

Abstract
AU-rich elements (AREs, usually containing repeated copies of AUUUA), when present in the 3′-untranslated regions (UTRs) of many mammalian mRNAs, confer instability on their host RNA molecules. The viral small nuclear RNA (snRNA) Herpesvirus saimiri U RNA 1 (HSUR 1) also contains an AUUUA-rich sequence. Here, we report that this ARE induces rapid degradation of HSUR 1 itself and of other snRNAs including HSUR 2 and cellular U1. Mutational analyses of the viral ARE establish that sequence requirements for mRNA and snRNA decay are the same, suggesting a similar mechanism. Moreover, the in vivo degradation activity of mutant AREs correlates with their in vitro binding to the HuR protein, implicated previously as a component of the mRNA degradation machinery. Our results suggest that ARE-mediated instability can be uncoupled from both ongoing translation and deadenylation of the target RNA.
Keywords: ARE, HSUR 1, RNA degradation, HuR, Herpesvirus saimiri
Targeted mRNA instability represents an important mode of post-transcriptional gene regulation in eukaryotic cells. Much attention has focused on AU-rich elements (AREs) that are present in the 3′-untranslated regions (3′ UTRs) of the mRNAs from many early response genes (ERGs), including cytokines, lymphokines, and proto-oncogenes (Caput et al. 1986; Shaw and Kamen 1988; for review, see Belasco and Brawerman 1993; Chen and Shyu 1995). The expression of ERGs is both sensitive to extracellular stimuli and transient, requiring rapid mRNA removal through destabilization following the cessation of transcription. The ARE is a major determinant for the rapid degradation of these mRNAs. Insertion of a 51-nucleotide AU-rich sequence derived from the GM-CSF (granulocyte–macrophage colony-stimulating factor) into the 3′ UTR of β-globin mRNA greatly shortens its normally long half-life (Shaw and Kamen 1988). Conversely, removal of the ARE stabilizes the labile proto-oncogene c-fos mRNA and confers a transforming phenotype (Miller et al. 1984; Meijlink et al. 1985; W. Lee et al. 1988). Depending on cell type, there appear to be different pathways regulating ARE-mediated mRNA decay. For instance, in a monocyte tumor cell line, c-fos mRNA is degraded constitutively, whereas the GM-CSF mRNA is stable (Schuler and Cole 1988). GM-CSF and interleukin-3 (IL-3) mRNAs have also been reported to be selectively stabilized in a human T cell line upon anti-CD28 antibody treatment (Lindsten et al. 1989).
The AREs in ERG mRNAs contain multiple copies of the sequence AUUUA (Caput et al. 1986; Shaw and Kamen 1988; for review, see Belasco and Brawerman 1993; Chen and Shyu 1995). Using a serum-inducible c-fos promoter and a reporter gene system in transiently transfected NIH-3T3 cells, two groups have performed extensive mutagenesis to identify the minimum sequence that directs mRNA degradation. Both found that an isolated single AUUUA is not active, whereas an AUUUA flanked by two Us (UUAUUUAUU) has weak destabilization activity and two separate or overlapping copies of UUAUUUAUU are highly destabilizing (Lagnado et al. 1994; Zubiaga et al. 1995). Using the same serum-inducible promoter system, Shyu and coworkers provided evidence that ARE-mediated mRNA decay proceeds in a biphasic manner, with shortening of the poly(A) tail followed by a first order degradation of the remaining RNA (Shyu et al. 1991; Chen et al. 1995). Whether the latter step involves exo- or endonucleases is not yet established. Also unknown is whether ARE-mediated mRNA decay is obligatorily coupled to ongoing translation, because evidence both in favor of and against this linkage has been reported (Koeller et al. 1991; Savant-Bhonsale and Cleveland 1992; Aharon and Schneider 1993; Chen et al. 1995; Curatola et al. 1995; Winstall et al. 1995; see below).
Although many proteins have been charaterized as factors recognizing AREs (Malter 1989; Bohjanen et al. 1991, 1992; Brewer 1991; Malter and Hong 1991; Vakalopoulou et al. 1991; Hamilton et al. 1993; Zhang et al. 1993; Katz et al. 1994; Nakagawa et al. 1995), we have concentrated on an apparent 32-kD protein, first identified by cross-linking to the c-fos ARE in HeLa cell extracts (Vakalopoulou et al. 1991). This protein also binds to several of the seven small nuclear RNAs (snRNAs) that are highly expressed in marmoset T cells transformed by Herpesvirus saimiri (Myer et al. 1992). These H. saimiri U RNAs (HSURs 1–7) are similar to cellular U RNAs in that they are transcribed by RNA polymerase II, possess 5′ trimethylated guanosine caps at their 5′ ends, and assemble into small nuclear ribonucleoprotein particles (snRNPs) of the Sm class (Murthy et al. 1986; S. Lee et al. 1988; Wassarman et al. 1989; Lee and Steitz 1990; Albrecht and Fleckenstein 1992). At their 5′ ends, HSUR 1, HSUR 2, and HSUR 5 contain four, two, and one copies of AUUUA, respectively. Characterization of the purified 32-kD protein demonstrated that it is identical to HuR (Myer et al. 1997), a ubiquitously expressed member of the ELAV family (Embryonic Lethal, Abnormal Vision; Ma et al. 1996). Like other ELAV proteins, HuR contains three RNA recognition motifs (RRMs) and binds in vitro to ARE sequences (Levine et al. 1993; Gao et al. 1994; Ma et al. 1996; Myer et al. 1997). Detailed analyses revealed a direct correlation between the in vitro affinity of an ARE sequence for HuR and its ability to direct in vivo degradation of a reporter mRNA, suggesting the involvement of HuR in the destabilization (or stabilization) of ARE-containing mRNAs (Myer et al. 1997).
We set out to test a model whereby the HSURs function to stabilize mRNAs normally targeted for rapid degradation by competitively binding cellular components of the mRNA degradation machinery (Myer et al. 1992). By transiently expressing HSUR 1, which has a strong affinity for HuR (Myer et al. 1992), in mouse L929 cells, we expected to observe elevated levels of ARE-containing mRNAs. Instead, we discovered that wild-type HSUR 1 is expressed at a much lower level than a mutant HSUR 1 with its AUUUA repeats converted to AGGUA, a mutation already known to stabilize mRNAs (Shaw and Kamen 1988; Vakalopoulou et al. 1991; Myer et al. 1997). After demonstrating that this is a post-transcriptional phenomenon, we have gone on to show that the HSUR 1 ARE can target the in vivo degradation of other snRNAs, as well as a reporter mRNA. Mutational analyses of the viral ARE reveal a correlation between binding of the HuR protein and in vivo degradation. We discuss the relevance of our findings to current understanding of the mechanism of ARE-mediated mRNA degradation and of the biological functioning of HSUR 1.
Results
The ARE of HSUR 1 confers rapid degradation in vivo
To test the biological function of the ARE at the 5′ end of HSUR 1, we mutated all four copies of AUUUA to AGGUA, an alteration that is known both to stabilize a β-globin reporter mRNA in NIH-3T3 cells and to abolish binding to HuR (Myer et al. 1997). The CUU sequence that follows the ARE was also mutated (to CGG) in this construct (Fig. 1A). HSUR promoters are similar to the well-characterized U1 snRNA promoter (S. Lee et al. 1988), whose transcription by RNA polymerase II is not affected by internal coding sequences (Dahlberg and Lund 1988). To achieve comparable transcription rates, we cloned wild-type and mutant HSUR 1 (Mut), as well as HSUR 3 (which does not have an ARE and serves as a transfection control; Fig. 1A), between the U1 promoter and 3′ termination box in a pUC plasmid (Fig. 1A; Yuo and Weiner 1989).
Figure 1.

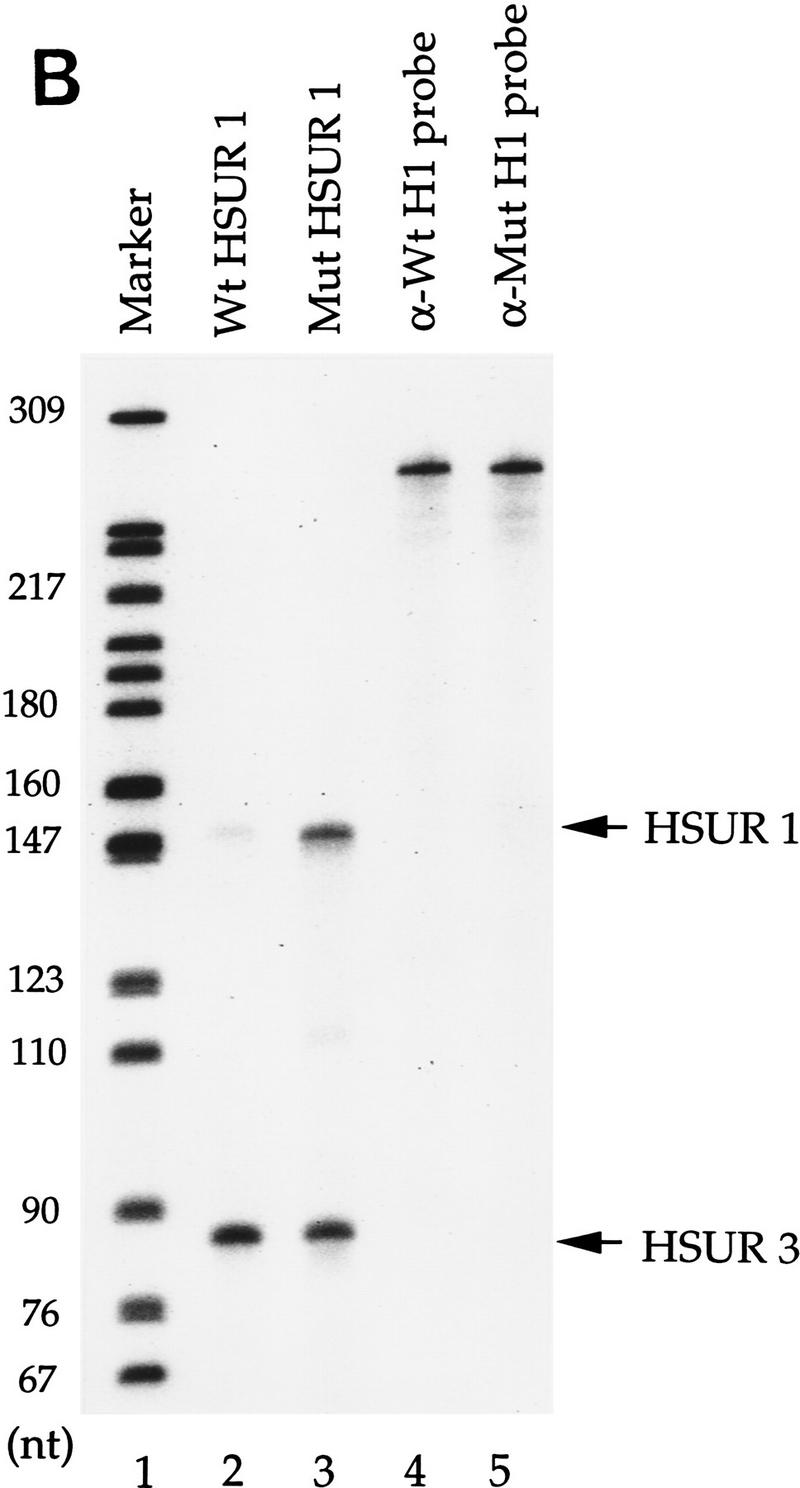


ARE-mediated HSUR 1 degradation in vivo. (A) HSUR RNA sequences and expression plasmid configuration. The HSUR 1 ARE and the nucleotides altered in Mut are underlined. The U1 promoter in the pUC–U1 vector is the 423-bp human genomic sequence upstream of U1 transcription initiation site, whereas the 3′ box is the 30-bp transcription termination signal downstream of the U1 coding sequence (Yuo and Weiner 1989). (B) T1 RNase protection analysis of wild-type and mutant HSUR 1 levels in transient transfection assays. The pUC–U1–HSUR 1 constructs were transiently cotransfected with a pUC–U1–HSUR 3 plasmid into mouse L929 cells (see Materials and Methods). Total RNA collected 48 hr after transfection was subjected to RNase T1 protection assays with wild-type and mutant HSUR 1 antisense RNA (lanes 2 and 3, respectively), together with antisense HSUR 3 RNA as an internal control. One-fiftieth of the amount of the anti-wild-type and anti-mutant HSUR 1 RNA probes used is shown in lanes 4 and 5. The data were quantitated with a Molecular Dynamics PhosphorImager and normalized to HSUR 3. Wild-type HSUR 1 levels were reproducibly one-eighth of those of mutant HSUR 1. (C) Whole-cell run-on assays of wild-type and mutant HSUR 1 transcription. The pUC–U1–HSUR 1 construct containing wild-type or mutant HSUR 1 sequences was cotransfected with pUC–U1–HSUR 3 into L cells, and whole-cell run-on assays were performed (see Materials and Methods). Total RNA was hybridized to nylon membranes that had been dot-blotted with wild-type (top) or mutant (bottom) HSUR 1 and HSUR 3 DNA fragments. Untransfected HSUR 4 DNA was also dotted as a negative control. The patterns of dots are illustrated at right; hybridizations with the run-on RNAs are at left. After quantitation and normalization against the cotransfected positive control HSUR 3 (also subtracting the untransfected negative control HSUR 4), the wild-type HSUR 1 (left dot, top) and the mutant (left dot, bottom) were found to have similar transcription rates (wild type:mutant = 0.95). (D) Immunoprecipitation of wild-type and mutant HSUR 1 from transfected mouse L929 cells. L cells were transiently transfected with the pUC–U1 constructs containing wild-type or mutant HSUR 1 genes, and whole-cell extracts were prepared by sonication. Equal amounts of extract were precipitated with anti-Sm monoclonal antibody Y12 or anti-U1 70K monoclonal antibody H111 as a control. RNA was harvested from the immunoprecipitation pellets (lanes 1,3,5,7) and supernatants (lanes 2,4,6,8), and wild-type (left) and mutant (right) HSUR 1 were assayed by T1 RNase protection. For both wild-type and mutant HSUR 1s, >90% of the RNA was in the anti-Sm precipitate (lanes 3,7), whereas >99% of the HSUR 1s remained in the supernatant with the anti-U1 70K antibody (lanes 2,6).
HSURs were shown previously to be efficiently expressed and assembled into Sm-precipitable snRNPs even in transiently transfected cells that do not serve as hosts for H. saimiri (Lee and Steitz 1990). Murine L929 cells were chosen for transient transfection in our study because they have been used previously to study AUUUA-mediated mRNA destabilization (Savant-Bhonsale and Cleveland 1992). Either wild-type or the mutant pUC–U1–HSUR 1 was cotransfected with pUC–U1–HSUR 3 into L929 cells with DEAE–dextran. To monitor the transfection efficiency, a plasmid encoding β-galactosidase was transfected in parallel. X-gal staining showed that transfection efficiencies ranged between 25% and 30% (data not shown). Total RNA was harvested 45–50 hr following transfection and was analyzed by T1 RNase protection.
We were surprised to observe that the wild-type HSUR 1 level (Fig. 1B, lane 2) was significantly lower (one-eighth) than that of the mutant (lane 3) when normalized to the cotransfected control HSUR 3. Comparable results were obtained in other cell lines of human, monkey, or mouse origin; HeLa cells, HEK-293 cells, Jurkat cells, COS cells, and NIH-3T3 cells all produced wild-type HSUR 1 at one-sixth to one-tenth the level of the mutant (data not shown).
To determine whether the lower level of wild-type compared with mutant HSUR 1 was attributable to greater instability or to some unexpected effect of the internal HSUR 1 sequence on promoter activity, we performed run-on transcription assays. Whole cell rather than the standard nuclear assay was used because of the low [α-32P]UTP incorporation into snRNAs in the nuclear system (Sheldon et al. 1993). Transfected mouse L929 cells were permeabilized and incubated with [α-32P]UTP at 30°C for 10 min (Sheldon et al. 1993). Total RNAs were isolated and hybridized to excess double-stranded DNA that had been denatured and immobilized on a nylon membrane. The cotransfected HSUR 3 and nontransfected HSUR 4 sequences served as positive and negative controls, respectively. Figure 1C shows that wild-type HSUR 1 has a transcription rate comparable (95%) with that of the mutant. Similar results (±10%) were obtained in repeated experiments (data not shown). We conclude that the lower level of wild-type HSUR 1 in transfected L cells is attributable to more rapid decay and that the ARE, which is altered in the mutant, is a cis-acting instability sequence.
We further investigated whether the transfected viral snRNAs assemble into Sm-precipitable snRNPs. Extracts of L cells transfected with wild-type or mutant HSUR 1 were precipitated either with anti-Sm monoclonal antibodies (Y12) or as a negative control, with anti-U1 70K monoclonal antibodies (H111). Comparison of the anti-Sm pellets (Fig. 1D, lanes 3,7) and supernatants (lanes 4,8) reveals that >90% of both the wild-type and mutant HSUR 1 assemble with Sm proteins. More than 99% remains in the supernatants in the control anti-U1 70K immunoprecipitations (lanes 2,6). We conclude that in transfected L cells, the more stable mutant as well as wild-type HSUR 1 become assembled into Sm snRNP particles.
Mutational analyses of the HSUR 1 ARE
Because detailed mutational studies have shed light on the exact sequence requirements for functional AREs in mRNA 3′ UTRs, we performed similar analyses of the HSUR 1 5′-end sequence. We first introduced two or three U → G substitutions into the HSUR 1 ARE, leaving either one intact copy of UUAUUUAUU in mutant M1 or one copy of AUUUA flanked by Gs in M2 (Fig. 2A). Both of these mutations yield high levels of HSUR 1 in mouse L cells (Fig. 2B, lanes 2,3) although the amount of M2 (10-fold relative to wild type) is slightly greater than that of M1 (7-fold relative to wild type). We then made single point mutations in the HSUR 1 5′ end, each disrupting only one copy of the AUUU repeat (M3–M6; see Fig. 2A). M3, with two copies of AUUUA flanked by Us, is the only mutant expressed at low levels (Fig. 2B, lane 4); yet its amount is reproducibly higher than that of wild-type HSUR 1 (1.8-fold; Fig. 2B, lane 1). M4, M5, and M6 each contain only one copy of UUAUUUAUU, as does M1; their RNA levels are similar to that of M1 (Fig. 2B, lanes 5–7; 5.8-, 6.4-, and 6.1-fold of wild type, respectively) and are lower than that of M2. Similar values were obtained in repeated experiments. These results suggest that at least two copies of AUUUA flanked by Us are required to target HSUR 1 for rapid degradation in L929 cells. Moreover, there appears to be a good correlation between the copy number of UUAUUUAUU and the activity of the ARE as an snRNA destabilization element in vivo.
Figure 2.


Mutational analysis of the HSUR 1 ARE. (A) Sequences of the six HSUR 1 point mutants, M1–M6, with the sites of U → G mutation underlined. (B) RNA levels of the HSUR 1 mutants. Wild-type HSUR 1 and mutants M1–M6, all driven by the same U1 promoter, were each transiently cotransfected with HSUR 3 into mouse L929 cells, and the RNA levels were assayed by T1 RNase protection (lanes 1–7, respectively). The antisense HSUR 1 probe covered a 120-nucleotide region at the 3′ end, which is common to wild-type HSUR 1 and all six mutants. When quantitated and normalized to HSUR 3, the mutant M1–M6 levels were found to be 7.0-, 10.1-, 1.8-, 5.8-, 6.4-, and 6.1-fold that of wild-type HSUR 1.
The HSUR 1 ARE can also target a β-globin reporter mRNA for degradation
Because the AU-rich 5′ end of HSUR 1 is similar to the AREs found in the 3′ UTRs of ERG mRNAs, we transplanted sequences from wild-type HSUR 1 and mutants M1, M2, and M3 into the 3′ UTR of a reporter construct to assess their effects on mRNA stability. We chose the β-globin reporter system with a serum-inducible c-fos promoter because it has been used in a number of transient transfection studies of mRNA AREs (Shyu et al. 1989; Schiavi et al. 1994; Zubiaga et al. 1995; Myer et al. 1997). The wild-type and mutant HSUR 1 sequences (+1 to +32) were each cloned into the parental pBBB construct (Fig. 3A) and transiently cotransfected into mouse L cells along with a control plasmid pEF–BOS–CAT, which constitutively expresses the CAT mRNA (Zubiaga et al. 1995; Myer et al. 1997). After 24 hr of serum starvation, cells were collected at 0, 1.0, 2.5, 4.0, and 5.5 hr following serum induction and total RNAs were isolated and analyzed by T1 RNase protection (Fig. 3B). The data, averaged from several experiments and standarized to the CAT mRNA internal control, are plotted in Figure 3C. The maximum signal in each case was considered 100%.
Figure 3.



Degradation of β-globin mRNA containing AREs from HSUR 1 and its mutants. (A) Schematic diagram of the β-globin reporter construct and the sequences inserted into its 3′ UTR. Transcription of the rabbit β-globin construct was driven by the serum-inducible human c-fos promoter (Zubiaga et al. 1995). (B) Transient transfection analyses. The β-globin plasmids were transiently transfected into mouse L929 cells with cotransfection of the pEF–BOS–CAT plasmid, which expresses CAT mRNA constitutively (Zubiaga et al. 1995). After serum starvation, the transcription of β-globin was stimulated by serum addition. Cells were harvested when serum stimulation was initiated (T = 0) and at 1.0, 2.5, 4.0, and 5.5 hr thereafter. Total RNA was isolated for T1 RNase protection analyses using both a β-globin and a CAT antisense probe. (C) Time course of β-globin mRNA decay. The β-globin signals in Fig. 3B were quantitated on a Molecular Dynamics PhosphorImager, standardized to the CAT internal control (the lower band of the doublet was quantitated as the major T1 digestion product), and plotted. One hundred percent RNA was arbitrarily assigned to the time point with the highest signal. These results are the average of several experiments.
As is evident from Figure 3, the positive control pB–ARE mRNA, which contains the c-fos ARE and has been shown previously to be functional in NIH-3T3 cells (Zubiaga et al. 1995; Myer et al. 1997), is unstable in L cells with levels below 17.5% 5.5 hr postinduction. On the other hand, the negative control mRNA from the parental pBBB plasmid is stable (Zubiaga et al. 1995; Myer et al. 1997), exhibiting an RNA level >80% after 5.5 hr. The sequence from the 5′ end of wild-type HSUR 1 is even more destabilizing than the c-fos ARE, reducing mRNA levels below 8% by the 5.5-hr time point. In contrast, both the M1 and M2 mutant sequences are ineffective as destabilization elements, with 74.5% and 88% RNA remaining 5.5 hr postinduction. Only the sequence from mutant M3 confers partial instability, with 37% of the RNA remaining after 5.5 hr. These results parallel the data shown in Figure 3: Wild-type HSUR 1 exhibits the lowest RNA level and M3 is the second lowest, whereas M1 and M2 levels are high and not detectably different. Hence, the viral ARE and its mutants appear to function equivalently in the 5′ end of HSUR 1 and in the 3′ UTR of a reporter mRNA, conferring rapid degradation on both RNA molecules.
Binding of the HuR protein correlates with the in vivo cis-acting degradation activity of the viral ARE
Because the AU-rich sequences from HSUR 1 and its mutants tested here are different from those analyzed previously (Myer et al. 1997), we performed UV cross-linking experiments to assess their binding to HuR. Relative affinities for the HuR protein were measured by the ability of each sequence to compete at 10-, 20-, and 50-fold molar excess with a radioactively labeled wild-type HSUR 1 transcript in HeLa cell nuclear extract. In the absence of competitor RNA (Fig. 4, lanes 1,14), the lowest band (arrow) in the UV cross-linking profile has been identified as HuR (Myer et al. 1992); its cross-linking is effectively competed by addition of unlabeled wild-type HSUR 1 (lanes 2–4). The results with HSUR 1 mutants, M1–M6, as well as Mut (the AUUU → AGGU mutant studied in Fig. 1), show that M3 alone competes for HuR binding (lanes 15–17). Thus, only the two sequences able to target the viral snRNA (Fig. 2) and mRNA (Fig. 3) for rapid degradation in vivo (wild-type HSUR 1 and M3) bind HuR. There appears to be a perfect correlation between the in vitro binding of the viral ARE sequences to the HuR protein and their in vivo cis-degradation activities.
Figure 4.

Binding of viral ARE sequences to HuR. The binding of the HuR protein to [α-32P]UTP-labeled wild-type HSUR 1 transcript in the presence of unlabeled wild-type or mutant HSUR 1 competitor RNA was assayed by UV cross-linking in HeLa cell nuclear extract. After RNase A treatment, label transfer was visualized by electrophoresis in a 12.5% SDS–polyacrylamide gel. The competitor RNAs, whose sequences are given in Figs. 1A and 2A, are indicated at top of the gel. (Lanes 1,14) Proteins cross-linked in the absence of specific competitor RNA. For each set of competitors, the first lane (lanes 2,5,8,11,15,18,21,24) contains a 10-fold molar excess, the second lane (lanes 3,6,12,16,19,22,25) a 20-fold molar excess, and the third lane (lanes 4,7,10,13,17,20,23,26) a 50-fold molar excess over the labeled HSUR 1 RNA. The cross-linked HuR protein is indicated with an arrow.
AU-rich sequences also target other snRNAs for rapid degradation
Because the four tandem copies of AUUU at its 5′ end direct rapid degradation of HSUR 1, we asked whether the effect of this sequence could be transferred to other snRNAs. HSUR 2, whose 5′ end is the most similar to HSUR 1 among the other six HSURs, has four tandem copies of AUUU interrupted only by a guanosine (G) (Fig. 5A). When transiently expressed in mouse L cells from the same U1 promoter, the level of HSUR 2 was found to be 10-fold higher than that of HSUR 1 (data not shown). We therefore designed a HSUR 2 mutant (M1) that substitutes the G with UA to generate four complete copies of AUUUA (Fig. 5A). We also made two other mutants as controls, M2 and M3 (Fig. 5A), which have substituted the G with CC and CA, respectively, at the same position. When transiently transfected into mouse L cells, HSUR 2 M1 (Fig. 5B, lane 2) exhibits a RNA level one-fifth that of the wild-type HSUR 2 (lane 1), whereas the control M2 and M3 levels are comparable with the wild-type HSUR 2 (lanes 3, 4, and 1, respectively). We conclude that tandem copies of AUUU can also target HSUR 2 for rapid degradation in vivo.
Figure 5.




AUUUA repeats target other snRNAs for rapid decay. (A) The 5′ ARE of wild-type HSUR 2 and its mutants. The single guanosine that interrupts the AUUUA repeat sequence in HSUR 2 was mutated to UA, CC, and CA (underlined) in HSUR 2 M1, HSUR 2 M2, and HSUR 2 M3, respectively. (B) RNA levels of wild-type and mutant HSUR 2s. Wild-type HSUR 2 or mutant M1, M2, or M3, all controlled by the same U1 promoter, were each transiently cotransfected with HSUR 3 into mouse L929 cells and analyzed by T1 RNase protection. The level of mutant M1 (lane 2), which has four tandem copies of AUUU, is one-fifth that of wild-type HSUR 2 (lane 1), whereas controls M2 and M3 (lanes 3 and 4, respectively), which have been mutated at the same two positions as M1, have levels 1.3- and 1.1-fold of the wild-type HSUR 2, respectively. (C) The 5′-end sequences of wild-type U1 and two U1 mutants. The 5′ splice site recognition sequence of U1 and the sequences replacing it in the mutants are underlined. The AU3–U1 mutant has four tandem copies of AUUU, whereas in the AGU–U1 mutant, there are four AUUU repeats interrupted by 3 Gs (the same sequence as in HSUR 1 M2). (D) Levels of AU3–U1 and AGU–U1 in duplicate transfection experiments. Each U1 mutant was transiently cotransfected with HSUR 3 into mouse L929 cells. U1 RNA levels were analyzed by primer extension using an oligonucleotide complementary to the most 3′ 20 nucleotides of U1 (nucleotides 155–164), whereas HSUR 3 was assayed by T1 RNase protection assay as above. When normalized to HSUR 3 and averaged between the duplicate transfection experiments, the AU3–U1 level was found to be one-fourth that of AGU–U1.
Because HSUR 1 and HSUR 2 are both viral snRNAs, we further investigated whether the (AUUU)4A sequence would confer instability on a cellular snRNA. It is known that the 5′ splice site recognition sequence of U1 (nucleotides 3–10) is dispensable and that substitution of this sequence does not affect U1 snRNA stability (Yuo and Weiner 1989). We therefore replaced this region with (AUUU)4A (mutant AU3–U1; Fig. 5C) and with the nonfunctional mutant ARE sequence of HSUR 1 M2 (mutant AGU–U1; Fig. 5C), which contains four copies of AUUUA interrupted by three Gs. These U1 constructs were transiently cotransfected with HSUR 3 into mouse L cells. In this case, U1 levels were analyzed by primer extension using a labeled oligonucleotide complementary to the 3′ end of U1 (nucleotides 155–164); the extension product from endogenous U1 is 164 nucleotides in length, whereas those for the two mutants are 176 nucleotides. HSUR 3, assayed by T1 RNase protection, served as an internal control. As shown in Figure 5D, AU3–U1 is expressed at much lower levels than AGU–U1. When normalized against HSUR 3 and averaged between the duplicate experiments, the AU3–U1 level was found to be one-fourth that of the AGU–U1. Hence, the HSUR 1 ARE is also transferable to a cellular snRNA, conferring rapid degradation.
Discussion
It has been known for some time that an ARE within the 3′ UTR confers instability on its host mRNA (for review, see Belasco and Brawerman 1993; Chen and Shyu 1995). Our study provides the first evidence that AREs are also able to destabilize snRNAs including viral HSUR 1, HSUR 2, and cellular U1 (Figs. 1 and 5). SnRNAs differ from mRNAs in that they are not polyadenylated and do not associate with ribosomes; the AREs we have analyzed are located in the snRNAs near the 5′ end, a region known to be accessible within the snRNP particles for interaction with trans-acting molecules (S. Lee et al. 1988). We show that the viral ARE and its mutants exhibit comparable destabilization activities when transferred from the snRNA to the 3′ UTR of a reporter mRNA (Figs. 2 and 3). Because an ARE sequence’s in vitro binding affinity for the HuR protein correlates with its ability to target in vivo degradation of both snRNAs (Figs. 2, 3, 4) and mRNAs (Myer et al. 1997), the HuR protein may participate in ARE-mediated decay of both snRNAs and mRNAs.
ARE-mediated snRNA decay and Sm snRNP biogenesis
Both mRNAs and the U snRNAs studied here are transcribed by RNA polymerase II. mRNAs then have their introns removed and become polyadenylated in the nucleus before transport to the cytoplasm, where they are translated on polysomes (for review, see Dreyfuss et al. 1996). The maturation and trafficking of U snRNAs, however, are quite different. After transcription, precursor snRNAs are exported to the cytoplasm, where they assemble with the core Sm proteins; the 5′ m7G cap becomes hypermethylated, and the 3′ end is often trimmed. Sm snRNPs are then transported back into the nucleus, where they participate in splicing or other nuclear functions (for review, see Dahlberg and Lund 1988; Parry et al. 1989).
Here, we have shown that the ARE at the 5′ end of wild-type HSUR 1 confers instability (Fig. 1B,C). Yet, in transiently transfected L cells, >90% of both the unstable wild-type and stabilized mutant HSUR 1 are assembled with Sm proteins (Fig. 1D), which appear to contribute to the stabilization of snRNAs (Jones and Guthrie 1990; Terns et al. 1993; Fischer et al. 1994). Thus, the instability conferred by the HSUR 1 ARE appears to occur rapidly, possibly even before assembly into Sm snRNPs. We speculate that there may be a competition for binding of HSUR 1 transcripts by RNA destabilization factors versus Sm proteins. Whether decay occurs in the nucleus or in the cytoplasm is a problem for future investigation.
Comparison of ARE-mediated snRNA and mRNA degradation
It has been demonstrated that ARE degradation signals within the 3′ UTR function independently of the host mRNA coding sequence and are transferable to other mRNAs (for review, see Belasco and Brawerman 1993; Chen and Shyu 1995). We have found that the 5′ ARE of HSUR 1 can also be transplanted to other snRNAs (Fig. 5), demonstrating the independence of the cis-degradation signal from the host snRNA sequence. Mutational analyses of ARE sequences located at the 5′ end of HSUR 1 (Fig. 2) or within the 3′ UTR of β-globin mRNA (Fig. 3) revealed comparable destabilization activities (Figs. 2 and 3), suggesting that common sequences orchestrate both decay processes.
Mutational analyses of the HSUR 1 ARE (Fig. 2A,B) further indicated that the shortest degradation sequence among those we tested in mouse L929 cells is two overlapping copies of UUAUUUAUU, namely UUAUUUAUUUAUU (as in mutant M3). This sequence targets β-globin mRNA weakly for destabilization in the same cell line (pB–H1 M3; Fig. 3A,B). In NIH-3T3 cells, the minimum mRNA degradation signal has been reported to be UUAUUUAUU (Zubiaga et al. 1995; Lagnado et al. 1994), which was confirmed in our previous studies (Myer et al. 1997). Our data therefore suggest some cell-type specificity, because UUAUUUAUU is unable to confer instability on either mRNA (data not shown) or snRNA (HSUR 1 M1, Fig. 2A,B) in L929 cells. At least two overlapping (pB–H1 M3; Fig. 3) or two separate copies of UUAUUUAUU sequences (data not shown) are required to destabilize the reporter β-globin mRNA in L929 cells.
We have shown previously that the in vitro affinity of an ARE sequence for the HuR protein correlates with its in vivo ability to target an mRNA for rapid degradation (Myer et al. 1997). Here, we report that the same correlation exists for snRNA destabilization and HuR binding (Figs. 3 and 6), arguing that HuR is likely to play an important role in ARE-mediated decay of snRNAs as well. Because the HuR protein has no detectable nuclease activity (Myer et al. 1997), it is possible that it recognizes and initiates the assembly of degradation complexes on ARE sequences. The observation that HuR protein is present in both nuclear (Vakalopoulou et al. 1991; Myer et al. 1992, 1997) and cytoplasmic extracts (Vakalopoulou et al. 1991) leaves open the question of whether decay occurs in the nucleus, the cytoplasm, or both. However, it is also conceivable that HuR may be a stabilizing factor that escorts mRNAs and snRNAs from the nucleus to the cytoplasm, providing protection from nucleases until its binding is actively reversed. Future studies involving altering HuR levels in vivo and producing monospecific anti-HuR antibodies will provide further insights into the role of HuR in RNA destabilization. Because ARE-mediated snRNA and mRNA degradation share the same cis-destabilization signals as well as a putative trans-acting factor, HuR, other components involved in the two degradation pathways may also be shared.
One controversial issue in ARE-mediated mRNA degradation is whether or not it is coupled to ongoing translation; there have been reports supporting both alternatives. Abolishing translation initiation by mutating the AUG translation start codon or introducing a strong stem–loop structure into the 5′ UTR has been shown to stabilize mRNAs containing an ARE (Savant-Bhonsale and Cleveland 1992; Aharon and Schneider 1993). Degradation can be re-established when translation is restored by introducing an internal ribosomal landing sequence downstream of the stem–loop (Aharon and Schneider 1993), providing evidence for translational coupling. However, it was demonstrated later that inserting a strong stem–loop structure anywhere upstream of an ARE sequence stabilizes the mRNA (Curatola et al. 1995). Similarly, placing the iron response element (IRE) in the 5′ UTR to regulate mRNA translation has produced conflicting results: Destabilization of an α-globin reporter mRNA by the c-fos or GM-CSF ARE appeared dependent on polysome loading and translation in growing NIH-3T3 cells (Winstall et al. 1995), whereas the destabilization of transferrin receptor reporter mRNA by the c-fos ARE was observed to be independent of its translation (Koeller et al. 1991). Finally, using a serum-inducible c-fos promoter and inserting a hairpin structure into the 5′ UTR to inhibit translation, Chen and coworkers (1995) observed that destabilization of a β-globin reporter mRNA by the c-fos or GM-CSF ARE can be uncoupled from their translation.
If ARE-mediated mRNA and snRNA decay indeed utilize the same or overlapping pathways, our data favor the idea that the mRNA decay is not obligatorily coupled to translation. Because snRNAs do not contain open reading frames, ARE-promoted snRNA degradation cannot require translation. Moreover, even though the first step of mRNA degradation is deadenylation (Shyu et al. 1991; Chen et al. 1995), our observation that nonpolyadenylated snRNAs are destabilized by the same sequences suggests that interactions with the poly(A) tail are not always essential to initiate decay. Additional evidence that AUUUA-containing nontranslated RNAs and mRNAs may be degraded by the same mechanism has been reported based on studies in murine macrophages (J. Kaszuba, D. Angerer, T. Said, A.V. Gabain, and M. Baccarini, in prep.).
Biological significance of HSUR 1 degradation
HSUR genes are conserved between oncogenic subgroups of H. saimiri (Biesinger et al. 1990) and are expressed only during latent infection (Murthy et al. 1986; S. Lee et al. 1988). Genetic analysis of subgroup A has identified a viral transforming protein, called STP-A11, that is required for viral-induced T-cell immortalization in culture (Desrosiers et al. 1985; Murthy et al. 1989; Jung et al. 1991). The ATP-A11 gene does not continue to be expressed in transformed T cells, suggesting that it may be important for the induction but not the maintenance of viral transformation (Kamine et al. 1984; Murthy et al. 1989). Viral deletion mutants lacking HSUR 1, HSUR 2, and HSUR 5 are still able to transform T cells in culture. However, the transformed cells grow much more slowly than those transformed by wild-type H. saimiri (doubling time of 1 week vs. 1.5 days), arguing that these HSURs may be important only for the maintenance of viral transformation (Murthy et al. 1989).
Why then does H. saimiri produce an snRNA (HSUR 1) that is quickly degraded, especially because viruses are known for evolving economical modes of gene expression? Our study suggests that rapid HSUR 1 degradation may be significant for maintaining viral transformation: HSUR 1 shares and may compete with mRNAs for the cellular degradation machinery, sacrificing itself, whereas host mRNAs with AREs are in turn stabilized. Elevated levels of these mRNAs, encoding proto-oncoproteins, lymphokines, and cytokines, would then enhance the viral transformed state. An alternative possibility is that HSUR 1, HSUR 2, and HSUR 5 bind HuR and retain it in the nucleus, as proposed previously (Myer et al. 1992). It will therefore be interesting to investigate whether expression of HSURs in either transformed T cells or transfected mammalian cell lines alters the localization of HuR and other putative components of the RNA degradation machinery, such as an RNase E-like activity that cleaves AUUUA sequences (Wennborg et al. 1995).
It is quite possible that in addition to HSUR 1, other viral products (either proteins or other HSURs) are required to elevate the levels of cellular ARE-containing mRNAs. This may explain why transiently expressing HSUR 1 alone in mouse L929 cells did not produce higher levels of β-globin mRNA with the GM-CSF ARE at its 3′ UTR (X.C. Fan, V.E. Myer, and J.A. Steitz, unpubl.). It is also possible that HSUR 1 may function to stabilize proto-oncogene and growth factor mRNAs only in certain host cells, such as marmoset T cells. Lack of a negative-control cell line (a non-viral-infected marmoset T cell line with similar growth rates to the transformed 1670 cells) makes it difficult to test this hypothesis directly. Future exploration of the model will involve analysis of point mutations within the HSUR 1 gene in the H. saimiri genome and testing the transformation efficiency and maintenance of these mutant viruses in marmoset T cells.
Materials and methods
Cell culture, nuclear extracts, and transient transfections
Murine L929 fibroblasts were passed in minimal essential medium (MEM, GIBCO) supplemented with 10% horse serum (HS; GIBCO). HeLa cells were maintained in RPMI 1640 medium (GIBCO) with 10% fetal bovine serum (FBS; GIBCO). NIH-3T3, COS, HEK-293, and Jurkat cells were grown in 10%FBS–Dulbecco’s modified Eagle medium (DMEM; GIBCO). HeLa cell nuclear extracts were prepared as described (Dignam et al. 1983), modified as in Myer and Steitz (1995).
L929 cells were grown in 100-mm dishes to between 50% and 70% confluence and then switched to MEM medium supplemented with 10% NuSerum (GIBCO) for DEAE–dextran transfection. Plasmids (4–6 μg) were resuspended in 40 μl of TBS, mixed with 80 μl of 10 mg/ml DEAE–dextran (Sigma)/TBS, and then overlaid on cells. After incubation for 4 hr, cells were shocked with 10% DMSO/Phosphate-buffered saline (PBS) for 1 min and allowed to recover in 10% HS–MEM. For investigation of β-globin mRNA stability, 3 μg of β-globin plasmid and 2 μg of control pEF–BOS–CAT plasmid were used. Transfected L929 cells were recovered in 10% HS–MEM overnight and serum-starved in 0.5% HS–MEM for 24 hr. Cells were then serum-stimulated by addition of 20% HS–MEM, and RNA was isolated at 0, 1, 2.5, 4, and 5.5 hr.
Oligonucleotides and plasmids
The plasmid pUC–U1 was a generous gift of Dr. Alan Weiner (Yuo and Weiner 1989). Wild-type HSUR 1, HSUR 2, and HSUR 3 sequences followed by the U1 3′-end box were generated by PCR using 5′ overhang oligonucleotides and cloned between the BglII and SalI sites in the pUC–U1 plasmid (Fig. 1A). Oligonucleotide-directed PCR mutagenesis was used to introduce mutations into the 5′ end of HSUR 1, HSUR 2, and U1, to generate mutants HSUR 1 Mut, M1–M6, HSUR 2 M1–M3, AU3–U1, and AGU–U1 (Figs. 1A, 2A, and 5A,C). All HSURs were also PCR-cloned between the EcoRI and HindIII sites of the pSP64 vector (Promega). After linearization with EcoRI restriction enzyme, antisense HSUR RNAs for RNase protection assays were generated by transcription with SP6 RNA polymerase in the presence of [α-32P]UTP. The DNA oligonucleotide used in the U1 primer extension assay was complementary to 20 nucleotides at the 3′ end of U1: CAGGGGAAAGCGCGAACGCA.
Plasmids for transient transfection contained a genomic copy of the rabbit β-globin gene driven by the serum-inducible human c-fos promoter. A 51-bp sequence from the c-fos 3′ UTR (bp 3099 to bp 3149 from the translation start site) followed by a test degradation sequence was inserted directly after the β-globin stop codon. The plasmids pB–ARE and pBBB, as well as the internal control plasmid pEF–BOS–CAT, have been described previously (Shyu et al. 1989; Zubiaga et al. 1995; Myer et al. 1997). Plasmids pB–Wt H1, pB–H1 M1, pB–H1 M2, and pB–H1 M3 were generated by replacing the c-fos AU-rich sequence in pB–ARE with the four different 32-bp sequences (+1 to +32) from the 5′ end of wild-type HSUR 1 and mutants M1, M2, and M3, respectively. The pGEM-3Z plasmids (Promega) transcribed to produce the antisense β-globin and CAT RNAs used in RNase protection assays have been described previously (Myer et al. 1997).
RNase protection and primer extension assays
Total RNA was collected from transfected cells using Trizol reagent (GIBCO) and treated with RQ1 DNase (Promega). T1 RNase protection assays were performed as described (S. Lee et al. 1988), with the following modifications: DNase-treated RNA (5–10 μg for snRNA, 20–30 μg for mRNA) was combined with 2 × 105 to 4 × 105 cpm of the appropriate [α-32P]UTP-labeled antisense probe, heated at 85°C for 5 min, incubated at 45°C overnight to allow annealing, and then digested with T1 RNase (1 U/10 μg of RNA; Calbiochem) at 30°C for 1 hr. Samples were electrophoresed on a 6% polyacrylamide/TBE gel. Results were quantitated with a Molecular Dynamics PhosphorImager.
Primer extension assays were performed as described by Tarn and Steitz (1994) with the following modifications: Five micrograms of total RNA was mixed with 2 × 105 to 5 × 105 cpm of [γ-32P]ATP kinase-labeled DNA primer, heated at 85°C for 5 min, and slow-cooled to 42°C to allow annealing. Reverse transcription was performed at 42°C for 30 min, and the samples were electrophoresed on a 6% polyacrylamide/TBE gel.
Whole cell run-on assays
Transiently transfected L929 cells in 100-mm dishes were permeabilized and collected according to Sheldon et al. (1993). After resuspension in 50 μl of buffer B, 15 μl of [α-32P]UTP (150 μCi) and 100 μl of reaction buffer were added and the mixture was incubated at 30°C for 10 min. One milliliter of Trizol solution (GIBCO) was added to terminate the reaction and to harvest total RNA, which was DNase treated, denatured in 0.16 m NaOH buffer on ice for 10 min, and neutralized in 0.3 m HEPES acid/0.3 m NaOAc.
All DNA probes were purified and denatured and corresponded to the full-length snRNAs. They were titrated to ensure that they were in excess of the amount of RNA being assessed by hybridization; 100 ng of each was dot-blotted and UV cross-linked onto a nylon filter (Bio-Rad), which was incubated in hybridization solution (50% formamide, 0.5% SDS, 200 μg/ml of salmon sperm DNA, 6× SSC, 1× Denhardt’s solution) at 55°C for 1 hr (Sheldon et al. 1993). One-third of the radioactive run-on RNA harvested from a 100-mm dish was hybridized to the filter in 1.2 ml of hybridization buffer at 55°C for 48 hr. The filter was then rinsed with 6× SSC, washed with 2× SSC, 0.5% SDS at 55°C twice for 15 min, at room temperature once for 5 min, and, finally, exposed to X-ray film.
Immunoprecipitation of HSURs
Immunoprecipitation was performed essentially as described previously (S. Lee et al. 1988) with the following modifications: Ten microliters of mouse ascites fluid containing Y12 monoclonal antibodies or hybridoma supernatant containing an equivalent amount of H111 antibodies was incubated with 2.5 mg of protein A–Sepharose (Pharmacia) in 500 μl of NET-2 (150 mm NaCl, 0.05% NP-40, 50 mm Tris-HCl at pH 7.5) at room temperature for 1 hr and washed with 1 ml of NET-2 twice. The antibody-bound beads were then incubated with sonicated extracts of 2 × 106 transfected cells at 4°C for 3 hr and washed with 1 ml of NET-2 five times. RNAs from the pellets and the supernatants were collected by phenol–chloroform extraction and ethanol precipitation.
UV cross-linking competition assays
The cross-linking mixture contained 200,000 cpm of [α-32P]UTP-labeled wild-type HSUR 1 transcript (specific activity 63,700 cpm/ng), 3 μg of Escherichia coli tRNA, 0.5 μl of 100 mm ATP, 5 μl of nuclear extract (from 5 × 106 HeLa cells) and competitor RNA at a 10-, 20-, or 50-fold molar excess over the labeled probe in a total of 20 μl. The HSUR 1 mutants, as well as the wild-type positive control, were transcribed with a trace amount of radioactive nucleotide to facilitate gel purification and quantitation. The cross-linking mixture was incubated at 30°C for 15 min, UV cross-linked with 254 nm of light for 15 min at 4°C, treated with RNase A for 15 min at 37°C, and fractionated on a 12.5% polyacrylamide/1% SDS gel as described (Myer et al. 1992).
Acknowledgments
We thank A.M. Weiner for providing DNA plasmid pUC–U1 and J. Belasco and A.-B. Shyu for providing DNA plasmids pBBB, pB–ARE, and pBOS–EF–CAT. We are also grateful to A.M. Weiner, I.G. Miller, and members of Steitz’s laboratory for helpful comments and supplying reagents and to A. Parker, K. Tycowski, and M. Frilander for helpful comments on the manuscript. This work was supported by grant CA 16038 from the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Joan.Steitz@Yale.edu; FAX (203) 624-8213.
References
- Aharon T, Schneider RJ. Selective degradation of short-lived mRNAs with the granulocyte-macrophage colony-stimulating factor AU-rich 3′ noncoding region is mediated by a cotranslational mechanism. Mol Cell Biol. 1993;13:1971–1980. doi: 10.1128/mcb.13.3.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht J-C, Fleckenstein B. Nucleotide sequence of HSUR 6 and HSUR 7, two small RNAs of herpesvirus saimiri. Nucleic Acids Res. 1992;20:1810. doi: 10.1093/nar/20.7.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belasco J, Brawerman G. Control of messenger RNA stability. San Diego, CA: Academic Press; 1993. [Google Scholar]
- Biesinger B, Trimble JJ, Desrosiers RC, Fleckenstein B. The divergence between two oncogenic herpesvirus saimiri strains in a genomic region related to the transforming phenotype. Virology. 1990;176:505–514. doi: 10.1016/0042-6822(90)90020-r. [DOI] [PubMed] [Google Scholar]
- Bohjanen PR, Petryniak B, June CH, Thompson CB, Lindsten T. An inducible cytoplasmic factor (AU-B) binds selectively to AUUUA multimers in the 3′ untranslated region of lymphokine mRNA. Mol Cell Biol. 1991;11:3288–3295. doi: 10.1128/mcb.11.6.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— AU RNA-binding factors differ in their binding specificities and affinities. J Biol Chem. 1992;267:6302–6309. [PubMed] [Google Scholar]
- Brewer G. An AU-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol Cell Biol. 1991;11:2460–2466. doi: 10.1128/mcb.11.5.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-YA, Shyu A-B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen C-YA, Xu N, Shyu A-B. mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: Different deadenylation kinetics and uncoupling from translation. Mol Cell Biol. 1995;15:5777–5788. doi: 10.1128/mcb.15.10.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatola AM, Nadal MS, Schneider RJ. Rapid degradation of AU-rich element (ARE) is activated by ribosome transit and blocked by secondary structure at any position 5′ to the ARE. Mol Cell Biol. 1995;15:6331–6340. doi: 10.1128/mcb.15.11.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg JE, Lund E. The genes and transcription of the major small nuclear RNAs. In: Birnsteil ML, editor. Structure and function of the major and minor small nuclear ribonucleoprotein particles. Heidelberg, Germany: Springer-Verlag; 1988. pp. 38–70. [Google Scholar]
- Desrosiers RC, Bakker A, Kamine J, Falk LA, Hunt RD, King NW. A region of the Herpesvirus saimiri genome required for oncogenicity. Science. 1985;228:184–187. doi: 10.1126/science.2983431. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1498. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Hentze M, Lamond AI. From transcription to protein (review) Cell. 1996;85:963–972. doi: 10.1016/s0092-8674(00)81298-2. [DOI] [PubMed] [Google Scholar]
- Fischer U, Heinrich J, Zee KV, Fanning E, Luhrmann R. Nuclear transport of U1 snRNP in somatic cells: Differences in signal requirement compared with Xenopus laevis oocytes. J Cell Biol. 1994;125:971–980. doi: 10.1083/jcb.125.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F-B, Carson CC, Levine T, Keene JD. Selection of a subset of mRNAs from combinatorial 3′ untranslated region libraries using neuronal RNA-binding protein Hel-N1. Proc Nat Acad Sci. 1994;91:11207–11211. doi: 10.1073/pnas.91.23.11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BJ, Nagy E, Malter JS, Arrick BA, Rigby WF. Association of heterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUA sequences. J Biol Chem. 1993;268:8881–8887. [PubMed] [Google Scholar]
- Jones MH, Guthrie C. Unexpected flexibility in an evolutionarily conserved protein-RNA interaction: Genetic analysis of the Sm binding site. EMBO J. 1990;9:2555–2561. doi: 10.1002/j.1460-2075.1990.tb07436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JU, Trimble JJ, King NW, Biesinger B, Fleckenstein BW, Desrosiers RC. Identification of transforming genes of subgroup A and C strains of Herpesvirus saimiri. Proc Natl Acad Sci. 1991;88:7051–7055. doi: 10.1073/pnas.88.16.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamine J, Bakker A, Desrosiers RC. Mapping of RNA transcribed from a region of the herpesvirus saimiri genome required for oncogenicity. J Virol. 1984;52:532–540. doi: 10.1128/jvi.52.2.532-540.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DA, Theodorakis NG, Cleveland DW, Lindsten T, Thompson CB. AU-A, an RNA-binding activity distinct from hnRNP A1, is selective for AUUUA repeats and shuttles between the nucleus and the cytoplasm. Nucleic Acids Res. 1994;22:238–246. doi: 10.1093/nar/22.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeller DM, Horowitz JA, Casey JL, Klausner RD, Harford JB. Translation and the stability of mRNAs encoding the transferrin receptor and c-fos. Proc Natl Acad Sci. 1991;88:7778–7782. doi: 10.1073/pnas.88.17.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado CA, Brown CY, Goodall GJ. AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: The functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A) Mol Cell Biol. 1994;14:7984–7995. doi: 10.1128/mcb.14.12.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Steitz JA. Herpesvirus saimiri U RNAs are expressed and assembled into ribonucleoprotein particles in the absence of other viral genes. J Virol. 1990;64:3905–3915. doi: 10.1128/jvi.64.8.3905-3915.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Murthy SC, Trimble JJ, Desrosiers RC, Steitz JA. Four novel U RNAs are encoded by a herpesvirus. Cell. 1988;54:599–607. doi: 10.1016/s0092-8674(88)80004-7. [DOI] [PubMed] [Google Scholar]
- Lee WM, Lin C, Curran T. Activation of the transforming potential of the human fos proto-oncogene requires message stabilization and results in increased amounts of partially modified fos protein. Mol Cell Biol. 1988;8:5521–5527. doi: 10.1128/mcb.8.12.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine TD, Gao F, King PH, Andrews LG, Keene JD. Hel-N1: An autoimmune RNA-binding protein with specificity for 3′ uridylate-rich untranslated regions of growth factor mRNAs. Mol Cell Biol. 1993;13:3494–3504. doi: 10.1128/mcb.13.6.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Ma W-J, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996;271:8144–8151. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- Malter JS. Identification of an AUUUA-specific messenger RNA binding protein. Science. 1989;246:664–666. doi: 10.1126/science.2814487. [DOI] [PubMed] [Google Scholar]
- Malter JS, Hong Y. A redox switch and phosphorylation are involved in the post-translational up-regulation of the adenosine-uridine binding factor by phorbol ester and ionophore. J Biol Chem. 1991;266:3167–3171. [PubMed] [Google Scholar]
- Mattaj IW. U snRNP assembly and transport. In: Birnstiel ML, editor. Struncture and function of major and minor small nuclear ribonucleoprotein particles. Heidelberg, Germany: Springer-Verlag; 1988. pp. 100–114. [Google Scholar]
- Meijlink F, Curran T, Miller AD, Verma IM. Removal of a 67-base-pair sequence in the noncoding region of protooncogene fos converts it to a transforming gene. Proc Natl Acad Sci. 1985;82:4987–4991. doi: 10.1073/pnas.82.15.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Curran T, Verma IM. c-fos protein can induce cellular transformation: A novel mechanism of activation of a cellular oncogene. Cell. 1984;36:51–60. doi: 10.1016/0092-8674(84)90073-4. [DOI] [PubMed] [Google Scholar]
- Murthy S, Kamine J, Desrosiers RC. Viral-encoded small RNAs in herpesvirus saimiri induced tumors. EMBO J. 1986;5:1625–1632. doi: 10.1002/j.1460-2075.1986.tb04405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy SC, Trimble JJ, Desrosiers RC. Deletion mutants of herpesvirus saimiri define an open reading frame necessary for transformation. J Virol. 1989;63:3307–3314. doi: 10.1128/jvi.63.8.3307-3314.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer VE, Steitz JA. Isolation and characterization of a novel, low abundance hnRNP protein: A0. RNA. 1995;1:171–182. [PMC free article] [PubMed] [Google Scholar]
- Myer VE, Lee SI, Steitz JA. Viral small nuclear ribonucleoproteins bind a protein implicated in messenger RNA destabilization. Proc Natl Acad Sci. 1992;89:1296–1300. doi: 10.1073/pnas.89.4.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer VE, Fan XC, Steitz JA. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 1997;16:2130–2139. doi: 10.1093/emboj/16.8.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa J, Waldner H, Meyer-Monard S, Hofsteenge J, Jeno P, Moroni C. AUH, a gene encoding an AU-specific RNA binding protein with intrinsic enoyl-CoA hydratase activity. Proc Natl Acad Sci. 1995;92:2051–2055. doi: 10.1073/pnas.92.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry HD, Scherly D, Mattaj I. “Snurpogenesis”: The transcription and assembly of U snRNP components. Trends Biochem Sci. 1989;14:15–19. [Google Scholar]
- Savant-Bhonsale S, Cleveland DW. Evidence for instability of mRNAs containing AUUUA motifs mediated through translation-dependent assembly of a >20S degradation complex. Genes & Dev. 1992;6:1927–1939. doi: 10.1101/gad.6.10.1927. [DOI] [PubMed] [Google Scholar]
- Schiavi SC, Shyu A-B, Wellington CY, Chen CY, Greenberg ME, Belasco JG. Multiple elements in the c-fos protein coding region accelerate mRNA deadenylation and decay by a mechanism coupled to translation. J Biol Chem. 1994;269:3441–3448. [PubMed] [Google Scholar]
- Schuler GD, Cole MD. GM-CSF and oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell. 1988;55:1115–1122. doi: 10.1016/0092-8674(88)90256-5. [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1988;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Ratnasabapathy R, Hernandez N. Characterization of the inducer of short transcripts, a human immunodeficiency virus type 1 transcriptional element that activates the synthesis of short RNAs. Mol Cell Biol. 1993;13:1251–1263. doi: 10.1128/mcb.13.2.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu A-B, Greenberg ME, Belasco JG. The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes & Dev. 1989;3:60–72. doi: 10.1101/gad.3.1.60. [DOI] [PubMed] [Google Scholar]
- Shyu A-B, Belasco JG, Greenberg ME. Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes & Dev. 1991;5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- Tarn W-Y, Steitz JA. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes & Dev. 1994;8:2704–2717. doi: 10.1101/gad.8.22.2704. [DOI] [PubMed] [Google Scholar]
- Terns MP, Lund E, Dahlberg JE. A pre-export U1 snRNP in xenopus laevis oocyte nuclei. Nucleic Acids Res. 1993;21:4569–4573. doi: 10.1093/nar/21.19.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakalopoulou E, Schaack J, Shenk T. A 32-kilodalton protein binds to AU-rich domains in the 3′ untranslated regions of rapidly degraded mRNAs. Mol Cell Biol. 1991;11:3355–3364. doi: 10.1128/mcb.11.6.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman DA, Lee SI, Steitz JA. Nucleotide sequence of HSUR 5 RNA from herpesvirus saimiri. Nucleic Acids Res. 1989;17:1258. doi: 10.1093/nar/17.3.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennborg A, Sohlberg B, Angerer D, Klein G, von Gabain A. A human RNase E-like activity that cleaves RNA sequences involved in mRNA stability control. Proc Natl Acad Sci. 1995;92:7322–7326. doi: 10.1073/pnas.92.16.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstall E, Gamache M, Raymond V. Rapid mRNA degradation mediated by the c-fos 3′ AU-rich element and that mediated by the Granulocyto-Macrophage Colony-Stimulating Factor 3′ AU-rich element occur through similar polysome-associated mechanisms. Mol Cell Biol. 1995;15:3796–3804. doi: 10.1128/mcb.15.7.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuo Y, Weiner AM. A U1 small nuclear ribonucleoprotein particle with altered specificity induces alternative splicing of an adenovirus E1A mRNA precursor. Mol Cell Biol. 1989;9:3429–3437. doi: 10.1128/mcb.9.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. Purification, characterization and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol. 1993;13:7652–7665. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]