

Abstract
The reelin (reln) and disabled 1(dab1) genes both ensure correct neuronal positioning during brain development. We have found that the intracellular Dab1 protein receives a tyrosine phosphorylation signal from extracellular Reln protein. Genetic analysis shows that reln function depends on dab1, and vice versa, as expected if both genes are in the same pathway. Dab1 is expressed at a higher level, yet phosphorylated at a lower level, in reln mutant embryo brains. In primary neuronal cultures, Dab1 tyrosine phosphorylation is stimulated by exogenous Reln. These results suggest that Reln regulates neuronal positioning by stimulating Dab1 tyrosine phosphorylation.
Keywords: Reeler, dab1, neuronal migration, extracellular matrix
Mammalian brain development requires the correct positioning of over 100 billion neurons, each of which makes thousands of connections. The majority of neurons migrate to their final destinations from proliferative zones in which they undergo their final mitoses. For instance, most pyramidal neurons of the cerebral cortex migrate radially from the proliferative zone adjacent to the ventricle before they differentiate (Hatten 1993; McConnell 1995; Lumsden and Gulisano 1997; Lambert de Rouvroit and Goffinet 1998; Pearlman et al. 1998). During differentiation the neurons extend axons that follow specific paths directed by growth cones on their tips. The mechanisms that control the timing and directions of migrations of neurons and of their growth cones are clearly critical for creating a functional nervous system.
Tyrosine kinases and phosphatases regulate many aspects of cell behavior, including proliferation, apoptosis, and differentiation. A growing body of evidence shows that tyrosine phosphorylation also regulates growth cone movements (Desai et al. 1997; Goodman and Tessier-Lavigne 1997; Holland et al. 1998). Similarly, tyrosine phosphorylation may also regulate migrations of neuronal precursors. Neural crest cells that express EphB kinases migrate preferentially into the rostral sclerotome, avoiding the caudal sclerotome in which their Ephrin ligand is expressed (Drescher 1997; Robinson et al. 1997). In Caenorhabditis elegans, the vab-1 gene, which encodes an Eph kinase, is required for migration of certain neuroblasts (George et al. 1998). Furthermore, undifferentiated cortical neurons utilize the protein tyrosine phosphatase (PTP) z/RPTPβ to migrate along radial glia (Maeda and Noda 1998), suggesting that the dephosphorylation of certain proteins may contribute to neuronal movement. However, proteins within neurons that are tyrosine phosphorylated or dephosphorylated in response to migration signals have not been identified.
Mutation of either the reelin (reln) (D’Arcangelo et al. 1995) or disabled (dab1) (Howell et al. 1997b; Sheldon et al. 1997; Ware et al. 1997) gene causes neuronal heterotopia or malpositioning. Neurons appear to be specified correctly and migrate out of their proliferative zones, but differentiate in the wrong locations. As a result, layered structures in the cerebral cortex, hippocampus, cerebellum and elsewhere are abnormal (Hoffarth et al. 1995; Ogawa et al. 1995; Gonzalez et al. 1997; Howell et al. 1997b; Sheppard and Pearlman 1997; Rice et al. 1998). A number of other mutations also cause heterotopias (Hatten 1993; Rakic and Caviness 1995; Pearlman et al. 1998). Mutation of LIS1 (Hattori et al. 1994), doublecortin (des Portes et al. 1998; Gleeson et al. 1998), cdk5 (Ohshima et al. 1996), or p35 (Chae et al. 1997) cause anomalous cortical lamina but the defects are distinct from those of dab1 and reln mutants (Lambert de Rouvroit and Goffinet 1998).
The similarity of the dab1 and reln phenotypes suggests that these two genes may be involved in the same process (Howell et al. 1997b; Sheldon et al. 1997; Gallagher et al. 1998; Rice et al. 1998). Reln is a secreted 400-kD glycoprotein that is restricted to specific parts of the brain (D’Arcangelo et al. 1995, 1997; Ogawa et al. 1995; Miyata et al. 1996; Nakajima et al. 1997). In the neocortex, Reln is expressed by Cajal-Retzius (CR) cells, adjacent to the region in which radially migrating neurons normally stop migrating and form the cortical plate (D’Arcangelo et al. 1995; Ogawa et al. 1995). Thus it seems likely that Reln signals to migrating cortical plate neurons, but its receptor and signaling mechanism are presently unknown. Dab1 is an 80-kD cytoplasmic protein that is expressed in many neurons, including cortical plate neurons (Howell et al. 1997a,b; Sheldon et al. 1997; Gallagher et al. 1998). The structure of Dab1 suggests it may be an intracellular signaling protein (Howell et al. 1997a). Thus it is possible that Dab1 is involved in relaying the Reln signal.
In this report we provide evidence that Reln and Dab1 act in the same signaling pathway, and that Reln stimulates Dab1 tyrosine phosphorylation in cortical neurons in vitro and in vivo. This suggests that tyrosine phosphorylation of Dab1 is a signal that a neuron has reached its final destination.
Results and Discussion
Genetic evidence for a common signaling pathway
The phenotypes of reln and dab1 mutants are extremely similar, suggesting that these genes may control the same process. dab1 could lie on a linear pathway upstream or downstream of Reln, or on a parallel pathway that is required, together with the Reln signal, for cessation of migration. If Dab1 and Reln are on parallel pathways, it is possible that each pathway may have partial function in the absence of the other. If the pathways are parallel, absence of both Dab1 and Reln may cause more severe neuronal malpositioning than the absence of either protein alone. To investigate whether there is a worsening of phenotype in dab1 reln double mutants, double heterozygous dab1−/+ reln−/+ animals were generated and interbred.
Double homozygous mutant progeny were obtained at expected frequency and resembled the single homozygotes. The cerebral cortex had lost lamination, the hippocampal scrolls were split, and the granule cells of the dentate gyrus were intermingled (data not shown). One characteristic of the neocortex in dab1 and reln homozygotes is invasion of the marginal zone by neurons that are normally found deep in the cortex (Fig. 1A, MZ). Increased neuron numbers were also detected in the marginal zone of compound homozygotes, but there was no significant quantitative difference between the marginal zone population in single and double homozygotes (Fig. 1C). The cerebella of the double mutant mice were also indistinguishable from the single mutants (Fig. 1B). Mutant cerebella were unfoliated and small. A molecular layer (M) was present but the granule cells (G) were reduced in number. Furthermore, the Purkinje cells (arrowheads), which normally express Dab1, were found clustered in central regions. In contrast, the Purkinje cells in a wild-type cerebellum form a monolayer above a dense granule cell layer. The compound mutants were also indistinguishable behaviorally from the single mutants. Thus it seems unlikely that either Reln or Dab1 has residual function in the absence of the other protein.
Figure 1.
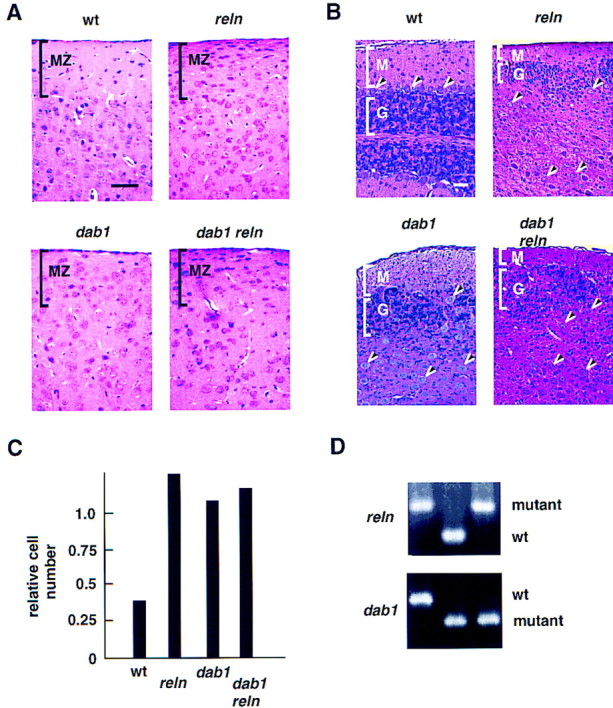
Histology of wild-type, single, and compound mutant brains. (A,B) Histology (hematoxylin and eosin stain) at postnatal day 23 (P23). The wild-type (wt) mouse was inbred strain 129/Sv. The single and compound homozygous mutant mice were siblings. (A) Coronal sections of cerebral cortex. The marginal zone (MZ) is cell poor in wild-type mice and densely packed in the mutants. Scale bar is 50 μm. (B) Sagittal sections of cerebella. (M) Molecular layer; (G) granule cell layer; (arrowheads), Purkinje cells. Scale bar is 50 μm. Note that the wild-type cerebellum is foliated, and the lower half of the section shown is a mirror image of the upper half. The mutant cerebella are not foliated. (C) Nuclei in the marginal zone (0.09 mm2) were counted and expressed as a ratio to the number of nuclei in layer 6 (0.13 mm2). Counts were within 15% on duplicate section. (D) PCR genotypes of dab1 reln, dab1, and reln samples.
The lack of additional defects in double mutants supports a model with Reln and Dab1 acting on the same signaling pathway. As the Reln protein and RNA are made appropriately in brains of dab1 mutants (Goldowitz et al. 1997; Gonzalez et al. 1997; Howell et al. 1997b; Sheldon et al. 1997; Yoneshima et al. 1997), it seems unlikely that Dab1 is required for Reln expression. Instead, Dab1 is likely to act within the migrating neurons responding to the Reln signal. For example, it may be part of a signal transduction pathway from a Reln receptor. Alternatively, Reln may stimulate the expression of another autocrine or paracrine factor that may signal via Dab1. Dab1 may also be involved in modifying the Reln protein so it can bind a receptor, or in expressing the Reln receptor or another component of the pathway. To distinguish some of these possibilities, biochemical studies were performed.
In vivo phosphorylation of Dab1 depends on reln gene expression
Dab1 is tyrosine phosphorylated in developing but not adult brains (Howell et al. 1997a), suggesting that Dab1 function may be regulated by tyrosine phosphorylation during development. Therefore, we tested whether Reln affects Dab1 protein expression or tyrosine phosphorylation. Embryonic and postnatal brains were collected from timed matings of reln−/+ mice and genotyped by PCR. Dab1 protein quantity and phosphotyrosine levels were determined by Western blot analysis of Dab1 immunoprecipitates prepared from equal quantities of total brain protein. Reln-dependent changes were observed in both Dab1 expression and tyrosine phosphorylation (Fig. 2).
Figure 2.

Comparison of Dab1 protein and phosphotyrosine levels between wild-type and reln mutant brains. Brains were collected either at E16 or at P21 from wild-type or mutant animals, weighed, and homogenized in RIPA buffer. Lysates were normalized for total protein concentration, then immunoprecipitated with either anti-Dab1 (+), or preimmune (−) antibodies. Immunoprecipitates were analyzed by SDS-PAGE, followed by Western blotting with either anti-phosphotyrosine (top) or anti-Dab1 antibody (middle). Relative levels of Dab1 protein were estimated by dilution of reln mutant samples (right). High Dab1 tyrosine phosphorylation was observed only in the embryonic samples. Equivalent protein concentration in cell lysates was confirmed by Coomassie stain (not shown) and blotting with anti-enolase antibody (bottom). Enolase is more highly expressed in P21 brain. (wt) Wild-type; (rl) reln; (arrow) position of Dab1.
Increased levels of Dab1 protein were observed in reln brain relative to wild-type or heterozygotes, both at embryonic day 16 (E16) and postnatal day 21 (P21) (Fig. 2). Analysis of different quantities of extract suggests that the increase is approximately threefold (right panel). The increase in Dab1 protein in reln brain suggests that Reln is regulating Dab1 expression, and has been reported recently by other investigators (Rice et al. 1998). However, it seems unlikely that changes in Dab1 protein level regulate neuronal position directly because the phenotype of reln mutants, which have excess Dab1, and dab1 mutants, which lack Dab1, are indistinguishable. Rather, it is possible that the reduced Dab1 protein level in embryos that express Reln is a down-regulation phenomenon.
Despite the increase in Dab1 protein content, the amount of phosphotyrosine in Dab1 was decreased slightly in E16 reln brain (Fig. 2). Based on three independent experiments, we estimate that the stoichiometry of Dab1 tyrosine phosphorylation was 3–6 times lower in reln than wild-type embryos. The decrease in Dab1 tyrosine phosphorylation in the mutant suggests that Reln stimulates Dab1 tyrosine phosphorylation directly or indirectly, and this phosphorylation may be important for the Reln- and Dab1-dependent positioning of neurons. However, the detection of some phosphotyrosine in Dab1 from reln mutant brain indicates that Reln-independent tyrosine phosphorylation of Dab1 also occurs. In contrast to embryonic brains, P21 wild-type and reln mutant brains had undetectable Dab1 phosphotyrosine (Fig. 2). This indicates that both the Reln-independent and -dependent mechanisms that regulate the phosphotyrosine content of Dab1 p80 act predominantly during embryogenesis, and is consistent with diminished signaling through Reln and Dab1 after birth. After birth the Reln expression declines in the prenatal sources such as the CR cells and increases in a number of interneurons throughout the neocortex and hippocampus (Alcántara et al. 1998; Pesold et al. 1998). The prenatal and postnatal roles for Reln are likely distinct and may operate through different signaling cascades.
Dab1 phosphorylation is stimulated directly by Reln in vitro
To test whether Reln would activate tyrosine phosphorylation of Dab1 directly or indirectly, we added Reln protein to embryonic brain cells. Reln was prepared by transfecting 293T cells with an expression vector (D’Arcangelo et al. 1997) and collecting conditioned media (Materials and Methods). Media from cells transfected with the expression vector but not with control DNA contained immunoreactive Reln at an estimated 350–700 pm concentration (Fig. 3A). Cell suspensions were prepared from E16 reln mutant forebrains and treated with fresh medium or conditioned media for 10 min. Protein phosphorylation was detected with antibodies to phosphotyrosine (Fig. 3B). Reln induced the tyrosine phosphorylation of an 80-kD protein specifically. To test whether this protein was Dab1, we immunoprecipitated Dab1 with affinity-purified anti-Dab1 (+) or control (−) antibodies, and analyzed the immunoprecipitates by Western blotting (Fig. 3C). Dab1 was detected in the immunoprecipitates and was specifically tyrosine phosphorylated in Reln-treated cultures. Reln-induced Dab1 tyrosine phosphorylation was observed in three independent experiments, using treatment times of 5, 10, 30, 60, or 240 min, and estimated Reln concentrations of 100–250 pm (data not shown).
Figure 3.

Tyrosine phosphorylation of Dab1 p80 is induced by the addition of Reln to dissociated embryonic brain cells. (A) Detection of Reln protein (triangle) by Western blot in conditioned media used to treat brain cells. (C) pCDNA3 vector; (R) Reln-expressing pCrl plasmid. (B) Cells isolated from E16 reln forebrains were treated for 0 or 10 min with fresh medium (−) or control (C) or Reln (R) conditioned media. Total cell lysates were analyzed using antiphosphotyrosine antibody. A Reln-induced 80-kD tyrosine phosphorylated protein is indicated by the arrow. (C) Cell lysates were immunoprecipitated with either anti-Dab1 (+) or preimmune (−) antibodies, and analyzed by Western blotting with anti-phosphotyrosine (top) or anti-Dab1 (bottom) antibodies. The Dab1 p80 doublet (arrow) may represent differential serine-threonine phosphorylation.
To characterize the response further, and facilitate the assay, we cultured embryonic forebrain cells before exposing them to Reln (Fig. 4). Culturing for 3 days did not interfere with detection of Reln-induced tyrosine phosphorylation of Dab1 (Fig. 4A). Analysis of different quantities of sample showed that 30 min exposure to 350–700 pm Reln induced an estimated fourfold increase in phosphotyrosine content of Dab1 (Fig. 4A, right panel). Sensitivity of the assay was greatly increased by using reln mutant embryo brain cells. When samples from wild-type or reln heterozygous animals were analyzed, basal tyrosine phosphorylation of Dab1 was higher, and the Reln-induced increase was lower (Fig. 4A; data not shown). This may reflect the decreased quantity of Dab1 that is available for phosphorylation in wild-type embryos (Fig. 2), or Reln-dependent down-regulation of the signaling cascade. In three experiments with cultured embryonic brain cells, tyrosine phosphorylation was increased strongly at 5, 10, 30, and 60 min, and declined by 4 hr (Fig. 4B; data not shown). The response requires extracellular divalent cations (Fig. 4C, EDTA), suggesting that the receptor or ligand requires divalent cations for interaction, or that uptake of an extracellular divalent cation is required to activate the phosphorylation event. Addition of the generic phosphotyrosine phosphatase inhibitor, sodium orthovanadate, does not increase the basal phosphorylation of Dab1 significantly (Fig. 4C, VO4), suggesting that Reln is more likely to induce Dab1 tyrosine phosphorylation by activating a tyrosine kinase than inhibiting a phosphotyrosine phosphatase. A high dose of orthovanadate actually inhibits Reln-induced phosphorylation, possibly indicating a phosphatase-activated tyrosine kinase is involved. Additional studies will be needed to identify the tyrosine kinases and phosphatases regulated by Reln.
Figure 4.




Cultured neurons respond to Reln: effect of genotype, time of exposure, and inhibitors. (A) E16 wild-type (wt) and reln (rl) mutant forebrain cells were dissociated and cultured for 3 days before treatment with fresh (−), control (C) or Reln (R) conditioned media for 30 min. Lysates were incubated with anti-Dab1 (+) or preimmune (−) antibodies, and immunoprecipitated proteins were Western blotted with anti-phosphotyrosine (top) and anti-Dab1 (bottom) antibodies. Anti-phosphotyrosine signal was quantitated by dilution of Reln-stimulated samples compared to undiluted control sample (right). (B) Reln mutant cells were treated for 0, 10, 30, or 240 min with fresh, control, or Reln media. (C) Reln mutant cells were treated for 30 min with control or Reln media in the presence or absence of EDTA (5 mm) or Na3VO4 (0.2 or 2 mm). (D) Cultured cells were fixed and stained with neuronal specific anti β-tubulin antibody Tuj1 (green, left), anti-Dab1 (red, right) and Dapi (blue) to determine the percentage of cells that were neurons and express Dab1.
Immunostaining of the cultured brain cells with antibody Tuj1, which recognizes a neuron-specific type III β tubulin, showed that 98% (n = 250) of the cells are neurons (Fig. 4D, left). Of these cells, 60% immunostain for Dab1 (Fig. 4D, right). Although it is possible that Reln induces the release of a secreted protein that induces Dab1 tyrosine phosphorylation by an autocrine or paracrine mechanism, the speed of the response and the lack of non-neuronal cells in the cultures indicates that tyrosine phosphorylation of Dab1 is probably induced by Reln acting on a specific receptor on the Dab1-expressing neurons.
Whereas the presence of Reln correlates with reduced Dab1 protein levels during embryo development (Fig. 2), Reln treatment of disaggregated embryonic neurons did not induce rapid loss of Dab1 protein (Fig. 4B). This contrasts with some tyrosine kinase substrates, which are degraded in the presence of the cognate tyrosine kinase (Dai et al. 1998). Indeed, Dab1 is not degraded when co-expressed with the tyrosine kinase Src in 293 cells, despite extensive tyrosine phosphorylation, and Dab1 is not degraded in an in vitro degradation system that depends on the tyrosine kinase Abl (Dai et al. 1998; data not shown). Thus, the reduction of Dab1 protein levels observed in E16 embryos might be a slower adaptive process than the rapid induction of Dab1 tyrosine phosphorylation.
Implications for neuronal positioning
These results show that Dab1 and Reln act on the same pathway to control neuronal positioning in the developing brain. The kinetics of tyrosine phosphorylation suggest that Reln interacts directly with a receptor on neurons that express Dab1, and the effects of a generic phosphotyrosine inhibitor suggest that the Reln receptor is linked to a tyrosine kinase. Reln may also stimulate other signaling pathways that may act in parallel to Dab1 tyrosine phosphorylation, but the genetics suggest that Dab1 tyrosine phosphorylation is one step in an essential, nonredundant pathway that regulates neuron position. Because Reln is also involved in the ingrowth of entorhinal afferents into the hippocampus (Del R’io et al. 1997), and Dab1 is in growth cones (data not shown), a Reln–Dab1 pathway may also regulate growth cone migration.
Increased tyrosine phosphorylation of Dab1 is expected to potentiate interactions with proteins that contain SH2 domains or other phosphotyrosine-dependent domains. These interactions may arrest neuronal migration by modulating the actin cytoskeleton, membrane flow, or adhesion complexes. Elucidation of the specific tyrosine residues on Dab1 p80 that are phosphorylated in response to Reln signaling should aid the identification of critical binding partners that may include proteins known to regulate adhesion. In addition, the biochemical assay for Reln signaling will facilitate the identification of a Reln receptor and other proteins in the signal-transduction pathway.
Materials and methods
Mice
Mice heterozygous for mutations in the dab1 gene, C57BL/6J-129/Sv hybrids, were bred with reln heterozygous mice (B6C3Fe-a/a-Relnrl; Jackson Labs). Progeny that were doubly heterozygous at the dab1 and reln loci were identified by PCR-based genotyping (D’Arcangelo et al. 1996; Howell et al. 1997b), and interbred to obtain single and compound mutant mice. All genotypes were obtained in ratios predicted by Mendelian inheritance including the reln dab1 double homozygous animals, which were identified 6 times out of 108 births (predicted: one out of 16).
Immunoprecipitation and Western blot analysis
Brains from E16 or P21 mice were homogenized in RIPA buffer (0.15 m NaCl, 1% Triton X100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 mm sodium phosphate (pH 7.0), 2 mm EDTA, 14 mm 2-mercaptoethanol, 50 mm NaF, 2 mm Na3VO4, 1 mm phenylarsine oxide, 20 μg aprotinin/ml, 10 μg pepstatin/ml, and 10 μg leupeptin/μl) at 70 mg tissue weight per ml. Lysates were clarified by centrifugation at 20,000g for 30 min after a 10-min incubation on ice. Samples were normalized for total protein concentrations, and incubated with anti-Dab1 (B3) (Howell et al. 1997a) or preimmune antibodies that were cross-linked to protein A sepharose with dimethyl pimelimidate at 4°C for 2 hr, followed by three washes with RIPA buffer. Proteins were eluted with twice-concentrated gel-loading buffer (4% SDS, 40% glycerol, 0.2 m Tris-HCl at pH 6.8, 5.6 m 2-mercaptoethanol, 5 mm EDTA, and 0.02% bromophenol blue) at 100°C for 10 min, followed by resolution by 7.5% SDS–polyacrylamide gel electrophoresis. Anti-Dab and anti-phosphotyrosine (4G10 monoclonal) Western blot analysis has been described previously (Howell et al. 1997a).
Reln was detected by Western blot with a mixture of three anti-Reln monoclonal antibodies (G10, 74, and 142; gifts from A. Goffinet, Lambert de Rouvroit and Goffinet 1998).
Dissociation of neurons and Reln treatment
Reln protein production was initiated 14 days after mating reln heterozygotes. 293T cells were transfected with the Reln-encoding plasmid, pCrl, or the control vector, pCDNA3 (D’Arcangelo et al. 1997). After 24 hr, 2 × 106 cells were transfered to 1 ml of Opti-MEM reduced serum media (Gibco-BRL) and incubated in suspension for 18 hr. The conditioned media were collected into siliconized tubes, and stored at 4°C. By labeling cultures with [35S] methionine and determining the percent of labeled, secreted protein that is Reln, we estimate that the concentration of Reln produced is 350–700 pm.
Embryos (E16) were collected from a pregnant dam and treated identically, but separately, pending genotyping. Forebrains were isolated and the meninges were discarded. The brains were then dissociated in 1 ml of Opti-MEM with a polished glass Pasteur pipet. Aliquots (200 μl) of dissociated brain cells, were rotated with 100 μl of fresh, Reln- or control-conditioned media at 37°C for times between 5 and 240 min. Reactions were stopped by centrifuging the cells at 1000g and washing with PBS. Cell pellets were snap frozen and stored at −70°C. After genotyping, samples derived from reln homozygous embryo brains were thawed and analyzed by immunoprecipitation and Western blotting as described above. In experiments with cultured neurons, cells were plated onto ECL cell attachment matrix (entactin, collagen IV, laminin; UBI), grown according to Maeda and Noda (1998) for 3 days, removed by aspiration, and dispersed by trituration. Tuj1 antibody was from BabCo.
Acknowledgments
We thank Gabriella D’Arcangelo and Tom Curran for the reelin cDNA, André Goffinet for the anti-Reln monoclonals, and A.M. Pendergast for cDNAs. We are grateful to Frank Gertler, Phil Soriano, Steve Tapscott, and Richard Hawkes for interesting discussion and advice on the manuscript, and to Tim Knight for assistance with the figures. This work was supported by National Cancer Institute grant CA 41072.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jcooper@fhcrc.org; FAX (206) 667-6522.
References
- Alcántara S, Ruiz M, D’Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelinmRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae T, Kwon Y, Bronson R, Dikkes P, Li E, Tsai L. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Curran T. Detection of the reelin breakpoint in reelermice. Brain Res Mol Brain Res. 1996;39:234–236. doi: 10.1016/0169-328x(96)00046-0. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Quackenbush RC, Courtney KD, Grove M, Cortez D, Reuther GW, Pendergast AM. Oncogenic Abl and Src tyrosine kinases elicit the ubiquitin-dependent degradation of target proteins through a Ras-independent pathway. Genes & Dev. 1998;12:1415–1424. doi: 10.1101/gad.12.10.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del R’io JA, Heimrich B, Borrell V, Forster E, Drakew A, Alcántara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrié A, Gelot A, Dupuis E, Motte J, Berwald NY, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Desai CJ, Sun Q, Zinn K. Tyrosine phosphorylation and axon guidance: Of mice and flies. Curr Opin Neurobiol. 1997;7:70–74. doi: 10.1016/s0959-4388(97)80122-5. [DOI] [PubMed] [Google Scholar]
- Drescher U. The Eph family in patterning neural development. Curr Biol. 1997;7:R799–R807. doi: 10.1016/s0960-9822(06)00409-x. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Howell BW, Soriano P, Cooper JA, Hawkes R. Cerebellar abnormalities in the disabled (mdab1-1) mouse. J Comp Neurol. 1998;402:238–251. [PubMed] [Google Scholar]
- George SE, Simokat K, Hardin J, Chisholm AD. The VAB-1 Eph receptor tyrosine kinase functions in neural and epithelial morphogenesis in C. elegans. Cell. 1998;92:633–643. doi: 10.1016/s0092-8674(00)81131-9. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Cushing RC, Laywell E, D’Arcangelo G, Sheldon M, Sweet HO, Davisson M, Steindler D, Curran T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of reelin. J Neurosci. 1997;17:8767–8777. doi: 10.1523/JNEUROSCI.17-22-08767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: A novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CS, Tessier-Lavigne M. Molecular mechanisms of axon guidance and target recognition. In: In: Cowan WM, Jessell TM, Zipursky SL, editors. Molecular and cellular approaches to neural development. New York, NY: Oxford University Press; 1997. pp. 108–178. [Google Scholar]
- Hatten ME. The role of migration in central nervous system neuronal development. Curr Opin Neurobiol. 1993;3:38–44. doi: 10.1016/0959-4388(93)90033-u. [DOI] [PubMed] [Google Scholar]
- Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- Hoffarth RM, Johnston JG, Krushel LA, Van der Kooy D. The mouse mutation reelercauses increased adhesion within a subpopulation of early postmitotic cortical neurons. J Neurosci. 1995;15:4838–4850. doi: 10.1523/JNEUROSCI.15-07-04838.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SJ, Peles E, Pawson T, Schlessinger J. Cell-contact-dependent signalling in axon growth and guidance: Eph receptor tyrosine kinases and receptor protein tyrosine phosphatase beta. Curr Opin Neurobiol. 1998;8:117–127. doi: 10.1016/s0959-4388(98)80015-9. [DOI] [PubMed] [Google Scholar]
- Howell BW, Gertler FB, Cooper JA. Mouse disabled (mDab1): A Src binding protein implicated in neuronal development. EMBO J. 1997a;16:1165–1175. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997b;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM. The reeler mouse as a model of brain development. Adv Anat Embryol Cell Biol. 1998;150:1–108. [PubMed] [Google Scholar]
- Lumsden A, Gulisano M. Neocortical neurons: Where do they come from? Science. 1997;278:402–403. doi: 10.1126/science.278.5337.402. [DOI] [PubMed] [Google Scholar]
- Maeda N, Noda M. Involvement of receptor-like protein tyrosine phosphatase ζ/RPTPβ and its ligand pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) in neuronal migration. J Cell Biol. 1998;142:203–216. doi: 10.1083/jcb.142.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell SK. Constructing the cerebral cortex: Neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- Miyata T, Nakajima K, Aruga H, Takahashi S, Ikenaka K, Mikoshiba K, Ogawa M. Distribution of a reelergene-related antigen in the developing cerebellum: An immunohistochemical study with an allogeneic antibody CR-50 on normal and reeler mice. J Comp Neurol. 1996;372:215–228. doi: 10.1002/(SICI)1096-9861(19960819)372:2<215::AID-CNE5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman AL, Faust PL, Hatten ME, Brunstrom JE. New directions for neuronal migration. Curr Opin Neurobiol. 1998;8:45–54. doi: 10.1016/s0959-4388(98)80007-x. [DOI] [PubMed] [Google Scholar]
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A, Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc Natl Acad Sci. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Caviness VSJ. Cortical development: View from neurological mutants two decades later. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D’Arcangelo G, Nakajima K, Goldowitz D, Curran T. Disabled-1 acts downstream of Reelinin a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- Robinson V, Smith A, Flenniken AM, Wilkinson DG. Roles of Eph receptors and ephrins in neural crest pathfinding. Cell Tiss Res. 1997;290:265–274. doi: 10.1007/s004410050931. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Sheppard AM, Pearlman AL. Abnormal reorganization of preplate neurons and their associated extracellular matrix: An early manifestation of altered neocortical development in the reeler mutant mouse. J Comp Neurol. 1997;378:173–179. doi: 10.1002/(sici)1096-9861(19970210)378:2<173::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert C, Russo CJ, Chua SC, Jr, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- Yoneshima H, Nagata E, Matsumoto M, Yamada M, Nakajima K, Miyata T, Ogawa M, Mikoshiba K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR–50 antigen/reelin. Neurosci Res. 1997;29:217–223. doi: 10.1016/s0168-0102(97)00088-6. [DOI] [PubMed] [Google Scholar]