

Abstract
Binding of the complement-downregulating protein factor H (fH) to the surface of the meningococcus is important for survival of the organism in human serum. The meningococcal vaccine candidate factor H binding protein (fHbp) is an important ligand for human fH. While some fHbp-specific monoclonal antibodies (MAbs) block binding of fH to fHbp, the stoichiometry of blocking in the presence of high serum concentrations of fH and its effect on complement-mediated bactericidal activity are unknown. To investigate this question, we constructed chimeric antibodies in which the human IgG1 constant region was paired with three murine fHbp-specific binding domains designated JAR 3, JAR 5, and MAb502. By surface plasmon resonance, the association rates for binding of all three MAbs to immobilized fHbp were >50-fold higher than that for binding of fH to fHbp, and the MAb dissociation rates were >500-fold lower than that for fH. While all three MAbs elicited similar C1q-dependent C4b deposition on live bacteria (classical complement pathway), only those antibodies that inhibited binding of fH to fHbp (JAR 3 and JAR 5) had bactericidal activity with human complement. MAb502, which did not inhibit fH binding, had complement-mediated bactericidal activity only when tested with fH-depleted human complement. When an IgG1 anti-fHbp MAb binds to sparsely exposed fHbp on the bacterial surface, there appears to be insufficient complement activation for bacteriolysis unless fH binding also is inhibited. The ability of fHbp vaccines to elicit protective antibodies, therefore, is likely to be enhanced if the antibody repertoire is of high avidity and includes fH-blocking activity.
INTRODUCTION
Neisseria meningitidis is an important cause of meningitis and sepsis. Meningococcal capsular polysaccharide-based vaccines are available for prevention of disease caused by strains with capsular group A, C, W-135, or Y (37). A broadly effective vaccine against group B strains, however, remains elusive (reviewed in references 19 and 28). The group B capsular polysaccharide cross-reacts with structures present in the host (14) and is poorly immunogenic (23). A vaccine that elicits autoreactive group B anticapsular antibodies also presents safety concerns.
Antibodies to noncapsular antigens can confer protection against group B meningococcal disease (9). A variety of protein antigens are being evaluated as vaccine candidates (19, 28, 40). One of the most promising antigens is a lipoprotein called factor H (fH) binding protein (fHbp) (21, 30, 47), which previously was referred to as genome-derived neisserial antigen 1870 (GNA1870) (33) or LP2086 (15, 32, 49). fHbp is present in nearly all disease-causing group B strains (2, 4, 36, 38) and binds human fH (21, 30, 43), which is a fluid-phase downregulator of the complement pathway (1, 26, 34, 39, 42). Binding of fH to the bacterial surface allows pathogens to escape innate host defenses (12, 21, 22, 27, 29–31, 42).
Two meningococcal vaccines that contain recombinant fHbp are currently in clinical development (13, 16, 24, 44). In preclinical studies with mice, antibodies elicited by fHbp vaccines bound to the bacterial surface, activated complement-mediated bactericidal activity directly (8, 16, 17, 33, 47), and inhibited binding of fH to fHbp (8, 30, 47). Inhibition of binding of fH to the bacterial surface would be expected to increase susceptibility of the bacteria to complement-mediated bacteriolysis. The studies demonstrating anti-fHbp inhibition of fH binding, however, were done under conditions that favored detection of inhibition (i.e., high concentrations of antibody and low concentrations of fH) (5, 8, 30, 41). Factor H concentrations in human sera typically range from 300 to 600 μg/ml (6), which would favor binding of fH to the bacteria in the presence of anti-fHbp antibody. Thus, the contribution of anti-fHbp antibody inhibition of fH binding to complement-mediated protective activity in human serum is unknown.
To investigate this question, we expressed the antigen binding domains of three murine anti-fHbp monoclonal antibodies (MAbs) as chimeric antibodies fused to the human IgG1 constant region. Having MAbs with different combining sites, some of which were known to inhibit fH binding (8, 41), and a common human Fc permitted us to investigate the effect of antigenic epitope on complement-mediated antibody functional activity independent of Fc-mediated differences in complement activation.
MATERIALS AND METHODS
Murine anti-fHbp MAbs.
The murine fHbp-specific monoclonal antibodies (MAbs) JAR 3 (IgG3), JAR 5 (IgG2b) (8, 47, 48), and MAb502 (IgG2a) (17, 41) have been previously described (Table 1). The JAR 3 and JAR 5 MAbs inhibit binding of each other to fHbp (47) and recognize overlapping epitopes that involve glycine and lysine at positions 121 and 122, respectively, of fHbp (5, 8). The respective epitopes recognized by the two paratopes were differentiated by dissimilar binding by Western blotting with different fHbp amino acid sequence variants. MAb502 recognizes a conformational epitope requiring an arginine at position 204 (41), and this MAb does not inhibit binding of JAR 3 or JAR 5 to fHbp. Control murine MAbs included SEAM 12 (20), which reacts with the group B capsule, and an anti-PorA P1.7 (NIBSC code 01/514), which was obtained from the National Institute for Biological Standards and Control, Potters Bar, United Kingdom.
Table 1.
Properties of the murine anti-fHbp MAbs
| MAb (references)c | IgG subclass | Bactericidal activity | Inhibition of fH binding | fHbp epitope |
|||
|---|---|---|---|---|---|---|---|
| Amino acid(s)a | Binding to fHbp amino acid sequence variantb |
||||||
| ID 1 | ID 9 | ID 74 | |||||
| JAR 3 (8, 48) | IgG3 | Human or Rabbit C | + | Gly121, Lys122 | ++ | ++ | + |
| JAR 5 (8, 48) | IgG2b | Rabbit C only | + | Gly121, Lys122 | ++ | + | ++ |
| MAb502 (17, 41) | IgG2a | Rabbit C only | − | Arg204 | ++ | ++ | − |
fHbp mutants with amino acid substitutions at these positions lost or gained MAb binding (5, 8). In addition, for MAb502, the amide signals of Gly148, Arg149, and Ala174 showed chemical shifts by nuclear magnetic resonance (NMR) upon binding with fHbp (41).
Relative binding compared to fHbp ID 1 measured by quantitative Western blotting with integrated intensities of the bands measured by an infrared scanner (38). ++, ≥100% compared to ID 1; +, 52 to 62% compared to ID 1; −, <3% compared to ID 1.
Human IgG1 chimeric mouse anti-fHbp MAbs.
RNA isolated from the murine hybridoma cells was converted into cDNA using an Omniscript reverse transcription (RT) kit (Qiagen) according to the manufacturer's instructions. cDNA was amplified using immunoglobulin heavy (H)- and light (L)-chain-specific primers (46) and inserted into the pGEM-T Easy vector (Promega) for sequencing. Based on the determined sequences, specific primers were designed to facilitate the insertion of the murine VH and VL sequences into a modified eukaryotic expression vector. pcDNA5/FRT from Invitrogen was modified to contain a bicistronic expression cassette inserted between the NheI and EagI restriction sites in the polylinker. The cassette contained insertion sites for the murine V regions preceded by a human VH or VL leader sequence and flanked by either the human kappa constant region or the complete human IgG1 constant region. The vector utilized an internal ribosomal entry segment (IRES) sequence between the VH and VL sequences to facilitate balanced translation of both chains. The DNA sequences of all constructs were verified prior to transfection.
Flp-In CHO cells (Invitrogen) were plated at 3.5 × 105 cells per well (in 2 ml Flp-In medium) in Nunclon Delta 6-well plates and incubated at 37°C with 5% CO2 overnight. Once cells reached 80% confluence, they were transfected with pOG44 (Invitrogen) and the FLP recombination target (FRT) vector containing the VH and VL inserts (9:1 ratio) using the TransFast transfection reagent (Promega). At 48 hours after transfection, the cells were trypsinized and placed in a fresh 6-well plate under drug selection with 600 μg/ml hygromycin. Transfected cells were adapted to serum-free suspension culture using Excell 302 medium (Sigma-Aldrich) and grown for approximately 2 weeks. Antibody from the cell culture supernatant was concentrated prior to purification using a 200-ml stirred cell (Amicon) and application of nitrogen gas pressure. Antibody was purified using HiTrap protein G columns (GE Healthcare). After extensive dialysis against phosphate-buffered saline (PBS), bovine serum albumin (BSA) was added to a final concentration of 1%, and aliquots were stored at −30°C.
ELISA.
Concentrations of the human IgG1-mouse chimeric MAbs were determined by a capture enzyme-linked immunosorbent assay (ELISA). Wells of a microtiter plate were incubated with goat anti-human κ-chain-specific antibody (Biosource) diluted 1:200 in PBS and incubated overnight at 4°C. The wells were blocked with PBS (pH 7.4) with 1% (wt/vol) bovine serum albumin (Equitech-Bio) (PBS-BSA) for 1 h at room temperature. Dilutions of the MAbs in PBS-BSA were added and incubated at 37°C for 2 h. After washing with PBS–0.05% Tween 20, bound human IgG was detected with goat anti-human IgG antibody conjugated with alkaline phosphatase (Invitrogen). The secondary antiserum was diluted 1:2,000 in the PBS-BSA and incubated for 1 h at room temperature. Antibody concentrations were assigned by comparison with a human IgG1 myeloma standard of known concentration (Sigma). Binding of the anti-fHbp MAbs to fHbp (amino acid sequence variant ID 1 as designated on the website http://pubmlst.org/neisseria/fHbp/) was measured by ELISA with recombinant fHbp on the plate, which was performed as previously described (7). The secondary detecting antibody was goat anti-human κ-chain-specific antibody conjugated with alkaline phosphatase (Biosource) diluted 1:2,000 in PBS-BSA and incubated for 1 h at room temperature.
Surface plasmon resonance.
The kinetics of binding of the human-mouse chimeric MAbs to fHbp were measured by surface plasmon resonance with immobilized recombinant fHbp (200, 500, and 1,000 response units [RU]) on CM5 chips (GE Healthcare, Piscataway, NJ). The immobilization was performed via amine coupling (amine coupling kit; GE Healthcare). Kinetics of binding were determined at MAb concentrations ranging from 0.016 to 50 μg/ml (0.1 nM to 333 nM) using a Biacore T/100 instrument (GE Healthcare, Piscataway, NJ). Kinetics of binding purified human fH were measured using immobilized fHbp (1,000, 2,000, and 4,000 responses units) on CM5 chips and fH concentrations ranging from 2.8 to 90 μg/ml (18 nM to 580 nM), each performed two to three times. The interaction binding (Ka) and dissociation (Kd) constants were determined locally by fitting to the kinetic simultaneous Kd/Ka model after first testing a model of equimolar stoichiometry. The analyses were performed using the Biacore T/100 evaluation software.
Determination of binding to N. meningitidis by flow cytometry.
Binding of the chimeric MAbs to the surface of live encapsulated bacteria was measured with strain H44/76 (B:15:P1.7,16; ST-32), which is a relatively high expresser of fHbp ID 1 (47, 48). In some experiments we also tested binding to isogenic knockout (KO) mutants of H44/76 in which fHbp, NspA, or both proteins were not expressed (27) (KO mutants were provided by Sanjay Ram, University of Massachusetts Medical School, Worcester, MA). The bacteria were grown to mid-log phase up to an optical density at 620 nm (OD620) of 0.6 in Mueller-Hinton broth supplemented with 0.25% glucose and 0.02 mmol/liter CMP N-acetyl neuraminic acid (Sigma-Aldrich), pelleted by centrifugation, and resuspended in Dulbecco's phosphate-buffered saline containing 0.1 g/liter CaCl2 and 0.1 g/liter MgCl2·6H2O (Mediatech) (pH 7.4) with 1% (wt/vol) bovine serum albumin (Equitech-Bio) (D-PBS-BSA). For the flow cytometric assays, a fixed concentration of anti-fHbp MAb (4 μg/ml) was incubated together with a bacterial cell density of ∼107 cells/ml for 1 h at room temperature. As a negative control, binding of 100 μg/ml of an irrelevant MAb was tested (monoclonal κ-chain antibody from human myeloma; Sigma). After two washes with D-PBS-BSA buffer, the cells were incubated with CF488 goat anti-human IgG (BioTium), diluted 1:500, for 1 h at room temperature. The bacteria were harvest by centrifugation, washed twice with D-PBS-BSA buffer, fixed with 0.5% (vol/vol) formaldehyde in PBS, and analyzed by flow cytometry. The dilutions of the primary and secondary antibodies were in D-PBS-BSA buffer.
Inhibition of binding of fH.
The bacterial cells were grown, harvested, and resuspended in D-PBS-BSA buffer as described above for measuring MAb binding to N. meningitidis by flow cytometry. For the inhibition assay, ∼107 cells/ml bacteria were incubated with anti-fHbp MAb (50 or 2 μg/ml in D-PBS-BSA buffer) for 30 min at room temperature, followed by the addition of 10 to 100 μg/ml of purified fH, which was incubated for an additional 30 min at room temperature. After the cells were washed with D-PBS-BSA buffer, bound fH was detected with a sheep polyclonal antiserum to fH (Life span Bioscience), followed by washing and incubation with a donkey anti-sheep IgG antibody (Invitrogen) conjugated with Alexa Fluor 488. Both of the antisera were diluted 1:500 in D-PBS-BSA buffer and incubated for 30 min at room temperature. In some experiments, 20% IgG-depleted human serum (see below), which contained ∼90 μg/ml of fH, was used as the source of human fH. To avoid bacteriolysis, the human serum was heated for 30 min at 56°C to inactivate heat-labile complement components essential for bacteriolysis. This heat treatment did not affect fH activity (25).
The ability of the anti-fHbp MAbs to inhibit binding of fH to fHbp was also measured by ELISA. Wells of a microtiter plate were incubated with fHbp (2 μg/ml in PBS) overnight at 4°C. The wells were blocked with PBS-BSA for 1 h at room temperature. Dilutions of the MAbs in PBS-BSA were added and incubated at 37°C for 2 h, followed by the addition of 2 μg/ml of human fH (Complement Technologies), which was incubated for an additional 1 h at room temperature. After washing with PBS–0.05% Tween 20, bound fH was detected with a sheep polyclonal antiserum to fH (Abcam), followed by washing and the addition of donkey anti-sheep IgG antibody (Sigma-Aldrich) conjugated with alkaline phosphatase. Both the primary and secondary antisera were diluted 1:5,000 in the PBS-BSA and incubated for 1 h. The inhibition results were expressed as the percentage of inhibition of fH binding in the presence of an anti-fHbp MAb compared with fH binding in the absence of the MAb.
Human complement sources.
Our primary complement source for measurement of bactericidal activity was serum from a healthy adult with normal total hemolytic complement activity and no detectable serum bactericidal antibodies against the tested strain. To eliminate nonbactericidal IgG antibodies, which might augment or inhibit complement-mediated activity of the test MAbs (45), the complement source was depleted of IgG using a protein G column (HiTrap protein G; GE Life Sciences, Piscataway, NJ) as previously described (6). Adequacy of IgG depletion was monitored by an IgG capture ELISA (see above), and maintenance of CH50 activity (as defined by 50% lysis of a red cell suspension) was assayed with a commercial assay (EZ Complement CH50 test kit; Diamedix Corp., Miami, FL). In some experiments, we used commercial human complement sources that had been depleted of fH, C1q, or C6 (Complement Technologies) and that were also depleted of IgG as described above.
Serum bactericidal assay.
Bactericidal activity was measured as previously described (3) using group B strain H44/76. In brief, the bacterial cells were grown, harvested, and resuspended in buffer as described above for measuring MAb binding to N. meningitidis by flow cytometry. The 40-μl bactericidal reaction mixture contained 1 to 100 μg/ml of MAb, ∼300 to 400 CFU of bacteria, and 20% human serum depleted of IgG as a complement source (see above). Immediately before the assay was performed, the anti-fHbp MAbs were centrifuged for 2 h at 100,000 × g to remove possible aggregates. Bactericidal activity (BC50) was defined by the MAb concentration that resulted in a 50% decrease in CFU/ml after 60 min of incubation in the reaction mixture compared with the CFU/ml in negative-control wells at time zero.
C4b and C3b deposition on N. meningitidis.
We used flow cytometry to measure deposition of human C4b and C3b on the surface of live bacteria of group B strain H44/76. The bacteria were grown as described above and resuspended in D-PBS-BSA buffer to a density of ∼108 cells/ml. The cells were than mixed with different concentrations of the chimeric human-mouse anti-fHbp MAbs diluted in D-PBS-BSA and commercial human complement (15% human serum that had been depleted of IgG as described above). In the experiments measuring C4b deposition as a marker for activation of the classical complement pathway, we used C1q-depleted human complement (Complement Technologies) that was tested with or without the addition of 30 μg/ml of purified C1q protein (Complement Technologies). In other experiments, we measured C4b or C3b deposition using C6-depleted human complement (Complement Technologies) to eliminate formation of a membrane attack complex and bacteriolysis. The reaction mixtures were incubated for 15 min at room temperature. Human C3b or C4b bound to bacteria was detected with a 1:100 dilutions of fluorescein isothiocyanate-conjugated anti-human C3c or C4b antibodies, respectively (Meridian Life Science) diluted in D-PBS-BSA buffer. These antibodies were incubated with the bacteria for 1 h at 4°C.
RESULTS
The three human IgG1 mouse chimeric anti-fHbp MAbs have similar binding avidities for fHbp.
As described in Materials and Methods, we constructed three human-mouse chimeric anti-fHbp antibodies in which each of the JAR 3, JAR 5, and MAb502 mouse paratopes were paired with a human IgG1 constant region. In an ELISA with recombinant fHbp adsorbed to the wells of a microtiter plate, the three MAbs showed similar respective binding (Fig. 1 A).
Fig. 1.

Binding of anti-fHbp MAbs. (A) ELISA. IgG bound to immobilized fHbp was detected with an anti-human kappa light-chain-specific alkaline phosphatase-conjugated antibody. Error bars represent the range in OD values observed in two independent experiments. (B and C) Flow cytometry. (B) Binding of anti-fHbp MAbs (4 μg/ml) with live bacterial cells of N. meningitidis group B strain H44/76. JAR 3, black dashed line; JAR 5, gray line; MAb502, black line. An irrelevant human MAb (100 μg/ml) served as a negative control (gray filled histogram). The binding curves of the three anti-fHbp MAbs are superimposed. (C) Same MAb concentrations as in panel B in the presence of heat-inactivated 20% IgG-depleted human serum as a source of human fH (∼90 μg/ml). (D and E) Surface plasmon resonance. (D) Representative data for binding of 0.25 μg/ml anti-fHbp MAbs (1.7 nM) to immobilized recombinant fHbp (1,000 RU). Lines are as in panel B. (E) Binding of purified human fH to immobilized recombinant fHbp (1,000 RU). fH concentrations of 12 to 90 μg/ml (71 to 580 nM) are shown: 12 μg/ml, thick black line; 23 μg/ml, dotted black line; 45 μg/ml, dashed black line; and 90 μg/ml, gray solid line. The flow cytometric data were replicated in two or three independent experiments.
We also measured MAb binding to the surface of live bacteria of group B strain H44/76 by flow cytometry. At MAb concentrations of between 0.8 and 40 μg/ml, the bindings of the three MAbs was indistinguishable from each other. The binding results obtained with 4 μg/ml are shown in Fig. 1B. Binding was not affected by the presence of heat-inactivated 20% IgG-depleted human serum, which contained ∼90 μg/ml of human fH (compare Fig. 1C, showing binding of the MAbs in the presence of serum, to Fig. 1B, showing binding without serum).
We measured the kinetics of binding of each of the chimeric MAbs with 200, 500, or 1,000 RU of immobilized fHbp. The MAb concentrations tested ranged from 0.016 to 50 μg/ml. Under any one condition, the respective binding kinetics of the three MAbs were similar. Representative data for 0.25 μg/ml (1.7 nM) of MAb and 1,000 RU of immobilized fHbp are shown in Fig. 1D. The three association constants (Ka) (1/ms) were similar (4.3E+06, 2.3E+06, and 1.2E+06 for JAR 3, JAR 5, and MAb 502, respectively). The dissociation rates were low for all three MAbs, which precluded calculation of accurate dissociation constants (Kd) (1/s).
The kinetics of binding of human fH to immobilized fHbp were measured at fH concentrations of 2.8 to 90 μg/ml and with 1,000, 2,000, or 4,000 RU of fHbp. Representative data for binding with 1,000 RU of immobilized fHbp are shown in Fig. 1E. The fH association rate constant, Ka, was 5.0E+04, and the dissociation rate constant, Kd, was 1.9E−02. These constants are similar to those previously reported in an assay that measured binding of fHbp to immobilized fH (14E+04 and 0.7E−02, respectively [11]). Collectively, the data showed that the association rate for binding of fH to fHbp was approximately 50- to 100-fold lower than that for binding of the anti-fHbp MAbs to fHbp, and the rate of dissociation of fH from fHbp was >500-fold higher than that for the MAbs.
All three human MAbs activate the human classical complement pathway, but only JAR 3 and JAR 5 have bactericidal activity.
Activation of the classical complement pathway is initiated when IgG binds to the bacterial surface and there is sufficient antigen-antibody complex to allow the proximate Fc regions of the antibodies to engage C1q, which in turn activates C4b. We therefore measured C4b binding to the surface of live N. meningitidis cells of group B strain H44/76 as a surrogate for C1q binding and C4b activation and as a marker of classical complement pathway activation.
When the source of complement was 15% C1q-depleted pooled human serum that had also been depleted of IgG (to remove the contribution of natural antibodies to meningococci), there was negligible C4b deposition elicited by the anti-fHbp MAbs (Fig. 2 A). When 30 μg/ml of purified C1q was added back to the reaction mixture, each of the MAbs gave similar activation of C4b deposition (Fig. 2B). Similar respective results were obtained with a second complement source that had been C6 depleted to prevent bacteriolysis (Fig. 2C). In contrast, JAR 3 and JAR 5 caused stronger C3b deposition than MAb502 (Fig. 2D). JAR 3 and JAR 5, but not MAb502, also elicited complement-mediated bactericidal activity (Fig. 2E). The mean concentrations ± standard errors (SE) for 50% killing in three assays were 9 ± 0.85 μg/ml for JAR 3, 15 ± 2 μg/ml for JAR 5 (P = 0.024 compared with JAR 3), and >100 μg/ml for MAb502 (P < 0.0003 compared with JAR 3 or JAR 5).
Fig. 2.

Complement activation on encapsulated group B bacteria of strain H44/76. (A to D) Flow cytometry. (A) Activation of C4b deposition by 4 μg/ml of MAb and 15% C1q-depleted complement that also had been depleted of IgG. Anti-fHbp MAb JAR 3, black dotted line; JAR 5, gray line; MAb502, black line; irrelevant human MAb (100 μg/ml), gray filled histogram (data for each are superimposed). (B) Same as in panel A except for the addition of 30 μg/ml of purified C1q protein to the reaction mixtures. Data were replicated in two independent experiments. (C) Activation of C4b deposition by 4 μg/ml of MAbs and 15% C6-depleted complement that also had been depleted of IgG. (D) Activation of C3b. The conditions and complement source were the same as for panel C. (E) Bactericidal activity. Survival of bacteria after incubation for 60 min at 37°C with each of the MAbs and complement (20% IgG-depleted human serum) is shown. With complement alone, survival was >195%.
Chimeric MAbs JAR 3 and JAR 5, but not MAb502, inhibit binding of fH to fHbp.
All three chimeric MAbs elicited similar classical complement activation as measured by C1q-dependent C4b deposition, while JAR 3 and JAR 5 elicited greater C3b deposition and greater bactericidal activity than MAb502. Factor H inhibits complement activation by several pathways (42). In previous studies, murine MAbs JAR 3 and JAR 5 inhibited binding of fH to fHbp (8, 30, 47), whereas murine MAb502 did not inhibit fH binding (41). By ELISA, the human IgG1 chimeric JAR 3 and JAR 5 MAbs also inhibited binding of fH to fHbp, while the chimeric MAb502 did not inhibit fH binding (Fig. 3 A). When 20% heat-inactivated IgG-depleted human serum was the source of fH, 50 μg/ml of chimeric JAR 3 or JAR 5, but not MAb502, inhibited binding of fH to the surface of live bacterial cells (Fig. 3B). As little as 2 μg/ml of JAR 3 or JAR 5 also inhibited binding of fH to the surface of live bacterial cells (Fig. 3C), although inhibition was less complete than with 50 μg/ml of the MAb (Fig. 3B).
Fig. 3.

Inhibition of fH binding by anti-fHbp MAbs. (A) ELISA. Inhibition of binding of fH (2 μg/ml) to recombinant fHbp immobilized on a microtiter plate is shown. ▿, JAR 3; ×, JAR 5; ○, MAb502. Error bars represent the OD range for two independent experiments. (B and C) Flow cytometry. (B) Inhibition of binding of fH (∼90 μg/ml in 20% IgG-depleted human serum) to live bacterial cells by 50 μg/ml of MAb. Black solid line, MAb502; dotted black line, JAR 3; gray solid line, JAR 5; dark gray filled area, bacteria without fH or MAb as a negative control; light gray filled area, bacteria with fH without a MAb as a positive control. (C) Same conditions as for panel B except that 2 μg/ml of each of the anti-fHbp MAbs was tested instead of 50 μg/ml. The ELISA and flow cytometry data were replicated in at least two independent experiments.
The correlation observed between bactericidal activity and the abilities of the different MAbs to inhibit fH binding did not necessarily prove that inhibition was responsible for the greater bactericidal activity of JAR 3 or JAR 5 than of MAb502. When fH was removed from the reaction by using fH-depleted pooled human complement, there was no bactericidal activity with the complement alone (the CFU/ml increased by >150% during the 1-h incubation). With this complement source, all three MAbs showed similar complement-mediated bactericidal activity (in two independent experiments, the mean BC50 values of the three MAbs ranged from 1.25 to 1.40 μg/ml [Table 2 ]). When purified human fH was added to the reaction mixture, MAb502 was no longer bactericidal (BC50 of >100 μg/ml, which was similar to the lack of activity when measured with the normal human complement source). The bactericidal activity of JAR 3 or JAR 5 measured with the fH-replete commercial complement pool also was similar to that measured with the normal human complement source (Fig. 2E). Note also that with fH-depleted complement, adding fH to the reaction mixture decreased the bactericidal activities of two control murine MAbs reactive with the group B capsule or PorA (Table 2). The effect of fH repleteness on the activities of these MAbs, however, was less pronounced than with the anti-fHbp MAbs.
Table 2.
Anti-fHbp MAb bactericidal activity measured with fH-depleted human complement
| MAb | Bactericidal activity (BC50, μg/ml)a with: |
|||
|---|---|---|---|---|
| fH-depleted complement |
fH-replete complement |
|||
| Mean | Range | Mean | Range | |
| Human IgG1 chimeric mouse anti-fHbp | ||||
| JAR 3 | 1.4 | 0.8–2.0 | 15.2 | 12.5–18 |
| JAR 5 | 1.25 | 1.0–1.5 | 23.5 | 22–25 |
| MAb502 | 1.25 | 0.75–1.5 | >100 | >100 |
| Positive-control mouse IgG2a | ||||
| Anti-PorA P1.7 | 0.5 | 0.3–0.7 | 1.05 | 1.0–1.1 |
| Anticapsular, SEAM 12 | 0.18 | 0.15–0.2 | 1.15 | 1.0–1.2 |
Data are means and respective ranges, for values observed in two independent assays, of MAb concentrations that gave 50% killing (BC50) after 1 h of incubation with bacteria and 20% fH-depleted pooled human serum as a complement source that also had been depleted of IgG (see Materials and Methods). Without added MAb, there was >150% survival of the bacteria with fH-depleted or -replete complement. For preparation of fH-replete complement, 50 μg/ml of human fH was added.
Elimination of fH binding to NspA enhances bactericidal activity of anti-fHbp MAbs JAR 3 and JAR 5 but not that of MAb502.
The much lower concentrations of anti-fHbp MAbs required for bacteriolysis when tested with fH-depleted complement than with fH-replete complement (Table 2) suggested that when fH was present, inhibition of fH binding by JAR 3 or JAR 5 may have been incomplete. For example, this could occur from binding of fH to a second meningococcal ligand such as NspA (27). To test this hypothesis, we used isogenic mutants of group B strain H44/76 in which genes encoding fHbp and NspA had been inactivated to measure fH binding.
By flow cytometry, a combined NspA/fHbp KO mutant and the fHbp KO-only mutant showed binding with a control murine anti-PorA P1.7 MAb that was similar to that of the parent strain (Fig. 4 A). As expected, there was much less binding of fH (100 μg/ml) with the fHbp-only KO mutant than with the wild-type (WT) strain (compare black line with gray line in Fig. 4B). In the absence of both fHbp and NspA expression (dashed line), there was a further decrease in bound fH, which was indistinguishable from that for the negative-control WT bacteria without added fH (light gray filled histogram). Similar respective results were obtained when 20% IgG-depleted human serum was used as the source of fH (Fig. 4C).
Fig. 4.
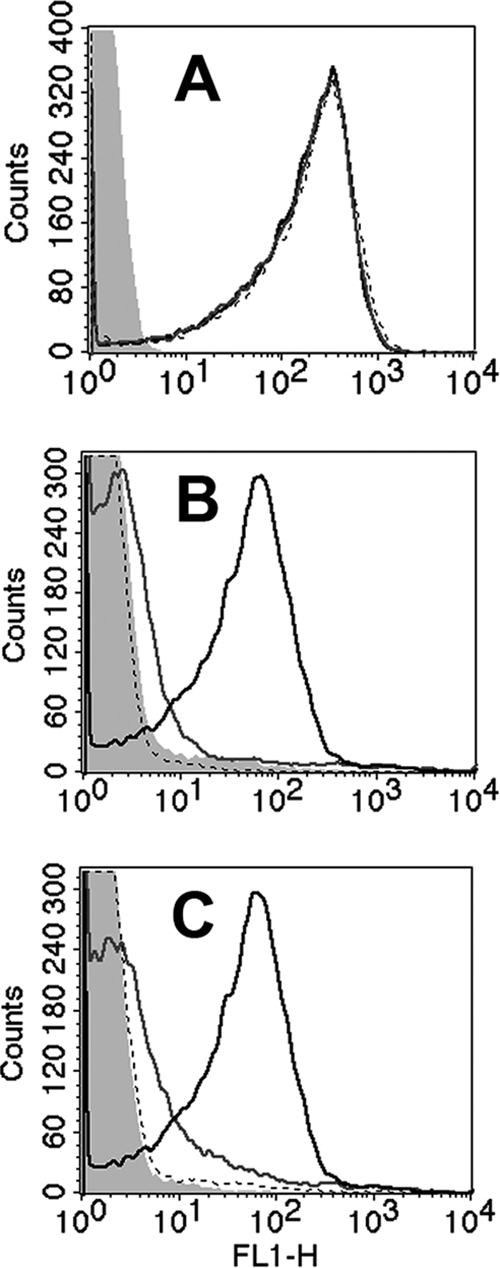
Binding of fH to fHbp and NspA KO mutants of group B strain H44/76 by flow cytometry. (A) Binding of anti-PorA MAb (P1.7, 20 μg/ml, positive control). Black line, wild-type strain; gray line, fHbp KO mutant; dashed line, double fHbp and NspA KO mutant; filled gray area, no MAb. The binding curves of the three MAbs are superimposed. (B) Binding of purified human fH (100 μg/ml). Designations are the same as in panel A except that the filled gray area is for no added fH. (C) Binding of fH in human serum (20%, IgG depleted). Designations are as in panel B. The results were replicated in two independent assays.
To determine a possible contribution of fH binding by NspA (and corresponding downregulation of complement activation) to the high anti-fHbp MAb concentrations required for bacteriolysis in the presence of fH, we measured anti-fHbp bactericidal activity with an isogenic NspA KO mutant strain (Fig. 5). By flow cytometry, the NspA KO and WT strains showed indistinguishable binding of purified fH (Fig. 5A). This result was expected since in a previous study deletion of NspA only in strains such as H44/76 with high fHbp expression had no discernible effect on fH binding (27). With chimeric JAR 3 or JAR 5 anti-fHbp MAbs, which inhibited binding of fH to fHbp, there was significantly greater complement-mediated killing of the NspA KO mutant than with the control WT strain (Fig. 5B and C, respectively). In contrast, with chimeric MAb502, which did not inhibit fH binding, there was no complement-mediated bactericidal activity detected against either strain (Fig. 5D). Two control mouse MAbs, anti-PorA and anticapsular, elicited similar respective complement-mediated bactericidal activities against the WT and mutant NspA KO strains (Fig. 5E and F, respectively).
Fig. 5.

Bactericidal activities of anti-fHbp MAbs measured against a mutant of group B H44/76 with genetic inactivation of NspA expression. (A) Binding of purified human fH (100 μg/ml) to live bacteria determined by flow cytometry. Gray line, NspA KO mutant; black line, wild-type control; dashed line, double fHbp and NspA KO control; light gray filled area, no fH. All three strains showed similar binding with a control anti-PorA P1.7 MAb (data not shown). (B to F) Survival of bacteria after incubation for 60 min at 37°C with each of the MAbs and 20% IgG-depleted human serum as a complement source. Open triangles, NspA KO mutant; closed triangles, control wild-type strain. (B) Chimeric anti-fHbp MAb JAR 3. (C) Chimeric anti-fHbp MAb JAR 5. (D) Chimeric anti-fHbp MAb502. (E) Control murine anti-PorA P1.7 MAb. (F) Control murine MAb, SEAM 12 reactive with group B capsule. Bactericidal activity results are from three independent experiments. Where indicated, survivals of the respective wild-type and NspA KO strains incubated at the MAb dilution were significantly different (*, P ≤ 0.02; **, P < 0.001).
DISCUSSION
As a vaccine candidate, recombinant fHbp elicits serum antibodies that activate complement-mediated bactericidal activity directly (48) and inhibit binding of fH to the bacterial surface (47). In human serum with high fH concentrations, however, the abilities of anti-fHbp antibodies to inhibit fH binding and increase susceptibility of the organism to complement-mediated bacteriolysis were unknown. Our results provide direct evidence of the ability of some anti-fHbp MAbs to inhibit fH binding over a wide range of antibody and fH concentrations and for the importance of this mechanism for eliciting anti-fHbp MAb bactericidal activity. Thus, all three chimeric anti-fHbp MAbs had similar avidities for binding to fHbp as measured by surface plasma resonance and had similar respective binding to the surface of live group B bacteria by flow cytometry. The three MAbs also showed similar activation of the classical complement pathway based on C1q-dependent C4b deposition. JAR 3 and JAR 5, however, which inhibited binding of fH to fHbp, caused stronger C3b deposition and exhibited complement-mediated bactericidal activity in the presence of human fH, whereas MAb502 lacked bactericidal activity (Fig. 2). Collectively these data are consistent with greater amplification of the alternative complement pathway by JAR 3 or JAR 5 than by MAb502 resulting from the ability of JAR 3 or JAR 5 to inhibit binding of fH. Further, these two MAbs could inhibit binding of fH to fHbp when fH was present in excess concentrations because of the much higher avidities of the MAbs for binding to fHbp than for binding of fH to fHbp (Fig. 1).
The amino acid residues affecting the fHbp epitopes recognized by the three murine anti-fHbp MAbs had been characterized in previous studies (8, 41). As expected, the spatial relationships of the respective epitopes were different for the anti-fHbp MAbs that inhibited fH binding (JAR 3 and JAR 5) and for the MAb that did not inhibit fH binding (MAb502). It is conceivable that when the individual MAbs are bound to the bacterial surface, these spatial differences could affect formation of a functional membrane attack complex and bacteriolysis independent of the ability of the MAbs to inhibit fH binding and decrease fH downregulation.
To determine whether differences in the locations of the respective epitopes affected bactericidal activity independent of fH inhibition, we measured anti-fHbp bactericidal activity using fH-depleted complement. Our results demonstrated that it was the inhibition of fH binding and not the relative positions of the epitopes that determined the functional activities of these MAbs, since when tested with fH-depleted complement, the three MAbs had similar bactericidal activities. Note also that in its murine IgG2b form, JAR 5 lacked human complement-mediated bactericidal activity despite its ability to inhibit fH binding (48), while the human IgG1 chimeric JAR 5 MAb elicited complement-mediated bactericidal activity. Thus, inhibition of fH binding is necessary but not sufficient for complement-mediated bactericidal activity by this fHbp-specific antibody. This observation highlights the importance of matching the isotype when comparing the relative efficacies of monoclonal antibodies in the complement-mediated bactericidal assay.
A recent study reported fH binding to fHbp knockout mutants of some meningococcal strains (27). The authors demonstrated that NspA was a second fH ligand and that, similar to the case for fHbp, binding to NspA occurred via fH domains 6 and 7 and was specific for human fH. The authors speculated that the ability of meningococci to bind fH via this second ligand might lessen the efficacy of fHbp vaccines. In the present study, an NspA KO mutant was found to be more susceptible to complement-mediated killing by the chimeric IgG1 anti-fHbp MAbs JAR 3 and JAR 5 than the parent wild-type strain. The greater bactericidal activity against the NspA KO mutant was not observed with chimeric anti-fHbp MAb502, which did not inhibit fH binding. These data provide direct evidence for the contribution of fH binding to NspA in enhancing resistance of the organism to anti-fHbp MAb complement-mediated bactericidal activity. Antibodies to NspA have been reported to block fH binding (27). Thus, one implication of our results is that a vaccine that elicits fH-blocking antibodies to both NspA and fHbp might have greater protective activity than vaccines that elicit fH-blocking antibodies to only one of the two proteins.
The presence of human fH had a relatively small effect on decreasing the complement-mediated bactericidal activities of two control murine MAbs directed at PorA or the group B capsule (Table 2). fHbp is present in the outer membrane as a monomer and is relatively sparsely exposed (47). Our hypothesis is that when an anti-fHbp MAb binds to fHbp, the ability of two IgG molecules to engage C1q is limited, which limits the amount of classical complement pathway activation (35). For anti-fHbp MAbs such as MAb502 that do not inhibit fH binding, the bound factor H (fH) downregulates complement activation, which limits formation of the membrane attack complex and bacteriolysis. If the anti-fHbp MAb can block binding of fH, then the classical pathway activation is sufficient to proceed to bacteriolysis. Monoclonal antibodies that bind to more abundant antigens such as the capsule or to PorA, which is a trimer, can more effectively engage C1q and elicit greater classical pathway activation, which is sufficient for bacteriolysis even with fH bound to the bacteria.
A limitation of the present study is that our conclusions relating inhibition of fH and anti-fHbp bactericidal activity are based on testing only two human-mouse chimeric anti-fHbp MAbs that inhibited fH binding and one chimeric MAb that did not inhibit fH binding. In a previous study, however, the magnitudes of the serum bactericidal titers of human fH transgenic mice immunized with recombinant fHbp vaccines correlated directly with the magnitudes of the serum dilutions that inhibited binding of fH to fHbp (6). Thus, inhibition of fH binding also appeared to be important for eliciting polyclonal anti-fHbp bactericidal activity (6).
In conclusion, complement-mediated serum bactericidal activity is the most important mechanism for protection of the host against meningococcal disease (reviewed in references 10, 18, and 19). The meningococcus has evolved a number of strategies for resisting killing by human complement. One of the most important is binding of fH and downregulation of complement activation. Our data indicate that the ability of fHbp vaccines to elicit protective antibodies is likely to be enhanced if the antibody repertoire is of high avidity and includes fH-blocking activity.
ACKNOWLEDGMENTS
This work was supported in part by Public Health Service grants R01 AI 046464 and AI 082263 (to D.M.G.) and AI57932 (to D.R.) from the National Institute of Allergy and Infectious Diseases, NIH. The work done by S.G. at Children's Hospital Oakland Research Institute was supported in part by a grant from Novartis Vaccines, Siena, Italy. S.G. is enrolled as a Ph.D. student at the University Vita Salute San Raffaele, Milan, Italy. The laboratory work at Children's Hospital Oakland Research Institute was performed in a facility funded by Research Facilities Improvement Program grant C06 RR 016226 from the National Center for Research Resources, NIH.
We are grateful to Yi De Sun, Novartis Diagnostic, Emeryville CA, for performing the plasmon resonance experiments, to Barbara Brogioni, Novartis Vaccines, Siena, Italy, for helpful discussions of the data, and to Sanjay Ram, University of Massachusetts Medical Center, for providing critical comments on the manuscript. Sanjay Ram also provided the H44/76 NspA KO and fHbpKO/NspA KO mutants used to measure fH binding and bactericidal activity.
Footnotes
Published ahead of print on 27 June 2011.
REFERENCES
- 1. Atkinson J. P., Frank M. M. 2006. Bypassing complement: evolutionary lessons and future implications. J. Clin. Invest. 116:1215–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bambini S., et al. 2009. Distribution and genetic variability of three vaccine components in a panel of strains representative of the diversity of serogroup B meningococcus. Vaccine 27:2794–2803 [DOI] [PubMed] [Google Scholar]
- 3. Beernink P. T., Caugant D. A., Welsch J. A., Koeberling O., Granoff D. M. 2009. Meningococcal factor H-binding protein variants expressed by epidemic capsular group A, W-135, and X strains from Africa. J. Infect. Dis. 199:1360–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beernink P. T., Leipus A., Granoff D. M. 2006. Rapid genetic grouping of factor H-binding protein (genome-derived neisserial antigen 1870), a promising group B meningococcal vaccine candidate. Clin. Vaccine Immunol. 13:758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beernink P. T., Lopasso C., Angiolillo A., Felici F., Granoff D. 2009. A region of the N-terminal domain of meningococcal factor H-binding protein that elicits bactericidal antibody across antigenic variant groups. Mol. Immunol. 46:1647–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beernink P. T., et al. 2011. A meningococcal factor H binding protein mutant that eliminates factor H binding enhances protective antibody responses to vaccination. J. Immunol. 186:3606–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beernink P. T., Shaughnessy J., Ram S., Granoff D. M. 2010. Impaired immunogenicity of a meningococcal factor H-binding protein vaccine engineered to eliminate factor H binding. Clin. Vaccine Immunol. 17:1074–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beernink P. T., et al. 2008. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate, factor H-binding protein. Infect. Immun. 76:4232–4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjune G., et al. 1991. Effect of outer membrane vesicle vaccine against group B meningococcal disease in Norway. Lancet 338:1093–1096 [DOI] [PubMed] [Google Scholar]
- 10. Borrow R., Balmer P., Miller E. 2005. Meningococcal surrogates of protection—serum bactericidal antibody activity. Vaccine 23:2222–2227 [DOI] [PubMed] [Google Scholar]
- 11. Dunphy K. Y., Beernink P. T., Brogioni B., Granoff D. M. 2011. Effect of factor H-binding protein sequence variation on factor H binding and survival of Neisseria meningitidis in human blood. Infect. Immun. 79:353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duthy T. G., et al. 2002. The human complement regulator factor H binds pneumococcal surface protein PspC via short consensus repeats 13 to 15. Infect. Immun. 70:5604–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Findlow J., et al. 2010. Multicenter, open-label, randomized phase II controlled trial of an investigational recombinant meningococcal serogroup B vaccine with and without outer membrane vesicles, administered in infancy. Clin. Infect. Dis. 51:1127–1137 [DOI] [PubMed] [Google Scholar]
- 14. Finne J., Leinonen M., Makela P. H. 1983. Antigenic similarities between brain components and bacteria causing meningitis. Implications for vaccine development and pathogenesis. Lancet ii:355–357 [DOI] [PubMed] [Google Scholar]
- 15. Fletcher L. D., et al. 2004. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect. Immun. 72:2088–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giuliani M. M., et al. 2006. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. U. S. A. 103:10834–10839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giuliani M. M., et al. 2005. The region comprising amino acids 100 to 255 of Neisseria meningitidis lipoprotein GNA 1870 elicits bactericidal antibodies. Infect. Immun. 73:1151–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Granoff D. M. 2009. Relative importance of complement-mediated bactericidal and opsonic activity for protection against meningococcal disease. Vaccine 27(Suppl. 2):B117–B125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Granoff D. M. 2010. Review of meningococcal group B vaccines. Clin. Infect. Dis. 50(Suppl. 2):S54–S65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Granoff D. M., et al. 1998. Bactericidal monoclonal antibodies that define unique meningococcal B polysaccharide epitopes that do not cross-react with human polysialic acid. J. Immunol. 160:5028–5036 [PubMed] [Google Scholar]
- 21. Granoff D. M., Welsch J. A., Ram S. 2009. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect. Immun. 77:764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haupt K., et al. 2007. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J. Infect. Dis. 196:124–133 [DOI] [PubMed] [Google Scholar]
- 23. Jennings H. J., Lugowski C. 1981. Immunochemistry of groups A, B, and C meningococcal polysaccharide-tetanus toxoid conjugates. J. Immunol. 127:1011–1018 [PubMed] [Google Scholar]
- 24. Jiang H. Q., et al. 2010. Broad vaccine coverage predicted for a bivalent recombinant factor H binding protein based vaccine to prevent serogroup B meningococcal disease. Vaccine 28:6086–6093 [DOI] [PubMed] [Google Scholar]
- 25. Kask L., et al. 2004. Structural stability and heat-induced conformational change of two complement inhibitors: C4b-binding protein and factor H. Protein Sci. 13:1356–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lambris J. D., Ricklin D., Geisbrecht B. V. 2008. Complement evasion by human pathogens. Nat. Rev. Microbiol. 6:132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lewis L. A., et al. 2010. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog. 6:e1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis S., Sadarangani M., Hoe J. C., Pollard A. J. 2009. Challenges and progress in the development of a serogroup B meningococcal vaccine. Expert Rev. Vaccines 8:729–745 [DOI] [PubMed] [Google Scholar]
- 29. Lu L., et al. 2008. Species-specific interaction of Streptococcus pneumoniae with human complement factor H. J. Immunol. 181:7138–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madico G., et al. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 177:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maruvada R., Blom A. M., Prasadarao N. V. 2008. Effects of complement regulators bound to Escherichia coli K1 and group B streptococcus on the interaction with host cells. Immunology 124:265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mascioni A., et al. 2009. Structural basis for the immunogenic properties of the meningococcal vaccine candidate LP2086. J. Biol. Chem. 284:8738–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masignani V., et al. 2003. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J. Exp. Med. 197:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meri S., Jordens M., Jarva H. 2008. Microbial complement inhibitors as vaccines. Vaccine 26(Suppl. 8):I113–117 [DOI] [PubMed] [Google Scholar]
- 35. Michaelsen T. E., Garred P., Aase A. 1991. Human IgG subclass pattern of inducing complement-mediated cytolysis depends on antigen concentration and to a lesser extent on epitope patchiness, antibody affinity and complement concentration. Eur. J. Immunol. 21:11–16 [DOI] [PubMed] [Google Scholar]
- 36. Murphy E., et al. 2009. Sequence diversity of the factor H binding protein vaccine candidate in epidemiologically relevant strains of serogroup B Neisseria meningitidis. J. Infect. Dis. 200:379–389 [DOI] [PubMed] [Google Scholar]
- 37. Pace D., Pollard A. J., Messonier N. E. 2009. Quadrivalent meningococcal conjugate vaccines. Vaccine 27:B30–B41 [DOI] [PubMed] [Google Scholar]
- 38. Pajon R., Beernink P. T., Harrison L. H., Granoff D. M. 2010. Frequency of factor H-binding protein modular groups and susceptibility to cross-reactive bactericidal activity in invasive meningococcal isolates. Vaccine 28:2122–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez de Cordoba S., Esparza-Gordillo J., Goicoechea de Jorge E., Lopez-Trascasa M., Sanchez-Corral P. 2004. The human complement factor H: functional roles, genetic variations and disease associations. Mol. Immunol. 41:355–367 [DOI] [PubMed] [Google Scholar]
- 40. Sadarangani M., Pollard A. J. 2010. Serogroup B meningococcal vaccines—an unfinished story. Lancet Infect. Dis. 10:112–124 [DOI] [PubMed] [Google Scholar]
- 41. Scarselli M., et al. 2009. Epitope mapping of a bactericidal monoclonal antibody against the factor H binding protein of Neisseria meningitidis. J. Mol. Biol. 386:97–108 [DOI] [PubMed] [Google Scholar]
- 42. Schneider M. C., Exley R. M., Ram S., Sim R. B., Tang C. M. 2007. Interactions between Neisseria meningitidis and the complement system. Trends Microbiol. 15:233–240 [DOI] [PubMed] [Google Scholar]
- 43. Schneider M. C., et al. 2009. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458:890–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Snape M. D., et al. 2010. Immunogenicity of two investigational serogroup B meningococcal vaccines in the first year of life: a randomized comparative trial. Pediatr. Infect. Dis. J. 29:e71–79 [DOI] [PubMed] [Google Scholar]
- 45. Vu D. M., Wong T. T., Granoff D. M. 2011. Cooperative serum bactericidal activity between human antibodies to meningococcal factor H binding protein and neisserial heparin binding antigen. Vaccine 29:1968–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., et al. 2000. Functional activities and immunoglobulin variable regions of human and murine monoclonal antibodies specific for the P1.7 PorA protein loop of Neisseria meningitidis. Infect. Immun. 68:1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Welsch J. A., Ram S., Koeberling O., Granoff D. M. 2008. Complement-dependent synergistic bactericidal activity of antibodies against factor H-binding protein, a sparsely distributed meningococcal vaccine antigen. J. Infect. Dis. 197:1053–1061 [DOI] [PubMed] [Google Scholar]
- 48. Welsch J. A., Rossi R., Comanducci M., Granoff D. M. 2004. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J. Immunol. 172:5606–5615 [DOI] [PubMed] [Google Scholar]
- 49. Zhu D., et al. 2005. Evaluation of recombinant lipidated P2086 protein as a vaccine candidate for group B Neisseria meningitidis in a murine nasal challenge model. Infect. Immun. 73:6838–6845 [DOI] [PMC free article] [PubMed] [Google Scholar]