

Abstract
Biochemical and genetic evidence suggests that the tyrosine kinase activity of c-Abl is tightly regulated in vivo by a cellular factor binding to the Src homology 3 (SH3) domain of Abl. We used the yeast two-hybrid system to identify a gene, PAG, whose protein product (Pag) interacts specifically with the Abl SH3 domain. Pag, also known as macrophage 23-kD stress protein (MSP23), is a member of a novel family of proteins with antioxidant activity implicated in the cellular response to oxidative stress and in control of cell proliferation and differentiation. In a co-expression assay, Pag associates with c-Abl in vivo and inhibits tyrosine phosphorylation induced by overexpression of c-Abl. Inhibition requires the Abl SH3 and kinase domains and is not observed with other Abl SH3-binding proteins. Expression of Pag also inhibits the in vitro kinase activity of c-Abl, but not SH3-mutated Abl or v-Abl. When transfected in NIH-3T3 cells, Pag is localized to nucleus and cytoplasm and rescues the cytostatic effect induced by c-Abl. These observations suggest Pag is a physiological inhibitor of c-Abl in vivo.
Keywords: c-Abl, Bcr-Abl, Philadelphia chromosome, two-hybrid system, SH3 binding protein
The c-abl proto-oncogene encodes a nonreceptor tyrosine kinase similar to the Src family of tyrosine kinases. It was originally identified as the cellular homolog of the transforming gene of Abelson murine leukemia virus (Goff et al. 1980; Wang et al. 1984). Structurally, the amino-terminal half of c-Abl is similar to many Src family kinases, consisting of a variable region followed by an SH3 domain, a Src homology 2 (SH2) domain, then the catalytic kinase or Src homology 1 (SH1) domain. Unique to the c-Abl tyrosine kinase, however, is a large carboxy terminus that is encoded by a single large exon. In this segment of >600 amino acids, there are three nuclear localization signals (Van Etten et al. 1989; Wen et al. 1996), several proline-rich sequences capable of binding to SH3-containing proteins such as Crk (Feller et al. 1994; Ren et al. 1994), a DNA-binding domain (Kipreos and Wang 1992), an actin-binding domain (McWhirter and Wang 1993; Van Etten et al. 1994), and sites for phosphorylation by protein kinase C (Pendergast et al. 1987) and cdc-2 kinase (Kipreos and Wang 1990). The physiological role of c-Abl is unknown. The ubiquitous expression pattern of c-Abl and its complex structure suggest that this tyrosine kinase may have a fundamental function in cell physiology. This is supported by the fact that mice with homozygous inactivation of the c-abl locus display a strong tendency for runted growth and neonatal death (Schwartzberg et al. 1991; Tybulewicz et al. 1991). Recently, it has been found that the nuclear form of c-Abl is an inhibitor of cell proliferation (Sawyers et al. 1994; Wen et al. 1996), and that c-Abl kinase activity can be activated by DNA damage (Kharbanda et al. 1995; Liu et al. 1996; Yuan et al. 1996) and is required for activation of the Jun kinase (JNK)/stress-activated protein kinase (SAPK) pathway, suggesting a role for c-Abl in the stress response to genotoxic insult.
In v-Abl- and Bcr/Abl-transformed cells, there are increased levels of phosphotyrosine-containing proteins (Sefton et al. 1981), and one of the most prominent phosphotyrosinated species is the Abl protein itself (Witte et al. 1981; Pendergast et al. 1993). Overexpression of c-Abl at levels 5- to 10-fold over the endogenous c-Abl, however, does not result in cell transformation or elevated tyrosine phosphorylation in cells, suggesting that the c-Abl tyrosine kinase activity is tightly regulated in vivo (Franz et al. 1989; Jackson and Baltimore 1989). In contrast, phosphotyrosine can be detected on c-Abl and many other cellular proteins after overexpression of c-Abl 100-fold or more in Cos cells or Sf9 insect cells (Pendergast et al. 1991a), suggesting that a cellular factor that regulates Abl tyrosine kinase activity may be titrated out. Abl proteins with deletion or point mutations of the SH3 domain induce transformation and increase phosphotyrosine levels in a variety of cell types (Franz et al. 1989; Jackson and Baltimore 1989; Van Etten et al. 1995), suggesting that the SH3 domain suppresses the intrinsic transforming ability of c-Abl. The negative regulatory effect of the SH3 domain, however, does not appear to act in cis, because SH3-deleted Abl and c-Abl have identical tyrosine kinase activity when assayed in vitro (Franz et al. 1989; Mayer and Baltimore 1994; Van Etten et al. 1995). Taken together, these observations suggest that c-Abl tyrosine kinase activity is reversibly inhibited in vivo by a cellular factor that interacts with the SH3 domain, such that mutations in SH3 lead to loss of inhibition of the Abl kinase by this factor, allowing expression of the latent kinase activity of Abl.
In this study, we have identified an Abl SH3-binding protein which, when overexpressed, can inhibit Abl kinase activity both in vivo and in vitro. This inhibitor associates with Abl through both the SH3 and the kinase (SH1) domains. Further, it can rescue the cytostatic effect induced by c-Abl. This protein, therefore, has many of the properties expected of a physiological inhibitor of c-Abl. Interestingly, this putative Abl inhibitor has been identified previously as a serum- and oxidative stress-induced protein with antioxidant and cell cycle regulatory properties.
Results
Identification of Abl SH3 domain binding proteins
To isolate candidate Abl inhibitors that interact with the Abl SH3 domain, we screened a HeLa cell cDNA library with the yeast two-hybrid system (Chien et al. 1991; Gyuris et al. 1993) with the murine Abl SH3 domain as a fusion bait. From a total of 10 million library transformants, we identified several positive clones that showed galactose-dependent growth on Leu− selective medium and galactose-dependent β-galactosidase activity. We rescreened these clones by use of an SH3 domain point mutant P131L as the bait in the yeast interaction trap. The P131L mutation, which substitutes leucine for a highly conserved proline residue at the hydrophobic-binding surface of SH3, blocks binding of the Abl SH3 domain to proline-rich ligands and potently activates transformation by c-Abl in vivo (Van Etten et al. 1995). Only those clones that had absent or reduced interaction with the P131L SH3 domain were analyzed further by DNA sequencing. Some of these remaining clones had proline-rich PXXP sequences, the canonical motif known to bind SH3 domains (Cicchetti et al. 1992; Ren et al. 1993), although others did not.
The most abundant clone, comprising nearly one-quarter of the final population, was recovered as two independent cDNA clones, one 650 nucleotides, the other 600 nucleotides. Both sequences were 100% identical to a previously cloned cDNA called human proliferation-associated gene (PAG) (Prosperi et al. 1993) (Fig. 1). The PAG gene is constitutively expressed in most human cells and is induced to higher levels on serum stimulation. The PAG cDNA codes for a 22-kD protein that is devoid of any consensus motif, is not particularly proline-rich, and lacks PXXP motifs. The murine homolog of Pag has been cloned as a macrophage protein (MSP23) induced by oxidative stress (Ishii et al. 1993) and as a protein (OSF-3) up-regulated during differentiation of osteoblasts (Kawai et al. 1994). Pag/MSP23 also shows significant homology to cell-surface antigens from Entamoeba histolytica and Helicobacter pylori, and to bacterial and yeast peroxiredoxin proteins (Chae et al. 1994a).
Figure 1.

Amino acid sequence of Pag and its murine and rat homologs. The sequence of human Pag (Prosperi et al. 1993) is given according to the single letter amino acid code. Letters above denote substitutions present in the murine homolog MSP23 (Ishii et al. 1993); letters below indicate substitutions in the rat homolog (HBP23) (Iwahara et al. 1995). Domains I and II are regions containing cysteine residues that are conserved among the family of peroxide reductases (Chae et al. 1994a). The arrows indicate the locations of the point of fusion of the two cDNAs recovered in the yeast two-hybrid screen with the B42 transactivation domain.
Pag interacts specifically with the Abl SH3 domain
To test the interaction specificity between Pag and the Abl SH3 domain, we repeated the yeast two-hybrid interaction with SH3 domains from c-Src, Grb-2, and the phosphatidylinositol 3-kinase (PI3K) p85 subunit. None of these SH3 domains showed any interaction with Pag (Fig. 2). Similarly, other domains of c-Abl including the carboxyl terminus and the kinase (SH1) domain, as well as several unrelated protein baits (Bicoid, cdc2 and β2-integrin; data not shown) did not interact with Pag in the yeast system.
Figure 2.

Pag interacts specifically with the Abl SH3 domain in the yeast two-hybrid system. Yeast strain EGY48, containing Pag fused to the B42 transactivation domain, was transformed with different LexA baits and β-galactosidase activity detected on galactose X-gal plates as blue color (dark). I3 and I7 are positive controls (Gyuris et al. 1993) with I3 demonstrating strong transactivation and I7 weaker transactivation. (A) Pag interacts with the Abl SH3 domain, but not with a mutant SH3 domain containing an activating point mutation (P131L) or the Abl kinase domain (SH1). (B) Pag fails to interact with SH3 domains from Grb2, PI3K p85 subunit, and c-Src.
Pag inhibits the tyrosine kinase activity of c-Abl in vivo
High-level expression of c-Abl by transient transfection induces tyrosine phosphorylation of Abl and other cellular proteins, possibly because a cellular inhibitor is present in limiting amounts (Pendergast et al. 1991a; Van Etten et al. 1995). We reasoned that an Abl SH3-binding protein that was a physiological inhibitor of c-Abl might suppress tyrosine phosphorylation when coexpressed with Abl. Accordingly, we tested clones identified in the two-hybrid screen for the ability to reduce or abrogate tyrosine phosphorylation induced by c-Abl in this assay. Coexpression of Pag in the sense, but not the antisense, orientation significantly suppressed the tyrosine phosphorylation induced by c-Abl, but not that induced by Abl proteins with SH3 deletion (ΔSH3) (Fig. 3A) or the activating SH3 point mutation (P131L) (data not shown). Interestingly, Pag was an excellent substrate for SH3-mutated Abl. None of the other clones we isolated in the two-hybrid screen had such an inhibitory effect (data not shown). Inhibition of Abl tyrosine kinase activity by Pag was sensitive to the level of expression of Pag (Fig. 3B).
Figure 3.

Pag inhibits the tyrosine phosphorylation induced by c-Abl in vivo. The indicated proteins were expressed by transient transfection of 293T cells, and lysates containing equal amounts of protein compared for their phosphotyrosine levels by immunoblotting with anti-phosphotyrosine antibody 4G10. Where indicated, the blot was then stripped and reprobed with anti-Abl mAb 8E9 to detect c-Abl or anti-hemagglutinin mAb 12CA5 to detect epitope-tagged Pag. (A) Pag inhibits c-Abl but not SH3-deleted Abl. Pag, in either sense (S) or antisense (AS) orientation, was cotransfected with wild-type c-Abl (c4) or SH3-deleted Abl into 293T cells. (B) Inhibition of Abl by Pag is dose-dependent. 293T cells were cotransfected with 5 ug of c-Abl (c4) and increasing amounts (1, 3, 5, 7, and 10 μg) of Pag. (C) Other SH3-binding proteins and pRb fail to inhibit c-Abl. 293T cells were cotransfected with Abl and different Abl SH3 domain binding proteins (Pag, Mena, Abi-1, Abi-2) or Rb SE, the C pocket of retinoblastoma gene product. Expression of these proteins at levels similar to that of Pag was confirmed by immunoblotting with anti-Mena or anti-epitope antibodies (data not shown). (D) Inhibition of Abl by Pag requires the Abl kinase domain. 293T cells were cotransfected with Pag in either sense (S) or antisense (AS) orientation and with the Abl c4-SrcTK chimera or c-Src.
We also tested several other known Abl SH3-binding proteins including Mena, the mouse homolog of Drosophila Ena (Gertler et al. 1996), and Abi-1 and Abi-2, SH3-containing proteins identified by two-hybrid screens (Dai and Pendergast 1995; Shi et al. 1995). When coexpressed with c-Abl, these SH3-binding proteins failed to inhibit tyrosine phosphorylation (Fig. 3C) but were instead good substrates for Abl kinase activity. Rb SE, the C pocket of the retinoblastoma gene product, has been shown to bind to the ATP-binding domain of nuclear Abl and inhibits its kinase activity in a cell-cycle-specific manner (Welch and Wang 1995). However, the Rb SE protein also failed to inhibit Abl-induced tyrosine phosphorylation in this hyperexpression assay (Fig. 3C). These observations show that inhibition of c-Abl kinase activity is a unique property of Pag.
In some experiments, coexpression of Pag appeared to slightly decrease the level of c-Abl protein in transfected cells (Fig. 3C, middle panel). This effect was never observed with SH3-mutated Abl or with expression of Pag in the antisense orientation. Because of the difficulty in comparing the relative expression of proteins by Western blotting, we labeled transfected cells with [35S]methionine and found a 20%–25% decrease in the level of c-Abl protein in cells cotransfected with Pag (Fig. 3C, top), suggesting that part of the reduction in phosphotyrosine mediated by Pag may be caused by decreased Abl expression. Increasing the level of Pag, however, resulted in further decreases in phosphotyrosine without affecting the expression of c-Abl (Fig. 2B), and coexpression of Pag inhibited the in vitro kinase activity of c-Abl (see below), showing that Pag also has a direct inhibitory effect on c-Abl kinase activity.
Inhibition of tyrosine phosphorylation might also be caused by some nonspecific effect of overexpression of Pag such as activation of a tyrosine phosphatase. To address this issue, we tested the effect of Pag on a closely related nonreceptor tyrosine kinase, c-Src, in the hyperexpression assay. Pag had no effect on phosphotyrosine levels induced by either overexpressed c-Src (Fig. 3D) or the c-Src mutant Y527F (data not shown). We also tested the effect of Pag on an Abl-SrcTK chimera in which the kinase domain from c-Src had been substituted for the Abl kinase domain, and could not detect any inhibition of the tyrosine phosphorylation induced by this chimera (Fig. 3D). These observations suggest that inhibition of tyrosine phosphorylation by Pag is specific to Abl and requires the Abl SH3 and kinase domains.
Pag and c-Abl associate in vivo
To investigate the in vivo association between Pag and c-Abl, the PAG gene product was epitope-tagged with the nine amino acid epitope from the influenza hemagglutinin (HA) protein recognized by monoclonal antibody 12CA5 (Wilson et al. 1984), and coexpressed with Abl in 293T cells. Pag coimmunoprecipitated with c-Abl (Fig. 4A), showing an in vivo association between Pag and Abl. Surprisingly, c4 ΔSH3, the SH3 deletion mutant of Abl, also coimmunoprecipitated with Pag, raising the possibility that the interaction between Pag and Abl in vivo may involve more than just the SH3 domain alone. To map a second interacting site, a series of Abl deletion mutant proteins with or without the SH3 domain were coexpressed with Pag. The c4 ΔSH3 ΔBcl protein, which is missing most of the carboxyl terminus, coimmunoprecipitated with Pag (Fig. 4B), indicating the carboxyl terminus of Abl is dispensable for this interaction. However, Abl proteins with deletion of the entire kinase domain (ΔSH1) or just the ATP-binding lobe (ΔATP) showed a drastic decrease in coimmunoprecipitation, suggesting that the ATP-binding lobe of the Abl kinase domain may be directly or indirectly involved in the interaction of Pag and Abl. Tyrosine phosphorylation of Abl is not required for the Pag-Abl interaction because the mutant c4 ΔBgl-Bcl, which lacks the carboxy-terminal catalytic lobe of the kinase domain and is not detectably tyrosine phosphorylated upon overexpression (data not shown), retained significant co-immunoprecipitation with Pag (Fig. 4B).
Figure 4.
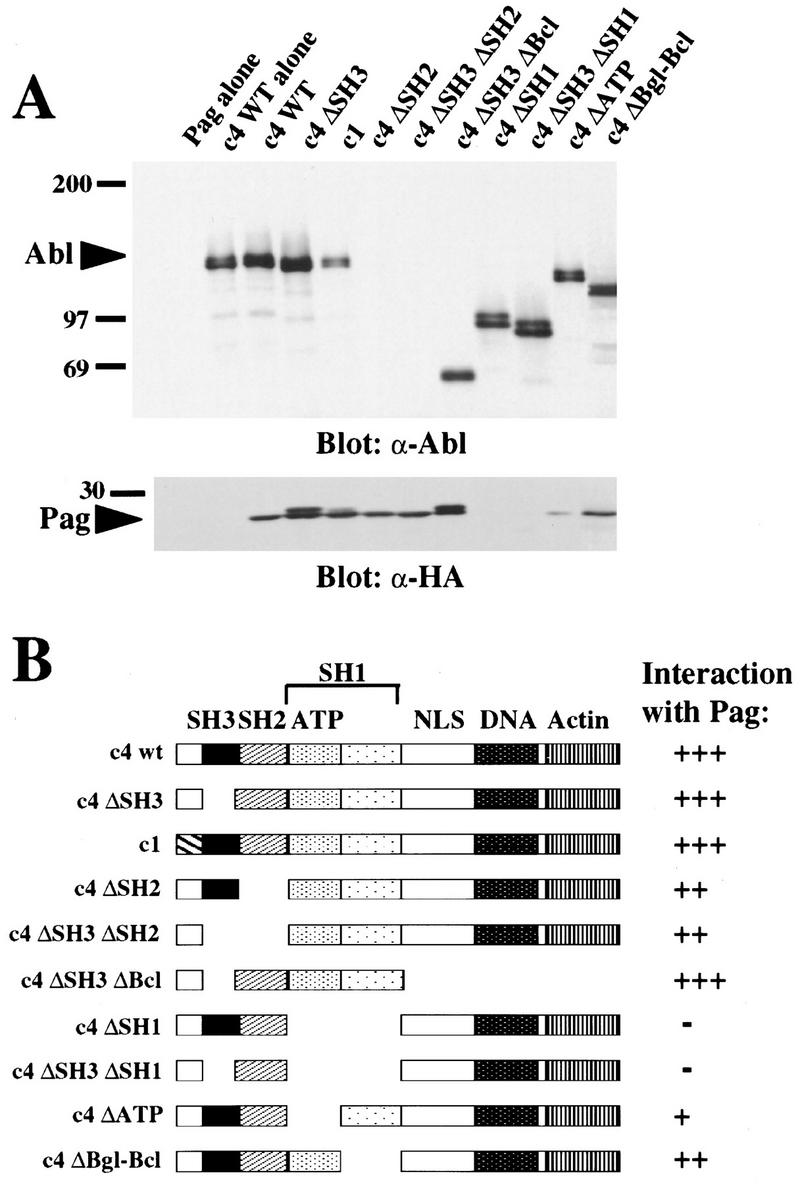
Pag interacts with Abl in vivo. (A) Epitope-tagged Pag and the indicated Abl proteins were coexpressed in 293T cells, cell extracts immunoprecipitated with anti-Abl antisera α-GEX4 (directed against the carboxyl terminus of Abl) (Jackson et al. 1993) or α-type Ib Abl antisera (directed against the Abl amino terminus, for the c4 ΔSH3 ΔBcl protein), and immunoblotted with anti-HA mAb 12CA5 for the detection of Pag or anti-Abl Ab 8E9 (directed against the SH2 domain of Abl). The ΔSH2 proteins were detected with a different anti-Abl Ab and found to be expressed at equivalent levels (data not shown). Coimmunoprecipitation of Abl with Pag was also observed when the anti-HA mAb was used for immunoprecipitation (data not shown). (B) Schematic representation of Abl deletion constructs and their interaction with Pag as measured by coimmunoprecipitation. The location of the SH3, SH2, and SH1 domains, the ATP-binding lobe of the kinase domain, the pentalysine nuclear localization signal, and the DNA-binding and actin-binding domains are indicated.
Pag is localized to nucleus and cytoplasm
To determine the subcellular localization of Pag, epitope-tagged Pag was coexpressed with c-Abl in both 293T and NIH-3T3 cells. Double immunofluorescence staining with anti-epitope monoclonal antibody (mAb) 12CA5 and anti-Abl antibodies was performed. Pag was expressed both in the nucleus and the cytoplasm in both cell types, a localization pattern similar to that of c-Abl (Fig. 5). Although Pag lacks a canonical nuclear localization signal, it could be translocated into the nucleus by association with other proteins such as Abl or enter passively because of its small size (Dingwall and Laskey 1991).
Figure 5.
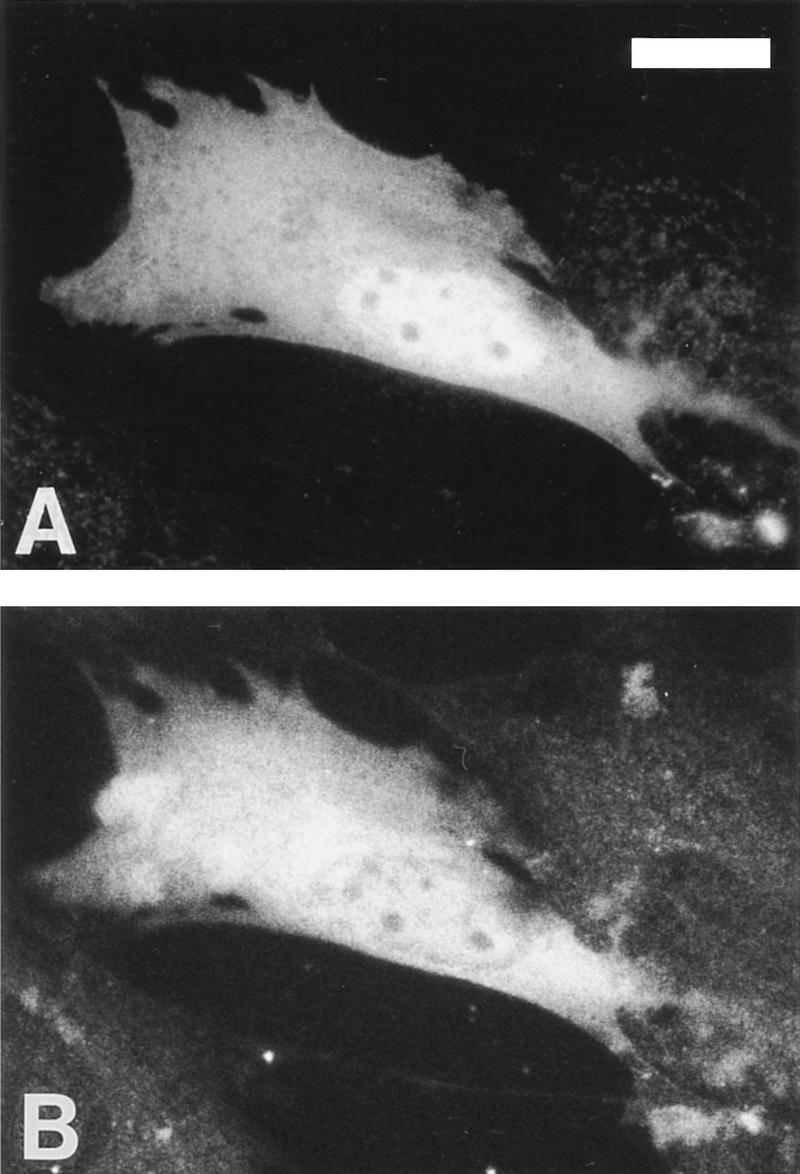
Pag is localized to nucleus and cytoplasm. NIH-3T3 cells were transfected with epitope-tagged Pag and type IV c-Abl and the expressed proteins were localized with indirect immunofluorescence with rabbit anti-Abl antibodies (A) and anti-HA mAb 12CA5 (B). Background staining levels are indicated by several nontransfected cells in the field. Cells transfected with Pag or Abl alone and stained with the opposite secondary antibody gave similar background staining (data not shown). Bar, 10 μm.
Pag inhibits phosphorylation by Abl in an in vitro kinase assay
Although c-Abl is not normally tyrosine phosphorylated in vivo, Abl tyrosine kinase activity, measured as autophosphorylation or phosphorylation of a physiological substrate such as c-Crk, can be readily detected after immunoprecipitation of c-Abl (Pendergast et al. 1991a). This suggests that cellular inhibitors of Abl may be partially or completely purified away during the immunoprecipitation process. We reasoned that overexpression of Pag might restore inhibition to Abl in an immune complex kinase assay. We immunoprecipitated c-Abl, SH3-deleted Abl, Bcr/Abl, and p160 v-Abl from 293T cells cotransfected with Pag, and tested their in vitro kinase activity with glutathione S-transferase (GST)–Crk as a substrate. Under conditions where significant coimmunoprecipitation of Pag and Abl was detected, Pag inhibited c-Abl autophosphorylation and phosphorylation of GST–Crk in vitro by 60%–70%, whereas it had no significant effect on the in vitro kinase activity of SH3-deleted Abl and v-Abl, which lacks the Abl SH3 domain (Fig. 6A). Interestingly, Pag did inhibit the auto- and transkinase activity of Bcr/Abl by ∼30% (Fig. 6A).
Figure 6.

Pag inhibits Abl kinase activity in vitro. (A) 293T cells cotransfected with Pag and wild-type c-Abl, SH3-deleted Abl (c4 ΔSH3), Bcr/Abl, or p160 v-Abl were immunoprecipitated with anti-Abl antibody and immune complex kinase assays performed with GST–Crk as a substrate. (Top) Western blot with anti-Abl antibody showing equivalent amounts of Abl proteins were immunoprecipitated. (Bottom) Autoradiograph showing32P incorporation. (B) NIH-3T3 cells were transfected with Pag in either sense (S) or antisense (AS) orientation and huICAM-1 as a cell-surface marker, selected with magnetic beads, immunoprecipitated with anti-Abl Ab, and immune complex kinase assays performed with GST–Crk as a substrate. (C) (His)6-purified Abl protein was tested for its in vitro kinase activity with GST–Crk as a substrate and increasing amounts of parental GST or GST–Pag fusion protein added.
By analogy to c-Src, tyrosine phosphorylation of c-Abl may stimulate Abl tyrosine kinase activity (Cooper and MacAuley 1988). Because coexpression of Pag and c-Abl in 293T cells suppresses tyrosine phosphorylation of Abl, the lower kinase activity of immunoprecipitated Abl might merely reflect decreased Abl tyrosine phosphorylation. To address this, we tested whether overexpression of Pag could inhibit the in vitro kinase activity of endogenous c-Abl after immunoprecipitation from NIH-3T3 cells. We overexpressed Pag transiently in NIH-3T3 cells together with human ICAM-1 as a cell surface marker, selected positively transfected cells with magnetic beads coated with antibody against huICAM-1, immunoprecipitated endogenous c-Abl, and performed an in vitro kinase assay with GST–Crk as a substrate. Overexpression of Pag had a significant (60%) inhibitory effect on the phosphorylation of GST–Crk by c-Abl, although it had less effect on the autophosphorylation of endogenous c-Abl (Fig. 6B).
These results showed that Pag had an inhibitory effect on Abl kinase activity in vitro when both Pag and Abl were purified from cells, but did not rule out the possibility that additional factor(s) coimmunoprecipitating with Abl and Pag were necessary for inhibition. To determine whether Pag protein could directly inhibit c-Abl, we purified hexahistidine-tagged c-Abl from transfected 293T cells by affinity chromatography on Nickel-agarose (B. Brasher and R.A. Van Etten, unpubl.), and tested whether the kinase activity of (His)6-c-Abl was inhibited by the addition of a GST–Pag fusion protein. When Pag was added at 10- to 15-fold molar excess over c-Abl, there was no inhibition of Abl kinase activity as measured by phosphorylation of GST–Crk (Fig. 6C). Only when Pag was added at higher (40- to 75-fold) molar excess over Abl was a modest decrease in GST–Crk phosphorylation observed (data not shown). These results suggest that Pag alone is unable to inhibit Abl kinase activity, but requires the presence of one or more additional cellular factors for efficient inhibition of c-Abl.
Pag rescues the cytostatic effect of c-Abl in NIH-3T3 cells
c-Abl has been reported to contribute to G1 arrest (Yuan et al. 1996) and apoptosis (Yuan et al. 1997) in response to ionizing radiation in fibroblasts. Overexpression of Abl in NIH-3T3 cells leads to growth arrest in G1 of the cell cycle (Jackson and Baltimore 1989; Sawyers et al. 1994), an effect that requires nuclear localization of Abl, Abl kinase activity, and the p53 and Rb tumor suppressor gene products (Wen et al. 1996). The mechanism of the Abl cytostatic effect is not known, but likely involves activation of c-Abl kinase in the nucleus of transfected cells. If Pag is a bona fide Abl inhibitor, it should be able to abrogate this G1/S cell cycle block. Importantly, cotransfection of Pag in the sense, but not antisense, orientation rescued the cytostatic effect induced in NIH-3T3 cells by c-Abl, but Pag had no effect on the cytostatic effect induced by SH3-mutated Abl, P131L (Fig. 7), nor did expression of Pag alone have any effect on the G1/S transition. Coexpression of Pag also dramatically increased the transfection efficiency of c-Abl in this assay (Fig. 7), suggesting that Pag also rescues the cytotoxic effect of c-Abl. These results suggest that Pag may normally function to inhibit negative growth and apoptotic responses associated with activation of nuclear c-Abl.
Figure 7.

Pag rescues the cytostatic and cytotoxic effects induced by c-Abl. NIH-3T3 cells were transfected with the indicated Abl constructs with or without PAG in the sense (S) or antisense (AS) orientation, and entry into the S phase of the cell cycle was measured as the percentage of transfected cells positive for BrdU incorporation relative to kinase-inactive Abl. (LacZ) Transfection of the β-galactosidase reporter construct alone; (c4) c-Abl type IV; (K290M) kinase-inactivating point mutant; (1Q2Q3Q) site-specific mutations in the first, second, and third Abl nuclear localization signals; (P131L) activated SH3 point mutant of c-Abl; (PAG-S and PAG-AS) cotransfection of a PAG expression vector in the sense or antisense orientation, respectively. The transfection efficiency (absolute percentage of β-galactosidase positive cells) is shown in parentheses after the construct name.
Discussion
A large body of experimental evidence suggests that Src-like tyrosine kinases are negatively regulated by intramolecular interactions. Src family kinases such as c-Src and Hck are inhibited by interaction of a phosphorylated tyrosine (residue 527 in c-Src) in the carboxy-terminal tail with the SH2 domain, with the SH3 domain also participating (Sicheri et al. 1997; Xu et al. 1997). Kinases of the Tec family, including Btk and Itk, are likely to be negatively regulated by intramolecular association of the SH3 domain with an adjacent proline-rich domain (Andreotti et al. 1997). It is unlikely that c-Abl is similarly regulated by intramolecular interactions, however. First, Abl has no Tyr 527 homolog in its carboxyl terminus and is not tyrosine-phosphorylated in the inactive state (Jackson and Baltimore 1989). Second, although mutation of the conserved FLVRES motif in the SH2 domain activates c-Src (Hirai and Varmus 1990a,b), the same mutation in the Abl SH2 domain abolishes fibroblast transformation by activated Abl (Mayer et al. 1992). Third, SH3-deleted or mutated Abl does not have increased tyrosine kinase activity in vitro (Mayer and Baltimore 1994; Van Etten et al. 1995), but Src proteins with deletions of the SH3 or SH2 domain have elevated in vitro kinase activity (Seidel-Dugan et al. 1992). Fourth, although we have detected binding of the Abl SH3 domain to a region in the carboxyl terminus of Abl (R.A. Van Etten, unpubl.), deletion of the carboxyl terminus does not activate Abl in vitro or in vivo. Taken together, these observations indicate that Abl and Src-like kinases are regulated by different mechanisms.
The existence of a cellular Abl inhibitor has been suggested by several lines of evidence, as mentioned previously. By use of a yeast two-hybrid system with the Abl SH3 domain fusion protein as a bait, we isolated several Abl SH3 domain interacting clones. One of these clones, the PAG gene, encodes a polypeptide that has many properties expected of a physiological inhibitor of c-Abl. First, Pag interacts specifically with Abl SH3 domain in the yeast system, and not with other SH3 domains from c-Src, Grb-2, or the PI3K p85 subunit. Second, when Pag and Abl are coexpressed in 293T cells, Pag inhibits the tyrosine phosphorylation induced by wild-type Abl but not that induced by Abl SH3 domain deletion (ΔSH3) and point (P131L) mutants. Further, the inhibition of tyrosine phosphorylation is both Abl-specific, as Pag has no effect on the closely related c-Src tyrosine kinase, and Pag-specific, as inhibition of c-Abl is not observed with other Abl SH3 domain-binding proteins such as Mena, Abi-1, and Abi-2. Third, Pag and Abl associate in vivo, as shown by coimmunoprecipitation. Fourth, Pag inhibits phosphorylation by Abl in an in vitro kinase assay by use of GST–Crk as a substrate. Finally, Pag rescues the cytostatic effect induced by c-Abl in NIH-3T3 cells, lending further support to the physiological significance of Pag.
Pag is the first Abl SH3 domain-interacting protein that can inhibit Abl kinase activity both in vivo and in vitro. Three other genes have been cloned by use of the two-hybrid screen in the search for Abl inhibitors. Abi-1 and Abi-2 are members of a family of SH3-containing proteins that have homology to homeobox transcription factors. Abi-1 binds to the carboxyl terminus of Abl and can suppress v-Abl transformation (Shi et al. 1995). Abi-2, identified with the Abl SH3 domain as a bait, also binds to the carboxyl terminus of Abl. Coexpression of a carboxy-terminal fragment of Abi-2 with c-Abl in fibroblasts appears to activate transformation by Abl (Dai and Pendergast 1995). Abi-1 and Abi-2, however, have not been shown directly to inhibit c-Abl kinase activity, but rather have some properties that suggest they may be effectors of Abl function. The AAP1 protein, isolated with the Abl SH3–SH2 domain as a bait, decreases the phosphorylation of GST–Crk by Abl in vitro (Zhu and Shore 1996); however, no in vivo role for AAP1 has been shown.
Pag appears to interact with Abl in vivo through multiple domains. Pag was initially identified in our two-hybrid screen as an SH3-interacting protein. Although some prolines are present in the Pag sequence, they do not form the consensus PXXP, a motif that has been shown to bind to SH3 domains (Cicchetti et al. 1992; Ren et al. 1993). The failure of Pag to interact with the P131L SH3 domain in yeast, however, strongly suggests Pag binds to SH3 via the same ligand-binding surface occupied by PXXP-containing ligands. The PXXP-binding motif was identified with combinatorial peptide (Feng et al. 1994; Yu et al. 1994) or phage display (Rickles et al. 1994, 1995) libraries to select SH3-binding sites, which may select for peptide ligands with prolines at i and i + 3 positions because of the requirement for polyproline II (PPII) helix formation for SH3 binding. Because polypeptides lacking proline can also assume a PPII conformation (Adzhubei and Sternberg 1993), it is possible that other classes of SH3 ligand exist. In support of this, in the three-dimensional crystal structure of c-Src, the SH3 domain and kinase domain are closely apposed by interaction of SH3 with a 14-residue polypeptide linker that contains only one proline, but adopts a PPII helical conformation in complex with the SH3 domain (Xu et al. 1997). The Pag protein may assume a similar conformation in making contact with the Abl SH3 domain. The location of the SH3-binding site in Pag is currently under investigation, but can be inferred to be in the carboxyl terminus, because a B42–Pag fusion protein containing only the carboxy-terminal 79 amino acids of Pag binds strongly to the Abl SH3 domain in yeast (Fig. 1).
Interestingly, Pag appears to interact with both the SH3 and the kinase domains of Abl in vivo. Indeed, the interaction of Pag with the kinase domain appears to be quantitatively more important than the SH3 interaction for coimmunoprecipitation of Pag and Abl. This might reflect the relatively low (micromolar) binding affinity observed for SH3-ligand interactions (Feng et al. 1994; Van Etten et al. 1995). Analysis of Abl deletion mutants suggests that Pag interacts with the amino-terminal portion of the kinase domain, which comprises the ATP-binding lobe. In this respect, the interaction of Pag with Abl is reminiscent of cyclin-dependent kinase inhibitors. Mutagenesis of the p21WAF1/CIP1 cyclin-dependent kinase inhibitor reveals independent binding sites for cyclin and cdk2, both of which are required for inhibitory activity (Fotedar et al. 1996); this type of interaction is confirmed in the structure of the the p27Kip1 cyclin-dependent-kinase inhibitor, which binds to the cyclin A–cdk2 complex by making contact with both the cyclin A subunit and the ATP-binding lobe of cdk2 (Russo et al. 1996). It is possible that Pag does not contact the kinase domain directly, however, because Pag failed to interact with the SH1 domain in yeast. Also, Pag was unable to efficiently inhibit c-Abl kinase activity when the two purified proteins were mixed in vitro. Failure of GST–Pag to inhibit purified c-Abl might again reflect a low binding affinity of Pag for Abl and the concentration of Pag (2–5 μm) that we used, but the strength of induction of the lacZ reporter gene by B42-Pag in yeast (Fig. 2) was suggestive of a considerably stronger Pag-SH3 association, on the order of 50–100 nm (Gyuris et al. 1993). Another possibility is that addition of the GST moiety to the amino terminus of Pag interferes with its function as an inhibitor, which seems unlikely because Pag can tolerate addition of an epitope tag (consisting of 22 amino acids) at its amino terminus and retain inhibitory function in vivo. The most plausible explanation is that Pag may require another cellular factor to inhibit Abl efficiently, and it is possible that this factor makes direct contact with the catalytic cleft of the Abl kinase domain. The use of Pag as the bait in a two-hybrid screen might help to identify this cofactor.
Pag only weakly inhibited the in vitro kinase activity of Bcr/Abl fusion protein of human chronic myelogenous leukemia (Fig. 6A), and coexpression of Pag did not decrease the level of tyrosine phosphorylation induced by Bcr/Abl in vivo (data not shown). This is interesting because the Bcr/Abl protein retains the Abl SH3 domain. The Bcr moiety of the fusion protein contains a coiled-coil oligomerization domain (McWhirter et al. 1993) and a region which mediates binding to the Abl SH2 domain (Pendergast et al. 1991b), both of which are required for Bcr/Abl transforming activity. In addition, Bcr/Abl exhibits elevated in vitro kinase activity relative to c-Abl (Lugo et al. 1990), whereas deletion of the Abl SH2 domain reduces the in vitro kinase activity of Bcr/Abl to that of c-Abl (Ilaria and Van Etten 1995). These observations suggest that the kinase activity of Bcr/Abl is stimulated by inter- or intramolecular interactions mediated by Bcr and Abl SH2, and it is plausible that the SH3 domain of Bcr/Abl may be inaccessible to ligands such as Pag as a consequence.
The PAG gene was cloned by differential screening of cDNA libraries from an untransformed and ras-transformed human mammary epithelial cell line (Prosperi et al. 1993). It is fairly ubiquitously expressed, as is c-Abl, and is induced to higher levels with serum stimulation. The murine homolog of Pag, MSP23, was cloned as a cDNA induced in mouse peritoneal macrophages by oxidative stress (Ishii et al. 1993). Like Pag, MSP23 is widely expressed among various tissues under normal conditions. Pag/MSP23 has homology to a growing family of proteins from prokaryotes and eukaryotes, many of which function as peroxide reductases (Chae et al. 1994a), and purified MSP23 has been shown to have thiol-specific antioxidant activity in vitro (Ishii et al. 1995). Additional functional roles are suggested for several members of this family. The MER5 gene was identified as a cDNA induced during early differentiation of murine erythroleukemia (MEL) cells (Yamamoto et al. 1989), and antisense-mediated inhibition of MER5 expression partially blocks MEL cell differentiation (Nemoto et al. 1990). A close relative of Pag, NKEF-A, was originally identified as a factor capable of enhancing natural killer cell cytotoxicity (Shau et al. 1994).
Our results suggest an additional function for Pag/MSP23: inhibition of c-Abl kinase activity. It is unlikely that antioxidant activity of Pag is responsible for this inhibition, because we have observed significant suppression of Abl-induced tyrosine phosphorylation with amino-terminal truncation mutants of Pag that lack the conserved Cys52 residue required for antioxidant activity (Chae et al. 1994b), (data not shown). Abl SH3-binding and kinase inhibition is, therefore, likely to represent a distinct function of Pag. Oxidative stress induces a pleiotropic physiologic response in mammalian cells, including induction of antioxidant proteins, protein-tyrosine phosphatases, and transcription factors such as c-Jun and NF-κB (Sies 1993). It is provocative that c-Abl kinase activity is stimulated by ionizing radiation and radiomimetic chemicals such as mitomycin C and cisplatin, both of which induce DNA damage via generation of free radicals. It is possible that the direct activation of c-Abl by these stimuli is mediated by free radicals, causing dissociation of Pag and increased enzymatic activity of both Abl and Pag, whereas the subsequent increase in Pag levels restores inhibition to Abl as part of a negative feedback response.
Pag might also play a role in regulating the cell cycle in the absence of oxidative stress. Dominant negative and antisense approaches suggest c-Abl may inhibit the G0 to G1 transition in fibroblasts (Sawyers et al. 1994; Daniel et al. 1995). It is possible that up-regulation of Pag with serum stimulation serves to relieve an Abl-dependent block to cell cycle entry. In addition, as an inhibitor of a proto-oncogene, PAG is also a candidate tumor suppressor gene. It is of interest that structural alterations of the chromosome 1p34.1, where PAG is located (Prosperi et al. 1994), are not infrequent in several types of human cancer, with translocations involving 1p34 in leukemia, particularly acute lymphoblastic leukemia (ALL), and deletions of distal 1p frequent in ALL, breast cancer, and neuroblastoma (Mitelman 1994).
Materials and methods
Cells and cell culture
NIH-3T3 and 293T cells were the kind gift of L. Klickstein (Brigham and Women’s Hospital, Boston, MA). NIH-3T3 cells were propagated in Dulbecco’s modified Eagle medium (DMEM) with 4.5 grams/liter of glucose, 10% calf serum, and penicillin/streptomycin. 293T cells were grown in DMEM, 10% heat-inactivated fetal calf serum, glutamine, nonessential amino acids, and penicillin/streptomycin.
DNA constructs
A cDNA fragment encoding the c-Abl SH3 domain (amino acids 72–138) was excised from pPLc4 Abl (Van Etten et al. 1989) by XmnI–HincII restriction enzyme digestion, then fitted with BamHI linkers and subcloned in frame into the plasmid Lex202+PL (kind gift of R. Brent, Massachusetts General Hospital, Boston). The resultant plasmid LexA–Abl SH3 directed the synthesis of a fusion protein containing the LexA DNA-binding domain and the SH3 domain of Abl when expressed in yeast strain EGY48. A similar strategy was used to generate yeast expression plasmids for the LexA-c-Src SH3 (amino acids 85–145), LexA–PI3K p85 SH3 (amino acids 1–85), LexA–Grb2 N-SH3 (amino acids 4–60), LexA–Abl SH1 (amino acids 239–540), and LexA–Abl carboxyl terminus.
The PAG cDNA was a generous gift of G. Goubin (Institut Curie, Paris, France). To express Pag in mammalian cells, the entire PAG cDNA was subcloned in-frame into pCGN (obtained from W. Herr, Cold Spring Harbor Laboratory, NY) at the BamHI site downstream of the sequence encoding the hemagglutinin (HA) tag.
The mammalian expression vector pcDNA-c4 Abl and its various internal deletion mutations have been described previously (Wen et al. 1996). To generate the kinase domain deletion mutants, the plasmid pGDN–Abl (kind gift of B. Mayer; Mayer and Baltimore 1994), in which a BamHI site had been created at the beginning of the kinase domain, was used. Constructs with deletion of the entire kinase domain or just the ATP-binding lobe were generated by BamHI–BclI or BamHI–BglII digestion respectively, fusing the ends in-frame, and subcloning the deleted cDNAs back into pcDNA vector. The GSC-Abl–SrcTK chimera plasmid (also a gift of B. Mayer; Mayer and Baltimore 1994) was similarly subcloned into the pcDNA vector.
For production of bacterial fusion protein, the PAG cDNA was subcloned in-frame into pGEX-4T-2 (Pharmacia) at the BamHI site. pGEX-2T-Crk (amino acids 120–225) and pGEX-4T-2-Pag were introduced into the Escherichia coli host strain NB42 F′, and GST fusion proteins purified by affinity chromatography on glutathione-agarose (Molecular Probes) as described (Van Etten et al. 1989).
The cDNA-encoding murine Mena was a generous gift of F. Gertler (Fred Hutchinson Cancer Research, Seattle, WA). The entire coding sequence of Mena was subcloned in-frame into pCGN mammalian expression vector. A cDNA encoding the full-length Abi-1 protein was the kind gift of S. Goff (Columbia University School of Medicine, New York, NY) and was cloned into pcDNAI. The pCGN-Abi-2 plasmid was a kind gift of A.M. Pendergast (Duke University Medical Center, Durham, NC). pFLAG-Rb SE was a gift of J. Wang (University of California, San Diego). The pBSK-c-Src and pBSK-c-Src Y527F were gifts of D. Morgan (University of California, San Francisco); the entire coding sequence of these two cDNAs were subcloned in frame into pcDNAI. The pCDM8-huICAM-1 was a gift of T. Springer (Harvard Medical School, Boston, MA).
Yeast two-hybrid screen
A genetic screen using the yeast interaction trap was performed as described (Gyuris et al. 1993; Zervos et al. 1993). The bait plasmid LexA–Abl SH3 was transformed into yeast strain EGY48 with a reporter plasmid (JK103) that contained the LexAop–LEU2 reporter gene. This strain was transformed with the HeLa interaction library, generated by fusion of cDNAs with the activation domain carried on the B42 acid blob (Zervos et al. 1993). Yeast transformation was performed by the lithium acetate method (Gietz et al. 1992). Ten million primary library transformants were amplified and screened. Seven hundred and fifty interacting clones were selected as meeting the following criteria: (1) They grew on Ura− His− Trp− Leu−–galactose plates but not on Ura− His− Trp− Leu−–glucose plates; and (2) they turned blue on Ura− His− Trp− X-gal–galactose plates but not on Ura− His− Trp− X-gal–glucose plates. Plasmids from these interacting clones were isolated by transformation of E. coli strain KC8. To test whether the library cDNAs encoded proteins that interacted specifically with the Abl SH3 domain, the library clones were transformed back into yeast along with either LexA–Abl SH3, LexA–Abl SH3 (P131L), LexA–Bicoid, LexA–cdc2, or LexA–Integrin. Those clones that interacted with SH3 (P131L) or the nonspecific baits were not considered further. The remaining specific interacting clones (∼200 clones) were DNA sequenced with dideoxy sequencing according to the manufacturer’s directions (U.S. Biochemical).
Transfections and Abl cytostatic assay
Calcium phosphate transfection of 293T cells was performed as described (Van Etten et al. 1995). LipofectAMINE was used to transfect NIH-3T3 cells according to manufacturer’s directions (GIBCO/Life Technologies). For the analysis of cell cycle arrest by Abl, the cells were labeled with 5-bromo-2-deoxyuridine (BrdU), then analyzed with immunofluorescence and histochemical staining as described previously (Wen et al. 1996). In some experiments, NIH-3T3 cells cotransfected with the huICAM-1 expression plasmid were positively selected by staining cells with the anti-huICAM-1 mAb R6-5 and anti-mouse microbeads, followed by selection on a MicroMACS column (Miltenyi Biotec).
Protein analysis
Proteins were analyzed by direct Western blotting or blotting after immunoprecipitation. 293T cell extracts containing equivalent amounts of Abl proteins were immunoprecipitated with anti-GEX4 antibody (directed against carboxy-terminal Abl sequences) as described (Van Etten et al. 1995), or with anti-exon Ib c-Abl antibody (gift of O. Witte, University of California, Los Angeles), then blotted with either anti-HA 12CA5 mAb (BabCo), anti-phosphotyrosine mAb 4G10 (Upstate Biotechnology, Inc.), or anti-Abl SH2 domain antibody 8E9 (Pharmingen), and developed with enhanced chemiluminescence (Amersham). For 35S-labeling of proteins, cells were labeled overnight with 35S-EXPRESS Protein labeling mix (125 μCi/plate, New England Nuclear), and extracts prepared and immunoprecipitated with anti-GEX4 antisera under conditions of quantitative immunoprecipitation of Abl. Immunoprecipitated proteins were resolved by SDS-PAGE and detected by autoradiography utilizing Amplify fluorescence enhancement (Amersham).
In vitro kinase assay
The immune complex kinase was performed as described (Konopka and Witte 1985; Jackson and Baltimore 1989). The kinase reaction was carried out in 50–100 μl of kinase buffer containing 20 mm PIPES at pH 7.2, 20 mm MnCl2, 0.5 μg GST–Crk, and 0.2 μl of [γ-32P]ATP (10 mCi/ml) at room temperature for 20 min. The proteins were eluted in Laemmli sample buffer and analyzed by SDS-PAGE and autoradiography. For the in vitro kinase assay with purified Abl, (His)6-tagged Abl was purified from 293T cells with nickel–agarose (Qiagen), eluted with 80 mm imidazole in high salt buffer (500 mm NaCl, 20 mm HEPES at pH 7.5), and concentrated with centricon-10 microfiltration unit (Amicon) (B. Brasher, unpubl.). GST or the GST–Pag fusion protein, purified from E. coli by affinity chromatography on glutathione–agarose, was added in 1.5- to 15-fold molar excess (corresponding to a Pag concentration of 0.2–2.0 μm) or 7.5- to 75-fold molar excess relative to Abl protein and the kinase reaction performed under similar conditions. Results were quantitated by densitometry and by PhosphorImager analysis.
Immunofluorescence
NIH-3T3 and 293T cells transfected with pCGN–PAG and pcDNA–c4 Abl were fixed with methanol and acetone and stained with anti-HA mAb 12CA5 and affinity-purified rabbit anti-GEX4 Abl antibodies, followed by rhodamine-conjugated donkey anti-rabbit IgG and fluorescein-conjugated donkey anti-mouse IgG (Jackson Immunoresearch), as described (Wen et al. 1996). Expressing cells were photographed on a Zeiss Axiophot microscope with Tmax ASA 400 black-and-white film (Kodak).
Acknowledgments
We thank Dr. Roger Brent for kindly providing the yeast two-hybrid cloning system; Dr. Gerard Goubin for the generous gift of the cDNA of the PAG gene; Dr. Frank Gertler for the cDNA of Mena, anti-Mena antibody, and sharing unpublished data; Bradley Brasher for the pcDNA-Abl (His)6 construct; Dr. Stephen Goff for the cDNA clone of Abi-1; Dr. Ann Marie Pendergast for the cDNA clone of Abi-2; Dr. Jean Wang for the pFLAG-Rb SE plasmid; Dr. David Morgan for the cDNA of c-Src and c-Src Y527F; Dr. Bruce Mayer for the Abl-SrcTK chimera and Abl ΔSH2 mutant; Dr. Timothy Springer for the cDNA of huICAM-1 and the anti-huICAM-1 antibody; Dr. Owen Witte for the anti-Abl type Ib antibody; and Dr. Peter Jackson for reading the manuscript and many helpful discussions. This work was supported in part by a grant from the Lucille P. Markey Charitable Trust. R.A.V. is a Lucille P. Markey Scholar in Biomedical Science and The Carl and Margaret Walter Scholar in Blood Research at Harvard Medical School.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rvanett@warren.med.harvard.edu; FAX (617) 278-3030.
References
- Adzhubei AA, Sternberg MJ. Left-handed poly-proline II helices commonly occur in globular proteins. J Mol Biol. 1993;229:472–493. doi: 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci. 1994a;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae HZ, Uhm TB, Rhee SG. Dimerization of thiol-specific antioxidant and the essential role of cystein 47. Proc Natl Acad Sci. 1994b;91:7022–7026. doi: 10.1073/pnas.91.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C-T, Bartel PL, Sternglanz R, Fields S. The two-hybrid system: A method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti P, Mayer BJ, Thiel G, Baltimore D. Identification of a protein that binds to the SH3 region of Abl and is similar to Bcr and GAP-rho. Science. 1992;257:803–806. doi: 10.1126/science.1379745. [DOI] [PubMed] [Google Scholar]
- Cooper JA, MacAuley A. Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proc Natl Acad Sci. 1988;85:4232–4236. doi: 10.1073/pnas.85.12.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes & Dev. 1995;9:2569–2582. doi: 10.1101/gad.9.21.2569. [DOI] [PubMed] [Google Scholar]
- Daniel R, Cai Y, Wong PMC, Chung S-W. Deregulation of c-abl mediated cell growth after retroviral transfer and expression of antisense sequences. Oncogene. 1995;10:1607–1614. [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Nuclear targeting sequences—a consensus? Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994;13:2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Chen JK, Yu H, Simon JA, Schreiber SL. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science. 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- Fotedar R, Fitzgerald P, Rousselle T, Cannella D, Doree M, Messier H, Fotedar A. p21 contains independent binding sites for cyclin and cdk2: Both sites are required to inhibit cdk2 kinase activity. Oncogene. 1996;12:2155–2164. [PubMed] [Google Scholar]
- Franz WM, Berger P, Wang JYJ. Deletion of an N-terminal regulatory domain of the c-abl tyrosine kinase activates its oncogenic potential. EMBO J. 1989;8:137–147. doi: 10.1002/j.1460-2075.1989.tb03358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425–1427. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the abelson murine leukemia virus genome and the homologous cellular gene: Studies with cloned viral DNA. Cell. 1980;22:777–785. doi: 10.1016/0092-8674(80)90554-1. [DOI] [PubMed] [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase phosphatase that associates with cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Hirai H, Varmus HE. Mutations in src homology regions 2 and 3 of activated chicken c-src that result in preferential transformation of mouse or chicken cells. Proc Natl Acad Sci. 1990a;87:8592–8596. doi: 10.1073/pnas.87.21.8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Site-directed mutagenesis of the SH2- and SH3-coding domains of c-src produces varied phenotypes, including oncogenic activation of p60c-src. Mol Cell Biol. 1990b;10:1307–1318. doi: 10.1128/mcb.10.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilaria RL, Jr, Van Etten RA. The SH2 domain of P210BCR/ABL is not required for transformation of hematopoietic factor-dependent cells. Blood. 1995;86:3897–3904. [PubMed] [Google Scholar]
- Ishii T, Yamada M, Sato H, Matsu M, Taketani S, Nakayama K, Sugita Y, Bannai S. Cloning and characterization of a 23-kDa stress-induced mouse peritoneal macrophage protein. J Biol Chem. 1993;268:18633–18636. [PubMed] [Google Scholar]
- Ishii T, Kawane T, Taketani S, Bannai S. Inhibition of the thiol-specific antioxidant activity of rat liver MSP23 protein by hemin. Biochem Biophys Res Comm. 1995;216:970–975. doi: 10.1006/bbrc.1995.2715. [DOI] [PubMed] [Google Scholar]
- Iwahara S, Satoh H, Song DX, Webb J, Burlingame AL, Nagae Y, Muller-Eberhard U. Purification, characterization, and cloning of a heme-binding protein (23 kDa) in rat liver cytosol. Biochemistry. 1995;34:13398–13406. doi: 10.1021/bi00041a017. [DOI] [PubMed] [Google Scholar]
- Jackson P, Baltimore D. N-terminal mutations activate the leukemogenic potential of the myristoylated form of c-abl. EMBO J. 1989;8:449–456. doi: 10.1002/j.1460-2075.1989.tb03397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PK, Paskind M, Baltimore D. Mutation of a phenylalanine conserved in SH3-containing tyrosine kinases activates the transforming ability of c-Abl. Oncogene. 1993;8:1943–1956. [PubMed] [Google Scholar]
- Kawai S, Takeshita S, Okazaki M, Kikuno R, Kudo A, Amann E. Cloning and characterization of OSF-3, a new member of the MER5 family, expressed in mouse osteoblastic cells. J Biochem. 1994;115:641–643. doi: 10.1093/oxfordjournals.jbchem.a124388. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Ren R, Pandey P, Shafman TD, Feller SM, Weichselbaum RR, Kufe DW. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature. 1995;376:785–788. doi: 10.1038/376785a0. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Wang JYJ. Differential phosphorylation of c-Abl in cell cycle determined by cdc2 kinase and phosphatase activity. Science. 1990;248:217–220. doi: 10.1126/science.2183353. [DOI] [PubMed] [Google Scholar]
- ————— Cell cycle-regulated binding of c-Abl tyrosine kinase to DNA. Science. 1992;256:382–385. doi: 10.1126/science.256.5055.382. [DOI] [PubMed] [Google Scholar]
- Konopka JB, Witte ON. Detection of c-abl tyrosine kinase activity in vitro permits direct comparison of normal and altered abl gene products. Mol Cell Biol. 1985;5:3116–3123. doi: 10.1128/mcb.5.11.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z-G, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JYJ. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- Lugo TG, Pendergast A, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol. 1994;14:2883–2894. doi: 10.1128/mcb.14.5.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, Jackson PK, Van Etten RA, Baltimore D. Point mutations in the abl SH2 domain coordinately impair phosphotyrosine binding in vitro and transforming activity in vivo. Mol Cell Biol. 1992;12:609–618. doi: 10.1128/mcb.12.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter JR, Wang JYJ. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of the Philadelphia chromosome-positive leukemias. EMBO J. 1993;12:1533–1546. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter JR, Galasso DL, Wang JYJ. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 1993;13:7587–7595. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F. Catalog of chromosome aberrations in cancer. New York, NY: Wiley-Liss; 1994. [Google Scholar]
- Nemoto Y, Yamamoto T, Takada S, Matsui Y, Obinata M. Antisense RNA of the latent period gene (MER5) inhibits the differentiation of murine erythroleukemia cells. Gene. 1990;91:261–265. doi: 10.1016/0378-1119(90)90097-b. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Traugh JA, Witte ON. Normal cellular and transformation-associated abl proteins share common sites for protein kinase C phosphorylation. Mol Cell Biol. 1987;7:4280–4289. doi: 10.1128/mcb.7.12.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast AM, Muller AJ, Havelik MH, Clark R, McCormick F, Witte ON. Evidence for regulation of the human ABL tyrosine kinase by cellular inhibitor. Proc Natl Acad Sci. 1991a;88:5927–5931. doi: 10.1073/pnas.88.13.5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON. BCR sequences esential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell. 1991b;66:161–171. doi: 10.1016/0092-8674(91)90148-r. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Gishizky ML, Havlik MH, Witte ON. SH1 domain autophosphorylation of P210 BCR/ABL is required for transformation but not growth factor independence. Mol Cell Biol. 1993;13:1728–1736. doi: 10.1128/mcb.13.3.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosperi MT, Ferbus D, Karczinski I, Goubin G. A human cDNA corresponding to a gene overexpressed during cell proliferation encodes a product sharing homology with amoebic and bacterial proteins. J Biol Chem. 1993;268:11050–11056. [PubMed] [Google Scholar]
- Prosperi MT, Apiou F, Dutrillaux B, Goubin G. Organization and chromosomal assignment of two human PAG gene loci: PAGA encoding a functional gene and PAGB a processed pseudogene. Genomics. 1994;19:236–241. doi: 10.1006/geno.1994.1053. [DOI] [PubMed] [Google Scholar]
- Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- Ren R, Zheng SY, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes & Dev. 1994;8:783–795. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- Rickles RJ, Botfield MC, Weng Z, Taylor JA, Green OM, Brugge JS, Zoller MJ. Identification of Src, Fyn, PI3K and Abl SH3 domain ligands using phage display. EMBO J. 1994;13:5598–5604. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickles RJ, Botfield MC, Zhou XM, Henry PA, Brugge JS, Zoller MJ. Phage display selection of ligand residues important for Src homology 3 domain binding specificity. Proc Natl Acad Sci. 1995;92:10909–10913. doi: 10.1073/pnas.92.24.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Sawyers CL, McLaughlin JL, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77:121–131. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- Schwartzberg PL, Stall AM, Hardin JD, Bowdish KS, Humaran T, Boast S, Harbison ML, Robertson EJ, Goff SP. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–1176. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- Sefton BM, Hunter T, Raschke WC. Evidence that the Abelson virus protein functions in vivo as a protein kinase that phosphorylates tyrosine. Proc Natl Acad Sci. 1981;78:1552–1556. doi: 10.1073/pnas.78.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel-Dugan C, Meyer BE, Thomas SM, Brugge JS. Effects of SH2 and SH3 deletions on the functional activities of wild-type and transforming variants of c-Src. Mol Cell Biol. 1992;12:1835–1845. doi: 10.1128/mcb.12.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shau H, Butterfield LH, Chiu R, Kim A. Cloning and sequence analysis of candidate human natural killer-enhancing factor genes. Immunogenetics. 1994;40:129–134. doi: 10.1007/BF00188176. [DOI] [PubMed] [Google Scholar]
- Shi Y, Alin K, Goff SP. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes & Dev. 1995;9:2583–2597. doi: 10.1101/gad.9.21.2583. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Sies H. Strategies of antioxidant defense. Eur J Biochem. 1993;215:213–219. doi: 10.1111/j.1432-1033.1993.tb18025.x. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VLJ, Crawford CE, Jackson PK, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl protooncogene. Cell. 1991;65:1153–1164. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Van Etten RA, Jackson P, Baltimore D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell. 1989;58:669–678. doi: 10.1016/0092-8674(89)90102-5. [DOI] [PubMed] [Google Scholar]
- Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. The C-terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol. 1994;124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten RA, Debnath J, Zhou H, Casasnovas JM. Introduction of a loss-of-function point mutation from the SH3 region of the Caenorhabditis elegans sem-5 gene activates the transforming ability of c-abl in vivo and abolishes binding of proline-rich ligands in vitro. Oncogene. 1995;10:1977–1988. [PubMed] [Google Scholar]
- Wang JYJ, Ledley F, Goff S, Lee R, Groner Y, Baltimore D. The mouse c-abl locus: Molecular cloning and characterization. Cell. 1984;36:349–356. doi: 10.1016/0092-8674(84)90228-9. [DOI] [PubMed] [Google Scholar]
- Welch PJ, Wang JYJ. Disruption of retinoblastoma protein function by coexpression of its C pocket fragment. Genes & Dev. 1995;9:31–46. doi: 10.1101/gad.9.1.31. [DOI] [PubMed] [Google Scholar]
- Wen S-T, Jackson PK, Van Etten RA. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals, and requires the p53 and Rb tumor suppressor gene products. EMBO J. 1996;15:1583–1595. [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Witte ON, Ponticelli A, Gifford A, Baltimore D, Rosenberg N, Elder J. Phosphorylation of the abelson murine leukemia virus transforming protein. J Virol. 1981;39:870–878. doi: 10.1128/jvi.39.3.870-878.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Matsui Y, Natori S, Obinata M. Cloning of a housekeeping-type gene (MER5) preferentially expressed in murine erythroleukemia cells. Gene. 1989;80:337–343. doi: 10.1016/0378-1119(89)90297-7. [DOI] [PubMed] [Google Scholar]
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- Yuan Z-M, Huang Y, Whang Y, Sawyers C, Weichselbaum R, Kharbanda S, Kufe D. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382:272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc Natl Acad Sci. 1997;94:1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervos AS, Gyuris J, Brent R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72:223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]
- Zhu J, Shore SK. c-Abl tyrosine kinase activity is regulated by association with a novel SH3-domain-binding protein. Mol Cell Biol. 1996;16:7054–7062. doi: 10.1128/mcb.16.12.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]