

Abstract
In yeast, four factors (CF I, CF II, PF I, and PAP) are required for accurate pre-mRNA cleavage and polyadenylation in vitro. CF I can be separated further into CF IA and CF IB. Here we show that CF IB is the 73-kD Hrp1 protein. Recombinant Hrp1p made in Escherichia coli provides full CF IB function in both cleavage and poly(A) addition assays. Consistent with the presence of two RRM-type motifs, Hrp1p can be UV cross-linked to RNA, and this specific interaction requires the (UA)6 polyadenylation efficiency element. Furthermore, the CF II factor enhances the binding of Hrp1p to the RNA precursor. A temperature-sensitive mutant in HRP1 yields mRNAs with shorter poly(A) tails when grown at the nonpermissive temperature. Genetic analyses indicate that Hrp1p interacts with Rna15p and Rna14p, two components of CF 1A. The HRP1 gene was originally isolated as a suppressor of a temperature-sensitive npl3 allele, a gene encoding a protein involved in mRNA export. Like Npl3p, Hrp1p shuttles between the nucleus and cytoplasm, providing a potential link between 3′-end processing and mRNA export from the nucleus.
Keywords: HRP1, hnRNP, mRNA polyadenylation, RNA processing, mRNA export
While in the nucleus, most mRNA precursors undergo a series of processing events prior to moving into the cytoplasm via the nuclear pore. These maturation events include cotranscriptional capping at the 5′ end (Lewis et al. 1995), splicing (Adams et al. 1996), and 3′-end cleavage followed by polyadenylation (Manley and Takagaki 1996; Wahle and Keller 1996). Proper execution of these steps is required to produce an export-competent mRNA species.
The poly(A) tail has been proposed to play a role in nearly every aspect of mRNA metabolism. These include maintaining the proper stability of transcripts (Beelman and Parker 1995), promoting export from the nucleus to the cytoplasm (Huang and Carmichael 1996), and recruiting mRNAs to the translation machinery (Sachs et al. 1997). Studies have proposed that it is the actual process of polyadenylation and not just the presence of the poly(A) tail itself, which is essential for mRNA export. When 3′ ends were artificially formed by cis-acting ribozyme cleavage in vivo, the resulting mRNA was not exported efficiently (Eckner et al. 1991; Liu and Smith 1994; Huang and Carmichael 1996).
A detailed picture of the biochemical requirements for mammalian mRNA 3′-end formation has started to emerge (Manley and Takagaki 1996; Ruegsegger et al. 1996; Wahle and Keller 1996). Six protein factors are required for this processing step. One set of factors is required for the proper recognition and cleavage of the pre-mRNA and consists of the cleavage and polyadenylation specificity factor (CPSF), cleavage stimulation factor (CstF), cleavage factors I and II (CF Im and CF IIm), and for most pre-mRNAs, poly(A) polymerase (PAP). The second set of biochemically defined factors participates in poly(A) addition and includes CPSF, PAP, and the nuclear poly(A)-binding protein II (PAB II). The polypeptide constituents of many of these factors are known, but their mechanisms of action have not yet been defined.
The development of a cell-free system for the accurate and efficient polyadenylation of yeast mRNAs indicated mechanistic similarities between mRNA 3′-end formation in Sacchacomyces cerevisiae and metazoans (Butler and Platt 1988). In yeast four separable protein factors in addition to PAP are required for mRNA 3′-end formation (Chen and Moore 1992; Kessler et al. 1996). Cleavage/polyadenylation factor IA (CF IA), CF IB, and CF II carry out cleavage, whereas specific polyadenylation occurs upon combination of yeast PAP, CF IA, CF IB, and polyadenylation factor I (PF I) (Chen and Moore 1992; Kessler et al. 1996). Identification of the genes encoding many of the protein subunits of these factors has led to further parallels between yeast and higher eukaryotes. For example, the Rna14p and Rna15p subunits of CF IA share homology with CstF subunits (Takagaki and Manley 1994). The Cft1p, Cft2p, and Brr5/Ysh1p subunits of CF II have sequence similarity to CPSF subunits (Chanfreau et al. 1996; Jenny et al. 1996; Stumpf and Domdey 1996; Zhao et al. 1997). The Yth1p component of PF I has structural similarity to the 30-kD subunit of CPSF (Barabino et al. 1997). Whereas the yeast proteins Pcf11p of CF IA and Fip1p of PF I are essential for 3′-end formation, they do not have known human counterparts (Preker et al. 1995; Amrani et al. 1997a).
Although significant progress has been made in identifying the proteins required for polyadenylation in both yeast and mammals, the set is not yet complete and the mechanisms by which these factors act remain poorly understood. Genetic studies with yeast afford a unique perspective as they can also reveal information regarding the biological impact of polyadenylation on other cellular functions. We recently identified the yeast HRP1 gene because of its genetic interactions with an mRNA-binding protein, Npl3p, which is required for proper mRNA nuclear export (Bossie et al. 1992; Russel and Tollervey 1992; Flach et al. 1994; Wilson et al. 1994; Henry et al. 1996; Lee et al. 1996). HRP1 encodes a protein with canonical RNA recognition motifs (RRMs) and is predicted to bind RNA (Henry et al. 1996). We now demonstrate that Hrp1p, as the CF IB factor, is necessary in vivo and in vitro for proper 3′-end formation. In the course of performing its essential function, Hrp1p recognizes the (UA)6 RNA element required for both the cleavage and poly(A) addition steps. Hrp1p also efficiently exits the nucleus, suggesting that it may have an additional role in the export of mRNAs to the cytoplasm.
Results
Hrp1p is the CF IB cleavage/polyadenylation factor
We recently purified and separated CF I into two components (CF IA and CF IB) and identified Rna14p, Rna15p, and Pcf11p as three of the four subunits of CF IA (Kessler et al. 1996; M. Kessler, J. Zhao, and C. Moore, unpubl.). CF IB is a single 73-kD polypeptide which, in combination with CF IA, reconstitutes CF I’s function in the in vitro cleavage and poly(A) addition reactions.
The identity of CF IB was determined by sequencing two tryptic peptides resulting from digestion of the 73-kD protein. The peptide sequences obtained were IFVGGIGPDVRPK and YGTVTDLK. Using these two amino acid sequences to search the current data base, Hrp1p (Henry et al. 1996) was identified as a perfect match. The first peptide corresponded to Hrp1p residues 245–257, whereas the second corresponded to residues 182–189.
Recombinant Hrp1 protein participates in 3′-end processing in vitro
To confirm that Hrp1p was indeed CF IB, we prepared recombinant protein from Escherichia coli as a glutathione S-transferase (GST) fusion and tested its ability to reconstitute CF I activity in the presence of CF IA. We first examined the function of GST–Hrp1p in the pre-mRNA cleavage reaction. In this assay, a mixture of CF II and nonseparated CF I, or CF IA and CF IB in combination, allows accurate cleavage of the test substrate GAL7 pre-mRNA (Fig. 1A, lanes 2,7). Efficient cleavage occurs when the GST–Hrp1p protein replaces CF IB (Fig. 1A, lane 8). Some stimulation of cleavage activity was also observed when the recombinant protein was mixed with nonseparated CF I (Fig. 1A, cf. lanes 2 and 3). Control reactions containing either CF IA, CF IB, or GST–Hrp1p alone or lacking CF I were unable to support cleavage (Fig. 1A, lanes 4–6,10). Likewise, reactions containing CF IB and GST–Hrp1p, or CF IA and GST, were also inactive (Fig. 1A, lanes 9,11). These results demonstrate that Hrp1p is required for in vitro cleavage activity and can fully replace CF IB.
Figure 1.


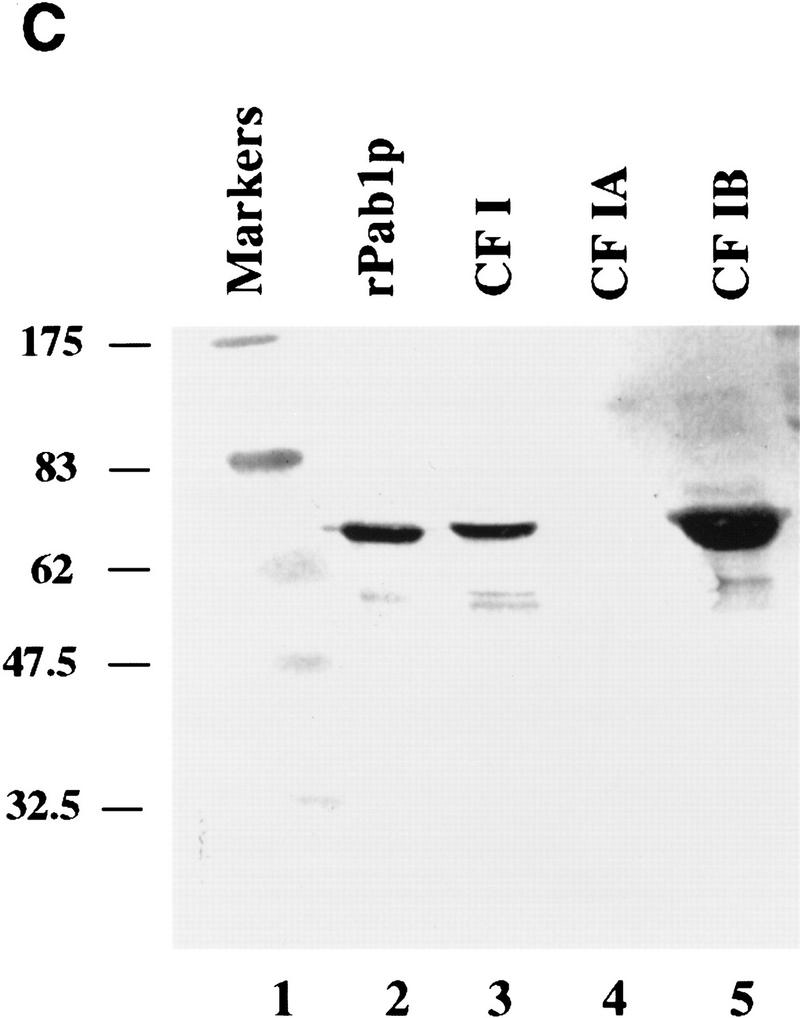
Reconstitution of CF I 3′-end processing activity by complementation with recombinant GST–Hrp1p. (A) Cleavage of GAL7 precursor RNA in vitro. Reactions contained the substrate RNA (lane 1) and 1 μl of partially purified CF II [poly(A)–Sepharose step, lanes 2–11]. Each was then supplemented with either CF I (Q–Sepharose step, lane 2), CF IA (gel filtration step, lane 4), CF IB (gel filtration step, lane 5), GST–Hrp1p (80 ng, lane 6), water (lane 10), combinations of these proteins (lanes 3,7–9), or a mixture of CF IA and GST–Hrp1p (80 ng, lane 11). The migration positions of the full-length RNA, upstream cleavage fragment (solid rectangle), and downstream cleavage fragment (open rectangle) are indicated at right. (B) Polyadenylation of the GAL7 precleaved precursor in vitro. All reactions contained substrate RNA (lane 1), PAP (30 ng), and 3 μl of a crude PF I fraction (Q–Sepharose). Reactions were then supplemented with either CF I (lane 2), CF IA (lane 4), CF IB (lane 5), GST–Hrp1p (lane 6), Pab1p (lane 11), water (lane 17), or a combination of these proteins (lanes 3,7–10,12–16). The migration positions of the precleaved (solid rectangle) and polyadenylated product are indicated at right. (C) Anti-Pab1 antibodies detect Pab1p in the crude CF IB fraction. The 3′-end processing fractions CF I (3 μg; lane 3), CF IA (0.2 μg; lane 4), and CF IB (0.2 μg; lane 5) were analyzed by immunoblotting. Recombinant Pab1p was assayed in parallel (lane 2). The sizes of protein markers (lane 1) are indicated in kilodaltons.
GST–Hrp1p also supports polyadenylation of a GAL7 precleaved substrate in the presence of CF IA, PF I, and PAP. Precleaved RNA contains only sequences upstream of the poly(A) site. A reaction containing CF I, PF I, and PAP results in efficient poly(A) addition to this substrate (Fig. 1B, lane 2). The polyadenylated RNA is seen as a discrete band of 50–70 residues in length. A reaction with a combination of CF IA and CF IB similarly results in poly(A) addition but with lower efficiency than with nonseparated CF I (Fig. 1B, lane 7). Addition of GST–Hrp1p to CF IA yields a polyadenylated product that is more diffuse and slower migrating than that observed with intact CF I or with the combination of CF IA and CF IB (Fig. 1B, lane 8). These tails range in size from 70 to 150 residues. The addition of GST–Hrp1p to CF IA and CF IB resulted in a significant increase of polyadenylated RNA that remains uniform in size (Fig. 1B, cf. lanes 9 and 7).
The CF IB used in these reactions was obtained from a gel filtration column, which is the step prior to the final purification of CF IB to a single 73-kD polypeptide (Kessler et al. 1996). We know that this CF IB fraction contains other proteins besides Hrp1p. One of these is the Pab1 protein (Fig. 1C, lane 5), which recently was shown to participate in poly(A) tail length control (Amrani et al. 1997b; Minvielle-Sebastia et al. 1997). We therefore tested whether recombinant Pab1p added to GST–Hrp1p and CF IA could regulate poly(A) tail length. When the two recombinant polypeptides and CF IA are added, accurate and efficient poly(A) addition is obtained (Fig. 1B, lane 10). No poly(A) addition was detected in reactions that lacked CF I or contained CF IA, CF IB alone, or GST combined with CF IA and Pab1p (Fig. 1B, lanes 4,5,15,17). Some polyadenylation occurs when GST–Hrp1p, or GST–Hrp1p and Pab1p, are combined with PF I and PAP (Fig. 1B, lanes 6,16), but the tails are very short and diffuse. A reaction containing Pab1p, PAP, and PF I; or a combination of Pab1p and CF IA results in no poly(A) addition (Fig. 1B, lanes 11,12). Addition of other RRM-containing RNA-binding proteins in place of Hrp1p, such as the human hnRNP A1 or the yeast Npl3p, also result in no poly(A) addition (Fig. 1B, lanes 13,14). Taken together, these results show that Hrp1p participates in both cleavage and polyadenylation reactions, whereas Pab1p is only required to maintain the accuracy of the final poly(A) tail length.
We had recently found that mutations in RNA15 and HRP1 can suppress the thermosensitive phenotypes of mutant NPL3 strains (Henry et al. 1996). Given this genetic interaction, it seemed plausible that the Npl3 protein might also play a role in 3′-end processing. However, Npl3p does not cofractionate with any of the four Q–Sepharose fractions (CF I, CF II, PF I, or PAP) required for mRNA polyadenylation (data not shown). Instead, it elutes in a nonoverlapping manner between the CF I and PF I components. Addition of recombinant Npl3p to processing reactions does not enhance either cleavage or poly(A) addition activities, indicating that it has no stimulatory role in vitro.
Isolation and characterization of HRP1 mutants
To examine the function of Hrp1 protein in vivo, we used random PCR-directed mutagenesis to generate a collection of recessive temperature-sensitive mutations in HRP1 (designated hrp1-1 through hrp1-8). These mutants, which are deleted for the chromosomal copy of HRP1 and carry a plasmid-borne temperature-sensitive allele, were isolated because of their failure to grow at 36°C. The corresponding mutations were initially defined by functional mapping to the central portion of HRP1 (Fig. 2A). The appropriate region of each mutant allele was then sequenced and compared with the wild-type HRP1 sequence (Fig. 2B). Each point mutation mapped either within the first RRM (RRM1) or to sequences immediately flanking both RRMs. These mutations also fell into regions of conservation between related hnRNPs (Fig. 2B). Five of the mutant alleles (hrp1-1, hrp1-2, hrp1-5, hrp1-6, hrp1-8) encode proteins that exhibit a single amino acid substitution compared with wild-type Hrp1p, whereas the remaining three alleles (hrp1-3, hrp1-4, and hrp1-7) encode proteins with two amino acid substitutions. The hrp1-4 and hrp1-7 alleles share an altered Met-191 (M191 → G,S) residue, whereas the hrp1-3 and hrp1-8 alleles both encode an altered Glu-158 (E158 → K,V). Although no systematic attempt was made to confirm which mutations contribute to the temperature-sensitive Ts− phenotype, the presence of overlapping mutations suggests that only one amino acid affects activity.
Figure 2.
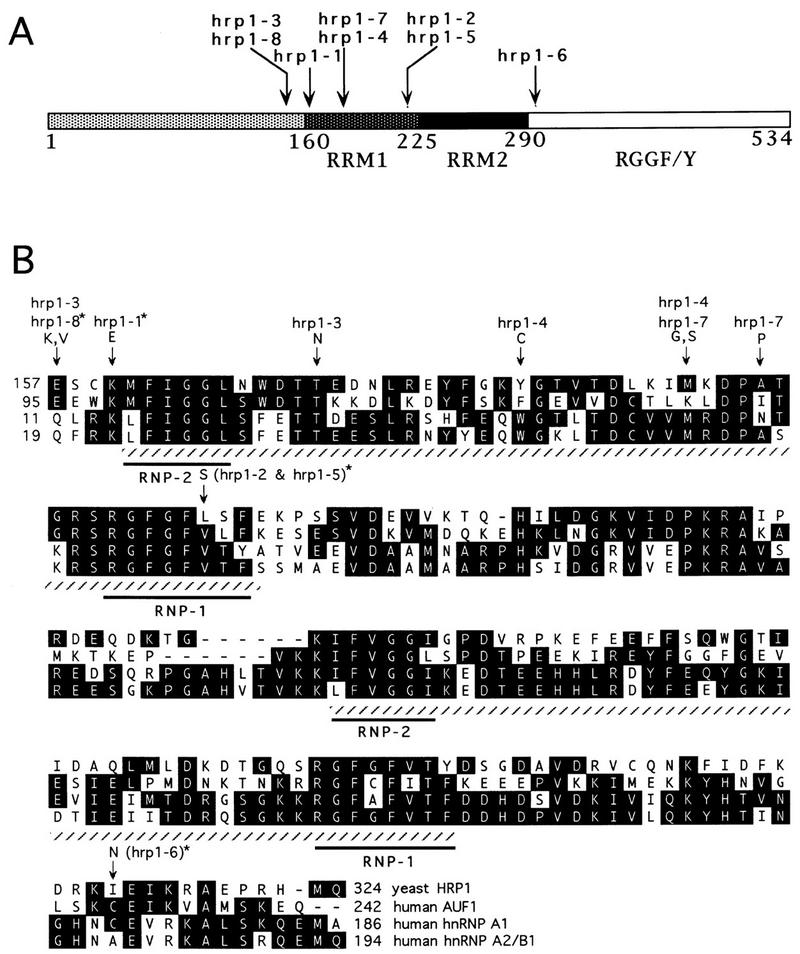
Mutations in eight temperature-sensitive (Ts−) hrp1 alleles map to the central portion of HRP1. (A) Localization of Ts− mutations with respect to the different regions of Hrp1p. The two RRMs are in the central portion of Hrp1p, whereas the carboxyl end contains repeats of the sequence RGGF/Y. (B) Amino acid changes within the two RRMs and homologies of Hrp1p with human AUF1 (Zhang et al. 1993), hnRNP A1 (Biamonti et al. 1989), and hnRNP A2/B1 (Burd et al. 1989). Amino acid changes are marked with an arrow, and the corresponding alleles are shown above the arrow. Those alleles representing single mutations are labeled with an asterisk. The two RRMs are shown by hatched underlining, with the RNP1 and RNP2 consensus sequences marked by an additional underline. If an amino acid occurs at a given position in two or more of the four sequences presented, it appears as a black-shaded box. The numbers refer to the position of the amino acid residue within each protein. Gaps are indicated with dashes.
An hrp1 TS− mutant displays poly(A) tail defects
TS− mutations in genes encoding known polyadenylation proteins, such as RNA14, RNA15, PAP1, FIP1, and PCF11, result in aberrantly short poly(A) tails in vivo (Minvielle-Sebastia et al. 1991; Patel and Butler 1992; Preker et al. 1995; Amrani et al. 1997a). To directly test an in vivo function for Hrp1p in polyadenylation, we examined the effect of the hrp1-5 mutation on poly(A) tail length. Total RNA was prepared from wild-type and hrp1-5 cells at various times after a shift from 23°C to 37°C. As a control, RNA was also isolated from the thermosensitive rna15-2 strain, which exhibits a poly(A) tail-shortening phenotype at 37°C (Minvielle-Sebastia et al. 1991). The RNA was labeled at the 3′ end and subjected to RNase digestion, leaving only the poly(A) tails intact (Fig. 3). The maximum length of poly(A) tails was decreased in the hrp1-5 mutant strain even at the permissive temperature (23°C), as compared to wild-type cells (Fig. 3, lanes 1,4), and this defect was enhanced following a 3- to 6-hr shift to 37°C (Fig. 3, lanes 5,6). A similar pattern was observed with the rna15-2 control strain (Fig. 3, lanes 7,8). In agreement with previous reports (Minvielle-Sebastia et al. 1991; Sachs and Deardorff 1992; Preker et al. 1995), the maximum length of poly(A) tails in wild-type cells increased slightly following the temperature shift (Fig. 3, lanes 2,3). Taken together, these results are consistent with a defect in the maturation of mRNA 3′ ends in the hrp1-5 mutant cells.
Figure 3.
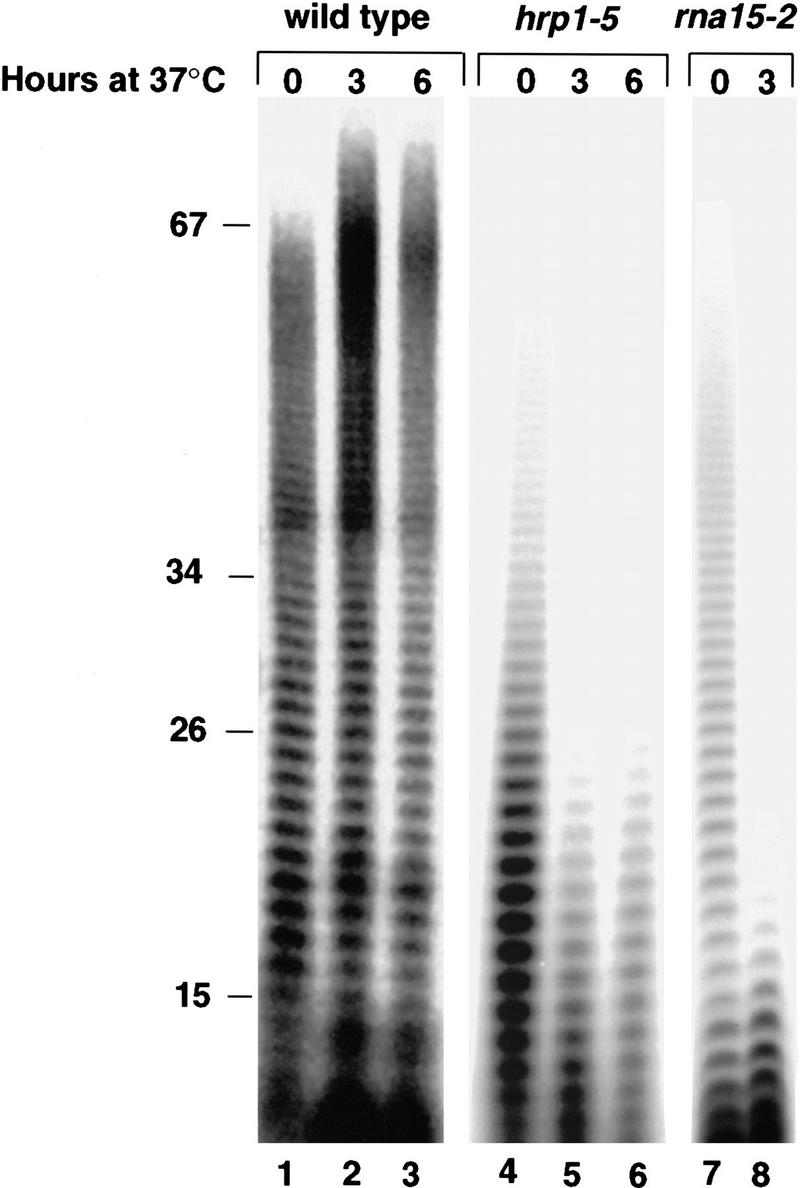
Length of poly(A) tails in the wild-type strain, and in the hrp1-5 and rna15-2 mutant strains after a shift to 37°C. Cells were grown at 24°C and then shifted to 37°C for the indicated times. Total RNA was 3′-end labeled with PAP and [α-32P]dATP, and digested with RNase T1 and RNase A to yield only intact labeled poly(A). After electrophoresis through a 16% acrylamide–8.3 m urea gel, the poly(A) tails were visualized by autoradiography.
Hrp1p interacts with both Rna14p and Rna15p in vivo
The purification of Hrp1p as a component of CF I, and the ability of this protein to support 3′-end processing with CF IA, suggests an association of Hrp1 with subunits of CF IA. To further examine this possibility, we tested for synthetic lethality between hrp1 mutations and rna14 or rna15 mutations. Physical or functional interactions between proteins can be assayed genetically through the analysis of double mutants of their genes; the combination of these mutations may show a synergistic interaction and lead to, for example, cell death (Guarente 1993). We thus crossed rna14-1, rna15-1, pap1-1, or npl3-1 mutants into a strain carrying a chromosomal disruption of the HRP1 gene, rescued by a plasmid encoding wild-type HRP1 and the URA3 marker. These strains were then transformed with a LEU2-marked plasmid carrying either wild-type HRP1 or one of the hrp1 temperature-sensitive alleles. Because of the presence of the chromosome-encoded mutant alleles of rna14-1, rna15-1, pap1-1, or npl3-1, these strains are strictly temperature-sensitive but can grow at the permissive temperature (24°C) on a selective medium that requires maintenance of the plasmids. However, the rna14-1 and rna15-1 strains were inviable even at this low temperature once the HRP1 URA3 plasmid was lost on selective medium containing 5-fluoro-orotic acid (5-FOA) (Fig. 4,D,E), indicating a synthetic lethal relationship. Interestingly, synthetic-lethal effects of the hrp1 rna15-1 and hrp1 rna14-1 combinations exhibited nonoverlapping allele specificity. The hrp1-6 and hrp1-8 alleles were synthetically lethal with rna15-1 (Fig. 4E, sectors 3 and 6), whereas the hrp1-1, hrp1-4, and hrp1-5 alleles were synthetically lethal with rna14-1 (Fig. 4D, sectors 4 and 5; and data not shown). Because the mutants map to different regions, one interpretation of these results is that the specificity represents interacting domains. No genetic interactions were observed with either the wild-type HRP1 control or the pap1-1 or npl3-1 alleles (Fig. 4A–C).
Figure 4.
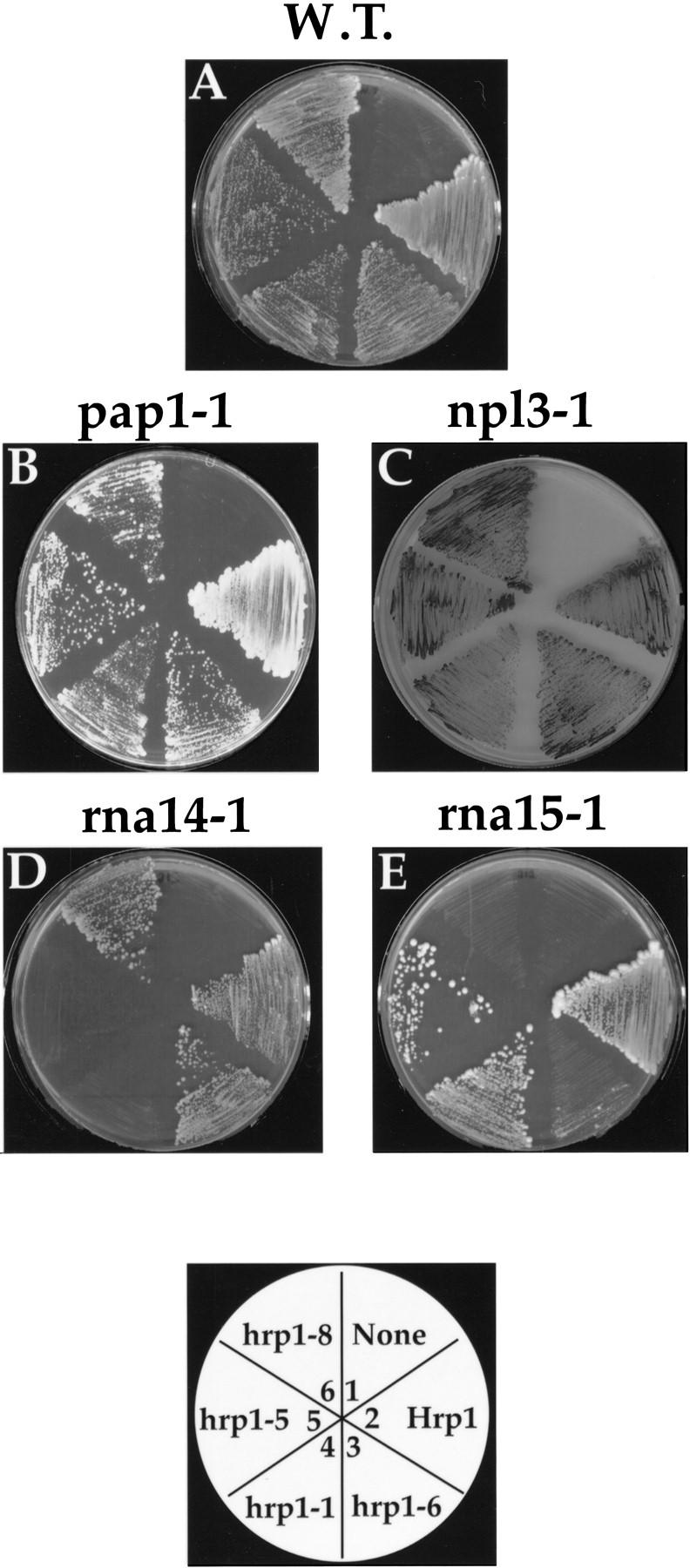
Synthetic lethality of hrp1 alleles. Δhrp1 cells bearing a CEN plasmid and either a chromosomal pap1-1 (B), npl3-1 (C), rna14-1 (D), or rna15-1 (E) allele are shown. The gene insert carried by each plasmid is indicated by the plate schematic. Transformants were grown on FOA medium lacking leucine supplemented with glucose at 25°C. Two independent colonies from each transformation were streaked onto each sector of the plate.
To confirm the interaction of Hrp1p with Rna14p and Rna15p, we used the two-hybrid system. In the version of the system that we employed, protein interactions are detected by activation of a β-galactosidase (lacZ) reporter gene through the interaction of a LexA (or Gal4) DNA-binding domain (BD) fusion protein with a Gal4 transcriptional activation domain (AD) fusion (Fields and Sternglanz 1994). A yeast reporter strain expressing a fusion of HRP1 to the AD was transformed with a plasmid encoding a fusion of the BD to each test protein. The Hrp1 fusion protein is functional in that it rescues the hrp1-5 Ts− phenotype. As a positive control, an AD fusion of Rna14p was tested against a BD fusion with Rna15p. Consistent with previous reports (Amrani et al. 1997a,b), a very strong interaction was detected between the Rna14 and Rna15 proteins of CF IA (Table 1). No interaction was observed between Rna14p and the human Cdc2 or Rfhm1 controls. Interaction between Hrp1p and either Rna14p or Rna15p was measured at a strength approximately one-tenth of that seen between Rna14p and Rna15p. However, the signal was significantly above that observed between Hrp1p and Pap1p, or between Hrp1p and the control proteins Fos, Cdc2, or Rfhm1. These results further support an interaction between Hrp1p and the CF IA components Rna14p and Rna15p.
Table 1.
Two-hybrid interactions
| Protein fused to Gal4 AD
|
Protein fused to LexA/Gal4 BD
|
Colony color
|
β-Galactosidase activity (units)
|
|---|---|---|---|
| Rna14 | Rna15 | dark blue | 824 ± 19 |
| Rna14 | Cdc2 | white | 1 ± 1 |
| Rna14 | Rfhm1 | white | 0 ± 0 |
| Hrp1 | Rna15 | light blue | 57 ± 9 |
| Hrp1 | Rna14 | light blue | 80 ± 4 |
| Hrp1 | Pap1 | white | 8 ± 1 |
| Hrp1 | Fos | white | 1 ± 1 |
| Hrp1 | Cdc2 | white | 0 ± 0 |
| Hrp1 | Rfhm1 | white | 0 ± 0 |
Quantitative β-galactosidase assays were performed on the EGY40 strain expressing the indicated constructs (Ausubel et al. 1987).
Numbers are the result of five pooled colonies assayed in triplicate.
Hrp1p binds the UA-rich efficiency element
Hrp1p contains two excellent matches to the RRM (Henry et al. 1996). In addition, the temperature-sensitive mutations clustered in and around the RRMs, suggesting that one function of Hrp1p may be the recognition of polyadenylation signal sequences. To test this possibility, radioactive RNAs were incubated with GST–Hrp1p in the presence of tRNA as a nonspecific competitor. The mixture was then irradiated with UV light, treated with RNase A, and resolved by SDS-PAGE. GST–Hrp1p can be cross-linked to full-length wild-type GAL7 RNA (Fig. 5, lane 3). The radioactively tagged polypeptide has a mass of 100 kD, which is consistent with a fusion protein of GST (26 kD) and Hrp1p (73 kD). No cross-linked product is generated when GST alone is used (Fig. 5, lane 2), indicating that the GST–Hrp1p cross-linking is Hrp1p specific. GST–Hrp1p does not cross-link to a mutant GAL7 transcript lacking the (UA)6 sequence (Fig. 5, lane 4). This cis-acting mutation completely inactivates polyadenylation in vitro (Chen and Moore 1992) and in vivo (Hyman and Moore 1993). Sequences of this type, called efficiency elements, are located upstream of the poly(A) site and form part of a yeast polyadenylation signal (Guo and Sherman 1996). Interestingly, GST–Hrp1p cross-links to the GAL7 precleaved substrate with a higher efficiency than it does to the full-length substrate (Fig. 5, cf. lanes 3 and 5). These results indicate that Hrp1p can bind full-length and precleaved GAL7 RNA in a manner that is dependent on the (UA)6 efficiency element and is consistent with the role for Hrp1p in cleavage and poly(A) addition. Moreover, addition of CF II significantly stimulated the binding of GST–Hrp1p to the full-length RNA (Fig. 5, lanes 10,11), whereas addition of CF IA had no effect (Fig. 5, lanes 7,8). CF II slightly increased the binding of Hrp1p to precleaved RNA (not shown).
Figure 5.

GST–Hrp1p can be specifically cross-linked to pre-mRNA. Recombinant GST–Hrp1p was cross-linked to either wild-type full-length RNA (lane 3), full-length RNA lacking the (UA)6 repeat (lane 4), or precleaved RNA (lane 5). Cross-linking of recombinant GST protein to full-length RNA was assayed in parallel (lane 2). In lanes 6–11, combinations of proteins were cross-linked to wild-type full-length precursor. Reaction products were electrophoresed through an 8% acrylamide gel, together with molecular size markers (lane 1), stained with silver, dried, and exposed to a PhosphorImager screen.
Hrp1p exits the nucleus
Many hnRNP proteins such as Npl3p exit the nucleus perhaps in association with mRNAs and rapidly re-enter in a process termed nucleocytoplasmic shuttling (Nakielny and Dreyfuss 1997). To test whether Hrp1p is capable of shuttling between the nucleus and the cytoplasm, we performed a nuclear export assay (Lee et al. 1996). This assay takes advantage of the properties of the thermosensitive nucleoporin mutant nup49-313. This mutant has the property that at the restrictive temperature, import into the nucleus is blocked whereas export out of the nucleus is unaffected (Sclenstedt et al. 1993; Doye et al. 1994). Thus, if Hrp1p is able to leave the nucleus, it should accumulate in the cytoplasm of the nup49-313 cells when these cells are shifted to 36°C. To monitor the localization of Hrp1p, the green fluorescent protein (GFP) was fused to HRP1 and placed under the control of the inducible GAL1 promoter. GFP–HRP1 is functional in that it can restore normal growth to the thermosensitive hrp1-5 mutant. The GFP–Hrp1 fusion protein remained entirely nuclear when nup49-313 cells were incubated at 25°C (Fig. 6B) but then accumulated in the cytoplasm when cells were shifted to 36°C (Fig. 6D). This localization parallels that observed for the Npl3 protein (Lee et al. 1996) in the nup49-313 strain and indicates that Hrp1p is able to exit the nucleus. In contrast, a nonshuttling control protein [NLS (nuclear localization signal)–GFP–β-galactosidase] remained nuclear at both the permissive and restrictive temperatures (Fig. 6F,H). Finally, cytoplasmic localization at 36°C is dependent on the nup49-313 mutation, as wild-type cells show nuclear localization of the fusion protein at the restrictive temperature (data not shown).
Figure 6.

Living cells were photographed after incubation in the export assay. Cells containing the nup49-313 allele were incubated at either 25°C (A,B,E,F), or at 36°C (C,D,G,H). Cells were photographed by either Nomarski optics (A,C,E,G), or for GFP fluorescence (B,D,F,H). (A–D) nup49-313 cells expressing GFP–Hrp1p; (E–H) nup49-313 cells expressing NLS–GFP–LacZ.
Discussion
In this report we identified Hrp1p as a novel component of the polyadenylation machinery in the yeast S. cerevisiae. The evidence that Hrp1p participates in both cleavage of the pre-mRNA and the subsequent addition of poly(A) tails is based on both in vivo and in vitro results. Cells bearing mutant HRP1 alleles contain mRNAs with aberrantly short poly(A) tails and show synthetic lethality in combination with mutations in RNA14 and RNA15, which encode proteins required for cleavage and polyadenylation. This interaction was confirmed by two-hybrid analysis. Hrp1p is the 73-kD protein of CF IB, which is also necessary for both steps of the in vitro processing reaction. Moreover, a recombinant GST–Hrp1 fusion protein can fully substitute for CF IB function.
In S. cerevisiae, three elements are sufficient to specify a poly(A) site (Guo and Sherman 1996). These include a PyA motif at the cleavage site, the UA-rich efficiency element, and the A-rich positioning element. Proper mRNA 3′-end formation requires recognition of all three cis-acting sequences. Currently, there are several RNA-binding proteins that could be recognizing these signals: Rna15p, Cft2p, and recently, a third one, Hrp1p. The CF IA subunit Rna15p can be cross-linked to GAL7 RNA, and this interaction does not require the efficiency element (Kessler et al. 1996). On the other hand, the cross-linking of the CF II subunit Cft2 (Zhao et al. 1997) and Hrp1p both require the efficiency element. However, Cft2p binding depends on ATP, whereas Hrp1p binding does not, and unlike Cft2p, Hrp1p binds to both uncleaved and cleaved substrates. The Yth1p component of PF I binds preferentially to poly(U), but its binding site on the precursor RNA remains unknown (Barabino et al. 1997).
These interactions suggest that assembly of a functional cleavage complex may be a dynamic step-wise process (Fig. 7). For example, the interaction of CF II may be an early step in the recognition of pre-mRNA and necessary for recruitment of the other cleavage factors, CF IA and Hrp1p. This possibility would be consistent with our observations that CF II enhances the binding of Hrp1p to the full-length RNA. CF II may then be displaced from the efficiency element by Hrp1p, which remains on the cleaved RNA during poly(A) addition, hence, the requirement of Hrp1p for both steps in the reaction. Many yeast sites have multiple efficiency elements. In this respect, it is also interesting that Hrp1p has two RRM motifs, which may allow it to contact more than one efficiency element simultaneously.
Figure 7.
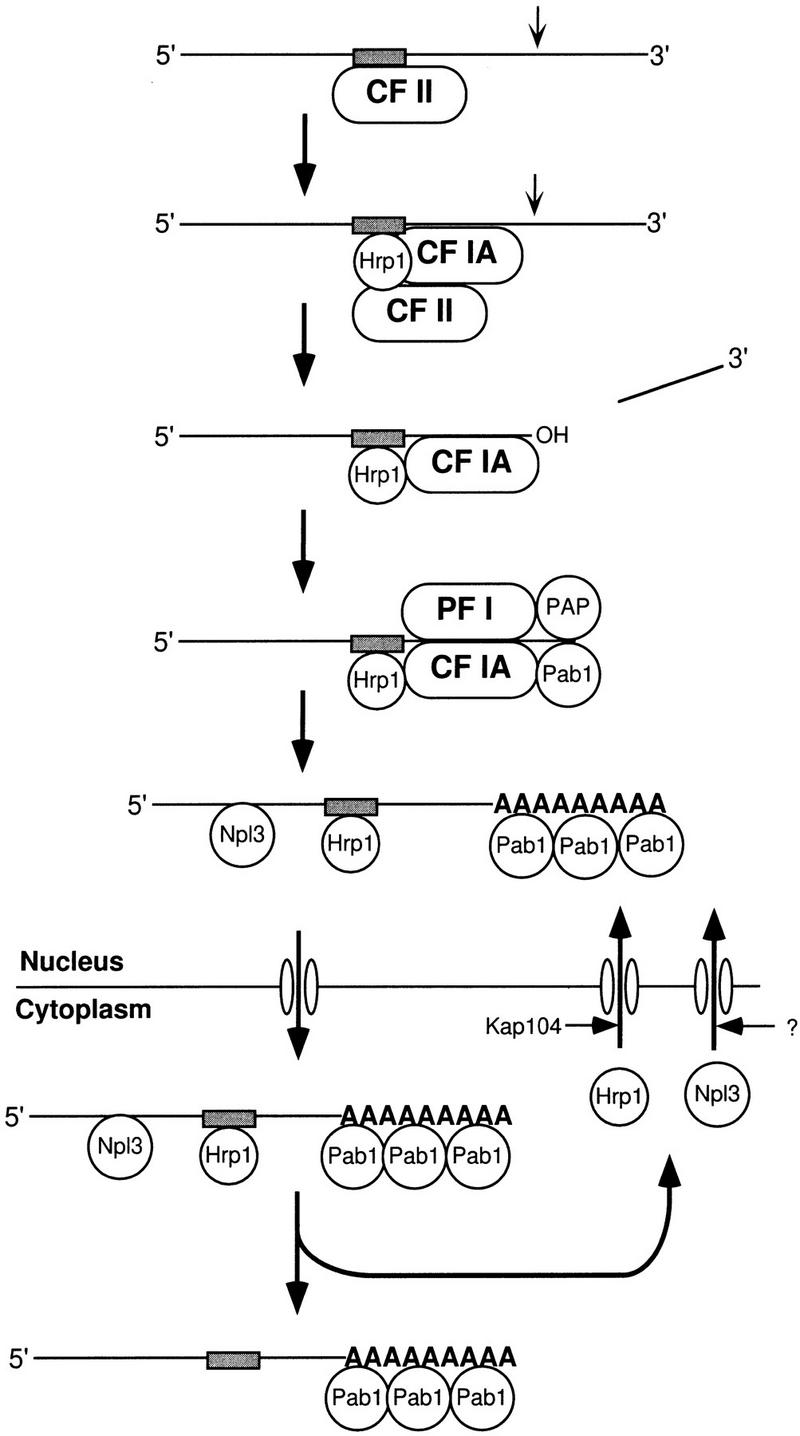
Schematic of Hrp1p dynamics. Pre-mRNA is initially recognized by CF II interacting at the UA-rich efficiency element. Recruitment of Hrp1p and CF IA to the complex displaces CF II, but continued association of CF II may stabilize Hrp1p binding. Hrp1p and CF IA remain bound to the cleaved RNA and promote poly(A) addition in combination with PF I, PAP, and Pab1p. After this step, Hrp1p remains bound to the mRNA along with other RNA-binding proteins such as Npl3p and Pab1p. Together with the processed mRNA, the proteins would move out of the nucleus at the nuclear pore, dissociate from the mRNA, and re-enter the nucleus via recognition by Kap104p in the case of Hrp1p and an as yet undefined nuclear transport receptor for Npl3p. The movements of Pab1p are not known.
We and others have found that the Pab1 protein cofractionates with CF I activity (Fig. 1C; Minvielle-Sebastia et al. 1997), in agreement with its interaction with Rna15p in two-hybrid assays and coimmunoprecipitation (Amrani et al. 1997b). Pab1p is associated with the poly(A) tails of mRNA in the cytoplasm, where it stimulates the initiation of translation (Sachs et al. 1997). Using a fully reconstituted system with purified fractions rather than complementation of Pab1p-depleted extracts, we were able to further dissect the role of Pab1p in mRNA 3′-end formation. A combination of Hrp1p, CF IA, PAP, and PF I are sufficient for poly(A) addition, but Pab1p acts to limit the size of the final poly(A) tail, and is not required for cleavage. This finding is consistent with the fact that cells bearing a temperature-sensitive pab1 mutation showed aberrantly long poly(A) tails in vivo (Sachs and Deardorff 1992) and in vitro (Amrani et al. 1997b; Minvielle-Sebastia et al. 1997). It also verifies the previous result that addition of anti-Pab1p antibodies to a processing extract causes an increase in poly(A) length but had no effect on the cleavage reaction (Amrani et al. 1997b).
HRP1 was originally identified because mutations suppressed the temperature sensitivity and mRNA export defect of npl3ts mutants (Henry et al. 1996). NPL3 encodes a major poly(A)+ RNA-binding protein and resides primarily in the nucleus (Bossie et al. 1992; Russel and Tollervey 1992; Wilson et al. 1994). However, Npl3p shuttles rapidly between the nucleus and the cytoplasm in a manner that depends on mRNA synthesis (Flach et al. 1994; Lee et al. 1996). This has led to the idea that Npl3p is a major mediator of mRNA export and may act in a manner similar to other heterogenous nuclear ribonucleoproteins (hnRNPs), like hnRNP A1. One model is that proteins such as Npl3p associate with the newly synthesized mRNA and contain “signals” that target the protein–RNA complex for export out of the nucleus (Nakielny and Dreyfuss 1997).
There are several possible explanations for why a mutant form of Hrp1p, a protein involved in polyadenylation (as we have demonstrated here), might compensate for loss of function of a nuclear transport factor like Npl3p. One possibility is that mRNA export can be considered as part of an orchestrated pathway, for example, transcription, capping, splicing, polyadenylation, and export. If Npl3p is required for the export step, perhaps by mutating proteins like Hrp1p, the cell can compensate for the presence of a defective Npl3p later in the pathway. Another possibility is that proteins such as Npl3p and Hrp1p package the RNA for export. Perhaps Hrp1p binds mRNA during the polyadenylation reaction and remains associated with it as it passes out of the nucleus into the cytoplasm. Our finding that Hrp1p is capable of exiting the nucleus is consistent with this view. Once in the cytoplasm, Hrp1p would have to be released from the mRNA and reimported into the nucleus. Interestingly, Hrp1p appears to use a special nuclear import receptor, termed Kap104p in yeast, which is similar to mammalian transportin (Aitchison et al. 1996). Transportin is required for import of hnRNP A1 protein into the nucleus. This view of the dynamics of Hrp1 protein is presented in Figure 7.
It is clear that Hrp1p resembles the mammalian hnRNP A1 in structure, with two RRMs and an RGG-rich carboxyl domain, and in the ability to shuttle from the nucleus to cytoplasm, even using a similar nuclear import receptor. HnRNP A1 can influence the choice of splice site but has no known role in mammalian polyadenylation (Weighardt et al. 1996). Our study raises questions as to why Hrp1p is necessary for polyadenylation in yeast and why there are no known counterparts in metazoan cells. Sequence similarity exists between many of the proteins found in the yeast and mammalian processing factors. Given the emerging level of conservation (Manley and Takagaki 1996), there may well be an Hrp1p-like protein waiting to be discovered. In this way, Hrp1p may be the first clue into a common mechanism linking polyadenylation and mRNA export in eukaryotes.
On the other hand, there may not be a mammalian equivalent of Hrp1p and it may represent a special adaptation to compensate in yeast for transport regulatory mechanisms that were lost during evolution. In general, unspliced RNA does not exit the nucleus unless it is complexed with proteins that allow it to bypass the normal mRNA transport machinery (Nakielny and Dreyfuss 1997). Furthermore, with the multiple introns of most mammalian pre-mRNAs, it is likely that the RNA becomes polyadenylated long before splicing is complete, such that the cell relies on the association of splicing factors to retain immature RNAs in the nucleus. In yeast, few genes have introns, and it may be necessary to monitor more closely 3′-end formation as the mark of a mature mRNA and couple this event to rapid transport.
In this study we have used both yeast genetics and biochemistry to show that the RNA-binding protein Hrp1p is an essential part of the polyadenylation machinery in yeast. As such, it is the first example of a constitutive mRNA processing factor that probably moves with the mRNA out of the nucleus. Future experiments will address the precise mechanism by which Hrp1p functions and how it serves to coordinate mRNA processing with transport.
Materials and methods
Strains, media, and genetic techniques
Yeast strains (Table 2) were grown and maintained on YPD (1% yeast extract, 2% peptone, 2% glucose) or on selective media (Sherman et al. 1986) supplemented with either 2% glucose, raffinose, or galactose. Temperature-sensitive mutants were grown at 24°C–26°C (permissive temperature) and shifted to 36°C–37°C (restrictive temperature). Yeast strains were transformed using the lithium acetate method (Ito et al. 1983) modified according to Gietz et al. (1995). Loss of URA3 plasmids from yeast cells was accomplished by plating on solid medium containing 5-FOA (Boeke et al. 1984).
Table 2.
Yeast strains
| Strain
|
Genotype
|
Source
|
|---|---|---|
| W303a | MATa ura3-1 leu2-3,112 his3-11,15 trp1-1 ade2-1 | A. Tzagoloff (Columbia University, New York, NY) |
| PSY773 | MATa npl3-1 his3-11,15 ade2-1 leu2-2 ura3-1 trp1-1 | M. Henry (University of Medicine and Dentistry of New Jersey, Stratford) |
| PSY818 | MATα hrp1::HIS3 ura3 ade2 ade8 his3 leu2 lys1 Trp− plus pRS316–HRP1 | M. Henry |
| rna15-1 | MATa rna15-1 his3-11,15 ade2-1 ura3-1 trp1-1 | F. Lacroute (University of Paris VI, Cedex, France) |
| rna15-2 | MATα ura3-1 trp1-1 ade2-1 leu2-3,112 his3-11,15 rna15-2 | F. Lacroute |
| rna14-1W | MATa rna14-1 his3-11,15 ade2-1 ura3-1 | F. Lacroute |
| LM21 | MATa rna14::TRP1 ura3-1 his3-11,15 ade2-1 trp1-1 can1-100 plus pLM28 | L. Minvielle-Sebastia (University of Basel, Switzerland) |
| LM31 | MATa rna15::TRP1 ade2-1 his3-11,15 ura3-1 trp1-1 can1-100 plus pLM13 | L. Minvielle-Sebastia |
| UR3148-6A | MATa pap1-1 leu ura | S. Butler (University of Rochester School of Medicine and Dentistry, NY) |
| EGY40 | ura3-1 his3-11 trp1-1 leu2-3,112 | R. Finley (Massachusetts General Hospital, Boston) |
| nup49-313 | MATa nup40::TRP1 ura3 leu2 his3 ade2 ade3 lys2 plus pUN100 nup49ts–LEU2 | V. Doye (Institut Curie, Cedex, France) |
| PSY1070 | MATa hrp1::HIS3 rna15-1 ura3 ade2 ade8 his3 leu2 Trp− plus pRS316–HRP1 | this study |
| PSY1071 | MATa hrp1::HIS3 rnal4-1 ura3 ade2 lys2 his3 leu2 Trp− plus pRS316–HRP1 | this study |
| PSY1072 | MATa hrp1::HIS3 npl3-1 ura3 ade2 lys2 his3 leu2 Trp− plus pRS316–HRP1 | this study |
| PSY1073 | MATa hrp1::HIS3 pap1-1 ura3 ade2 ade8 lys1 his3 leu2 Trp− plus pRS316–HRP1 | this study |
| PSY1076 | MATa hrp1-5 ura3-1 trp1-1 ade2-1 leu2-3,112 his3-11,15 | this study |
RNA synthesis
Capped and uniformly labeled RNAs used for the cleavage and poly(A) addition assays were prepared by in vitro transcription of linearized DNAs as described (Chen and Moore 1992) from the following plasmid templates: (1) pJCGAL7-1 (Chen and Moore 1992) for full-length wild-type RNA containing both the GAL7 poly(A) site and flanking sequences; (2) pJCGAL7-3 (Chen and Moore 1992) for a mutant version of the full-length RNA, lacking the (UA)6 repeat sequence upstream of the poly(A) site; and (3) pJCGAL7-9 (Zhelkovsky et al. 1995) for precleaved wild-type GAL7 RNA lacking sequences downstream of the poly(A) site.
HRP1 plasmids
Plasmids used in this study are listed in Table 3. To create pGAL–HRP1, HRP1 was amplified by PCR from plasmid pMHY40 (Henry et al. 1996). Digestion of the PCR product with BamHI and ApaI yielded an ∼1420-bp HRP1 fragment lacking the carboxy-terminal 61 codons. The HRP1 coding region was completed by replacement of a SacI–HindIII fragment from pMHY40. To create pGAL–GFP–HRP1, a fragment containing the entire HRP1 open reading frame (ORF) was ligated into linearized pCGF-1C (Kahana and Silver 1996). The E. coli GST–HRP1 expression vector (pGEX–HRP1) was created by subcloning an ∼1.6-kb fragment from pGAL–HRP1 into pGEX-3X (Pharmacia Biotech., Inc.).
Table 3.
Plasmid constructions
| Plasmid
|
Construction
|
|---|---|
| pGAL–HRP1 | HRP1 placed under control of the GAL1 promoter in the CEN LEU2 vector pPS311 |
| pGEX–HRP1 | Hrp1 fused to the carboxyl terminus of GST in the vector pGEX-4T-1 |
| YIp5–hrp1-5 | yeast integrating plasmid carrying hrp1-5 |
| pMHY23 | CEN LEU2 vector carrying HRP1 |
| pCGF-1C–HRP1 | Hrp1 fused to the carboxyl terminus of pCGF-1C |
| pEG202–RNA14 | Rna14 fused to the LexA DNA-binding domain of pEG202 |
| pEG202–RNA15 | Rna15 fused to the LexA DNA-binding domain of pEG202 |
| pACTII–RNA14 | Rna15 fused to the Ga14 activation domain of pACTII |
| pACTII–HRP1 | Hrp1 fused to the Ga14 activation domain of pACTII |
| Yeplac112–PAP1/GBD | Pap1 fused to the Ga14-binding domain and placed in vector Yeplac112 |
| pEG202–Fos | human Fos fused to the LexA DNA-binding domain of pEG202 |
| pEG202–hncdc | human Cdc2 fused to the LexA DNA-binding domain of pEG202 |
| pEG202–rfhm1 | human rfhm1 fused to the LexA DNA-binding domain of pEG202 |
Protein isolation
Fractions containing either CF I or combined CF II/PF I were obtained from Q–Sepharose chromatography of yeast crude whole cell extracts as described (Kessler et al. 1996). CF I was further purified and then separated into C F IA and CF IB by gel filtration (Kessler et al. 1996). CF II fractions from the poly(A)–Sepharose step were obtained as described previously (Zhao et al. 1997). Recombinant PAP was expressed and purified as described (Kessler et al. 1995). GST-tagged recombinant Hrp1 was purified by glutathione affinity chromatography as specified by the supplier (Pharmacia Biotech, Inc.). Pab1 protein and anti-Pab1 antibodies were a gift from Dr. Alan Jacobson (University of Massachusetts, Worcester). HnRNP A1 protein was a gift from Dr. Adrian Krainer (Cold Spring Harbor Laboratory).
Peptide microsequencing and sequence analysis
Following S–Sepharose chromatography, purified CF IB (0.3 μg) was dialyzed (0.1 m NH4HCO3, 0.1% SDS) for 6 hr at room temperature and dried (Jenny et al. 1994). The dried protein pellet was resuspended in 2× laemmli dye (Laemmli 1970), electrophoresed on an 8% acrylamide–SDS gel, and copper stained (Bio-Rad) (Lee et al. 1987). The 73-kD protein band was excised, destained, and submitted for microsequencing to Dr. William Lane at the Harvard University Microchemistry Facility (Cambridge, MA). Peptide sequence was determined by collisionally activated dissociation (CAD) on a Finnigan TSO 7000 triple quadrupole mass spectrometer. Homology searches were performed using the BLAST program (Altschul et al. 1990).
Isolation of hrp1 thermosensitive alleles
Temperature-sensitive alleles of HRP1 were generated by random PCR mutagenesis and plasmid shuffling. An internal segment of the HRP1 ORF was removed from pMHY23 (CEN LEU2 HRP1) and, together with the PCR HRP1 product, was cotransformed into PSY818 (Henry et al. 1996). Transformants were selected on synthetic complete (SC) medium lacking leucine, and replica-plated to SC plates lacking leucine and containing FOA and the dye erythrocin B (7.5 mg/liter) and incubated at both 25°C and 36°C to identify potential Ts− alleles. Of 12 potential clones, 8 remained temperature sensitive after rescue of the plasmid and transformation back into PSY818. PSY1076 (chromosomal hrp1-5) was generated using the two-step pop-in/pop-out replacement method (Scherer and Davis 1979).
Mapping of hrp1 thermosensitive alleles
The plasmid-borne temperature-sensitive mutant alleles (hrp1-1 to hrp1-8) were initially mapped by swapping restriction fragments from each mutant plasmid (hrp1 LEU2) into the wild-type parent plasmid (HRP1 LEU2) to generate a hybrid hrp1/HRP1 gene. The hrp1-1, hrp1-3, hrp1-4, and hrp1-5 mutations were mapped to a 590-bp BglII–SacI fragment encoding the amino terminus of HRP1. In contrast, the hrp1-2, hrp1-5, and hrp1-6 mutations mapped to a 370-bp NdeI–BglII fragment encoding the central portion of the gene. The corresponding base changes were identified by DNA sequencing.
RNA analyses
For the analysis of poly(A) tails, wild-type (W303a), and mutant strains PSY1076 (hrp1-5) and rna15-2 were grown in YPD to an OD600 of 26.0. A 1.6-ml aliquot was taken as time zero, the culture was diluted with an equal volume of YPD prewarmed to 45°C, and incubated at 37°C for the indicated times. Cells were harvested by centrifugation and frozen in dry ice/ethanol. Total yeast RNA was isolated from 2 ml of cells using a modification of the hot phenol method (Köhrer and Domdey 1991).
RNAs were labeled at their 3′ ends using a modification of the method developed by Preker et al. (1995) employing yeast PAP and [α-32P]dATP (A.M. Zhelkovsky and C.L. Moore, unpubl.). Labeled RNAs were digested essentially as described (Preker et al. 1995), in an 80-μl reaction containing 10 mm Tris-HCl (pH 7.9), 0.3 m NaCl, 20 μg of yeast tRNA, 25 units of RNase T1 (Boehringer Mannheim), and 1 μg of RNase A (Sigma) for 40 min at 37°C. The digestion was stopped by adding 20 μl of termination buffer (1 mg/ml of proteinase K, 5% SDS, and 50 mm EDTA) and incubating for 30 min at 37°C. The nuclease-resistant poly(A) tails were then extracted with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with 2.5 m ammonium acetate, 15 mm MgCl2, 2.5 volumes of ethanol, 32 μg of yeast tRNA, and 125 μg of glycogen. RNAs were recovered by centrifugation at 16,000g for 30 min at 4°C, rinsed in 70% ethanol, then 100% ethanol, and finally resuspended in 6 μl of RNA loading dye (98% formamide, 10 mm EDTA, 0.2% bromphenol blue, 0.2% xylene cyanol). The RNA (2–3 μl) was then electrophoresed on a 16% acrylamide–8.3 m urea gel and visualized by either autoradiography or by exposure to a PhosphorImager screen (Molecular Dynamics).
Processing assays
Cleavage assays were conducted as described (Chen and Moore 1992; Kessler et al. 1996; Zhao et al. 1997). Each 16-μl reaction contained full-length GAL7 RNA in 20 mm HEPES–KOH (pH 7.0), 1 mm magnesium acetate, 75 mm potassium acetate, 2% polyethylene glycol (PEG-8000, Fisher), 2 mm ATP, 20 mm creatine phosphate, 10 nm labeled precleaved RNA (8 ng), 1.5 μm (0.6 μg) tRNA, 1 mm DTT, 0.4 units of RNasin (Promega), and 0.1 mg/ml of purified BSA (New England Biolabs). Each reaction was supplememted with either 3 μl of CF I or 2 μl of CF IA + 2 μl CF IB, or 80 ng of GST–Hrp1p (or different combinations as described) and 1 μl of purified CF II. Reactions were assembled on ice, started by placement at 30°C, incubated for 20 min, and terminated by the addition of proteinase K and SDS (Kessler et al. 1996).
Specific poly(A) addition to precleaved RNA was assayed in 16-μl reactions assembled as described for cleavage but using precleaved GAL7 RNA, 2 μl of CF II/PF I (8 μg), and 1 μl of freshly diluted PAP (13–20 ng). In reactions containing Pab1p, 80 ng was used.
UV cross-linking
UV cross-linking assays were performed essentially as described (Zhao et al. 1997). Reactions (10–16 μl) were assembled on ice and contained 1 mm magnesium acetate, 75 mm potassium acetate, 1.5 mm tRNA, 20 nm radioactive RNA substrates, and proteins as indicated. Samples were incubated for 5 min at 30°C and then irradiated at room temperature with UV light for 10 cycles (each for 30 sec at 120 mJ/cm2 in a UV Strataliker 1800, Stratagene) on Parafilm on a glass plate 15 cm from the UV source. The samples were digested with RNase A (Sigma, 0.9 mg/ml) for 30 min at 37°C, and proteins were separated on 7% polyacrylamide–SDS gels, silver stained (Gottlieb and Chavko 1981), dried, and exposed to a PhosphorImager screen (Molecular Dynamics) for 2 days.
Two-hybrid assays
LexA–DNA binding domain fusions were constructed in pEG202. Gal4-activating domain fusions were constructed in pACTII. pLexA–RNA15 and pLexA–Rna14 were created by PCR amplification from pYEP352-15X and pYEP352-14H (Bonneaud et al. 1994). pAS–HRP1 was created by the PCR amplification of yeast genomic DNA from strain EGY40. PAP-Gal4 BD was provided by A. Zhelkovsky (Tufts University School of Medicine, Boston, MA) and the GBD reporter plasmid pBS-GAL by J. Strasswimmer (Tufts University School of Medicine, Boston, MA). Negative control plasmids pLexA–Fos, pLexA–hncdc2, pLexA–rfhm1, and LexA reporter plasmid pSH18-34 were a gift of R. Brent (Massachusetts General Hospital, Boston).
To confirm that the attachment of binding or activation domains to the amino terminus of Rna14p, Rna15p, or Hrp1p did not adversely affect their function, we demonstrated the ability of the fusion construct to rescue a lethal defect. pLexA–RNA14 was transformed into strain LM21 and pLexA-15 was transformed into LM31. These strains contain chromosomal disruptions of the RNA14 and RNA15 genes, respectively, and are covered by URA3 plasmids, which carry the wild-type genes (Bonneaud et al. 1994). The transformants were confirmed for growth on complete medium lacking tryptophan and histidine and containing 5-FOA, indicating the presence of pLexA fusions, the disrupted genes, and the loss of the covering plasmids. pAS–HRP1 was transformed into strain PSY1076, and transformants were confirmed for growth on complete media lacking leucine at the nonpermissive temperature.
For two-hybrid assays, yeast manipulations were performed according to standard techniques (Ausubel et al. 1987). Strain EGY40, containing reporter plasmid pSH18-34 or pBS–GAL, were first transformed with BD plasmids and selected on complete medium lacking uracil and histidine or complete medium lacking uracil and tryptophan. Five colonies were pooled and transformed with either pAS–HRP1 or two negative control plasmids, obtaining ∼500 colonies after selection on complete medium lacking uracil, histidine, and leucine or complete medium lacking uracil, tryptophan, and leucine. Transformants were replica plated to minimal media containing X-gal supplemented with either tryptophan or histidine and examined for blue color development after incubation for 24 hr at 30°C. Interactions were quantitated by outgrowing five colonies and assaying soluble β-galactosidase activity (Ausubel et al. 1987).
Synthetic lethal interactions
Synthetic lethal interactions between the plasmid-borne hrp1ts alleles with temperature-sensitive alleles of related genes were tested using a shuffling strategy. Each strain, in addition to each gene to be tested (npl3-1, rna15-1, rna14-1, pap1-1), carries a disrupted chromosomal hrp1::HIS3 gene that is complemented by pRS315–HRP1. These strains were then transformed with the hrp1ts plasmid collection and transformants were selected on complete plates lacking leucine at 25°C. Those transformants unable to grow on 5-FOA (Boeke et al. 1984) plates lacking leucine were scored as synthetically lethal. The strains (PSY1070–PSY1073) were generated by crossing PSY818 to strains rna15-1, rna14-1W, PSY773, and UR3148-6A, respectively, sporulating the resultant diploid, and screening for Ts−, His+, and Ura+ spore clones.
Nuclear export assay
Nup49-313 cells (Doye et al. 1994) were transformed with pPS811 (GFP-NPl3) (Lee et al. 1996), pPS817 (NLS–GFP–LacZ) (Lee et al. 1996), or pCGF-1C–HRP1 (GFP–HRP1). The nuclear export assay was performed as described previously (Lee et al. 1996). Cells were examined for GFP fluorescence signal in the FITC channel of a fluorescence microscope.
Acknowledgments
We are grateful to Drs. David Mangus and Alan Jacobson for the recombinant Pab1 protein and anti-Pab1p antibodies, Dr. Adrian Krainer for hnRNP A1, Dr. François Lacroute for the rna14-1, rna15-1, and rna15-2 strains, Dr. Scott Butler for the pap1-1 strain, and Alexander Zhelkovsky for plasmid Yeplac112-PAP1/Gal4 BD. We thank Alexander Zhelkovsky, Neptune Mizrahi, Steffen Helmling, and Chuck Cole for critically reading the manuscript. This work was supported by U.S. Public Health service grant R01 GM41752 from the National Institutes of Health (NIH) (to C.L.M.), RO1 GM36373 (to P.A.S.), an American Cancer Society Postdoctoral Fellowship (to M.F.H.), and the NIH Cancer Genetics Training grant (to E.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL cmoore@opal.tufts.edu; FAX (617) 636-0337.
References
- Adams MD, Rudner DZ, Rio DC. Biochemistry and regulation of pre-messenger-RNA splicing. Curr Opin Cell Biol. 1996;8:331–339. doi: 10.1016/s0955-0674(96)80006-8. [DOI] [PubMed] [Google Scholar]
- Aitchison JD, Blobel G, Rout MP. Kap104p: A karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science. 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amrani N, Minet M, Wyers F, Dufour M-E, Aggerbeck LP, Lacroute F. PCF11 encodes a third protein component of yeast cleavage and polyadenylation factor I. Mol Cell Biol. 1997a;17:1102–1109. doi: 10.1128/mcb.17.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Minet M, Le Gouar M, Lacroue F, Wyers F. Yeast Pab1 interacts with Rna15 and participates in the control of the poly(A) tail length in vitro. Mol Cell Biol. 1997b;17:3694–3701. doi: 10.1128/mcb.17.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA. Current protocols in molecular biology. New York, NY: John Wiley/Greene; 1987. pp. 13.6.1–13.6.4. [Google Scholar]
- Barabino SML, Hübner W, Jenny A, Minvielle-Sebastia L, Keller W. The 30-kD subunit of mammalian cleavage and polyadenylation specificity factor and its yeast homolog are RNA-binding zinc finger proteins. Genes & Dev. 1997;11:1703–1716. doi: 10.1101/gad.11.13.1703. [DOI] [PubMed] [Google Scholar]
- Beelman C, Parker R. Degradation of mRNA in eukaryotes. Cell. 1995;81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- Biamonti GM, Buvoli MT, Bassi C, Morandi F, Cobianchi F, Riva S. Isolation of an active gene encoding human hnRNP protein A1: Evidence for alternative splicing. J Mol Biol. 1989;207:491–503. doi: 10.1016/0022-2836(89)90459-2. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Lacroute F, Fink GR. A positive selection for mutants lacking orotidine-5′ phosphate decarboxylase activity in yeast: 5-Fluoroorotic acid resistance. Mol & Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Bonneaud N, Minvielle-Sebastia L, Cullin C, Lacroute F. Cellular localization of RNA14p and RNA15p, two yeast proteins involved in mRNA stability. J Cell Sci. 1994;107:913–921. doi: 10.1242/jcs.107.4.913. [DOI] [PubMed] [Google Scholar]
- Bossie MA, Dehoratius C, Barcelo G, Silver P. A mutant nuclear protein with similarity to RNA binding proteins interferes with nuclear import in yeast. Mol Biol Cell. 1992;3:875–893. doi: 10.1091/mbc.3.8.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Swanson MS, Gorlach M, Dreyfuss G. Primary structures of the heterogenous nuclear ribonucleoprotein A2, B1, and C2 proteins: a diversity of RNA binding proteins is generated by small peptide inserts. Proc Natl Acad Sci. 1989;86:9788–9792. doi: 10.1073/pnas.86.24.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JS, Platt T. RNA processing generates the mature 3′ end of yeast CYC1 messenger RNA in vitro. Science. 1988;242:1270–1274. doi: 10.1126/science.2848317. [DOI] [PubMed] [Google Scholar]
- Chanfreau G, Noble SM, Guthrie C. Essential yeast protein with unexpected similarity to subunits of mammalian cleavage and polyadenylation specificity factor (CPSF) Science. 1996;274:1511–1514. doi: 10.1126/science.274.5292.1511. [DOI] [PubMed] [Google Scholar]
- Chen J, Moore CL. Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol Cell Biol. 1992;12:3470–3481. doi: 10.1128/mcb.12.8.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V, Wepf R, Hurt EC. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. EMBO J. 1994;13:6062–6075. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R, Ellmeier W, Birnstiel ML. Mature mRNA 3′ end formation stimulates RNA export from the nucleus. EMBO J. 1991;10:3513–3522. doi: 10.1002/j.1460-2075.1991.tb04915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Sternglanz R. The two-hybrid system: An assay for protein-protein interactions. Trends Genet. 1994;10:286–292. doi: 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- Flach J, Bossie M, Vogel J, Corbett A, Jinks T, Willins DA, Silver P. A yeast RNA-binding protein shuttles between nucleus and cytoplasm. Mol Cell Biol. 1994;14:8399–8407. doi: 10.1128/mcb.14.12.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- Gottlieb M, Chavko M. Silver staining of native and denatured eucaryotic DNA in agarose gels. Anal Biochem. 1981;165:33–37. doi: 10.1016/0003-2697(87)90197-7. [DOI] [PubMed] [Google Scholar]
- Guarente L. Synthetic enhancement in gene interaction: A genetic tool come of age. Trends Genet. 1993;9:362–366. doi: 10.1016/0168-9525(93)90042-g. [DOI] [PubMed] [Google Scholar]
- Guo Z, Sherman F. 3′-end-forming signals of yeast mRNA. Trends Biochem Sci. 1996;21:477–481. doi: 10.1016/s0968-0004(96)10057-8. [DOI] [PubMed] [Google Scholar]
- Henry M, Borland CZ, Bossie M, Silver PA. Potential RNA binding proteins in Saccharomyces cerevisiae identified as suppressors of temperature-sensitive mutations in NPL3. Genetics. 1996;142:103–115. doi: 10.1093/genetics/142.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Carmichael GG. Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol Cell Biol. 1996;16:1534–1542. doi: 10.1128/mcb.16.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman LE, Moore CL. Termination and pausing of RNA polymerase II downstream of yeast polyadenylation sites. Mol Cell Biol. 1993;13:5159–5167. doi: 10.1128/mcb.13.9.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukado Y, Murata K, Kimura A. Transformation of yeast cells with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny A, Hauri H-P, Keller W. Characterization of cleavage and polyadenylation specificity factor and cloning of its 100-kilodalton subunit. Mol Cell Biol. 1994;14:8183–8190. doi: 10.1128/mcb.14.12.8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenny A, Minvielle-Sebastia L, Preker PJ, Keller W. Sequence similarity between the 73-kilodalton protein of mammalian CPSF and a subunit of yeast polyadenylation factor I. Science. 1996;274:1514–1517. doi: 10.1126/science.274.5292.1514. [DOI] [PubMed] [Google Scholar]
- Kahana J, Silver P. Current protocols in molecular biology. New York, NY: Jon Wily/Greene; 1996. Use of the A. victoria green fluorescent protein to study protein dynamics in vivo; pp. >9.7.22–9.7.28. [DOI] [PubMed] [Google Scholar]
- Kessler MM, Zhelkovsky AM, Skvorak A, Moore CL. Monoclonal antibodies to yeast poly(A) polymerase (PAP) provide evidence for association of PAP with cleavage factor I. Biochemistry. 1995;34:1750–1759. doi: 10.1021/bi00005a032. [DOI] [PubMed] [Google Scholar]
- Kessler MM, Zhao J, Moore CL. Purification of the Saccharomyces cerevisiae cleavage/polyadenylation factor I. J Biol Chem. 1996;271:27167–27175. doi: 10.1074/jbc.271.43.27167. [DOI] [PubMed] [Google Scholar]
- Köhrer K, Domdey H. Preparation of high molecular weight RNA. Methods Enzymol. 1991;194:398–405. doi: 10.1016/0076-6879(91)94030-g. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee C, Levin A, Branton D. Copper staining: A five minute protein stain for sodium dodecyl sulfate-polyacrylamide gels. Anal Biochem. 1987;166:308–312. doi: 10.1016/0003-2697(87)90579-3. [DOI] [PubMed] [Google Scholar]
- Lee MS, Henry M, Silver PA. A protein that shuttles between the nucleus and the cytoplasm is an important mediator of RNA export. Genes & Dev. 1996;10:1233–1246. doi: 10.1101/gad.10.10.1233. [DOI] [PubMed] [Google Scholar]
- Lewis, J.D., S.I. Gunderson, and I.W. Mattaj. 1995. The influence of 5′ and 3′ end structures on pre-mRNA metabolism. J. Cell Sci. (Suppl.) 19: 13–19. [DOI] [PubMed]
- Liu C, Smith LD. Differential accumulation of mRNA and interspersed RNA during Xenopus oogenesis and embryogenesis. Zygote. 1994;2:307–316. doi: 10.1017/s0967199400002136. [DOI] [PubMed] [Google Scholar]
- Manley JL, Takagaki Y. The end of the message—another link between yeast and mammals. Science. 1996;274:1481–1482. doi: 10.1126/science.274.5292.1481. [DOI] [PubMed] [Google Scholar]
- Minvielle-Sebastia L, Winsor B, Bonneaud N, Lacroute F. Mutations in the yeast RNA14 and RNA15 genes result in an abnormal mRNA decay rate; sequence analysis reveals an RNA-binding domain in the RNA15 protein. Mol Cell Biol. 1991;11:3075–3087. doi: 10.1128/mcb.11.6.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minvielle-Sebastia L, Preker PJ, Wiederkehr T, Strahm Y, Keller W. The major yeast poly(A)-binding protein is associated with cleavage factor IA and functions in pre-messenger RNA 3′-end formation. Proc Natl Acad Sci. 1997;94:7897–7902. doi: 10.1073/pnas.94.15.7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G. Nuclear export of proteins and RNAs. Curr Opin Cell Biol. 1997;9:420–429. doi: 10.1016/s0955-0674(97)80016-6. [DOI] [PubMed] [Google Scholar]
- Patel D, Butler JS. Conditional defect in mRNA 3′ end processing caused by a mutation in the gene for poly(A) polymerase. Mol Cell Biol. 1992;12:3297–3304. doi: 10.1128/mcb.12.7.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker PJ, Lingner J, Minvielle-Sebastia L, Keller W. The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell. 1995;81:379–389. doi: 10.1016/0092-8674(95)90391-7. [DOI] [PubMed] [Google Scholar]
- Ruegsegger U, Beyer K, Keller W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J Biol Chem. 1996;271:6107–6113. doi: 10.1074/jbc.271.11.6107. [DOI] [PubMed] [Google Scholar]
- Russel ID, Tollervey D. NOP3 is an essential yeast protein which is required for pre-rRNA processing. J Cell Biol. 1992;119:737–747. doi: 10.1083/jcb.119.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB, Deardorff JA. Translation initiation requires the PAB-dependent poly(A) ribonuclease in yeast. Cell. 1992;70:961–973. doi: 10.1016/0092-8674(92)90246-9. [DOI] [PubMed] [Google Scholar]
- Sachs AB, Sarnow P, Hentze MW. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Acad Sci. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclenstedt G, Hurt E, Doye V, Silver PA. Reconstitution of nuclear protein transport with semi-intact yeast cells. J Cell Biol. 1993;123:785–798. doi: 10.1083/jcb.123.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink GR, Hicks JB. Laboratory course manual for methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1986. [Google Scholar]
- Stumpf G, Domdey H. Dependence of yeast pre-mRNA 3′-end processing on CFT1: A sequence homolog of the mammalian AAUAAA binding factor. Science. 1996;274:1517–1520. doi: 10.1126/science.274.5292.1517. [DOI] [PubMed] [Google Scholar]
- Takagaki Y, Manley JL. A polyadenylation factor subunit is the human homologue of the Drosophila suppressor of forked protein. Nature. 1994;372:471–474. doi: 10.1038/372471a0. [DOI] [PubMed] [Google Scholar]
- Wahle E, Keller W. The biochemistry of polyadenylation. Trends Biochem Sci. 1996;21:247–250. [PubMed] [Google Scholar]
- Weighardt F, Biamonti G, Riva S. The roles of heterogeneous nuclear ribonucleoproteins (hnRNP) in RNA metabolism. BioEssays. 1996;18:747–756. doi: 10.1002/bies.950180910. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Datar KV, Paddy MR, Swedlow JR, Swanson MS. Characterization of nuclear polyadenylated RNA-binding proteins in S. cerevisiae. J Cell Biol. 1994;127:1173–1184. doi: 10.1083/jcb.127.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol. 1993;13:7652–7665. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kessler MM, Moore CL. Cleavage factor II of S. cerevisiae contains homologues to subunits of the mammalian cleavage/polyadenylation specificity factor and exhibits sequence-specific, ATP-dependent interaction with precursor RNA. J Biol Chem. 1997;272:10831–10838. doi: 10.1074/jbc.272.16.10831. [DOI] [PubMed] [Google Scholar]
- Zhelkovsky AM, Kessler MM, Moore CL. Structure-function relationships in the Saccharomyces cerevisiae poly(A) polymerase. J Biol Chem. 1995;270:26715–26720. doi: 10.1074/jbc.270.44.26715. [DOI] [PubMed] [Google Scholar]