

Abstract
Kinase A is the sensor histidine kinase responsible for processing postexponential phase information and providing phosphate input to the phosphorelay that activates developmental transcription via phosphorylated Spo0A. A protein inhibitor, KipI, of kinase A was discovered encoded in an operon of genes of unknown function but regulated by the availability of fixed nitrogen. KipI is a potent inhibitor of the autophosphorylation reaction of kinase A but does not inhibit phosphate transfer to the Spo0F response regulator once kinase A is phosphorylated. KipI is an inhibitor of the catalytic domain of kinase A affecting the ATP/ADP reactions and not the phosphotransferase functions of this domain. The inhibitory activity of KipI is counteracted by the product of another gene in the operon, KipA. This protein may bind to KipI, preventing its function as an inhibitor of kinase A. KipI may be the first representative of a new class of signal transduction inhibitors that function by direct interaction with the catalytic domain of histidine kinases to counteract signals influencing the “sensor” domain of such kinases. This inhibitor represents yet another way by which the phosphorelay signal transduction system is affected by negative regulators under the control of metabolic, environmental, or cell cycle influences antithetical to the initiation of developmental transcription.
Keywords: Phosphorelay, sporulation, histidine kinase inhibitor, signal transduction, kinase A, Bacillus subtilis
The initiation of developmental transcription in sporulation of Bacillus subtilis represents a cellular commitment to a process that requires the coordination of a myriad of cellular events to assure that they occur in the correct order and at the correct time. Commitment to initiate this complex process and abandon vegetative growth and division is not made lightly and involves analysis of many signals that communicate the status of metabolism, the environment, and the cell cycle (Hoch 1993). How a cell interprets this information and how it is used to decide between vegetative growth and sporulation is only now being revealed. Many of the signals, both positive and negative, that affect this decision are interpreted through the phosphorelay signal transduction system (Burbulys et al. 1991). The phosphorelay is an extended version of the familiar two-component signal transduction systems used extensively in bacteria to perceive and transduce a variety of signals (Parkinson and Kofoid 1992). Perception is the province of a histidine kinase that acts as a signal receptor and promotes the transduction of information to chemical energy by its regulation of the autophosphorylation activity of the kinase (Ninfa and Magasanik 1986). The kinase-bound phosphate is transferred to a response regulator protein mated specifically and usually exclusively to the kinase. Phosphorylation of the response regulator activates its functions—normally transcription regulation. The phosphorelay differs from this paradigm in that the response regulator Spo0F receives phosphate from two different kinases, KinA and KinB, and Spo0F is not a transcription factor but only an intermediate in the ultimate activation of a transcription factor (Burbulys et al. 1991; Trach and Hoch 1993). This factor, Spo0A, is the recipient of the phosphate from Spo0F by means of a response regulator phosphotransferase, Spo0B, unique to the phosphorelay. Since originally discovered in the sporulation system of B. subtilis (Burbulys et al. 1991), phosphorelays have been described in other bacteria, yeast, and fungi (Posas et al. 1996; Uhl and Miller 1996).
Why use a multicomponent phosphorelay in place of a two-component system when the end product, an activated transcription factor, is the same in both? The rationale originally proposed was that a multicomponent system provided more targets for regulation of the final phosphorylation level of the transcription factor (Burbulys et al. 1991). Subsequent events have shown that this is likely to be true. Regulation of the phosphorelay is now known to occur not only at the level of phosphate input by control of the kinases but also at the level of the response regulators Spo0F and Spo0A by regulated dephosphorylation (Perego and Hoch 1996b; Perego et al. 1996). Spo0A∼P is subject to dephosphorylation by the Spo0E phosphatase (Ohlsen et al. 1994) and Spo0F∼P is the substrate for two of the Rap family of phosphatases RapA and RapB (Perego et al. 1994). Because Spo0F∼P and Spo0A∼P are connected by the Spo0B phosphotransferase, which is freely reversible, dephosphorylation of one component rapidly results in lowered phosphate levels in the other.
The transcription of the genes for these phosphatases is tightly regulated by physiological processes inimical to sporulation (Perego and Hoch 1996a). RapB is induced by glucose in exponential growth, and RapA is regulated by the ComA transcription factor that induces competence, a physiological state in which sporulation is not desirable. Therefore, signals from cellular processes that are not compatible with sporulation prevent its occurrence by dephosphorylating the phosphorelay. The phosphorelay is best thought of as a signal integration circuit, in which positive signals regulating kinases and negative signals regulating phosphatases compete to influence the output of the system, the phosphorylation level of the Spo0A transcription factor (Ohlsen et al. 1994). The phosphorelay is not unlike a mitogen-activated protein (MAP) kinase cascade, where kinases and phosphatases compete in a like manner to regulate the output from the system.
Are all of the signals that influence the sporulation phosphorelay acting via known channels of phosphate ingress or egress? Have we exhausted the means by which two-component systems and phosphorelays may be regulated? The experiments described in this communication show that a new type of inhibitor of such systems can be found that acts directly on the catalytic domain of the kinase rather than on its signal perception domain. Such an inhibitor may be a paradigm for new mechanisms of signal transduction control.
Results
Cloning a kinA Inhibitory DNA fragment
Negative regulators of the phosphorelay (e.g., phosphatases) that act by dephosphorylating the signal transduction components are sporulation inhibitors if overexpressed by means of placing the gene coding for them on a multicopy plasmid. This observation suggested to us that expression of a gene encoding an inhibitor of phosphate input, that is, a kinase inhibitor, should show the same phenotype when expressed from a multicopy vector. To search for such inhibitors, a library of B. subtilis chromosomal DNA in the shuttle vector pHT315 was produced and yielded a series of sporulation-defective mutants when transformed into the sporulating B. subtilis strain JH642 (Table 1). One of these mutants carried a plasmid, pRM90, that gave sporulation-defective transformants when purified and retransformed into the sporulation-proficient strain JH642, and the transformants were identical in phenotype to those of a kinA-defective mutant. Comparison of the sporulation frequency of strains bearing pRM90 to the same strains carrying the parental plasmid pHT315 showed that pRM90 reduced sporulation of the wild-type strain to the level of a kinA mutant (Table 2). PRM90 did not further reduce the sporulation in a kinA strain, whereas when pRM90 was transformed into a kinB mutant strain (JH19980), the transformants showed a stage 0 sporulation defect typical of a double mutant kinA, kinB strain (Table 2). These two kinases account for virtually all of the sporulation under these conditions. These results suggest that KinA was the target of pRM90 inhibition.
Table 1.
B. subtilis strains used in this study
| Strain
|
Genotypea
|
|---|---|
| JH642 | wild type |
| JH19980 | kinB::tet |
| JH19087 | kinA::spc |
| JH19108 | kinB::tet, kipA::catb |
| JH19112 | kinB::tet, kipI::catb |
| JH19115 | kinB::tet, amyE::pRM111spc |
| JH19117 | kinB::tet, kipR::catb, amyE::pRM111spc |
| JH19168 | kinB::tet, ΔkipI |
| JH19190 | kinB::tet, tnrA, amyE::pRM111spc |
| JH19192 | kinB::tet, tnrA, kipR::cat, amyE::pRM111spc |
All strains also carry the trpC2, phe-1 markers.
Presumed polar insertions on downstream genes in the operon.
Table 2.
Effect of multicopy plasmid pRM90 on sporulation
| Straina (relevant genotype)
|
Plasmid
|
Viable cells/ml
|
Spores/ml
|
Percent spores
|
|---|---|---|---|---|
| JH642 (wild type) | pHT315 | 2.0 × 108 | 3.0 × 107 | 15.0 |
| pRM90 | 3.6 × 108 | 1.4 × 106 | 0.4 | |
| JH19087 (kinA) | pHT315 | 3.9 × 108 | 2.3 × 106 | 0.6 |
| pRM90 | 3.6 × 108 | 1.8 × 106 | 0.5 | |
| JH19980 (kinB) | pHT315 | 3.6 × 108 | 5.0 × 107 | 14.0 |
| pRM90 | 4.8 × 108 | 4.0 × 103 | 0.0008 |
Strains carrying the multicopy plasmids were grown in 3 ml of SSM containing erythromycin at 25 μg/ml for 36 hr before plating.
When the nucleotide sequence of the chromosomal DNA fragment cloned in plasmid pRM90 was determined, the results revealed a portion of the B. subtilis genome previously identified by the B. subtilis genome sequencing project (Fig. 1). This region is organized in an operon (see below) originally defined by six genes (see Materials and Methods) designated ycsF, ycsG, ycsI, ycsJ, ycsO, and ycsK (Akagawa et al. 1995).
Figure 1.

Restriction map of the chromosomal region carrying the operon containing the kip genes. Arrows indicate the lengths of the various genes. The position of putative promoters are indicated by small arrows. Putative transcription terminators are also shown. The fragments used in plasmid constructions are indicated by the lines. (Δ) Internal deletions. Abbreviations for restriction enzymes are (A) ApaI; (B) BbsI (not unique); (C) ClaI; (E) EcoRI; (Eg) EagI; (H) HindIII; (N) NotI; (P) PflMI; (S) SstI; and (Su) Sau3A (not unique).
The first gene, ycsF, encodes a protein of 257 amino acids, similar (34% identity, 57% similarity) to the LamB protein (262 amino acids) of Aspergillus nidulans (Richardson et al. 1992). LamB in fungi seems to be required for the utilization of lactam rings as a nitrogen source. The second gene, ycsG, codes for a highly hydrophobic protein of 404 amino acids with 11 potential transmembrane domains. This protein shows similarity (21% identity, 49% similarity) to bacterial permeases such as BraB (branched chain amino acid transport system II) of Pseudomonas aeruginosa (437 amino acids) (Hoshino et al. 1990). Gene three, ycsI, encodes a product of 263 amino acids with similarity (45% identity, 62% similarity) to a transmembrane protein of 301 amino acids from Schizosaccharomyces pombe whose function is unknown (accession no. Q09674).
Genes four and five, kipI and kipA, were designated as ycsJ in the original sequencing study (Akagawa et al. 1995). Our sequencing and gene expression results indicate that ycsJ is actually two genes that produce two proteins. Sequencing of this region from two wild-type Bacillus strains (see Materials and Methods) also gave two genes that expressed two proteins. The products of the kipI (240 amino acids) and kipA (337 amino acids) genes are both distantly related to the ureamidolyase enzyme of Saccharomyces cereviseae (Genbauffe and Cooper 1991), which is a single protein in yeast and is much bigger (1835 amino acids) than the sum of kipI and kipA (577 amino acids). Two genes (HI1731 and HI1730) whose products are highly similar to kipI and kipA have been identified in Haemophilus influenzae, and they are closely linked to LamB and BraB homologs (Fleischmann et al. 1995).
The sixth gene, ycsO, now renamed kipR, codes for a protein of 247 amino acids whose sequence contains a helix–turn–helix motif typical of DNA-binding proteins. Results reported below show KipR to be a regulator of the kip gene-containing operon.
The seventh gene, ycsK, codes for a protein of 213 amino acids with no significant homology to any other protein in the databank. This gene is followed by a putative transcription terminator.
Gene amplification and inactivation analyses
Plasmid pRM90 contains the carboxyl end of the ycsI gene, the kipI gene, and a truncated kipA gene (Fig. 1). To determine the gene responsible for the sporulation phenotype, deletions were made in the plasmid. A deletion of kipI in plasmid pRM92 (Fig. 1) was 1200-fold less effective at preventing sporulation than pRM90 (Table 3). Deletion of a large portion of kipA in pRM91 was without effect on sporulation inhibition. However, if a complete copy of the kipA gene was added to pRM90, the plasmid pRM94 was much less effective in preventing sporulation. Deletion of kipI from such a plasmid, pRM93, resulted in enhanced sporulation relative to strains bearing pHT315. This enhancement is likely attributable to KipA neutralization of the chromosomally encoded KipI. Thus, the sporulation inhibitor is encoded in kipI, and its effects are neutralized in strains bearing kipA in addition to kipI on multicopy plasmids.
Table 3.
Effect of deletions in pRM90 on sporulation of a kinB strain
| Plasmida
|
Sporulation phenotype (plates)
|
Relative sporulation frequency (liquid)
|
|---|---|---|
| pHT315 | Spo+ | 1 |
| pRM90 | Spo0 | 0.0008 |
| pRM91 | Spo0 | 0.0008 |
| pRM92 | Spo+ | 1 |
| pRM93 | Spo++ | 2.27 |
| pRM94 | Spo± | 0.56 |
Parental strain for all plasmids was JH19980 kinB::tet.
Deletion of the chromosomal copy of the kipI gene with or without deletion of the kipA gene enhances sporulation (strain JH19168; Table 4). However, deletion of kipA alone decreased sporulation 4- to 5-fold and 300-fold if glucose was added to the culture medium (strain JH19108; Table 4). Data presented below will show that glucose is an inducer of the kip gene-containing operon. Under glucose conditions, the kipA insertional inactivation strain JH19108 mimics a strain with a multicopy kipI. Thus, the kipI gene product is likely to be the inhibitor of sporulation, and the genetics are consistent with the product of the kipA gene product being an inhibitor of KipI or at least preventing or reversing the sporulation inhibition by KipI. Note that the insertional inactivation strains JH19108 and JH19112 may also prevent expression of the kipR regulator gene (described below), but this regulator has little effect in the nitrogen-rich medium used in the experiments described in Table 4. Compare strains JH19112 and JH19168 where the latter strain has a nonpolar deletion of kipI and the results are the same. To test whether kipI was a direct inhibitor of KinA, the KipI and KipA proteins were purified and tested for their effects on the phosphorelay reactions.
Table 4.
Effect of kipI and kipA chromosomal genotype and glucose on sporulation
| Strain
|
Relevant genotype
|
− Glucose
|
Percent spores
|
+ Glucose
|
Percent spores
|
|||
|---|---|---|---|---|---|---|---|---|
|
kipI
|
kipA
|
viable cells/ml
|
spores/ml
|
viable cells/ml
|
spores/ml
|
|||
| JH19980 | + | + | 4.9 × 108 | 1.2 × 108 | 24.5 | 1.6 × 109 | 9.8 × 105 | 0.06 |
| JH19108 | + | − | 4.6 × 108 | 2.4 × 107 | 5.2 | 2.2 × 109 | 5.0 × 103 | 0.0002 |
| JH19112 | − | − | 4.5 × 108 | 1.6 × 108 | 35.5 | 1.4 × 109 | 1.2 × 106 | 0.08 |
| JH19168 | − | + | 4.9 × 108 | 1.9 × 108 | 38.8 | 1.6 × 109 | 2.8 × 106 | 0.17 |
Cultures were grown in SSM at 37°C with or without glucose at 2% (wt/vol) final concentration. Samples were tested for spore formation after ∼16 hr of growth. Results are the average of two independent experiments.
Effect of KipI and KipA on KinA autophosphorylation
The kipI gene and a complete copy of the kipA gene were amplified by PCR from chromosomal DNA and subcloned independently into the pET vector expression system with His-tagged extensions. After the sequence of the genes was verified, each protein was expressed in Escherichia coli and purified by Ni–NTA affinity chromatography. KipI was obtained as a soluble protein in this system, whereas KipA was only partially soluble and mostly present in inclusion bodies. The purified proteins gave the expected molecular masses when analyzed by SDS-PAGE: KipI was 26,720 daltons, and KipA was 36,931 daltons.
The effect of the proteins on KinA activity was analyzed in the standard phosphorylation reaction with [γ-32P]ATP (Fig. 2A). KipI at 4 μm concentration strongly inhibited the autophosphorylation of 0.5 μm KinA. The addition of 6 μm KipA to this reaction partially overcame the inhibition by KipI. When 2 μm Spo0F was added to the reaction along with KinA, both Spo0F and KinA were labeled. However, when added, KipI strongly inhibited the accumulation of Spo0F∼P and no labeled KinA was observed. The addition of KipA alone to the autophosphorylation of KinA or to the phosphorylation of Spo0F had no effect. However, KipA addition partially reversed the inhibition by KipI of Spo0F∼P accumulation. These results suggested that KipI inhibits the autophosphorylation of KinA and this inhibition may be partially prevented by KipA. These data are entirely consistent with the in vivo genetic data.
Figure 2.


Effect of KipI and KipA on KinA and KinA-C autophosphorylation. Reactions were performed as described in Materials and Methods. KinA (A) and KinA-C (B) were used at 0.5 μm while Spo0F, KipI, and KipA were added at final concentrations of 2, 4, and 6 μm, respectively. (+) The proteins present in each reaction.
To gain some insight into the site of action of KipI, a kinase domain fragment of KinA consisting of the last 217 carboxy-terminal residues (L. Wang and J.A. Hoch, in prep.), was tested for inhibition by KipI. The kinase A catalytic domain is deficient in autophosphorylation, but the residual activity in the truncated protein is inhibited by KipI (Fig. 2B). Therefore, the site of KinA inhibition by KipI is the catalytic domain. The inhibition of Spo0F∼P accumulation by KipI may be only related to the effect of KipI on the autophosphorylation of KinA, but the present data do not exclude an effect on the phosphotransferase reaction. Titration of KipI against 0.5 μm KinA showed half-maximal inhibition at 1–2 μm KipI (Fig. 3). The inhibition is noncompetitive with respect to ATP (data not shown).
Figure 3.

Titration of inhibition of KinA autophosphorylation by KipI. The assay was performed in standard conditions as described in Materials and Methods. KipI was added at increasing concentrations, as shown. The reactions were loaded on a 15% SDS–polyacrylamide gel, which was exposed to X-ray film and subjected to quantitative analysis by a Molecular Dynamics PhosphorImager. The percentage of labeled KinA compared to the labeled KinA obtained in the control in the absence of KipI was plotted against the concentrations of KipI used in each reaction.
KipI does not inhibit NRII kinase of E. coli
To determine whether KipI was specific for KinA or whether it was a general kinase inhibitor, the effect of KipI was tested on purified NRII kinase of E. coli. KipI at a concentration sufficient to inhibit the autophosphorylation of KinA was without effect on the autophosphorylation of NRII. Titration of KipI against NRII at a higher concentration had no effect on the NRII reaction (Fig. 4).
Figure 4.

Specificity of KipI inhibition toward KinA. Autophosphorylation conditions were as described in Materials and Methods. KinA and NRII were used at 0.5 μm final concentration. Increasing concentrations of KipI resulted in inhibition of KinA autophosphorylation, but no effect was observed on NRII.
Effect of KipI on reverse reactions of KinA
It seemed possible that KipI could have an apparent effect on the autophosphorylation of KinA either by inhibiting this reaction or by being a direct phosphatase of KinA∼P. To decide between these possibilities, labeled KinA∼P was produced and purified from residual ATP. When KinA∼P was incubated with ADP, some loss of labeled KinA∼P was observed (Fig. 5A, lane 2) and this can all be accounted for by conversion to ATP (Fig. 5B). No free phosphate was formed. Addition of KipI prevents this loss of KinA∼P and inhibits formation of ATP (Fig. 5A,B, lane 3). No free phosphate was observed. Addition of KipA alone or with KipI has little apparent effect (Fig. 5A,B, lanes 4,5). Rather than stimulate the loss of KinA∼P through a phosphatase reaction, the addition of KipI prevented the ADP-dependent loss of KinA∼P. Therefore, KipI inhibits both the ATP-dependent autophosphorylation and its reverse reaction from ADP. Thin-layer chromatography (TLC) of several other reactions revealed that the [γ-32P]ATP was not hydrolyzed by KipI, and the addition of KinA to KipI did not stimulate ATP hydrolysis (data not shown). The results are consistent with KipI being a specific inhibitor of KinA and its effect does not reside in an activity that destroys ATP or dephosphorylates KinA∼P.
Figure 5.
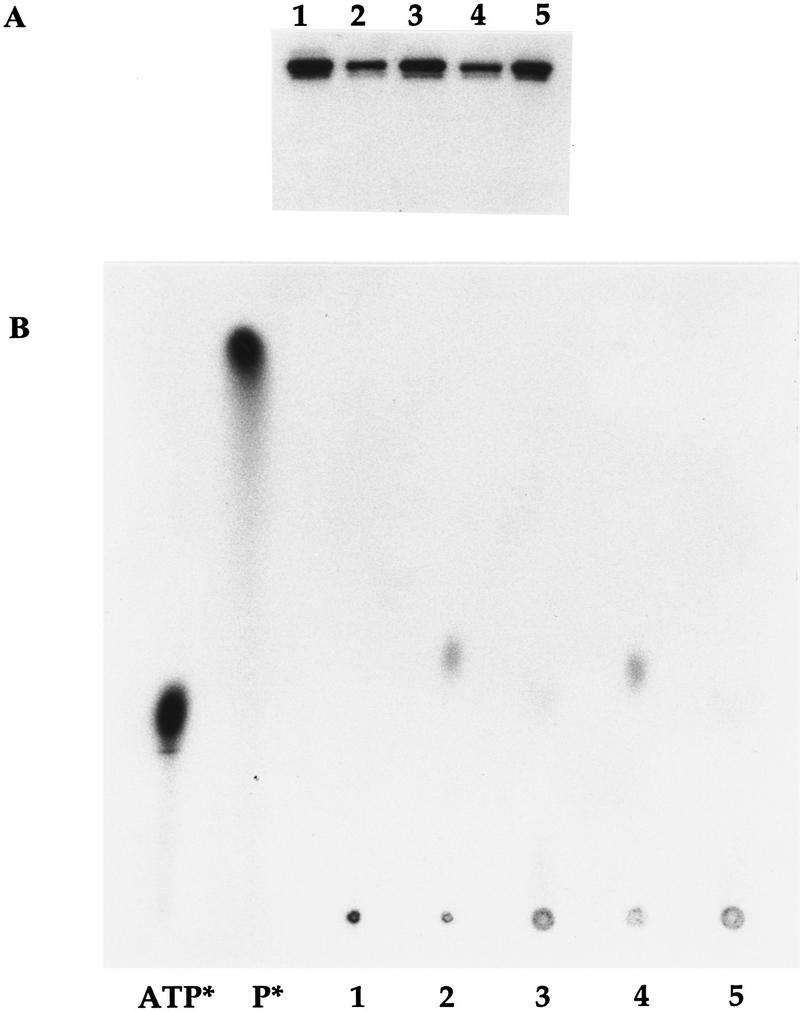
Mechanism of KipI inhibition of KinA autophosphorylation. Purified KinA∼P (30 pmoles) was incubated in the standard reaction buffer for 3 min at room temperature in the presence of nothing (lane 1); 300 μm ADP (lane 2); 300 μm ADP and 5 μm KipI (lane 3); 300 μm ADP and 5 μm KipA (lane 4); 300 μm ADP and 5 μm each KipI and KipA (lane 5). Final volume was 30 μl. Reactions were stopped by the addition of 10 μl of 5× SDS loading buffer. Reaction mixtures (39 μl) were loaded on a 15% SDS–polyacrylamide gel (A) while 1 μl was applied to a TLC plate (B). Markers used were purified [γ-32P]ATP (ATP*) and 32P carrier-free (Amersham) (P*).
Effect of KipI on phosphotransferase reactions
To determine whether KipI affected the phosphotransferase reaction of KinA, labeled KinA∼P and Spo0F∼P were prepared and purified. Purified KinA∼P incubated with KipI is not dephosphorylated, and addition of Spo0F results in uninhibited transfer of phosphate to Spo0F (Fig. 6A). The same results were obtained with purified labeled KinA-C∼P, which consists of only the catalytic domain of KinA (Fig. 6A). When purified Spo0F∼P was incubated with KipI, there was no loss of phosphate from Spo0F∼P, and the back reaction to KinA was uninhibited (Fig. 6B). The identical results were obtained with KinA-C as the phosphate acceptor (Fig. 6B).
Figure 6.


KipI does not prevent the phosphotransferase reaction from KinA/KinA-C∼P to Spo0F and from Spo0F∼P to KinA/KinA-C. (A) Purified KinA∼P or KinA-C∼P (30 pmoles) was incubated for 3 min at room temperature in standard reaction buffer. The presence of Spo0F (5 μm) and/or KipI (5 μm) in the reaction is indicated (+). (B) Spo0F∼P (50 pmoles) was incubated in standard reaction buffer with KinA (5 μm) or KinA-C (5 μm) in the presence of KipI (5 μm) as indicated (+).
Therefore, KipI affects the KinA autophosphorylation reaction and its reverse reaction. It is not an inhibitor of the reverse phosphotransferase reaction and likely does not inhibit the forward reaction. It is not a phosphatase of KinA∼P or of Spo0F∼P and does not stimulate a latent autophosphatase activity of KinA.
Catabolite induction of kip genes
Transcriptional analysis of the kip gene-containing operon was carried out by assaying β-galactosidase activity of a promoter–lacZ fusion construction (pRM111; Fig. 1) cloned in the transcriptional fusion vector pJM116 and integrated ectopically in the amyE locus (see Materials and Methods). In the nutrient broth-based Schaeffer’s sporulation medium (SSM), kip transcription increased during the logarithmic phase, reaching a maximum level at T0 (15 Miller units), thereafter decreasing to ∼7 Miller units between T3 and T4 (Fig. 7). Because the sporulation defect of a kipA deletion strain was enhanced by the addition of glucose, transcription of the operon was analyzed in the presence of sugars. The addition of glucose at 1% final concentration to SSM strongly induced transcription of the operon in early exponential phase, but a sharp decrease reproducibly occurred at T−1 (Fig. 7). Similar induction was also observed when fructose at 1% or glycerol at 2% was added to SSM. The same construction was also placed isotopically on the chromosome by means of a pJM783 integrative plasmid, and the same patterns of transcription were observed.
Figure 7.

Expression of the operon containing the kip genes in sporulation medium. The wild-type strain JH19115 harboring the lacZ fusion construct plasmid pRM111 was grown in SSM (○), in SSM supplemented with 1% glucose (▴), and in SSM supplemented with 1% glucose and 0.3% glutamine (▾). Time 0 represents the transition from vegetative to sporulation phase. β-Galactosidase was expressed as Miller units (Miller 1972).
The induction of the operon by glucose provided a means to determine the boundaries of the operon. Transcriptional lacZ fusions to kipI, kipR, and ycsK were constructed in pJM783 and isotopically integrated in the B. subtilis chromosome as described in Materials and Methods. β-Galactosidase assays were carried out in SSM in the presence of 1% glucose. These lacZ fusions showed the same pattern of induction observed with plasmid pRM111, although the maximum level of transcription gradually decreased the farther the distance from the promoter (data not shown). No transcriptional activity was obtained from plasmid pRM116 carrying a lacZ fusion to the 3′ end of a putative Rho-independent terminator of transcription, located downstream of ycsK (Fig. 1). Furthermore, transcription of ycsL, located downstream of the terminator, showed a totally different regulatory pattern (data not shown). This analysis suggests that the operon containing the kip genes comprises seven cistrons in a single transcriptional unit induced by several carbon sources.
Nitrogen control of kip gene expression
When the B. subtilis strain JH19115 carrying the promoter–lacZ fusion construct pRM111 was grown in SSM in the presence of 0.3% glutamine, induction by glucose was prevented (Fig. 7). Strain JH19115 was grown in Spizizen salts minimal medium in the presence of different nitrogen sources to examine the basis for this nitrogen effect. In a good nitrogen source, such as NH4+, very little transcription was observed. However, poor nitrogen sources such as allantoin, allowed higher levels of transcription from the promoter–lacZ construct pRM111. Expression in different nitrogen sources increased as follows: NH4+ < arginine < glutamine < urea < proline = γ amino butyric acid = asparagine < allantoin < glutamate (data not shown). When a good and a poor nitrogen source were both present in the medium, the good nitrogen source effect was dominant.
Many nitrogen-regulated genes in B. subtilis are transcriptionally activated by the TnrA regulator, which responds to poor nitrogen conditions (Wray et al. 1996). We tested whether the enhanced transcription of the kip gene-containing operon under these conditions was a result of TnrA action by studying transcription in a tnrA deletion strain. Cells were grown in minimal glucose media with glutamate as a nitrogen source, and β-galactosidase activity driven by a single-copy chromosomal promoter–lacZ construct (pRM111) was assayed. Deletion of kipR increased the final level of transcription >10-fold (Fig. 8). Deleting tnrA in this kipR mutant prevented most of this increase, indicating that TnrA is an activator of kip operon transcription and the loss of repression regulation by KipR allows TnrA to activate transcription of the operon. In the absence of TnrA, KipR repression of kip transcription is essentially complete.
Figure 8.

Regulation of expression by KipR and TnrA. β-Galactosidase activity of the lacZ fusion construct carried by plasmid pRM111 was assayed in the wild-type strain JH19115 (○), the kipR mutant JH19117 (□), in the tnrA mutant JH19190 (•), and in the kipR, tnrA double mutant JH19192 (▪). Cells were grown in Spizizen salts minimal medium supplemented with glutamate, as described in Materials and Methods. Time and units as in Fig. 7.
Discussion
The regulation of the activity of KinA is of paramount importance for the initiation of developmental transcription at the onset of sporulation. KinA is functional at the end of exponential growth and must respond to those cellular signals that would promote sporulation at the expense of further growth. Essentially all of the phosphate that is sequestered by the phosphorelay components at this time arises from the action of KinA. Many of the mechanisms by which KinA is regulated still remain obscure, although the proteins described here must play a role in controlling KinA activity under some physiological conditions. The operon in which KipI and KipA reside is induced by glucose when readily available sources of nitrogen, such as glutamine or ammonia, are scarce. This operon is regulated by TnrA activation and by KipR repression. TnrA positively regulates a number of genes and operons coding for proteins that degrade nitrogen-containing compounds (Ferson et al. 1996; Wray et al. 1996). TnrA-dependent promoters are characterized by a common upstream sequence (TGTNAN7TNACA), two of which are present in the promoter for the kip gene-containing operon, from the putative −60 to −30 region. Nitrogen-limited TnrA activation is clearly antagonized by KipR repression, which responds to some other function of the operon. In addition, there is some induction by catabolites that has an unknown basis. It seems likely that the genes of this operon must code for a system to take up and degrade some nitrogenous compound, perhaps with a lactam ring, but its identity is obscure. Regardless, some physiological situations responsible for induction must be contrary to sporulation, as the product of induction, KipI, is a potent inhibitor of KinA activity.
KipI is an inhibitor of the autophosphorylation of KinA by ATP. It is also an inhibitor of the reverse reaction from KinA∼P to KinA and ATP. KipI does not inhibit by dephosphorylating the substrate or the product. Inhibition is not attributable to stimulation of a phosphatase activity of the kinase similar to that of the action of PII on NRII of nitrogen regulation (Ninfa et al. 1995). It also does not stimulate the reverse reaction, giving an apparent inhibition. KipI targets the carboxy-terminal catalytic domain where it interferes with the ATP-dependent reactions of the kinase. However, the inhibition is noncompetitive with respect to ATP. Because KipI is an inhibitor of the autophosphorylation of the isolated catalytic domain of KinA, its activity does not depend on the amino-terminal “sensor” domain of this kinase. Whether the amino-terminal domain of this kinase plays any role in the binding of KipI cannot be determined. A detailed kinetic comparison of the effects of KipI on intact and amino-terminal truncated KinA would be fruitless, as all amino-terminal truncated versions of KinA are defective in the autophosphorylation reaction (L. Wang and J.A. Hoch, unpubl.). It seems most likely that the sensor domain recognizes something other than KipI and acts to regulate the kinase in response to other signals.
In contrast to its effects on the autophosphorylation reaction, KipI appears to have little effect on the phosphotransferase reaction of KinA. Purified KinA∼P transfers its phosphate to Spo0F unimpeded by KipI. Similarly, purified Spo0F∼P transfers phosphate back to KinA or to an amino-terminal truncated KinA regardless of the presence of KipI. Thus, KipI does not interfere with the capacity of the kinase to recognize its cognate response regulator or to transfer phosphate on and off the reactive histidine of KinA. This suggests that the structure of the catalytic domain is such that the autophosphorylation and response regulator recognition portions of the molecule are independent and are not integrated or overlapping.
Genetic studies indicated that KipA modulated the inhibitory activity of KipI on sporulation. Overexpression of KipI inhibited sporulation, whereas overexpression of KipA enhanced sporulation and a strain producing both proteins from a plasmid was only slightly sporulation defective. KipA produced from the expression vector in E. coli was only partially effective in reversing the KipI effects in vitro. This discrepancy between in vivo and in vitro results is likely to be a result of expressing KipA in E. coli, where it is mainly present in inclusion bodies. Although KipA was purified from the soluble fraction, it seems likely that most of this protein is not in a native conformation and does not have native activities. Both KipI and KipA have been produced in the same cell from an expression vector (L. Wang, R. Grau, M. Perego, and J.A. Hoch, unpubl.). In this case, a His-tagged KipI and KipA copurified on a Ni–NTA column, indicating that KipI and KipA may form a complex. Regardless of whether these data explain the activity of KipA on KipI inhibition in vitro, the genetic evidence that KipA counteracts KipI inhibition is compelling.
What the interaction of KipI with both KinA and KipA has to do with the actual role of KipI in vivo is still a mystery. KipI and KipA were identified in the B. subtilis sequencing project as homologous to ureamidolyase of S. cereviseae (Akagawa et al. 1995). Homologs of KipI and KipA were also found in the H. influenzae genome and identified with the yeast enzyme (Fleischmann et al. 1995). Ureamidolyase catalyzes the ATP-dependent degradation of urea to NH3 and CO2 via allophanic acid (Whitney and Cooper 1973). The available evidence is not in agreement with KipI and KipA being a bacterial form of ureamidolyase. Neither KipI nor KipA is biotinylated whether assayed in B. subtilis or when expressed in E. coli. No ureamidolyase activity can be detected in purified KipI and KipA. A urease-defective mutant of B. subtilis cannot be made to use urea as a nitrogen source even if the operon bearing KipI and KipA is induced. These unpublished experiments are not consistent with a urea-degrading role for KipI and KipA. The function of the operon encoding KipI and KipA is obscure, but the operon is regulated by available nitrogen in a manner very similar to that of ureamidolyase in S. cereviseae (Magasanik 1992; Cunnigham et al. 1994).
KipI acts as an anti-kinase and KipA could be thought of as an anti-anti-kinase. Because kipI and kipA are transcriptionally coregulated, it seems unlikely that expression controls determine the activity of the anti-kinase. It is reasonable to assume that KipI–KipA interaction regulates anti-kinase activity, and this interaction may be subject to effector molecules and/or covalent modification.
It is generally believed that the catalytic domain of histidine kinases is controlled by the sensor domain, which acts as the information recognition and processing site. To our knowledge, KipI is the first inhibitor of two-component histidine kinases directed toward the catalytic domain. The effects of KipI on KinA suggest that the catalytic domain may not be entirely passive in signal transduction. Inhibitors of the catalytic domain may act to block or counteract signals that influence the sensor domain. Perhaps two-component systems designed to recognize the cellular level of a single metabolite such as available nitrogen may only need a sensor domain to interact with a single effector. The phosphorelay is a signal integration circuit that regulates a cellular developmental event, and many signals can and do influence the decision to initiate development. The cell has evolved several mechanisms to cope with these signals other than regulation of the sensor domain. Some signals manifest in the activation of phosphatases that dephosphorylate the response regulator components of the phosphorelay (Perego et al. 1994). The signals transduced by phosphatases are believed to be negative influences on the developmental process that function by counteracting phosphate input into the phosphorelay by KinA (Ohlsen et al. 1994). Because phosphatases play this role, it should not be surprising that the cell would use inhibitors such as KipI to counteract phosphate input by KinA.
The rationale for a cell using the phosphorelay to control development has been postulated to stem from the potential of this type of signal transduction system for regulation by a variety of signals promoting either growth or sporulation (Burbulys et al. 1991). Because sporulation initiation is seemingly only dependant on raising the level of phosphorylation of the Spo0A transcription factor, inhibition by phosphatases or enzyme inhibitors should prevent the process. It should not be unexpected that other components of the phosphorelay are regulated by dephosphorylation or inhibition of function.
Materials and methods
Bacterial strains and growth conditions
The B. subtilis strains used in this study are described in Table I [the tnrA deletion strain SF706T was obtained from Susan Fisher (Ferson et al. 1996)]. For sporulation efficiency, B. subtilis strains were grown in Schaeffer’s sporulation medium (Schaeffer et al. 1965) and then treated with CHCl3 or heated at 80°C for 10 min before plating. Transformation of B. subtilis was carried out as described (Anagnostopoulos and Spizizen 1961).
β-Galactosidase in B. subtilis strains harboring lacZ fusions were assayed as described previously, and the specific activity was expressed in Miller units (Miller 1972). Cells were grown in Schaeffer’s sporulation medium or Spizizen’s minimal medium (Spizizen 1958) without (NH4)2SO4 and supplemented with 20 mm MgSO4, 1% glucose, 0.005% each tryptophan and phenylalanine, and 20 mm nitrogen source.
Plasmids and strains constructions
Plasmid pRM90 was obtained from a B. subtilis JH642 chromosomal library constructed by ligating partial Sau3A restriction fragments in the multiple cloning site of the pHT315 shuttle vector (Arantes and Lereclus 1991; V. Dartois, T. Djavakhishvili, and J.A. Hoch, unpubl.). Plasmid pRM91 was derived from pRM90 by deletion of the fragment comprised of the ApaI site in kipA and the EcoRI site in the multiple cloning site. Plasmid pRM92 was obtained by religating plasmid pRM90 digested previously with EagI to create a 500-bp internal deletion in the KipI coding gene. The multicopy vector pRM94 carrying both kipI and kipA coding sequences was obtained by replacing the ApaI–EcoRI fragment of pRM90 with a ApaI–EcoRI fragment containing the entire kipA gene obtained by PCR amplification of chromosomal DNA. The kipA multicopy plasmid pRM93 was obtained by digestion of pRM94 with EagI and religation to delete the EagI fragment internal to kipI.
The entire operon containing the kip genes and its boundaries were amplified by PCR using the Expand High Fidelity PCR system (Boehringer Mannheim) from chromosomal DNA of strain JH642 with the oligonucleotides 5′-AGACATGGTACCAGGAAACGGCTGATTATATCACG-3′ KpnI and 5′-AAATATCTGCAGCTTGTGGAGTGGGAAACACTTG-3′ PstI. The fragment obtained (6908 bp) was digested with KpnI, EcoRI, and PstI to generate four fragments of 2, 1.45, 1.6, and 1.8 kbp, respectively. Each fragment was cloned in the shuttle vector pHT315 (Arantes and Lereclus 1991) to generate plasmids that were subjected to partial nucleotide sequence.
Plasmids pRM107, pRM108, and pRM109 were constructed in the integrative vector pJM103 (Perego 1993). Their linearization and transformation in B. subtilis-competent cells resulted in integration by a double crossover event that yielded strains JH19112, JH19108, and JH19117, respectively. In these strains, all of the genes in the operon located downstream of the site of integration may also be inactivated because of a polar effect.
For the construction of strain JH19168 (kipI−kipA+), plasmid pRM106, constructed in the integrative vector pJM103, was integrated in the chromosome of JH19980 via single crossing-over by transformation and selection for Cm resistance (CmR) (5 μg/ml). Two Spo+ transformants were grown for several days in Luria Bertani (LB) medium without antibiotic to allow spontaneous excision of the plasmid from the chromosome. The cultures were grown to stationary phase and then diluted 1:100 in fresh medium every 12 hr for 5 days before plating serial dilutions on LB agar plates. The colonies obtained were screened for Cm sensitivity (CmS). Three colonies out of 200 checked were CmS. Their chromosomal DNA was used in PCR reactions using oligonucleotides 5′-TGAGAATCATGACTGTACGATATCAAATCGAAC-3′ and 5′-TCCCGTCTCGAGTGTCAGCCATTCCATCTTCATTCTCGTTTTCAGTTC-3′ to amplify the kipI region and verify the presence of the internal deletion of ∼0.6 kb. The three CmS colonies analyzed yielded a PCR product 0.6 kb smaller than the control chromosomal DNA from JH19980, (kipI+, kipA+). One of these CmS colonies was designated JH19168 and used in further studies.
Transcriptional lacZ fusion plasmids were constructed in vectors pJM783 for isotopic integrations and pJM116 for ectopic integrations in the amyE locus (Perego 1993). Plasmid pRM111 was constructed by cloning a PCR-generated KpnI–HindIII fragment into pJM116. Plasmid pRM112 carries the same fragment but in the lacZ fusion vector pJM783.
Nucleotide sequence
Extensive nucleotide sequence of the operon containing the kip genes was carried out to define the precise end points of each open reading frame. The regions comprising nucleotides 7300–9060 and 10350–13000 in the original sequence (GenBank accession no. d38161) were resequenced from fragments obtained by PCR amplification of the chromosomal DNA of strain JH642. Corrections have been submitted to the databank as an update to the original sequence. To ensure that kipI and kipA were two separate genes, the chromosomal region containing both genes was amplified by PCR reaction from chromosomal DNA extracted from two natural Bacillus isolates (Bacillus natto from a commercial Natto and B. subtilis polish, a strain in our collection). The fragments were cloned in the pET16b expression vector, sequenced, and then transferred to the E. coli BL21 (DE3) strain for protein expression and purification. The results (not shown) confirmed that in these Bacillus natural isolates, KipI and KipA are two distinct proteins coded by two separate genes.
Expression and purification of KipI and KipA
The coding sequences for kipI and kipA were amplified by PCR reaction from chromosomal DNA of the wild-type strain JH642. kipI was cloned as a NcoI–XhoI fragment in the NcoI–XhoI sites of the pET28a expression vector (Novagen), which promotes the fusion of the carboxy-terminal end of the protein with six histidine residues. The kipA gene was cloned in the NdeI–BamHI sites of the pET16b vector (Novagen), thereby adding 10 histidine codons to the 5′ end of the gene. Plasmids were constructed in E. coli DH5α and, after sequence verification, transferred in the E. coli expression host BL21(DE3) plysS. Cells carrying the pET28a–KipI or the pET16b–KipA expression vectors were grown in LB medium supplemented with kanamycin (30 μg/ml) or ampicillin (100 μg/ml), respectively. At an OD600 of 0.6, cells were induced by the addition of 1 mm IPTG and allowed to grow for an additional 3 hr at 37°C before harvesting. The His-tagged proteins were purified by affinity chromatography on Ni–NTA agarose columns (Qiagen), in 20 mm Tris (pH 8.0), 10 mm KCl, and 1 mm PMSF using a 0–150 mm gradient of imidazole.
Phosphorylation assay conditions
Phosphorylation assays were performed in 30 μl volume in the standard reaction buffer (50 mm EPPS at pH 8.0, 0.1 mm EDTA, 20 mm MgCl2, 5% glycerol). KinA was added to a final concentration of 0.5 μm. The reactions were initiated by the addition of 5 μl of a mixture of [γ-32P]ATP and unlabeled ATP to reach a final concentration of 300 μm. Reactions were carried out for 3 min at room temperature and stopped by the addition of 12 μl of 5× SDS loading buffer (25 mm Tris-HCl at pH 6.8, 1.5% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.02% Bromophenol blue). Reactions were placed in dry ice before loading onto 15% SDS–polyacrylamide gels. Electrophoresis was carried out at constant voltage (200 V) for ∼2.5 hr. Where necessary, the lower portion of the gel was removed to reduce the background, which is attributable to unincorporated [γ-32P]ATP. Gels were dried and exposed for 1 hr to Kodak AR films.
Purification of KinA∼P and Spo0F∼P
Phosphorylated KinA and Spo0F were purified as follows: 5 μm KinA or KinA-C and 25 μm Spo0F were labeled by the standard phosphorylation assay in 1 ml volume. After 30 min incubation at room temperature, the mixtures were applied to an FPLC S-100 Sephacryl column (1.6 × 64 cm). Gel filtration-purified KinA∼P, KinA-C∼P, and Spo0F∼P were checked on a 15% SDS–polyacrylamide gel that had been exposed to autoradiography. KinA-C, spanning from residue 389 to 606, was expressed in the pET16b expression vector (L. Wang, in prep.). KinA and Spo0F were purified as described previously (Perego et al. 1989; Zapf et al. 1996).
Analytical TLC
TLC was performed according to Randerath and Randerath (1967), with some modifications. KinA∼P (30 pmoles) was incubated with 5 μm KipI and/or KipA in the standard assay buffer for 5 min at room temperature. ADP at 300 μm final concentration was added to initiate the reactions. Two microliters of stop buffer (5× loading buffer) was added after 3 min incubation, and 1 μl from each reaction was applied to PEI cellulose F layer plates (Alltech). Plates were developed in 1.6 m LiCl for ∼45 min and then dried and exposed overnight to X-ray films. Reactions were also loaded on 15% SDS–polyacrylamide gels and exposed to X-ray films as well.
Acknowledgments
This research was supported in part by grant GM19416 from the National Institute of General Medical Sciences (National Institutes of Health, U.S. Public Health Service). L.W. and R.G. contributed equally to this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hoch@scripps.edu; FAX (619) 784-7966.
References
- Akagawa E, Kurita K, Sugawara T, Nakamura K, Kasahara Y, Ogasawara N, Yamane K. Determination of a 17484 bp nucleotide sequence around the 39° region of the Bacillus subtilis chromosome and similarity analysis of the products of putative ORFs. Microbiology. 1995;141:3241–3245. doi: 10.1099/13500872-141-12-3241. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:741–746. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes O, Lereclus D. Construction of cloning vectors for Bacillus thuringiensis. Gene. 1991;108:115–119. doi: 10.1016/0378-1119(91)90495-w. [DOI] [PubMed] [Google Scholar]
- Burbulys D, Trach KA, Hoch JA. The initiation of sporulation in Bacillus subtilis is controlled by a multicomponent phosphorelay. Cell. 1991;64:545–552. doi: 10.1016/0092-8674(91)90238-t. [DOI] [PubMed] [Google Scholar]
- Cunnigham TS, Dorrington RA, Cooper TG. The UGA4 UASNTR site required for GLN3-dependent transcriptional activation also mediates DAL80-responsive regulation and DAL80 protein binding in Saccharomyces cerevisiae. J Bacteriol. 1994;176:4718–4725. doi: 10.1128/jb.176.15.4718-4725.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferson AE, Wray LV, Fisher SH. Expression of the Bacillus subtilis gabP gene is regulated independently in repsonse to nitrogen and amino acid availability. Mol Microbiol. 1996;22:693–701. doi: 10.1046/j.1365-2958.1996.d01-1720.x. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb J, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Genbauffe FS, Cooper TG. The urea amidolyase (DUR1,2) gene of Saccharomyces cerevisiae. J DNA Seq Map. 1991;2:19–32. doi: 10.3109/10425179109008435. [DOI] [PubMed] [Google Scholar]
- Hoch JA. Regulation of the phosphorelay and the initiation of sporulation in Bacillus subtilis. Annu Rev Microbiol. 1993;47:441–465. doi: 10.1146/annurev.mi.47.100193.002301. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Kose K, Uratani Y. Cloning and nucleotide sequence of the gene braB coding for the sodium-coupled branched-chain amino acid carrier in Pseudomonas aeruginosa PAO. Mol & Gen Genet. 1990;220:461–467. doi: 10.1007/BF00391754. [DOI] [PubMed] [Google Scholar]
- Magasanik B. Regulation of nitrogen utilization. In: Stratherm, et al., editors. The molecular and cellular biology of the yeast Saccharomyces cerevisiae: Metabolism and gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 283–317. [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. pp. 352–355. [Google Scholar]
- Ninfa AJ, Magasanik B. Covalent modification of the glnG product, NRI, by the glnL product, NRII, regulates the transcription of the glnALG operon in Escherichia coli. Proc Natl Acad Sci. 1986;83:5909–5913. doi: 10.1073/pnas.83.16.5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninfa AJ, Atkinson MR, Kamberov ES, Feng J, Ninfa EG. Two-component signal transduction. In: Hoch JA, Silhavy TJ, editors. Control of nitrogen assimilation by the NRI-NRII two-component system of enteric bacteria. Washington, D.C.: ASM Press; 1995. pp. 67–88. [Google Scholar]
- Ohlsen KL, Grimsley JK, Hoch JA. Deactivation of the sporulation transcription factor Spo0A by the Spo0E protein phosphatase. Proc Natl Acad Sci. 1994;91:1756–1760. doi: 10.1073/pnas.91.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JS, Kofoid EC. Communication modules in bacterial signaling proteins. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- Perego M. Bacillus subtilis and other Gram-positive bacteria: Biochemistry, physiology, and molecular genetics. In: Sonenshein AL, Hoch JA, Losick R, editors. Integrational vectors for genetic manipulation in Bacillus subtilis. Washington, D.C.: American Society for Microbiology; 1993. pp. 615–624. [Google Scholar]
- Perego M, Hoch JA. Cell-cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc Natl Acad Sci. 1996a;93:1549–1553. doi: 10.1073/pnas.93.4.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Protein aspartate phosphatases control the output of two-component signal transduction systems. Trends Genet. 1996b;12:97–101. doi: 10.1016/0168-9525(96)81420-x. [DOI] [PubMed] [Google Scholar]
- Perego M, Cole SP, Burbulys D, Trach KA, Hoch JA. Characterization of the gene for a protein kinase which phosphorylates the sporulation-regulatory proteins Spo0A and Spo0F of Bacillus subtilis. J Bacteriol. 1989;171:6187–6196. doi: 10.1128/jb.171.11.6187-6196.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M, Hanstein CG, Welsh KM, Djavakhishvili T, Glaser P, Hoch JA. Multiple protein aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in Bacillus subtilis. Cell. 1994;79:1047–1055. doi: 10.1016/0092-8674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Perego M, Glaser P, Hoch JA. Aspartyl-phosphate phosphatases deactivate the response regulator components of the sporulation signal transduction system in Bacillus subtilis. Mol Microbiol. 1996;19:1151–1157. doi: 10.1111/j.1365-2958.1996.tb02460.x. [DOI] [PubMed] [Google Scholar]
- Posas F, Wurgler-Murphy SM, Maeda T, Witten EA, Thai TC, Saito J. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 “two-component” osmosensor. Cell. 1996;86:865–875. doi: 10.1016/s0092-8674(00)80162-2. [DOI] [PubMed] [Google Scholar]
- Randerath K, Randerath E. Thin-layer separation methods for nucleic acid derivatives. Methods Enzymol. 1967;12:323–347. [Google Scholar]
- Richardson IB, Katz ME, Hynes MJ. Molecular characterization of the lam locus and sequences involved in regulation by the AmdR protein of Aspergillus nidulans. Mol Cell Biol. 1992;12:337–346. doi: 10.1128/mcb.12.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer P, Millet J, Aubert J. Catabolic repression of bacterial sporulation. Proc Natl Acad Sci. 1965;54:701–711. doi: 10.1073/pnas.54.3.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spizizen J. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci. 1958;44:1072–1075. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trach KA, Hoch JA. Multisensory activation of the phosphorelay initiating sporulation in Bacillus subtilis: Identification and sequence of the protein kinase of the alternate pathway. Mol Microbiol. 1993;8:69–79. doi: 10.1111/j.1365-2958.1993.tb01204.x. [DOI] [PubMed] [Google Scholar]
- Uhl MA, Miller JF. Integration of multiple domains in a two-component sensor protein: The Bordetella pertussis BvgAS phosphorelay. EMBO J. 1996;15:1028–1036. [PMC free article] [PubMed] [Google Scholar]
- Whitney PA, Cooper TG. Urea Carboxylase from Saccharomyces cerevisiae. J Biol Chem. 1973;248:325–330. [PubMed] [Google Scholar]
- Wray LV, Ferson AE, Rohrer K, Fisher SH. TnrA, a transcription factor required for global nitrogen regulation in Bacillus subtilis. Proc Natl Acad Sci. 1996;93:8841–8845. doi: 10.1073/pnas.93.17.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapf JW, Hoch JA, Whiteley JM. A novel phosphorylation activity of the Bacillus subtilis sporulation protein Spo0F that employs phosphoramidate substrates. J Biol Chem. 1996;35:2926–2933. doi: 10.1021/bi9519361. [DOI] [PubMed] [Google Scholar]