

Abstract
Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli are noninvasive attaching and effacing (A/E) bacterial pathogens that cause intestinal inflammation and severe diarrheal disease. These pathogens utilize a type III secretion system to deliver effector proteins into host epithelial cells, modulating diverse cellular functions, including the release of the chemokine interleukin-8 (IL-8). While studies have implicated the effectors NleE (non-locus of enterocyte effacement [LEE]-encoded effector E) and NleH1 in suppressing IL-8 release, by preventing NF-κB nuclear translocation, the impact of these effectors only partially replicates the immunosuppressive actions of wild-type EPEC, suggesting another effector or effectors are involved. Testing an array of EPEC mutants, we identified the non-LEE-encoded effector C (NleC) as also suppressing IL-8 release. Infection by ΔnleC EPEC led to exaggerated IL-8 release from infected Caco-2 and HT-29 epithelial cells. NleC localized to EPEC-induced pedestals, with signaling studies revealing NleC inhibits both NF-κB and p38 mitogen-activated protein kinase (MAPK) activation. Using Citrobacter rodentium, a mouse-adapted A/E bacterium, we found that ΔnleC and wild-type C. rodentium-infected mice carried similar pathogen burdens, yet ΔnleC strain infection led to worsened colitis. Similarly, infection with ΔnleC C. rodentium in a cecal loop model induced significantly greater chemokine responses than infection with wild-type bacteria. These studies thus advance our understanding of how A/E pathogens subvert host inflammatory responses.
INTRODUCTION
Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC) are among the most widespread bacterial causes of infantile diarrhea in both developing and developed countries (47). Despite the health risks posed by these attaching and effacing (A/E) pathogens, there are few effective therapies to prevent or treat their infection. To develop improved therapies, we need a clearer understanding of how these bacteria successfully infect their hosts, particularly in the face of the varied host defenses found within the mammalian gastrointestinal (GI) tract. Like other bacteria, EPEC and EHEC are recognized by innate host receptors such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain protein (NOD)-like receptors that detect conserved microbial molecular patterns (1, 32, 43). This initial recognition enables the host to quickly respond to infectious threats by producing inflammatory cytokines and antimicrobial peptides (6, 31, 40, 57). In fact, these host inflammatory and immune responses to infection lead to intestinal tissue damage, including inflammatory cell infiltration into the infected mucosa and damage to the gut epithelium. Much of this pathology has been linked to inflammatory responses initiated by the infected epithelium, including the production of the chemokine interleukin-8 (IL-8), an important neutrophil chemoattractant. Not surprisingly, enteric pathogens have evolved diverse and elegant strategies to avoid and suppress intestinal defenses in order to colonize and survive within the host's GI tract. A common strategy involves the injection of bacterial proteins into host cells through a type III secretion system (T3SS) (7, 22, 26, 34), with these effector proteins subverting key aspects of host cell function (47, 48).
The mouse pathogen Citrobacter rodentium bears many similarities to EPEC (28), being noninvasive, infecting its hosts by attaching to the apical surface of intestinal epithelial cells (IECs), effacing their microvilli, and creating pedestal-like structures, as well as causing colitis and mild diarrhea (21, 50). It also shares a similar array of translocated effectors with EPEC and EHEC. The main pathogenicity island of EPEC and C. rodentium, termed the locus of enterocyte effacement (LEE) region, encodes six different T3SS-dependent effectors, including translocated intimin receptor (Tir), mitochondrial-associated protein (Map), and EPEC-secreted proteins F, G, H, and Z (EspF, -G, -H, and -Z, respectively). Additional effector proteins have also been identified outside the LEE-encoded region. These non-LEE encoded effector (Nle) genes are located within six pathogenicity islets scattered throughout the EPEC and C. rodentium genomes (13, 23). The complete effector repertoire and their currently identified virulence functions have recently been reviewed elsewhere (8, 11) and involve pedestal formation, actin rearrangement, and microtubule and epithelial barrier disruption, as well as the modulation of inflammatory responses.
Several reports have shown that EPEC suppresses innate immune responses from infected epithelial cells through the actions of its effectors (25, 42, 46). These studies have demonstrated that intestinal epithelial cells infected with wild-type (WT) EPEC produce lower levels of IL-8 as well as other inflammatory mediators compared to cells infected by EPEC strains lacking a functional T3SS. The reduction in IL-8 release occurred in association with impaired NF-κB and p38 mitogen-activated protein kinase (MAPK) signaling. In part the suppression of IL-8 release appears to be mediated by the effectors NleE and NleB, which were recently shown to inhibit NF-κB activation during EPEC infection (36), whereas the effector NleH1 was also found to subvert NF-κB function by preventing translocation of the NF-κB subunit ribosomal protein S3 (15, 52). Since these effectors have only been shown to affect NF-κB signaling, we suspected that additional EPEC effectors were involved in inhibiting proinflammatory responses by targeting p38 MAPK signaling pathways.
Recently several studies have identified NleC as another effector that suppresses inflammatory response through inhibition of NF-κB (3, 35, 38, 56). These studies showed that NleC also suppressed IL-8 release and NF-κB activation by cultured epithelial cells, specifically through the degradation of the NF-κB p65 subunit. NleC is a zinc protease that cleaves NF-κB p65 subunit (3, 35, 38, 56). Curiously, despite the prominent immunosuppressive role attributed to NleC in vitro, no studies have yet examined the potential for NleC to modulate inflammation in vivo. In addition, despite considerable evidence that ectopic NleC suppresses IL-8 release by degrading the NF-κB p65 subunit, at least in vitro (3, 35, 38, 56), attempts to localize the NleC protein within host cells have yielded conflicting results. While several studies identified ectopically expressed NleC as predominantly localized to the nucleus, where it could potentially access the NF-κB p65 subunit (3, 35, 38), another study found that EPEC-derived NleC was predominantly localized to the site of bacterial attachment, at the host cell membrane (56). This discrepancy in NleC's localization raised the possibility that NleC may impact other host signaling pathways, along with NF-κB.
In this study, by screening an array of EPEC and C. rodentium mutants, we confirmed NleC as a novel effector protein involved in restraining epithelial inflammatory responses during infection, by suppressing NF-κB. We also found that it impacts p38 MAPK signaling, and we demonstrate that NleC plays a significant role in suppressing the colitis triggered by C. rodentium infection, as well as having an impact on the competitiveness of this pathogen in vivo.
MATERIALS AND METHODS
Cell culture, bacterial strains, growth conditions.
Caco-2 intestinal epithelial cells and HT-29 intestinal epithelial cells were obtained from the American Type Culture Collection (ATCC) and grown in Dulbecco's modified Eagle's minimal essential medium (DMEM) with 4.5 g/liter d-glucose, 1× nonessential amino acids, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml), and 10% fetal bovine serum (Gibco). Cells were seeded at high density in polystyrene T75 culture flasks, 6-well plates (6WP), or 12-well plates (12WP) and used for experiments at confluence 4 days after seeding. Cultured cells were used between passages 6 and 20. The bacterial strains used in this study are listed in Table 1 . The EPEC in-frame deletion mutants were generated using the suicide vector pRE112 via sacB-based allelic exchange (14), while the C. rodentium nleC deletion mutant was created using the lambda Red recombinase system (10). All strains were grown from single colonies on Luria-Bertani (LB) plates in LB broth at 37°C overnight with shaking.
Table 1.
EPEC and Citrobacter rodentium strains used in this study
| Strain or genotype | Deficiency | Source or reference |
|---|---|---|
| EPEC E2348/69 (serotype O127:H6) | ||
| WT | None | 27 |
| ΔescN | T3SS | 16 |
| ΔescF | T3SS | 14 |
| ΔescU | T3SS | 14 |
| ΔescV | T3SS | 14 |
| Δtir | Tir | 26 |
| ΔespF | EspF | 53 |
| ΔespG | EspG | 21 |
| ΔespH | EspH | 41 |
| ΔespZ | EspZ | 45 |
| Δmap | Map | 24 |
| ΔnleA | NleA | 20 |
| ΔnleB2 | NleB2 | 14 |
| ΔnleC | NleC | This study |
| ΔnleD | NleD | 14 |
| ΔnleE | NleE | This study |
| ΔnleH1 | NleH1 | This study |
| ΔnleC/nleC (MCE003) | None | This study |
| ΔnleC/nleC-HA (MCE004) | NleC | This study |
| ΔfliC | Flagella | 28 |
| ΔfliC ΔescN | T3SS and flagella | 28 |
| ΔfliC ΔnleC | FliC and NleC | This study |
| ΔfliC ΔnleC/nleC | Flagella | This study |
| C. rodentium DBS 100 | ||
| WT | None | 44 |
| ΔnleC | NleC | 51 |
| ΔnleC/nleC-2HA | NleC | This study |
Generation of nleC complemented and nleC-HA strains.
Complementation of nleC was achieved by chromosomal insertion with Tn7 into an EPEC ΔnleC mutant. A chloramphenicol-marked Tn7 delivery vector was created by subcloning a SacI-frt-cat-frt, Klenow-end-filled fragment from pFCM1 into the EcoRV site of pUC18R6KT-mini-Tn7T (5), yielding pMAC5, and transformed into DH5α λpir. The orientation of the cat cassette was confirmed with EcoRI/NcoI restriction digests. The nleC complementation construct was made by fusing the nleBCD operon promoter to nleC that contained 19 nucleotides (nt) upstream of the predicted start site. A 348-bp region containing the nleBCD promoter was PCR amplified with oligonucleotides nleBCD-f (TCAGAATTCCCAAGCTATATGTTAACTGC) and nleBCDp-r (GTTTATCCATATTTTCTTCACAAC). A 1,033-bp PCR product that contains the nleC gene, with 19 bp of sequence upstream of the ATG, was amplified with oligonucleotides nleC19-f (GTTG TGAAGAAAATATGGATAAACCAGGGTATTAGATATAAACATG) and nleC-r (CGACGGATCCTCATCGCTGATTGTGTTTGTC). The promoter and nleC gene were fused by splicing by overlap extension (SOEing) PCR, using 1 ng of each PCR product as a template in a single PCR as previously described (9), using oligonucleotides nleBCD-f and nleC-r. A hemagglutinin (HA)-tagged nleC gene (nleC-HA) was also amplified using the same templates and the SOEing PCR protocol, except using oligonucleotide nleCHA-r (CGACGGATCCTAAGCGTAATCTGGAACATCGTATGGGTATCGCTGATTGTGTTTGT CCAC) instead of nleC-r. Both PCR products were sequenced at Nucleic Acid Protein Service (NAPS) Unit (University of British Columbia, Vancouver, British Columbia, Canada) and subcloned as an EcoRI/BamHI fragment into a similarly cut plasmid, pMAC5, yielding pMAC5/19nleC and pMAC5/19nleCHA, and transformed into EC100Dpir+. The complementing constructs were mobilized into the EPEC ΔnleC strain by mating with ω7249 (2) harboring the Tn7 helper plasmid, pTNS2 (5) and either pMAC5/19nleC or pMAC5/19nleCHA. Transconjugants were selected on LB containing chloramphenicol, and the proper Tn7 insertion was checked as previously described (5). The resulting complementing strains were designated MCE003 (nleC) and MCE004 (nleC-HA).
Infection protocol.
Caco-2 and HT-29 cells cultured in 6WP and 12WP were infected by different strains of EPEC in DMEM nutrient mixture F-12 (DMEM F-12) for 4 h. At least 30 min before infection, the medium in 6WP or 12WP was replaced by DMEM F-12 (without supplements). Overnight bacterial culture with an optical density at 600 nm (OD600) of approximately 0.5 was used for infection. After infection, cells were washed twice with 2 ml of phosphate-buffered saline (PBS) and the medium was replaced by DMEM growth medium containing gentamicin (1 μg/ml) in order to prevent host cell death and the overgrowth of extracellular bacteria. At different time points postinfection, supernatants were collected for sandwich enzyme-linked immunosorbent assay (ELISA) (24 h), or cell lysates (2 to 6 h) were prepared for Western blotting. For infections using ΔfliC EPEC strains, Caco-2 cells were infected with the bacterial cultures for 3 h as described above. Cells were washed with PBS, which was replaced by 2 ml of DMEM F-12. Purified FliC (100 μg) was added for 30 min to 1 h, and cell lysates were prepared for Western blotting.
Measurement of IL-8 secretion levels.
The level of IL-8 released into the supernatant was assessed using an ELISA kit (BD Bioscience) following the manufacturer's instructions.
Assessment of bacterial adherence to Caco-2 cells.
To assess bacterial adhesion, Caco-2 cells were infected with bacteria for 4 h. Cells were then washed with warm PBS three times and finally scraped into warm PBS and mixed by pipetting. Serial dilutions were performed, aliquots of the scraped cells were streaked onto agar plates, and the plates were incubated in 37°C overnight. Bacterial colonies (CFU) were counted the next day.
NF-κB activity assay.
Caco-2 cells were seeded into 24WP at 2.5 × 105 cells/well in 1 ml of medium and incubated overnight in a 37°C 5% CO2 incubator. One hour prior to transfection, cells were washed with sterile PBS, and 0.5 ml of prewarmed low-serum medium (0.5% fetal bovine serum [FBS], 1% nonessential amino acids, 1% GlutaMax) was added to each well. Per well, 25 ng pNF-κB Luc (luciferase) vector (Clontech Labs) was mixed with 75 ng phRL-TK (Renilla) vector (Promega Corporation) and topped up to 500 ng of total DNA with pCMV4a vector (Stratagene). DNA was diluted in 50 μl DMEM, and 3 μl of GenJet for Caco-2 cells (SignaGen Laboratories) was diluted in 50 μl DMEM in a separate tube. Diluted GenJet was added directly to diluted DNA and mixed. The transfection mixture was incubated at room temperature for 15 min prior to addition of 100 μl to each well of Caco-2 cells. Twenty-four hours posttransfection, medium was aspirated from Caco-2 cells and replaced with 0.5 ml prewarmed serum-free DMEM. Cells were infected with 1 μl of overnight-standing EPEC cultures as indicated. Infections were performed in triplicate for each strain used. For uninfected cells, 1 μl of uninoculated LB medium was added to Caco-2 cells. At 2, 3, or 4 h postinfection, medium was aspirated and replaced with fresh DMEM supplemented with 100 μg/ml gentamicin and 5 ng of recombinant human IL-1β per well for 3 h. The dual-luciferase reporter assay (Promega Corporation) was used according to the manufacturer's instructions. Briefly, infected Caco-2 cells were washed with 500 μl of sterile PBS −/−, and 100 μl of passive lysis buffer was added, followed by 15 min of incubation at room temperature with rocking. Twenty microliters of cell lysate was used for luciferase quantification. Data were plotted as a ratio of firefly/Renilla luciferase activity to enumerate NF-κB activity relative to a constitutive CMV promoter, and NF-κB activities resulting from all infections were plotted relative to that in uninfected cells, which was set at 100%.
Western blotting.
Following infection with EPEC, Caco-2 cells were washed twice with 2 ml of PBS (Gibco). Cells were then lysed in 100 μl of lysis buffer (Promega) on ice for 5 to 10 min and then scraped into microcentrifuge tubes. The tubes were centrifuged at 13,000 × g for 12 min to pellet debris, and the supernatant was transferred to another tube for Western blots. Caco-2 proteins (50 μg in cleared cell lysate) were resolved by 9 to 11% SDS-PAGE and transferred to 0.2 μm polyvinylidene fluoride (PVDF) membranes. Blots were then blocked for 1 h with 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.05% Tween 20 (TBST). Membranes were then incubated with primary antibody in TBST overnight at 4°C and probed with the respective secondary antibody for 1 h at room temperature. Rabbit polyclonal phospho-p38 MAPK (Cell Signaling) and phospho-NF-κB p65 (Cell Signaling) antibodies were used in this study.
Mouse infections and the cecal loop experiment.
Female C57BL/6 mice (8 to 12 weeks) were purchased from Charles River and housed at the Child and Family Research Institute. All mouse experiments were performed according to Canadian Council on Animal Care (CCAC) guidelines (3a). Cecal loop experiments were adopted from the ileal loop experiments previously described (4). In brief, mice were anesthetized by intraperitoneal injection of ketamine and xylazine diluted in sterile PBS. Following a midline abdominal incision, the cecum was exposed, and the proximal colon close to the cecum was ligated twice, while the ileocecal valve prevented backflow into the ileum, thus isolating the cecum. Bacteria were prepared from overnight cultures in LB medium and diluted (1:50) in DMEM and left at 37°C in a 5% CO2 atmosphere for 3 h to preactivate the bacteria (12). Three hundred microliters of the preactivated cultures containing approximately 1 × 107CFU of C. rodentium was injected into the cecal loop. The intestine was then returned to the abdominal cavity, and the incision was closed with discontinuous sutures. The mice were euthanized at 12 h, and tissues and stool contents were collected for bacterial counts and immunofluorescence.
Tissue collection and bacterial counts.
Tissue collection and bacterial counts were performed as described previously (28). Briefly, mice were euthanized over the course of infection and dissected, and their large intestines, including the cecum, were collected in 10% neutral buffered formalin (Fisher) for histological analyses or processed for tissue pathology assays. For viable cell counts, colon tissues and stool pellets were collected separately and homogenized in PBS (pH 7.4), with dilutions plated onto LB agar or MacConkey agar plates.
RNA extraction and semiquantitative and quantitative real-time PCR.
Following the euthanizing of the mice, colonic tissues were immediately transferred to RNA-later (Qiagen), frozen in liquid N2, and stored at −20°C. Total RNA was purified using Qiagen RNeasy kits (Qiagen) according to the manufacturer's instructions. cDNA was synthesized with the Omniscript reverse transcription (RT) kit (Qiagen) and oligo(dT) (ABM, Inc.) followed by quantitative real-time PCR techniques. Quantitative PCR was carried out on a Bio-Rad MJ Mini-Opticon real-time PCR system (Bio-Rad) using IQ SYBR green supermix (Bio-Rad) and macrophage inflammatory protein 2 (MIP-2), MIP-3α, and β-actin primers with the sequences and conditions previously described (25). Quantification was carried out using Gene Ex Macro OM 3.0 software (Bio-Rad), with which PCR efficiencies for each of the primer sets were incorporated into the final calculations.
Immunofluorescence staining.
Immunofluorescence staining of control and infected tissues was performed using previously described procedures (28). In brief, tissues were rinsed in ice-cold PBS, embedded in OCT (optimal cutting temperature) compound (Sakura, Finetech), frozen with isopentane (Sigma) and liquid N2, and stored at −70°C. Serial sections were cut at a thickness of 6 μm and directly blocked with an endogenous biotin blocking kit (Invitrogen) and 1% bovine serum albumin, followed by the addition of polyclonal rat antisera generated against Tir (1:8,000); the biotinylated anti-HA antibody (Covance) was used at 1:500. Following extensive washing with Tris-buffered saline, Alexa 488- or Alexa 568-conjugated goat anti-rat IgG antibodies, or Alexa 488- or Alexa 568-conjugated streptavidin (1:300 dilutions; Molecular Probes) was added. Following extensive washing, the cells were then stained with 1 μg ml−1 of 4′,6′-diamidino-2-phenylindole (DAPI; Sigma) at 1:15,000 for 10 min and washed with PBS before the coverslips were mounted on glass slides in Mowiol (Aldrich) and sealed with nail polish. Sections were viewed at 350, 488, and 568 nm with a Zeiss AxioImager microscope, and images were obtained using an AxioCam HRm camera operating through AxioVision software (version 4.4).
Statistical analysis.
All of the results are expressed as the mean value ± standard error of the mean (SEM). Nonparametric Mann-Whiney t tests were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). A P value of 0.05 or less was considered significant.
RESULTS
Suppression of IL-8 secretion by EPEC is T3SS dependent.
EPEC is known to suppress IL-8 secretion by epithelial cells in a T3SS-dependent manner (25, 42). We confirmed this observation using the ΔescN, ΔescF, ΔescU, and ΔescV T3SS mutants, which lack critical T3SS components and are unable to secrete translocator and effector proteins. After infecting Caco-2 cells with WT EPEC, we recovered 775 ± 70 pg/ml of IL-8, whereas all T3SS mutants induced more than 2-fold-higher (P < 0.05) levels of IL-8 (ΔescN, 1,591 ± 190 pg/ml; ΔescV, 1,816 ± 203 pg/ml; and ΔescU, 2,119 ± 600 pg/ml) (Fig. 1A) that were all statistically similar.
Fig. 1.
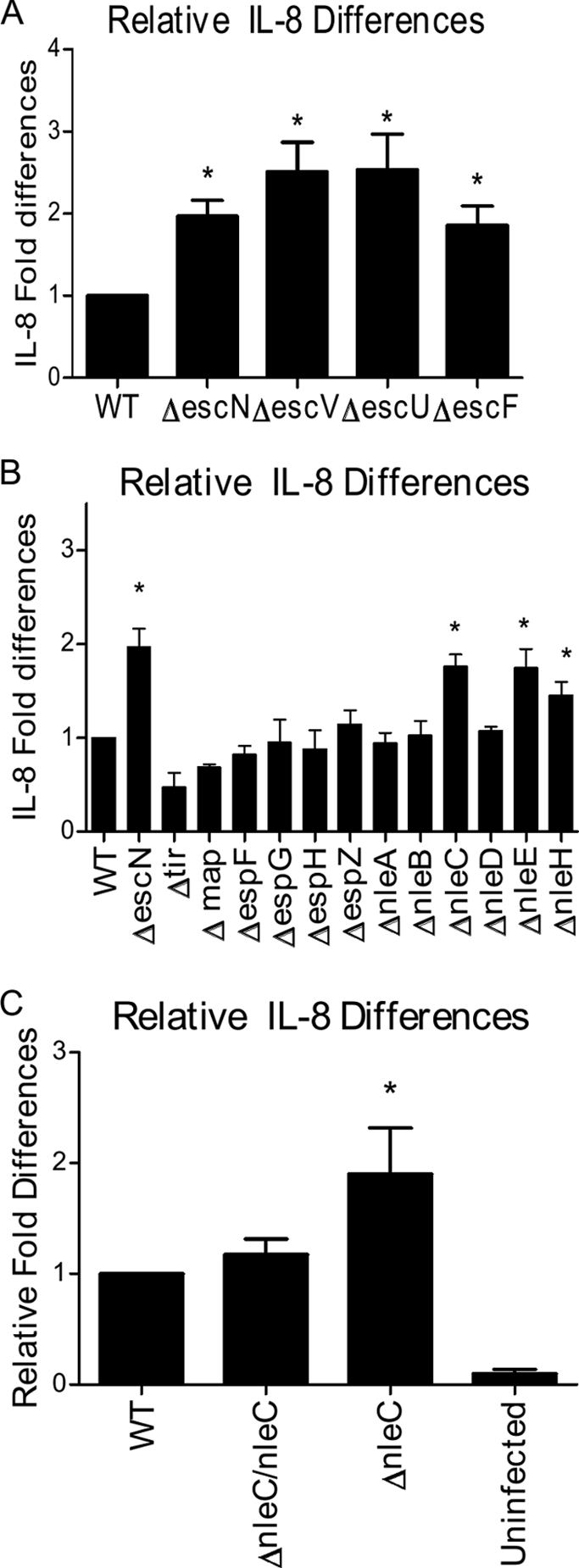
NleC suppresses EPEC-induced intestinal epithelial IL-8 responses. (A) Caco-2 cells were infected with wild-type (WT) EPEC and T3SS EPEC mutants (ΔescN, ΔescF, ΔescU, and ΔescV) for 4 h. Supernatants were collected 24 h later and subjected to IL-8 ELISA. All T3SS mutants induced significantly higher IL-8 release than WT-infected cells. (B) Caco-2 cells were infected with mutants with single-gene deletions in the LEE- and non-LEE-encoded regions as described above. ΔescN, ΔnleC, ΔnleE, and ΔnleH mutants induced significantly higher IL-8 release than WT-infected cells. (C) Complementation of ΔnleC EPEC by chromosomal insertion is seen in Caco-2 cells infected as described above. Complementation of ΔnleC restores the suppressive phenotype. There is no significant differences between WT-infected cells and cells infected with complemented strain. Error bars for all panels represent the standard error of the mean from at least three independent experiments. *, P <0.05.
NleC inhibits IL-8 secretion by EPEC-infected cells.
Given the T3SS dependency, EPEC mutants with single gene deletions within the LEE (Δtir, ΔespF, ΔespG, ΔespH, ΔespZ, and Δmap) and non-LEE-encoded regions (ΔnleA, ΔnleB2, ΔnleC, ΔnleD, ΔnleE, and ΔnleH1) were screened. We confirmed (Fig. 1B) that most mutants were as effective as WT EPEC at inhibiting IL-8 secretion (36), whereas the ΔnleE and ΔnleC mutants induced significantly greater levels of IL-8 (P < 0.05 and P < 0.01, respectively). TheΔnleH1 strain also induced elevated IL-8 levels (P < 0.05), reflecting the ability of NleH1 to prevent translocation of the newly identified NF-κB subunit RPS3 (15, 52).
We previously showed that β-defensin-2 expression was also suppressed by EPEC's T3SS (25), and we found it was significantly elevated in Caco-2 cells infected with ΔnleC EPEC compared to WT EPEC (data not shown). To address whether the exaggerated IL-8 response to ΔnleC EPEC could be seen in another intestinal epithelial cell line, we infected HT-29 colonic epithelial cells. Similar to Caco-2 cells, ΔnleC and ΔescN EPEC infection led to significantly higher levels of IL-8 (3,853 ± 238 pg/ml and 3,397 ± 211 pg/ml, respectively) compared to WT EPEC (2,366 ± 206 pg/ml) (P < 0.05).
We next tried to complement the nleC mutation in EPEC by introducing the nleC gene on the pACYC184 vector (pnleC) to create the ΔnleC pnleC strain. Unfortunately complementation was not achieved, potentially due to changes in the secretion of other effector proteins from EPEC. Since the nleC gene is located in an operon along with nleB, nleH, and nleD (30), its overexpression could alter its transcriptional control, disrupting normal T3SS functioning, as we recently found with the effector EspZ (45). To overcome these issues, chromosomal insertion of the full nleC gene was used to create the ΔnleC/nleC EPEC strain (Fig. 1C). Chromosomal insertion of the nleC gene did not alter effector secretion profiles (data not shown), but statistically lower IL-8 release was seen with Caco-2 cells infected with the ΔnleC/nleC strain (808 ± 50 pg/ml) compared to cells infected by ΔnleC EPEC (1,181 ± 94 pg/ml) (P = 0.02), whereas the response was not significantly different from that seen with WT EPEC (777 ± 70 pg/ml).
EPEC NleC inhibits NF-κB activation.
NleC has been proposed to directly cleave the NF-κB p65 subunit (3, 35, 38, 56). To test this in our system, Caco-2 cells were infected for 3 h with WT, ΔescN, ΔnleC, or ΔnleC/nleC EPEC. Signaling proteins were quantified by Western blot analysis. NF-κB phosphorylation was inhibited by WT EPEC compared to that with the ΔescN strain (Fig. 2A). In contrast, NF-κB phosphorylation in cells infected by ΔnleC EPEC was similar to that in WT EPEC cells. Compared to earlier findings, this lack of an effect on NF-κB was surprising; however, we decided to use a stronger NF-κB stimulation by infecting Caco-2 cells for 3 h with several EPEC strains followed by 3 h of gentamicin treatment and then stimulation with interleukin (IL-1β). We measured NF-κB activity using Caco-2 cells transfected with plasmids containing firefly and Renilla luciferase genes, which provided us with a quantitative readout of NF-κB activity. Data are shown as firefly/Renilla luciferase levels and plotted relative to uninfected and IL-1β-stimulated cells set to 100% NF-κB activity. As shown in Fig. 2B, NF-κB activation decreased following WT EPEC infection, yet NF-κB activation was significantly greater in cells infected with either ΔescN or ΔnleC EPEC (P < 0.01 and P < 0.001, respectively) 3 h postinfection. Moreover, complementation of the nleC deletion, as assessed by the ΔnleC/nleC and ΔnleC/nleC-HA EPEC strains, normalized NF-κB activation to WT levels.
Fig. 2.

NleC suppresses epithelial inflammatory responses by impairing phosphorylation of NF-κB and p38 MAPK. (A) Caco-2 cells were infected with the WT or ΔescN, ΔnleC, or ΔnleC/nleC strain for 3 h. Cells infected with bacteria washed in PBS to remove flagellin in the supernatant are marked with “+” in the “Washed” column. At the end of the infection, cells were lysed, and NF-κB p65 subunit phosphorylation was examined by Western blotting. Lane 9 contains the uninfected control. At the same time point, ΔescN (lane 3, bottom) strain-infected cells had greater I-κB degradation. (B) NF-κB activity assay at 3 h postinfection using a dual-luciferase reporter system. Caco-2 cells transfected with the constitutively expressed Renilla luciferase gene and firefly luciferase gene under the control of the NF-κB enhancer were infected with the indicated strains or left uninfected (UI). Luciferase activity was quantified postinfection and following treatment with IL-1β (see Materials and Methods). Data are plotted as firefly/Renilla luciferase activity, and 100% NF-κB activity was set for uninfected, IL-1β-treated cells. Stars denote statistical significance compared to NF-κB activity from cells infected with WT EPEC at the same time point *, P < 0.01; and **, P < 0.001.
NleC localizes to the EPEC-induced pedestal.
We next examined the cellular localization of NleC within infected epithelial cells, using a strain of EPEC (ΔnleC/nleC-HA) that contained a chromosomal insertion of NleC tagged with two hemagglutinin (HA) epitopes at the C terminus of the NleC protein. The resulting strain produced high levels of HA-tagged NleC that was secreted in a T3SS-dependent manner (data not shown). Following standard EPEC infection and effector staining protocols (45), we identified strong HA staining within the EPEC-induced pedestal (Fig. 3A and B) underlying the adherent EPEC, similar to the observation by Yen et al. (56). In fact, the HA staining partially overlapped with staining for the translocated effector Tir, indicating NleC is translocated into host cells, where it predominantly resides at the tip of the EPEC-induced pedestal. The specificity of the staining for HA was confirmed, as no HA staining was detected in cells infected with WT EPEC (lacking HA-tagged NleC) (Fig. 3A and B). Since the localization of NleC near the host cell membrane would not fit with NleC's role in degrading the p65 subunit of NF-κB, this prompted us to examine whether other inflammatory signaling pathways were also suppressed by NleC.
Fig. 3.

The HA-tagged effector NleC localizes to the host cell membrane underlying adherent EPEC. (A). Immunofluorescence staining of HeLa cells infected with either WT EPEC or ΔnleC/nleC-HA EPEC. (B) Inset from panel A. Bacteria were preactivated in DMEM, and cells were infected for 3 h after preactivation. NleC-2HA is stained in red, Tir is stained in green, and DAPI is blue. Note in the merged image that the HA staining overlaps with the Tir staining, and both are found underlying the adherent ΔnleC/nleC-HA EPEC.
EPEC NleC inhibits p38 MAPK phosphorylation.
Aside from NF-κB, WT EPEC has been shown to also affect inflammatory signaling by inhibiting the phosphorylation of p38 MAPK (42). Through infection with Caco-2 cells as described above, our blotting revealed a modest increase in p38 MAPK phosphorylation, although densitometry analysis revealed it was not significantly different from WT EPEC (Fig. 4A). Based on this response, we addressed whether NleC could also be playing a role modulating p38 MAPK signaling, as this pathway is known to play an important role in the production of IL-8 in our system. Notably, TLR5 recognition of FliC causes phosphorylation of p38 MAPK within 30 min of stimulation, possibly prior to the actions of T3SS effectors like NleC and thereby limiting our ability to assess the roles of specific T3SS effectors (25). To overcome this obstacle and better define the signaling pathways altered by NleC, we washed the EPEC cultures before adding them to the epithelial cell cultures. While this would remove shed flagella within the culture, the bacteria would remain flagellated. As shown in Fig. 4A, we detected a stronger and significant effect of NleC on p38 MAPK signaling in these samples. To further address whether the initial stimulation with FliC was interfering with the function of NleC, we infected Caco-2 cells with EPEC mutants lacking FliC (ΔfliC, ΔfliC ΔescN, ΔfliC ΔnleC, and ΔfliC ΔnleC/nleC EPEC). Cells were infected for 3 h followed by 30 min of FliC stimulation. After stimulation, cells were lysed and proteins were quantified by Western blotting followed by densitometric analysis. At 30 min post-FliC stimulation, NF-κB phosphorylation following ΔfliC ΔnleC infection was similar to that seen following ΔfliC infection, suggesting that this approach did not alter general proinflammatory signaling (Fig. 4B). In contrast, both ΔfliC ΔescN and ΔfliC ΔnleC EPEC induced significantly (P < 0.05) higher levels of p38 MAPK phosphorylation compared to cells infected with the ΔfliC and ΔfliC ΔnleC/nleC EPEC strains (Fig. 4C). Densitometry revealed approximately 2-fold-higher phosphorylation of p38 MAPK in cells infected with ΔfliC ΔnleC EPEC compared to cells infected with ΔfliC EPEC (Fig. 4C). Overall, these data suggest that NleC affects p38 MAPK signaling in host cells.
Fig. 4.

NleC suppresses epithelial inflammatory responses by impairing phosphorylation of p38 MAPK. (A) Caco-2 cells were infected with the WT or ΔescN, ΔnleC, or ΔnleC/nleC mutant for 3 h. Cells infected with bacteria that were washed in PBS to removed flagellin are marked with + in the “Washed” column. At the end of the infection, cells were lysed, and p38 MAPK phosphorylation was examined through Western blotting. Lane 9 contains the uninfected control. At 3 h postinfection, ΔescN (Washed −) and ΔnleC (Washed −) induced significantly higher p38 MAPK phosphorylation. (B) Caco-2 cells were infected with ΔfliC, ΔfliC ΔescN, ΔfliC ΔnleC, ΔfliC ΔnleC/nleC EPEC for 3 h, followed by 30 min of FliC stimulation or just received the FliC stimulation (FliC). At the end of the infection, phosphorylation of NF-κB and p38 MAPK was probed in the lysate through Western blotting. Ctrl, control. (C) Densitometric analysis of phosphorylated p38 MAPK (phospho p38/p38) following infection and stimulation as outlined in panel C. Stimulation with the ΔfliC ΔescN and ΔfliC ΔnleC strains as well as FliC stimulation on its own induced significantly higher phosphorylation of p38 MAPK compared to that in the ΔfliC and ΔfliC ΔnleC/nleC-infected cells. *, P < 0.05.
The C. rodentium ΔnleC mutant shows no defects in colonization but causes exaggerated colitis.
Based on our observation that EPEC NleC plays an important role in suppressing epithelial inflammatory responses in vitro, we next tested whether NleC expressed by C. rodentium had similar effects in vivo. We infected C57BL/6 mice with either WT or ΔnleC C. rodentium and enumerated the pathogen burdens found within the ceca and colons of infected mice at day 6 (D6) and D10. Interestingly, C. rodentium numbers were similar between the two groups in both the cecum and the colon at both time points (in the cecum at D6, WT, 2.0 ± 1.7 × 107 CFU/g, and ΔnleC mutant, 4.3 ± 1.4 × 106 CFU/g; at D10, WT, 6.6 ± 3.6 × 108 CFU/g, and ΔnleC mutant, 9.1 ± 5.2 × 108 CFU/g; in the colon at D6, WT, 6.3 ± 1.8 × 108 CFU/g, and ΔnleC mutant, 2.9 ± 0.9 × 108 CFU/g, at D10, WT, 5.2 ± 1.7 × 108 CFU/g, and ΔnleC mutant, 1.8 ± 0.6 × 108 CFU/g) (Fig. 5A). We also assessed the tissue pathology suffered by these mice, blindly scoring histological sections for edema, goblet cell depletion, hyperplasia, and tissue integrity, as well as inflammatory cell infiltration, as previously described (17). We found that at D6, the mean histological score for the ΔnleC C. rodentium-infected ceca was 2.58 ± 0.20, a level significantly higher than the score of 0.68 ± 0.28 obtained for WT C. rodentium-infected cecal tissues (Fig. 5B). The ΔnleC score reflected worsened edema, hyperplasia, and increased inflammatory cell infiltration. At D10, the elevated pathology scores were maintained in the ceca of ΔnleC C. rodentium-infected mice at 2.60 ± 0.40, a level still significantly higher than the score of 1.10 ± 0.40 obtained for WT C. rodentium-infected cecal tissues (Fig. 5B). Specifically, at D10 we observed more inflammatory cell infiltration and goblet cell depletion in the ΔnleC C. rodentium-infected cecal tissues. While the scores for the colons of the ΔnleC C. rodentium-infected mice were also elevated at both D6 and D10, they did not reach statistical significance (data not shown). As bacterial colonization did not differ between the infection groups at either D6 or D10 postinfection (p.i.), the increased damage suffered following ΔnleC C. rodentium infection appears to reflect the actions of NleC.
Fig. 5.

Impact of NleC on C. rodentium colonization and colitis. C57BL/6 mice were infected with WT or ΔnleC C. rodentium for 6 and 10 days. ΔnleC C. rodentium outcompeted WT C. rodentium in competitive index studies. (A) Bacterial numbers were enumerated, and there were no significant differences in bacterial colonization in the cecum and colon at these time points. (B) Histological scoring analysis of hematoxylin-and-eosin-stained tissues revealed worsened histology scores in the ceca of mice infected with ΔnleC C. rodentium at both time points.
The C. rodentium ΔnleC mutant outcompetes WT C. rodentium in CI studies.
Considering the impact of NleC in limiting IL-8 release in vitro as well as its role in controlling host pathology in vivo, we found it surprising that ΔnleC C. rodentium showed no defects in colonization levels compared to WT C. rodentium. However, comparing colonization levels in separate hosts is a relatively insensitive measurement of virulence, and in fact, previous studies using this approach found only modest colonization defects in C. rodentium strains lacking NleE or NleH (3, 15, 54). This may reflect that C. rodentium is a highly adapted murine pathogen and can readily colonize mice, even when it is lacking important virulence factors. We therefore tested the ΔnleC mutant using the competitive index (CI) assay, a more sensitive measure of virulence that we have repeatedly used in the past. After infecting C57BL/6 mice with equal numbers of WT and ΔnleC C. rodentium cells and collecting tissues at D4, we found the ΔnleC strain to have a surprisingly high CI of 4.41 ± 0.98 (Fig. 6A), indicating that the mutant strain significantly outcompeted WT C. rodentium in colonizing the murine GI tract. Two different wild-type strains transformed with the same plasmid with resistance to different antibiotics had a CI of 1.01 ± 0.08. The determination that the C. rodentium ΔnleC strain can outcompete WT C. rodentium when coinfected in vivo was highly reproducible, and interestingly, the competitive advantage displayed by the ΔnleC strain was maintained, albeit at a slightly lower level at D7 (Fig. 6A). To better define the basis for the high CI, in additional mice, we separated the luminal stool contents from the gut tissues, and we collected the stool as well as the cecal and colonic tissues separately. When the CI was calculated for these different isolates, the CI for tissue adherent ΔnleC C. rodentium in the cecum was 1.06 ± 0.19. In contrast, the CI in the stool was 2.68 ± 0.91, while the CI in the distal colon was 9.54 ± 4.06 (Fig. 6B). Overall, these data show that ΔnleC C. rodentium colonizes the cecum at roughly equal efficiency with the WT strain, but the mutant outcompetes WT C. rodentium in spreading down the GI tract to colonize the distal colon.
Fig. 6.

(A) C57BL/6 mice were infected with WT C. rodentium and ΔnleC C. rodentium for 4 and 7 days, and cecal plus colonic tissues were collected for bacterial enumeration. At both time points, ΔnleC C. rodentium outcompeted WT C. rodentium with CIs of 4.41 ± 0.98 (P < 0.05) and 2.13 ± 0.88, respectively. The dotted line indicates a CI of 1. Error bars indicate the standard error of the mean values obtained independently from at least six mice. (B) CI for luminal content, cecum, and colon at D4 postinfection. Shown are the CIs for ΔnleC C. rodentium in the cecum (1.06 ± 0.19), stool (2.68 ± 0.91), and distal colon (9.54 ± 4.06).
Cecal loop infection shows NleC translocates into intestinal epithelial cells in vivo.
The increase in inflammatory cell infiltration and tissue damage seen in the intestines of mice infected by ΔnleC C. rodentium (alone) at D6 and D10 demonstrates that NleC modulates inflammatory responses in vivo; however, the severity of the resulting colitis was less than expected, compared to the dramatic effect that NleC plays in suppressing IL-8 release from IECs. In part, the impact of NleC on the colitic response may have been mitigated by the involvement of inflammatory responses from uninfected IECs, as well as macrophages and other cell types that were not directly infected, but instead were stimulated by shed bacterial products that activate innate receptors, including TLR2 and TLR4 (18–20). To more accurately assess the impact of NleC or other T3SS effectors on epithelial cell-derived inflammatory responses in vivo, we hypothesized that the analysis needed to occur very early during infection, to limit the contribution of uninfected IECs and other cells. Unfortunately, very few C. rodentium cells initially colonize the murine GI tract following oral gavage, with heavy colonization of the epithelium not occurring until D4 or D6 (28), too late for our purposes. To overcome this problem, we utilized a recently developed cecal loop model, by which we inject a large number of preactivated C. rodentium cells into the cecum. C. rodentium cells were incubated with DMEM for 3 h to induce type III secretion (13). This approach allows us to synchronize their arrival and subsequent direct infection of large regions of the cecal epithelium (see Materials and Methods).
To test the efficacy of the model, we first assessed whether we could detect NleC within the intestinal epithelium of mice following infection of the cecal loop. We generated a plasmid expressing NleC tagged with two hemagglutinin (HA) epitopes at the C terminus of NleC protein. After incorporating this construct into ΔnleC C. rodentium, the resulting strain (ΔnleC/nleC-2HA) produced high levels of HA-tagged NleC that was secreted in a T3SS manner (data not shown). We infected cecal loops with ΔnleC/nleC-2HA C. rodentium or WT C. rodentium, and after 12 h, the tissues were collected and stained for HA, as well as the translocated effector Tir, to identify the sites of C. rodentium attachment to host cells (Fig. 7A). Tir staining was seen on the apical surface of infected epithelial cells, lying beneath the adherent bacteria (Fig. 7B). Similar to our in vitro assessment, the HA signal was found to overlap with Tir on the surface of cells infected with ΔnleC/nleC-2HA C. rodentium. Notably, HA staining was not seen in uninfected (Tir-negative) cells or in IECs infected by WT C. rodentium (Fig. 7A and B).
Fig. 7.

(A) Cecal loop model permits observation of NleC translocated into intestinal epithelial cells in vivo. Ceca of C57BL/6 mice were surgically ligated and injected with preactivated ΔnleC/nleC-2HA C. rodentium for 12 h. At the end of the infection, ceca were collected for immunostaining. (A) ΔnleC/nleC-2HA C. rodentium-infected cecal epithelium (original magnification, 1,000×). (B) Inset from panel A showing that HA signal (green) overlaps with Tir (red) on the surface of cells infected with ΔnleC/nleC-2HA C. rodentium. Nuclei of cells and bacteria were stained with DAPI (blue).
C. rodentium ΔnleC strain infection induces significantly higher chemokine expression.
Since the cecal loop model permitted effective and rapid infection of the cecal epithelium, with subsequent translocation of C. rodentium effectors into infected host cells, we next used this model to compare the inflammatory responses elicited by WT and ΔnleC C. rodentium. Twelve hours following injection, cecal loops were dissected, and tissue samples were homogenized and plated to enumerate C. rodentium, while other samples were collected for RNA extraction. C. rodentium colonization in the cecum did not differ significantly between mice infected with the WT ([7.5 ± 2.9] × 107 CFU/g) and ΔnleC C. rodentium ([8.7 ± 4.6] × 107 CFU/g). After isolation of mRNA, quantitative RT-PCR was performed to examine the expression of MIP-2, MIP-3α, and keratinocyte chemoattractant (KC). All three of the MIP-2, MIP-3α, and KC genes encode chemokines that are expressed by IEC and can putatively attract macrophages, dendritic cells, and/or neutrophils to the site of infection. In comparison to cecal tissues taken from mice infected with WT C. rodentium, cecal tissues taken from mice infected with ΔnleC C. rodentium expressed significantly higher mRNA levels (P < 0.05) of MIP-2 (5.2-fold), MIP-3α (2.9-fold), and KC (5.8-fold) compared to mice infected with WT C. rodentium (Fig. 8), confirming that ΔnleC C. rodentium triggers a significantly greater inflammatory response than WT C. rodentium.
Fig. 8.
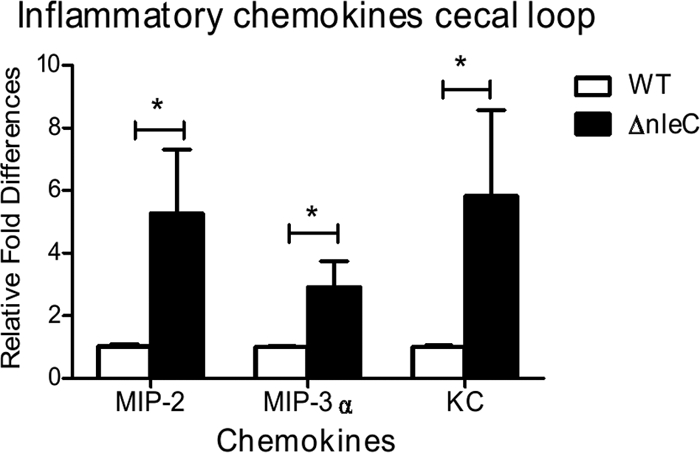
Impact of NleC on bacterial colonization and inflammatory cytokine production in the cecal loop model. qPCR was performed to examine the gene expression of the chemokines MIP-2, MIP-3α, and KC. Tissues infected with ΔnleC C. rodentium expressed significantly higher mRNA levels of MIP-2, MIP-3α, and KC compared to those of mice infected with WT C. rodentium. Error bars represent the standard error of mean from at least three independent experiments. *, P<0.05.
DISCUSSION
Following their ingestion, enteric bacterial pathogens must not only survive the harsh environment of the host's GI tract but also avoid or overcome an array of mucosal defenses in order to replicate and successfully colonize their hosts. Unlike the pathogens Shigella and Salmonella, which invade the intestinal epithelium, thereby escaping the gut and its luminal defenses, the diarrheagenic pathogens EPEC and EHEC colonize the mucosal surface by attaching to the apical surface of epithelial cells. How these pathogens remain at the mucosal surface, surviving and multiplying within the luminal environment while being exposed to all of the antimicrobial responses elicited by the underlying epithelial cells, remains obscure. However, recent studies have found that aside from subverting the host cell cytoskeleton and modulating host cell apoptosis, A/E pathogens also suppress inflammatory signaling, presumably as a means to create a protected niche on the surface of the infected epithelium.
This virulence strategy has been demonstrated through several studies showing that WT EPEC infection suppresses the expression of the chemokine IL-8 and several other epithelial cell-derived mediators, including MIP3α and β-defensin-2. Moreover, this suppression was shown to occur in a T3SS-dependent manner and was linked to the inhibition of several signaling pathways, including NF-κB and p38 MAPK. Similarly, in an earlier study examining IEC responses during C. rodentium infection in mice, we found that expression of the inducible nitric oxide synthase (iNOS) enzyme was dramatically upregulated in the colonic epithelium during later stages of C. rodentium infection (49). Interestingly, further examination revealed that the iNOS expression was almost solely localized to uninfected IECs, whereas infected cells showed little if any expression of this proinflammatory enzyme. Since C. rodentium strains lacking a T3SS are unable to colonize the intestines of mice, we were unable to address whether the inhibition of iNOS was T3SS dependent; however, these findings indicate that A/E pathogens do subvert epithelial inflammatory responses in vivo.
Until recently, the T3SS-dependent effectors that mediate this suppression have remained undefined; in this study, we confirmed the T3SS effector NleC plays an important role in suppressing IL-8 release from infected IECs. NleC is encoded on the same operon as the effectors NleG, NleB, NleH, and NleD, all of which are found on the PP4 lambda-like prophage in the EPEC genome (23). A 330-amino-acid protein with a molecular mass of 36 kDa (30), NleC is a homolog of the apoptosis-inducing effector AIP56, encoded by Photobacterium damselae. EPEC's NleC shares 100% and 95% identity with NleC from EHEC and C. rodentium, respectively, suggesting the effector's function is likely conserved among A/E pathogens. Modulation of inflammatory signaling is the first role attributed to NleC, as it was not found to be involved in either A/E lesion formation or adherence (30).
We and others have previously shown that EPEC induces IL-8 expression in IEC through MAPK and NF-κB activation in response to TLR5-dependent recognition of FliC. In this study, we demonstrated that NleC interferes with both NF-κB and p38 MAPK signaling, resulting in heightened IL-8 release from infected IECs. Interestingly, while the impact of NleC on IL-8 release was dramatic, its effects on the proinflammatory signaling pathways were subtle. We were unable to detect an overt difference in NF-κB activation following infection by the ΔnleC EPEC strain alone and only detected such an effect after stimulation by IL-1β. Similarly, Baruch et al. recently demonstrated that NleC suppresses NF-κB activation in IECs in response to tumor necrosis factor alpha (TNF-α)-induced IL-8 secretion; however, they did not examine whether NleC had an impact on MAPK activation, perhaps because MAPKs do not play a major role in TNF-α-induced signaling (3). In contrast, we detected significantly elevated p38 MAPK phosphorylation following ΔnleC EPEC infection, but only by delaying FliC stimulation until 3 h postinfection. The subtle nature of these changes may reflect the actions of other T3SS effectors playing compensatory roles, as well as the nature of the assays and the proinflammatory stimuli used. We also showed for the first time that NleC impacts on intestinal inflammatory responses in vivo. While little is known about the role of NF-κB in determining the contribution of IECs to C. rodentium colitis, we and others recently showed that IEC-specific p38 MAPK signaling plays a major role in the immune and inflammatory response to C. rodentium infection (25, 42), in keeping with our findings that the ΔnleC strain triggered greater chemokine responses in vivo. It has been recently reported that p38 MAPK modulates NF-κB signaling during C. rodentium infection (4), and both of these signaling pathways are important in MIP-2, MIP-3α, and KC production by epithelial cells (33, 39, 51). By suppressing both NF-κB and p38 MAPK activation, C. rodentium likely suppresses the local inflammatory response, creating a protected niche in the intestine.
Previously, NleC was shown to translocate into host cells but its localization within the host cell remain controversial (30), as Baruch et al. recently showed ectopically expressed NleC localizing to the host cell nucleus (3), whereas Yen et al. showed NleC localized near the pedestal (56). Through the use of HA tags, our data align with the observations made by Yen et al. (56), as we localized NleC primarily to the sites of pathogen attachment, both in vitro and in vivo. This was seen in both EPEC- and C. rodentium-induced pedestals. The staining of the HA tag partially overlapped with staining for the T3SS effector Tir, potentially indicating that it is found at the apical cell membrane, just beneath the bacterium, although it is certainly possible that NleC ultimately reaches other sites in the host cell, including the nucleus, where it may be too diffuse to detect by immunofluorescence. While the binding partner of NleC has not been identified, recent studies have indicated NleC functions as a metalloprotease and can cleave p65 (3), potentially explaining how it impacts NF-κB function. In support of this finding, we also observed an increase in total p65 abundance following ΔnleC EPEC infection (data not shown); however, our findings that bacterially delivered NleC has an impact on MAPK signaling and predominantly localizes to the pedestal suggest it may have other roles, potentially at the host membrane. By localizing to the host membrane, NleC could potentially suppress inflammatory responses by interfering with upstream events involved in multiple signaling pathways.
Over the last 2 years, other EPEC T3SS-dependent effectors have been identified as suppressing IL-8 release and modulating NF-κB activation in epithelial cells. Both NleE and NleB were shown to bind to NF-κB, and in their absence, NF-κB signaling in HeLa cells was increased 20-fold. This was associated with increased levels of IL-8 mRNA in infected HeLa cells (36, 37). Similarly, NleH1 has been reported to bind ribosomal protein S3 to subvert NF-κB function and infection by an nleH1 mutant induced higher levels of IL-8 gene transcription than WT EPEC (15). Curiously, none of these effectors were tested using epithelial cells of intestinal origin. Therefore, we tested EPEC strains lacking these effectors and confirmed both NleE and NleH1 suppress IL-8 secretion in IEC lines, in concert with NleC. Interestingly, the ΔnleB mutant was not found to induce more IL-8 from infected cells, whereas the ΔnleE mutant did induce a greater response. This difference may reflect previous studies in that, in the absence of both effectors, restoration of NleE but not NleB expression partially restored EPEC's capacity to stabilize IκB (36). This suggests that unlike NleB, NleE is both required and sufficient to suppress NF-κB activation and IL-8 release. Taken together, it appears that A/E pathogens use at least these four effectors to inhibit inflammatory responses by suppressing both NF-κB and p38 MAPK signaling pathways.
In keeping with its immunosuppressive role in vitro, NleC also inhibited C. rodentium-induced colitis following oral gavage. However, this route of infection is not an ideal system to address C. rodentium effector functions as it has frequently proven difficult to show significant attenuation in colonic pathology, even with bacterial strains lacking important virulence factors. We believe this lack of concordance between in vitro and in vivo actions reflects the initially low pathogen burden seen during the first 4 days of C. rodentium infection, resulting in very little intestinal inflammation. By 4 to 6 days postinfection, when the infection has widely spread throughout the lower bowel and inflammation is evident, we estimate at most, only 20% of IECs are actually directly infected, and presumably A/E pathogens can only suppress the responses of cells they directly infect. Moreover, the resulting inflammatory responses seen during these infections are not solely arising from the infected epithelium, but also from uninfected IECs and immune cells exposed to shed bacterial products such as flagellin. These complications likely explain why NleC was found to play only a modest role in modulating the colitis caused by C. rodentium infection, yet had a striking effect on chemokine responses in a cecal loop model, where we maximized IEC infection, prior to the recruitment of significant numbers of inflammatory cells. Our in vivo results also raise a question concerning our in vitro findings that over a 4-h time course, NleC was unable to block flagellin-induced epithelial p38 MAPK signaling that occurred prior to the injection of NleC. This finding is likely of little relevance to an in vivo setting, where the epithelium targeted by A/E pathogens is covered by a mucus layer that would limit exposure to shed bacterial products prior to direct infection. Moreover, in an in vivo setting, A/E pathogen infection of specific epithelial cells could possibly last for days, meaning that translocated effectors like NleC would be present and presumably abrogating inflammatory signaling for hours or days after the initial infection.
While we demonstrated the impact of NleC on host inflammatory responses and pathology, curiously we saw no significant attenuation in the ability of the ΔnleC strain to colonize the colonic epithelium of murine hosts when given as a single infection. This may not be surprising, since comparing single strain infections is not a highly sensitive measure of virulence, as C. rodentium is a highly successful pathogen with a multitude of compensatory virulence strategies. Strikingly, however, we did identify a robust and significant CI of 4.4 for the mutant, indicating that the ΔnleC strain outcompeted the WT strain by more than 4-fold during the infection. This advantage was most evident at D4 p.i., with the advantage modestly reduced by D7 of the infection. The basis for this advantage is unclear, but host inflammatory responses have been previously shown to aid C. rodentium, as well as other enteric pathogens, including Salmonella enterica serovar Typhimurium, in their colonization of the murine lower bowel (29, 55). Since C. rodentium appears to initially colonize the intestine in a clonal fashion, localized differences in inflammation and pathological changes in the gut mucosa could provide the mutant with a competitive advantage over the WT strain, promoting its spread through the host's intestines. Alternatively, aside from inflammation, NF-κB and p38 MAPK-dependent signaling also affects other host processes such as cell death and tissue repair. Localized changes in epithelial cell sloughing, for example, could also prove selectively beneficial to the mutant strain. In contrast, previous studies have shown that C. rodentium mutants lacking either of the other two effectors (i.e., NleE and NleH1) are outcompeted by WT C. rodentium. It is unclear why such divergent competitiveness is seen between ΔnleC, ΔnleE, and ΔnleH mutants, since they all cause exaggerated release of IL-8 in vitro. However, it may reflect that NleE and NleH1 primarily target NF-κB signaling, whereas NleC also targets p38 MAPK. Aside from affecting inflammation, NF-κB also plays an antiapoptotic role in the intestinal epithelium; therefore, while NleC, NleE, and NleH may all suppress inflammatory responses, the loss of NleE or NleH may limit C. rodentium's ability to subvert other key functions of epithelial cells, such as apoptosis, which could significantly impair their competitive fitness. Taken together, this study suggests that NleC is a unique virulence factor that impacts several aspects of inflammatory signaling. Additional studies are needed to address the probable multivalent actions of NleC and how it and other T3SS effectors work together to modulate host inflammatory responses.
ACKNOWLEDGMENTS
We thank Tina Huang for technical assistance and Laura Sly for helpful suggestions.
This work was supported by grants to B.A.V. and B.B.F. from the Canadian Institutes of Health Research and the Crohn's and Colitis Foundation. H.P.S. was funded by a graduate studentship from the Child and Family Research Institute. S.R.S. was funded by a NSERC graduate studentship. M.A.C. is supported by a Canadian Association of Gastroenterology/CIHR/Ferring Pharmaceuticals fellowship. J.M.C. was funded by a CIHR master's award. B.B.F. is an HHMI International Research Scholar, and the University of British Columbia Peter Wall Distinguished Professor. B.A.V. is the Children with Intestinal and Liver Disorders Foundation Chair in Pediatric IBD Research and the Canada Research Chair in Pediatric Gastroenterology (Tier 2).
Footnotes
Published ahead of print on 11 July 2011.
REFERENCES
- 1. Artis D. 2008. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8:411–420 [DOI] [PubMed] [Google Scholar]
- 2. Babic A., Lindner A. B., Vulic M., Stewart E. J., Radman M. 2008. Direct visualization of horizontal gene transfer. Science 319:1533–1536 [DOI] [PubMed] [Google Scholar]
- 3. Baruch K., et al. 2011. Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a. Canadian Council on Animal Care 2011. CCAC guide to the care and use of experimental animals. Canadian Council on Animal Care, Ottawa, Ontario, Canada: http://www.ccac.ca/en_/standards/guidelines [Google Scholar]
- 4. Chandrakesan P., et al. 2010. Novel changes in NF-{kappa}B activity during progression and regression phases of hyperplasia: role of MEK, ERK, and p38. J. Biol. Chem. 285:33485–33498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi K. H., et al. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448 [DOI] [PubMed] [Google Scholar]
- 6. Chuang T. H., Ulevitch R. J. 2000. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur. Cytokine Netw. 11:372–378 [PubMed] [Google Scholar]
- 7. Cornelis G. R. 2002. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat. Rev. Mol. Cell Biol. 3:742–752 [DOI] [PubMed] [Google Scholar]
- 8. Croxen M. A., Finlay B. B. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8:26–38 [DOI] [PubMed] [Google Scholar]
- 9. Croxen M. A., Sisson G., Melano R., Hoffman P. S. 2006. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 188:2656–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dean P., Kenny B. 2009. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr. Opin. Microbiol. 12:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dean P., Maresca M., Schuller S., Phillips A. D., Kenny B. 2006. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc. Natl. Acad. Sci. U. S. A. 103:1876–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng W., et al. 2010. A comprehensive proteomic analysis of the type III secretome of Citrobacter rodentium. J. Biol. Chem. 285:6790–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edwards R. A., Keller L. H., Schifferli D. M. 1998. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207:149–157 [DOI] [PubMed] [Google Scholar]
- 15. Gao X., et al. 2009. Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog. 5:e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gauthier A., Puente J. L., Finlay B. B. 2003. Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect. Immun. 71:3310–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gibson D. L., et al. 2008. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10:618–631 [DOI] [PubMed] [Google Scholar]
- 18. Gibson D. L., et al. 2008. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10:388–403 [DOI] [PubMed] [Google Scholar]
- 19. Gibson D. L., et al. 2010. Interleukin-11 reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinal STAT3 signaling. Gastroenterology 139:1277–1288 [DOI] [PubMed] [Google Scholar]
- 20. Hausmann M., et al. 2002. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology 122:1987–2000 [DOI] [PubMed] [Google Scholar]
- 21. Hecht G. 2001. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G1–G7 [DOI] [PubMed] [Google Scholar]
- 22. Hilbi H., et al. 1998. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 273:32895–32900 [DOI] [PubMed] [Google Scholar]
- 23. Iguchi A., et al. 2009. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191:347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenny B., Jepson M. 2000. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell. Microbiol. 2:579–590 [DOI] [PubMed] [Google Scholar]
- 25. Khan M. A., et al. 2008. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect. Immun. 76:1410–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D. W., et al. 2005. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U. S. A. 102:14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levine M. M., et al. 1985. The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J. Infect. Dis. 152:550–559 [DOI] [PubMed] [Google Scholar]
- 28. Luperchio S. A., Schauer D. B. 2001. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3:333–340 [DOI] [PubMed] [Google Scholar]
- 29. Lupp C., et al. 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:204. [DOI] [PubMed] [Google Scholar]
- 30. Marches O., et al. 2005. Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infect. Immun. 73:8411–8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Medzhitov R., Preston-Hurlburt P., Janeway C. A., Jr 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397 [DOI] [PubMed] [Google Scholar]
- 32. Miao E. A., et al. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 107:3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mihaescu A., Santen S., Jeppsson B., Thorlacius H. 2010. p38 mitogen-activated protein kinase signalling regulates vascular inflammation and epithelial barrier dysfunction in an experimental model of radiation-induced colitis. Br. J. Surg. 97:226–234 [DOI] [PubMed] [Google Scholar]
- 34. Monack D. M., Mueller A., Falkow S. 2004. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2:747–765 [DOI] [PubMed] [Google Scholar]
- 35. Muhlen S., Ruchaud-Sparagano M. H., Kenny B. 2011. Proteasome-independent degradation of canonical NF{kappa}B complex components by the NleC protein of pathogenic Escherichia coli. J. Biol. Chem. 286:5100–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nadler C., et al. 2010. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6:e1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Newton H. J., et al. 2010. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6:e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pearson J. S., Riedmaier P., Marches O., Frankel G., Hartland E. L. 2011. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-kappaB for degradation. Mol. Microbiol. 80:219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reibman J., Hsu Y., Chen L. C., Bleck B., Gordon T. 2003. Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am. J. Respir. Cell Mol. Biol. 28:648–654 [DOI] [PubMed] [Google Scholar]
- 40. Rock F. L., Hardiman G., Timans J. C., Kastelein R. A., Bazan J. F. 1998. A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. U. S. A. 95:588–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodriguez-Escudero I., et al. 2005. Enteropathogenic Escherichia coli type III effectors alter cytoskeletal function and signalling in Saccharomyces cerevisiae. Microbiology 151:2933–2945 [DOI] [PubMed] [Google Scholar]
- 42. Ruchaud-Sparagano M. H., Maresca M., Kenny B. 2007. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell. Microbiol. 9:1909–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sansonetti P. J. 2004. War and peace at mucosal surfaces. Nat. Rev. Immunol. 4:953–964 [DOI] [PubMed] [Google Scholar]
- 44. Schauer D. B., Falkow S. 1993. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect. Immun. 61:2486–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shames S. R., et al. 2010. The pathogenic E. coli type III effector EspZ interacts with host CD98 and facilitates host cell prosurvival signalling. Cell. Microbiol. 12:1322–1339 [DOI] [PubMed] [Google Scholar]
- 46. Sharma R., et al. 2006. Balance of bacterial pro- and anti-inflammatory mediators dictates net effect of enteropathogenic Escherichia coli on intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 290:G685–G694 [DOI] [PubMed] [Google Scholar]
- 47. Spears K. J., Roe A. J., Gally D. L. 2006. A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol. Lett. 255:187–202 [DOI] [PubMed] [Google Scholar]
- 48. Vallance B. A., Chan C., Robertson M. L., Finlay B. B. 2002. Enteropathogenic and enterohemorrhagic Escherichia coli infections: emerging themes in pathogenesis and prevention. Can. J. Gastroenterol. 16:771–778 [DOI] [PubMed] [Google Scholar]
- 49. Vallance B. A., et al. 2002. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect. Immun. 70:6424–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vallance B. A., Deng W., Jacobson K., Finlay B. B. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 71:3443–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Walton K. L., Holt L., Sartor R. B. 2009. Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 296:G601–G611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wan F., et al. 2011. IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nat. Immunol. 12:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Warawa J., Finlay B. B., Kenny B. 1999. Type III secretion-dependent hemolytic activity of enteropathogenic Escherichia coli. Infect. Immun. 67:5538–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wickham M. E., et al. 2007. Citrobacter rodentium virulence in mice associates with bacterial load and the type III effector NleE. Microbes Infect. 9:400–407 [DOI] [PubMed] [Google Scholar]
- 55. Winter S. E., et al. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yen H., et al. 2010. NleC, a type III secretion protease, compromises NF-kappaB activation by targeting p65/RelA. PLoS Pathog. 6:e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang D., et al. 2004. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 303:1522–1526 [DOI] [PubMed] [Google Scholar]