. A positive control for release of plasmid into the cytosol was an L. monocytogenes strain expressing PSA bacteriophage holin and lysin under the control of the actA promoter (holin-lysin) designed to lyse in the cytosol. Pgd− infection stimulated an approximately 2.5-fold increase in luciferase expression compared to wild-type infection (Fig. 4C), while Pgd− Oat− infection stimulated an 11-fold increase, establishing that Pgd− and Pgd− Oat− strains underwent cytosolic bacteriolysis and released DNA to the cytosol. This led us to hypothesize that additional cytosolic receptors could be activated by Pgd− and Pgd− Oat− L. monocytogenes strains.
Lysozyme-sensitive L. monocytogenes strains induce AIM2-dependent pyroptosis.
To determine the role of lysozyme activity in induction of pyroptosis, we assayed wild-type and LysM− BMM infected with lysozyme-sensitive strains for release of lactate dehydrogenase (LDH) and secretion of IL-1β. To determine the pathway of activation, we assayed BMM lacking ASC or NLRP3. Holin-lysin and the L. monocytogenes Δ2473 strain were recently identified as hyperinducers of pyroptosis and were therefore used as positive controls (51).
Infection of wild-type BMM with Pgd− and Pgd− Oat− L. monocytogenes strains stimulated 2- and 2.5-fold more LDH release than wild-type infection, respectively (Fig. 5A). Pgd− and Pgd− Oat− L. monocytogenes infections stimulated 3- and 5-fold more IL-1β secretion than wild-type infection, respectively (Fig. 5B). As expected, lysozyme-sensitive strains in the LLO− background stimulated no cell death or IL-1β secretion (Fig. 5A and B). The increase in LDH release and IL-1β secretion induced by Pgd− and Pgd− Oat− L. monocytogenes infections in wild-type BMM was not observed in LysM− BMM (Fig. 5A and B), indicating a lysozyme-dependent mechanism of inflammasome induction. Pgd− and Pgd− Oat− L. monocytogenes infections induced increased LDH release and IL-1β secretion in NLRP3− BMM, but no increase was observed in ASC− BMM. This indicates that lysozyme-sensitive L. monocytogenes strains activate an ASC-dependent pathway of inflammasome activation independent of NLRP3.
Fig. 5.

Pgd− and Pgd− Oat− L. monocytogenes strains induce increased inflammasome activation. (A) LDH release in wt, LysM−, ASC−, or NLRP3− Pam3CSK4-stimulated BMM. A total of 5 × 105 BMM were infected for 6 h at an MOI of 5. (B) IL-1β secretion detected by ELISA in wt, LysM−, ASC−, or NLRP3− Pam3CSK4-stimulated BMM at 6 h postinfection at an MOI of 5. Error bars represent standard deviations of the means. Statistical significance was evaluated compared to wt L. monocytogenes infection using Student's t test. Data are representative of more than 3 independent experiments with similar results. *, P < 0.01.
L. monocytogenes strains that lyse in the cytosol activate AIM2 (51), a cytosolic DNA receptor that stimulates inflammasome activation in an ASC-dependent manner (25, 65). To test the hypothesis that AIM2 detects lysozyme-sensitive L. monocytogenes, we measured cell death and IL-1β release in immortalized BMM (iBMM) with shRNA-mediated AIM2 stably knocked down. LDH release and IL-1β secretion were completely dependent on the presence of AIM2 (Fig. 6A and B). A control strain ectopically expressing Legionella pneumophila flagellin (49a) was used to stimulate AIM2-independent cell death mediated by Nlrc4 (IPAF) activation. These data indicate that lysozyme-sensitive strains stimulated the AIM2 inflammasome following lysozyme-induced bacteriolysis and subsequent release of bacterial DNA into the host cell cytosol.
Fig. 6.

Pgd− and Pgd− Oat− L. monocytogenes strains activate the AIM2 inflammasome. (A) Pam3CSK4-stimulated immortalized shRNA AIM2 knockdown or shRNA scrambled macrophages were infected at an MOI of 5, and LDH was measured at 6 h. (B) IL-1β secretion was measured by ELISA in Pam3CSK4-stimulated immortalized shRNA AIM2 knockdown or shRNA scrambled immortalized macrophages (iBMM) infected for 6 h. Error bars represent standard deviations of the means determined in triplicate. Statistical significance was evaluated using Student's t test. Data are representative of more than 3 independent experiments with similar results. **, P < 0.0001. Lp fla, Legionella flagellin.
Bacterial replication is required for induction of Pgd− and Pgd− Oat− strain-mediated cell death.
We hypothesized that during infection with Pgd− and Pgd− Oat− L. monocytogenes strains, vacuolar lysozyme could generate increased amounts of bacterial ligands, potentially released to the cytosol following dissolution of the phagosome. An alternative hypothesis was that phagosomal lysozyme might compromise the cell wall integrity of lysozyme-sensitive strains, resulting in bacteriolysis in the cytosol. If either hypothesis were correct, we would observe rapid induction of pyroptosis. However, we were unable to detect significant LDH release induced by Pgd− or Pgd− Oat− L. monocytogenes in wild-type BMM until 4.5 h postinfection (Fig. 7A).
Fig. 7.

Bacterial replication but not cell-to-cell spread is required for release of LDH. LDH release over time is shown. (A) Pam3CSK4-stimulated wt BMM were infected at an MOI of 5, and LDH release was assayed at 3, 4.5, and 6 h postinfection. (B) A total of 0.1 mg/ml chloramphenicol (cm) was added at 1, 2, 3, 4, or 5 h postinfection, and LDH release was measured at 6 h postinfection. Error bars represent standard deviations of the means. Data are representative of more than 3 independent experiments with similar results.
An alternative hypothesis was that lysozyme, encountered in secondary vacuoles following cell-to-cell spread, could generate ligands that resulted in pyroptosis. However, inhibition of cell-to-cell spread by treatment of macrophages with cytochalasin D, an inhibitor of actin polymerization, at 4 h postinfection resulted in a slight drop in LDH levels in all samples (57; data not shown). Pgd− and Pgd− Oat− L. monocytogenes strains induced increased overall pyroptosis, suggesting that cell-to-cell spread was not required and that increased bacterial ligands were not likely generated by lysozyme in secondary vacuoles.
We then considered that cytosolic bacterial replication might be required to induce pyroptosis. Bacterial translation was blocked at 1, 2, 3, 4, or 5 h after infection using 100 μg/ml chloramphenicol (Cm), a bacteriostatic antibiotic, and LDH release was measured at 6 h (Fig. 7B). No significant LDH release was detected in macrophages treated with Cm at 1 h. However, a strain ectopically expressing Legionella flagellin (49a) that stimulates the Nlrc4 inflammasome induced rapid LDH release. An L. monocytogenes strain expressing holin-lysin, which expresses phage holin and lysin under the actA promoter, did not induce LDH release when treated with Cm at 1 or 2 h, presumably because the actA promoter was not activated until 2 h postinfection. When allowed to replicate for 2 h, Pgd− L. monocytogenes stimulated 6% total LDH release and Pgd− Oat− L. monocytogenes stimulated 16% total LDH release, while wild-type bacteria stimulated only 5% total LDH release, indicating that increased bacteriolysis of lysozyme-sensitive strains occurs soon after escape. However, LDH release increased with the amount of time that Pgd− and Pgd− Oat− L. monocytogenes strains replicated in the cytosol. These data suggest that bacterial intracellular replication was required to stimulate lysozyme-dependent cell death and that L. monocytogenes strains were subject to lysozyme activity in the cytosol.
Extracellular lysozyme acts on Pgd− and Pgd− Oat− cytosolic bacteria.
To determine whether lysozyme could gain access to the macrophage cytosolic compartment, we assayed for bacteriolysis using the delivery of plasmid DNA as described above. In the presence of extracellular lysozyme, lysozyme-sensitive strains delivered plasmid, while delivery by the wild type was not significantly affected (Fig. 8A).
Fig. 8.

Extracellular lysozyme accesses the BMM cytosol and results in increased degradation of lysozyme-sensitive L. monocytogenes strains and subsequent host cell death. (A) IFN-α/βR− BMM were infected with L. monocytogenes bearing a plasmid-containing luciferase under a CMV promoter, and 1 mg/ml extracellular lysozyme was added at 1 h postinfection. Luciferase expression was detected at 6 h postinfection. (B) Pam3CSK4-stimulated LysM− BMM were infected at an MOI of 5, and 1 mg/ml extracellular lysozyme was added at 1 h postinfection. LDH release was measured at 6 h. (C) Pam3CSK4-stimulated LysM− BMM were treated with cycloheximide and infected at an MOI of 5, and LDH release was measured at 6 h. Error bars represent standard deviations of the means. (A and B) Statistical significance was evaluated using Student's t test. *, P < 0.05; ***, P < 0.0001. (C) One-way ANOVA was performed to analyze the statistical significance of LDH release following various treatments of BMM infected with individual L. monocytogenes strains. *, P < 0.05; **, P < 0.001. The means of LDH values from infections treated with lysozyme compared to those treated with lysozyme plus cycloheximide were determined to be not significant (ns) using Student's t test. The means of LDH values from infections with Pgd−, Oat−, or Pgd− Oat− L. monocytogenes were significant by one-way ANOVA. Data are representative of at least 3 independent experiments with similar results. LysM− BMM were infected with either wt (D, E) or Pgd− Oat− (F, G) L. monocytogenes for 2 h. A total of 1 mg/ml extracellular lysozyme was added at 1 h postinfection (E, G), and cells were scored for evidence of bacterial degradation (depicted in inset). Size bars represent 10 μm. Data are representative of more than 3 independent experiments with similar results.
To visualize whether extracellular lysozyme could induce bacteriolysis, LysM− BMM were infected with wild-type or Pgd− Oat− L. monocytogenes and 1 mg/ml lysozyme was added extracellularly after 1 h, which is within the biological range (6, 7, 19–21, 38, 48, 61). Infected LysM− BMM were visualized at 2 h postinfection for bacterial degradation (Fig. 8D to G). A total of 19.2% of Pgd− Oat− strain-infected LysM− BMM treated with extracellular lysozyme showed evidence of bacterial degradation (Fig. 8G), whereas no degradation was observed in untreated Pgd− Oat− strain-infected LysM− BMM (Fig. 8F). Similarly, LysM− BMM infected with LLO− Pgd− Oat− L. monocytogenes and treated with extracellular lysozyme had evidence of bacterial degradation, while untreated BMM did not (data not shown), suggesting that extracellular lysozyme accesses L. monocytogenes-containing vacuoles.
Extracellular lysozyme also resulted in induction of increased LDH release but did not affect lysozyme-resistant strains (Fig. 8B). Neither the addition of extracellular mutanolysin nor the transfer of supernatant from Pam3CSK4-treated uninfected wild-type BMM (data not shown) had any affect on LDH release. As a control to verify that extracellular lysozyme was not activating host cells, Pam3CSK4-stimulated LysM− BMM were treated with cycloheximide to prevent host translation and were infected with L. monocytogenes following treatment with extracellular lysozyme. No significant difference was observed in the presence of cycloheximide, suggesting that lysozyme did not activate a pathway that induced cytosolic bacteriolysis (Fig. 8C). These data suggest that extracellular lysozyme accessed the host cytoplasm and can act on lysozyme-sensitive L. monocytogenes, resulting in bacteriolysis and generation of cytosolic ligands, which stimulate cell death.
Lysozyme-sensitive L. monocytogenes strains demonstrate in vivo defects that are not rescued in the absence of LysM.
To determine whether the in vitro phenotype of lysozyme-sensitive strains affected virulence in vivo, we infected B6 and LysM− mice by intravenous (i.v.) injection and quantified CFU at 48 h. Pgd− L. monocytogenes had an approximate 2-log defect in both the spleen and liver, and the Pgd− Oat− strain had a 3.5-log defect in the spleen and liver (Fig. 9). Experiments testing for rescue in LysM− mice were initially inconsistent; however, we did not observe a significant rescue in LysM− mice. This could be due to the presence of residual lysozyme activity detected in the serum of LysM− mice (see Fig. S1 in the supplemental material).
Fig. 9.
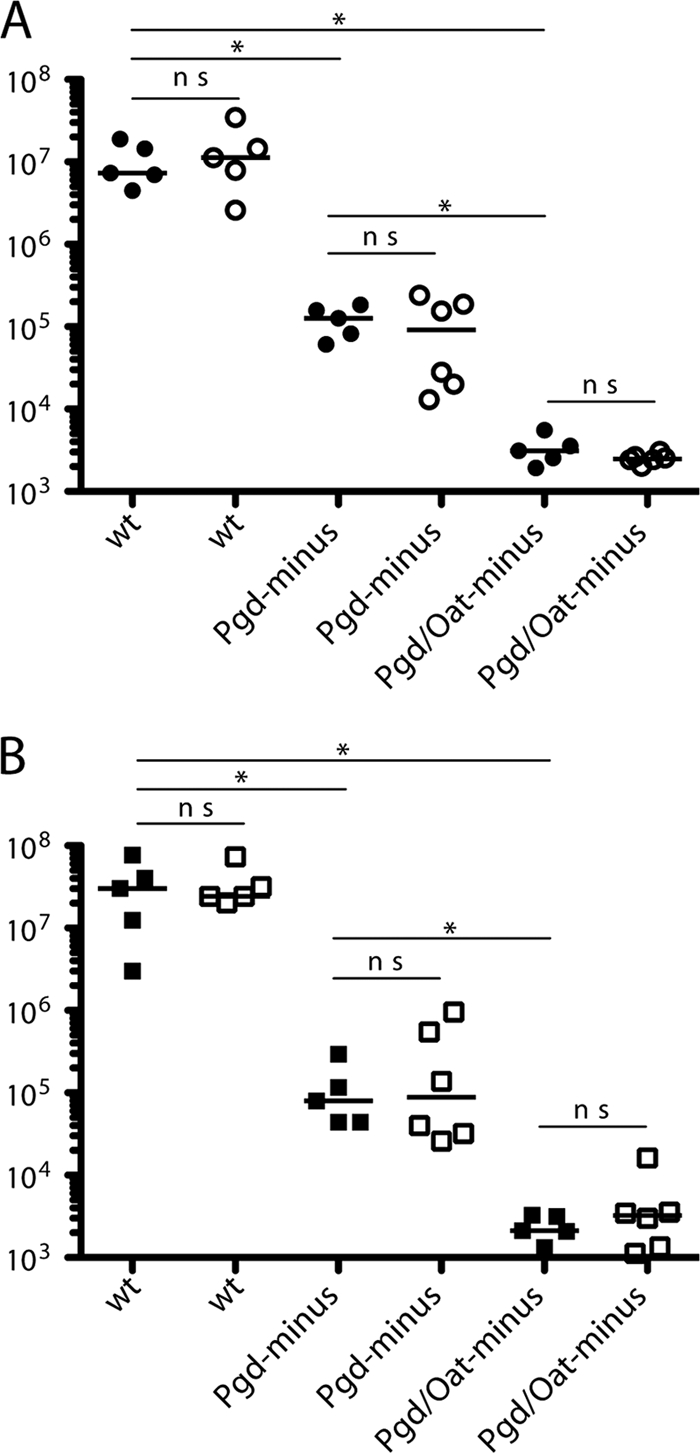
Pgd− and Pgd− Oat− L. monocytogenes strains are defective in vivo but are not rescued in LysM− mice. C57BL/6 or LysM− mice were infected with 1 × 105 bacteria by i.v. injection, and CFU were quantified in the spleen (A) and liver (B) at 48 h postinfection. Spleen and liver are represented by circles and squares, respectively. Filled and open shapes represent C57BL/6 and LysM− mice, respectively. Statistical significance was evaluated using a Mann-Whitney test. Data are representative of more than 3 independent experiments with similar results. *, P < 0.02; ns, not significant.
DISCUSSION
The precise role of lysozyme during innate immunity to microbial infection has yet to be fully appreciated. The aim of this study was to assess the role of host lysozyme resistance by comparing L. monocytogenes strains that were either resistant or sensitive to lysozyme. The results of this study showed that lysozyme acts extracellularly, within phagocytic vacuoles, and surprisingly in the cytosol of infected cells. In phagosomes, lysozyme activity led to the release of bacterial ligands that activated a vacuole-specific program of cytokine induction. In the cytosol, lysozyme led to bacteriolysis, thereby activating two distinct cytosolic innate immune pathways, one leading to the expression of IFN-β and another leading to DNA-dependent, AIM2-dependent pyroptosis. All of the phenotypes associated with lysozyme sensitivity were reversed during infection of macrophages lacking LysM. Surprisingly, all of the above-mentioned phenotypes were restored by simply adding lysozyme to the extracellular medium.
Transcription of IL-12 and IL-1β is stimulated by L. monocytogenes trapped in phagosomes (43). Lysozyme-sensitive L. monocytogenes stimulated increased expression of these cytokines in LLO− and LLO-expressing backgrounds. TLR2 is a candidate receptor involved in this pathway since it is both localized at the cell surface and recruited to phagosomes upon internalization of microbes (44). TLR2 has been reported to detect various cell wall-associated ligands, including peptidoglycan (although there is controversy over whether it is a bona fide ligand), and has been shown to form heterodimers with TLR1 or TLR6 to detect lipopeptides and lipoteichoic acid (LTA) (29, 59). It is likely that lysozyme activity generates increased cell wall fragments that are detected by TLR2. However, although vacuolar cytokines were dependent on MyD88, infection with Pgd− L. monocytogenes induced similar levels of IL-1β and IL-12 in TLR2− and wild-type BMM (data not shown). Therefore, the precise nature of the bacterial ligand(s) or host receptor(s) is not known.
Cytosolic L. monocytogenes strains induce a MyD88-independent response that is monitored by IFN-β expression. Here, IFN-β expression is elevated approximately 2-fold by lysozyme-sensitive mutants. One possible explanation is that cytosolic bacteria are lysed, resulting in the release of DNA and cyclic di-AMP, two ligands that activate IFN-β expression (31, 54a, 66). A second possibility is that N-deacetylated peptidoglycan shed from growing bacteria has enhanced stimulatory activity. Indeed, Boneca et al. reported that N-deacetylated peptidoglycan had increased NOD1 stimulatory activity on peritoneal elicited macrophages and that digested Pgd− peptidoglycan induced increased NF-κB activity in both a NOD1- and NOD2-dependent manner (3). However, using NOD1−, NOD2−, or NOD1− NOD2− BMM, we still observed an approximate 3-fold difference in IFN-β expression levels between wild-type and Pgd− bacteria (data not shown). While it is possible that NOD1 and NOD2 expression levels were different, our data favor the hypothesis that increased bacteriolysis and release of nucleic acids led to enhanced IFN-β expression.
This study also revealed that lysozyme acts in the cell cytosol, causing bacteriolysis and subsequent host cell death. These results support the recently emerging concept that cytosolic bacteriolysis is linked to activation of the AIM2-dependent inflammasome (51). Indeed, using strains differentially susceptible to lysozyme, we showed that increased lysozyme sensitivity correlates with increased bacteriolysis and AIM2-dependent pyroptosis. This is the first study to show a link between lysozyme and AIM2 stimulation. However, a previous study has linked lysozyme activity to the NLRP3 inflammasome. Shimada et al. have shown that vacuolar digestion of S. aureus peptidoglycan by lysozyme is required for NLRP3 stimulation (54). However, we found no significant role for NLRP3 in response to lysozyme-sensitive L. monocytogenes strains despite distinct lysozyme activity in the vacuole, suggesting that L. monocytogenes peptidoglycan is not sensed via the same pathway. It is unknown whether S. aureus lysozyme-digested peptidoglycan is the ligand detected directly by NLRP3 or whether activation is stimulated by its feature as a small crystalline molecule (30). Nevertheless, similar to S. aureus, L. monocytogenes specifically subverts inflammasome activation by modification of its peptidoglycan.
All of the phenotypes associated with lysozyme sensitivity could be restored simply by adding lysozyme to the culture medium. In our experiment, we used 1 mg/ml hen egg white lysozyme (HEWL), which is within the biological range (6, 7, 19–21, 38, 48, 61). Although HEWL has the same enzymatic activity as mammalian lysozyme, it may have properties that differ in specificity for deacetylated peptidoglycan or access to BMM cytosol compared to endogenous mouse lysozyme M. There is evidence that macrophages internalize other extracellular peptides in addition to lysozyme. For example, Tan et al. have reported that macrophages can acquire antimicrobial peptide defensins from apoptotic neutrophil granules to become increasingly resistant to Mycobacterium tuberculosis (55). The Lehrer lab has shown that macrophages can take up alpha-defensins (23; R. I. Lehrer, personal communication). It has also been shown that anti-LLO antibodies can act in a phagosome to inhibit L. monocytogenes escape (13). Therefore, soluble innate immune factors, including secreted lysozyme, may also concentrate in macrophages.
How lysozyme enters the cytosol is less obvious. However, several other small cationic peptides have been reported to translocate from the extracellular milieu to the cytosol. HIV-1 Tat, herpes simplex virus 1 VP22 transcription factor, and a Drosophila protein, Antp, have been shown to enter cells from the culture medium and localize in the cytoplasm. Of these translocated proteins, HIV-1 Tat has been the best studied. Tat is secreted from infected cells and subsequently enters adjacent cells and accumulates in the cytosol (16, 32). Tat is a 101-amino-acid cationic peptide with a calculated isoelectric point of 9.61 and has been used as a fusion protein to deliver ovalbumin, β-galactosidase, peroxidase, and green fluorescent protein into cells (14, 22, 49). A basic domain rich in arginine and lysine residues has been identified in Tat as the region required for translocation (41, 62). Although lysozyme does not share a homologous domain, it too is a small cationic peptide of 14 kDa, with a calculated isoelectric point of 9.11. Although speculative, its similar features in size and cationic quality are properties that could suggest a mechanism for its access to the cytosol.
Lysozyme-sensitive L. monocytogenes strains were severely attenuated in the mouse model of listeriosis. However, the loss of virulence was not rescued in LysM− mice. The most likely explanation was a compensatory and redundant mechanism of LysP. LysP compensation in LysM− mice has been reported separately by Ganz et al. and Markart et al. (17, 34). Markart et al. showed that LysP mRNA is typically not expressed in the alveolar space in wild-type mice but is upregulated significantly in LysM− mice (34). Ganz et al. showed that lysozyme was detected with an antibody that recognized both M and P isoforms in alveolar and peritoneal macrophages (17). Certainly, LysP compensatory expression in relative tissues could explain the lack of rescue. Lysozyme P circulating in the blood may also be a contributing factor. Pgd− Oat− L. monocytogenes had a log decrease in CFU in liver by 30 min (data not shown), suggesting an early encounter with lysozyme. Also, based on our results, circulating lysozyme P may enter macrophages and act intracellularly in a vacuole and the host cell cytosol.
In addition, lysozyme-sensitive L. monocytogenes may be susceptible to other host cell hydrolytic enzymes or stresses not present in BMM. Popowska et al. have shown that Pgd− L. monocytogenes is more sensitive to autolysis-inducing agents, Triton X-100 and EDTA, and various antibiotics (46). Indeed, it is predicted that Pgd− L. monocytogenes bearing N-acetylated glucosamine residues would have a more negative surface charge due to lack of an exposed primary amine residue and would thus be more susceptible to cationic peptides (63). However, Pgd− and Pgd− Oat− L. monocytogenes strains were not sensitive to cationic peptides, including LP9, a human lysozyme-derived 9-mer, in comparison to a strain lacking MprF, which is known to be sensitive to cationic agents (see Fig. S2 in the supplemental material) (24, 56, 67). In the future, it will be of interest to determine whether it is the lytic activity of mouse lysozyme or its cationic properties that act on Pgd− and Pgd− Oat− L. monocytogenes strains.
We conclude that L. monocytogenes resistance to lysozyme is an essential determinant of pathogenesis. Indeed, many bacterial pathogens are lysozyme resistant (12, 63). The role of lysozyme appears to be multifaceted. We and others have shown that lysozyme activity can kill bacteria and stimulate inflammation, but in other contexts, it has been shown to inhibit inflammation. Ganz et al. showed that Micrococcus luteus induced increased inflammation in LysM− mice, suggesting that peptidoglycan degradation by lysozyme is required to downregulate inflammation (17). Thus, one role of lysozyme may be to degrade potentially proinflammatory peptidoglycan fragments associated with commensal or nonpathogenic bacteria, which may be a strategy to avoid mounting an inappropriate inflammatory response. This may be similar to a mechanism whereby host enzymes inactivate bacterial lipopolysaccharide to avoid prolonged tolerance and increased inflammation (39). However, lysozyme also serves to generate peptidoglycan fragments that are important immunostimulatory ligands during infection. Davis and Weiser suggest that modified peptidoglycan that is more resistant to hydrolysis by lysozyme results in the generation of larger fragments, which might be differentially detected by a host (12). The ability of lysozyme to act from within cells (in both phagosomes and the cytosol) and extracellularly highlights its role as a versatile antimicrobial factor that contributes to multiple aspects of host defense during bacterial infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. D. Sauer, Josh Woodward, and Nicholas Arpaia for thoughtful discussions and critiques. We thank J. D. Sauer, Chelsea Witte, Pete Lauer, and Adam Williamson for technical experimental advice.
This research was supported by National Institutes of Health grants A1063302 and AI27655.
D. A. Portnoy consults with and has a financial interest in Aduro BioTech, a company that may stand to benefit from the results of this research.
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 18 July 2011.
REFERENCES
- 1. Bera A., Biswas R., Herbert S., Gotz F. 2006. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect. Immun. 74:4598–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bera A., Herbert S., Jakob A., Vollmer W., Gotz F. 2005. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 55:778–787 [DOI] [PubMed] [Google Scholar]
- 3. Boneca I. G., et al. 2007. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc. Natl. Acad. Sci. U. S. A. 104:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Callewaert L., Michiels C. W. 2010. Lysozymes in the animal kingdom. J. Biosci. 35:127–160 [DOI] [PubMed] [Google Scholar]
- 5. Camilli A., Tilney L. G., Portnoy D. A. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8:143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohn Z. A. 1978. Activation of mononuclear phagocytes: fact, fancy, and future. J. Immunol. 121:813–816 [PubMed] [Google Scholar]
- 7. Cole A. M., Dewan P., Ganz T. 1999. Innate antimicrobial activity of nasal secretions. Infect. Immun. 67:3267–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cole A. M., et al. 2005. Decreased clearance of Pseudomonas aeruginosa from airways of mice deficient in lysozyme M. J. Leukoc. Biol. 78:1081–1085 [DOI] [PubMed] [Google Scholar]
- 9. Crisostomo M. I., et al. 2006. Attenuation of penicillin resistance in a peptidoglycan O-acetyl transferase mutant of Streptococcus pneumoniae. Mol. Microbiol. 61:1497–1509 [DOI] [PubMed] [Google Scholar]
- 10. Cross M., Mangelsdorf I., Wedel A., Renkawitz R. 1988. Mouse lysozyme M gene: isolation, characterization, and expression studies. Proc. Natl. Acad. Sci. U. S. A. 85:6232–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis K. M., Akinbi H. T., Standish A. J., Weiser J. N. 2008. Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae. PLoS Pathog. 4:e1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis K. M., Weiser J. N. 2011. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect. Immun. 79:562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Edelson B. T., Unanue E. R. 2001. Intracellular antibody neutralizes Listeria growth. Immunity 14:503–512 [DOI] [PubMed] [Google Scholar]
- 14. Fawell S., et al. 1994. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. U. S. A. 91:664–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Franchi L., Kanneganti T. D., Dubyak G. R., Nunez G. 2007. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282:18810–18818 [DOI] [PubMed] [Google Scholar]
- 16. Frankel A. D., Pabo C. O. 1988. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55:1189–1193 [DOI] [PubMed] [Google Scholar]
- 17. Ganz T., et al. 2003. Increased inflammation in lysozyme M-deficient mice in response to Micrococcus luteus and its peptidoglycan. Blood 101:2388–2392 [DOI] [PubMed] [Google Scholar]
- 18. Ginsburg I. 2002. The role of bacteriolysis in the pathophysiology of inflammation, infection and post-infectious sequelae. APMIS 110:753–770 [DOI] [PubMed] [Google Scholar]
- 19. Gordon S., Todd J., Cohn Z. A. 1974. In vitro synthesis and secretion of lysozyme by mononuclear phagocytes. J. Exp. Med. 139:1228–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hankiewicz J., Swierczek E. 1974. Lysozyme in human body fluids. Clin. Chim. Acta 57:205–209 [DOI] [PubMed] [Google Scholar]
- 21. Hansen N. E., Karle H. 1971. Blood and bone-marrow lysozyme in neutropenia: an attempt towards pathogenetic classification. Br. J. Haematol. 21:261–270 [DOI] [PubMed] [Google Scholar]
- 22. Hauber J., Malim M. H., Cullen B. R. 1989. Mutational analysis of the conserved basic domain of human immunodeficiency virus tat protein. J. Virol. 63:1181–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hazrati E., et al. 2006. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 177:8658–8666 [DOI] [PubMed] [Google Scholar]
- 24. Herbert S., et al. 2007. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog. 3:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hornung V., et al. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishikawa H., Ma Z., Barber G. N. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones S., Portnoy D. A. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 62:5608–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S., et al. 2010. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40:1545–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kumar H., Kawai T., Akira S. 2009. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388:621–625 [DOI] [PubMed] [Google Scholar]
- 30. Lamkanfi M., Kanneganti T. D. 2010. Nlrp3: an immune sensor of cellular stress and infection. Int. J. Biochem. Cell Biol. 42:792–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leber J. H., et al. 2008. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 4:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mann D. A., Frankel A. D. 1991. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 10:1733–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mariathasan S., et al. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232 [DOI] [PubMed] [Google Scholar]
- 34. Markart P., et al. 2004. Comparison of the microbicidal and muramidase activities of mouse lysozyme M and P. Biochem. J. 380:385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Markart P., Korfhagen T. R., Weaver T. E., Akinbi H. T. 2004. Mouse lysozyme M is important in pulmonary host defense against Klebsiella pneumoniae infection. Am. J. Respir. Crit. Care Med. 169:454–458 [DOI] [PubMed] [Google Scholar]
- 36. Martinon F., Mayor A., Tschopp J. 2009. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27:229–265 [DOI] [PubMed] [Google Scholar]
- 37. Medzhitov R., Janeway C., Jr 2000. Innate immune recognition: mechanisms and pathways. Immunol. Rev. 173:89–97 [DOI] [PubMed] [Google Scholar]
- 38. Mendiondo O., Suit H., Fixler H. 1978. Lysozyme levels and macrophage content of tumor tissue in C3H mice bearing fibrosarcoma transplants treated by radiation and Corynebacterium parvum. Int. J. Radiat. Oncol. Biol. Phys. 4:829–834 [DOI] [PubMed] [Google Scholar]
- 39. Munford R. S., Varley A. W. 2006. Shield as signal: lipopolysaccharides and the evolution of immunity to gram-negative bacteria. PLoS Pathog. 2:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Myers J. T., Tsang A. W., Swanson J. A. 2003. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J. Immunol. 171:5447–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagahara H., et al. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4:1449–1452 [DOI] [PubMed] [Google Scholar]
- 42. Nash J. A., Ballard T. N., Weaver T. E., Akinbi H. T. 2006. The peptidoglycan-degrading property of lysozyme is not required for bactericidal activity in vivo. J. Immunol. 177:519–526 [DOI] [PubMed] [Google Scholar]
- 43. O'Riordan M., Yi C. H., Gonzales R., Lee K. D., Portnoy D. A. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U. S. A. 99:13861–13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ozinsky A., Smith K. D., Hume D., Underhill D. M. 2000. Co-operative induction of pro-inflammatory signaling by Toll-like receptors. J. Endotoxin Res. 6:393–396 [PubMed] [Google Scholar]
- 45. Ozoren N., et al. 2006. Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J. Immunol. 176:4337–4342 [DOI] [PubMed] [Google Scholar]
- 46. Popowska M., Kusio M., Szymanska P., Markiewicz Z. 2009. Inactivation of the wall-associated de-N-acetylase (PgdA) of Listeria monocytogenes results in greater susceptibility of the cells to induced autolysis. J. Microbiol. Biotechnol. 19:932–945 [DOI] [PubMed] [Google Scholar]
- 47. Portnoy D. A., Jacks P. S., Hinrichs D. J. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prixova J. 1975. Serum lysozyme in mice subjected to combined immunosuppression with 6-mercaptopurine and hydrocortisone. Folia Microbiol. (Praha) 20:509–512 [DOI] [PubMed] [Google Scholar]
- 49. Ruben S., et al. 1989. Structural and functional characterization of human immunodeficiency virus tat protein. J. Virol. 63:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49a. Sauer J. D., et al. 11 July 2011. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc. Natl. Acad. Sci. U. S. A. [Epub ahead of print.] doi: 10.1073/pnas.1019041108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sauer J. D., et al. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79:688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sauer J. D., et al. 2010. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schnupf P., Portnoy D. A. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 9:1176–1187 [DOI] [PubMed] [Google Scholar]
- 53. Shimada J., et al. 2008. Lysozyme M deficiency leads to an increased susceptibility to Streptococcus pneumoniae-induced otitis media. BMC Infect. Dis. 8:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shimada T., et al. 2010. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7:38–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54a. Stetson D. B., Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 1:93–103 [DOI] [PubMed] [Google Scholar]
- 55. Tan B. H., et al. 2006. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J. Immunol. 177:1864–1871 [DOI] [PubMed] [Google Scholar]
- 56. Thedieck K., et al. 2006. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol. Microbiol. 62:1325–1339 [DOI] [PubMed] [Google Scholar]
- 57. Tilney L. G., Portnoy D. A. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Torres D., et al. 2004. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72:2131–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Travassos L. H., et al. 2004. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 5:1000–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vazquez-Boland J. A., et al. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vinding T., Eriksen J. S., Nielsen N. V. 1987. The concentration of lysozyme and secretory IgA in tears from healthy persons with and without contact lens use. Acta Ophthalmol. 65:23–26 [DOI] [PubMed] [Google Scholar]
- 62. Vives E., Brodin P., Lebleu B. 1997. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 272:16010–16017 [DOI] [PubMed] [Google Scholar]
- 63. Vollmer W. 2008. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 32:287–306 [DOI] [PubMed] [Google Scholar]
- 64. Vollmer W., Tomasz A. 2000. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 275:20496–20501 [DOI] [PubMed] [Google Scholar]
- 65. Warren S. E., et al. 2010. Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185:818–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Woodward J. J., Iavarone A. T., Portnoy D. A. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zemansky J., et al. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. 191:3950–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zipperle G. F., Jr., Ezzell J. W., Jr., Doyle R. J. 1984. Glucosamine substitution and muramidase susceptibility in Bacillus anthracis. Can. J. Microbiol. 30:553–559 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.