

Abstract
Cyclic di-GMP (c-di-GMP) is a broadly conserved, intracellular second-messenger molecule that regulates biofilm formation by many bacteria. The synthesis of c-di-GMP is catalyzed by diguanylate cyclases (DGCs) containing the GGDEF domain, while its degradation is achieved through the phosphodiesterase activities of EAL and HD-GYP domains. c-di-GMP controls biofilm formation by Pseudomonas fluorescens Pf0-1 by promoting the cell surface localization of a large adhesive protein, LapA. LapA localization is regulated posttranslationally by a c-di-GMP effector system consisting of LapD and LapG, which senses cytoplasmic c-di-GMP and modifies the LapA protein in the outer membrane. Despite the apparent requirement for c-di-GMP for biofilm formation by P. fluorescens Pf0-1, no DGCs from this strain have been characterized to date. In this study, we undertook a systematic mutagenesis of 30 predicted DGCs and found that mutations in just 4 cause reductions in biofilm formation by P. fluorescens Pf0-1 under the conditions tested. These DGCs were characterized genetically and biochemically to corroborate the hypothesis that they function to produce c-di-GMP in vivo. The effects of DGC gene mutations on phenotypes associated with biofilm formation were analyzed. One DGC preferentially affects LapA localization, another DGC mainly controls swimming motility, while a third DGC affects both LapA and motility. Our data support the conclusion that different c-di-GMP-regulated outputs can be specifically controlled by distinct DGCs.
INTRODUCTION
Upon contacting a surface, bacteria can initiate behavioral adaptations that result in the colonization of that surface. This process, known as biofilm formation, occurs on surfaces in environmental, industrial, and medical settings (7). While there has long been evidence that bacteria regulate adhesion to surfaces in response to internal and external cues, such as the nutritional status of the cell (15, 35) or the availability of nutrients in the environment (2, 24, 39, 48), we are just beginning to understand how bacteria integrate these signals to control the process of attachment (27).
Recent studies have highlighted the dinucleotide second messenger cyclic di-GMP (c-di-GMP) as a central player in the signaling networks that control biofilm formation. c-di-GMP is synthesized from two GTP molecules by diguanylate cyclases (DGCs), which contain the GGDEF domain (38, 42). The degradation of c-di-GMP is achieved by phosphodiesterases (PDEs) containing either the EAL (5, 44) or HD-GYP domain (41). A number of proteins contain both GGDEF and EAL domains but typically show either DGC or PDE activity only.
Individually or in tandem, GGDEF and EAL domains appear in combination with diverse regulatory domains commonly found in bacterial signaling proteins (9, 45). Regulatory domains can control the synthesis or degradation of c-di-GMP in response to light (49), oxygen (43, 50), or through posttranslational modifications, such as phosphorylation by histidine kinases (37, 40). Thus, many DGCs and PDEs are poised to respond directly to environmental signals or cellular signaling pathways.
Biofilm research has utilized P. fluorescens as a model, because growth on surfaces is a typical lifestyle for this bacterium in nature and because its attachment to plant roots can promote plant growth (10). Exploring the environmental cues that control the transition between planktonic and biofilm growth for P. fluorescens has provided general insights into how bacteria regulate the switch between motile and sessile lifestyles. An early study from our group identified a large adhesive protein, LapA, required for the attachment of P. fluorescens to surfaces (13). Subsequent investigations have detailed a c-di-GMP signaling system that regulates biofilm formation via the posttranslational modification of the LapA adhesin. Briefly, the periplasmic protease LapG inhibits attachment by cleaving the LapA protein and promoting the release of this adhesin from the cell surface (31). LapG's activity is regulated by an inner membrane protein, LapD, which binds c-di-GMP in the cytoplasm (33). c-di-GMP binding to LapD results in a conformational change that is propagated across the inner membrane by a HAMP signaling module to LapD's periplasmic domain (29). When bound to c-di-GMP, LapD is capable of binding the periplasmic protease LapG, inhibiting LapA cleavage and thereby promoting the retention of LapA on the cell surface.
While we have a detailed picture of how the LapD-LapG system regulates surface attachment by P. fluorescens Pf0-1 in response to c-di-GMP, much less is known about inputs that modulate c-di-GMP in this signaling system. The one input characterized to date is the depletion of c-di-GMP by RapA, a PDE that is conditionally expressed in response to phosphate starvation (24). No DGCs had been described in the Pf0-1 strain prior to this study, leaving open the issue of which signaling proteins produce c-di-GMP that is sensed by LapD. We set out to identify the DGCs required for biofilm formation by this organism and determined their contribution(s) to biofilm formation. Here, we report that of 30 predicted DGCs in P. fluorescens Pf0-1, mutations to 4 cause significant reductions in biofilm formation under the conditions tested. One DGC preferentially affects LapA localization, another DGC mainly controls swimming motility, while a third affects both LapA and motility. The data presented argue for specificity in the outputs controlled by certain DGCs.
MATERIALS AND METHODS
Strains and media.
Strains and plasmids used in this study are listed in Table S1. P. fluorescens Pf0-1 and Escherichia coli S17-1 (λpir) strains were used throughout this study. The SMC 4798 strain of Pf0-1, which carries three hemagglutinin (HA) epitope tags within a fully functional, chromosomally encoded LapA, is designated wild-type (WT) P. fluorescens in this study. Bacteria were routinely cultured with liquid LB medium in a test tube or on solidified LB with 1.5% agar. Gentamicin (Gm) was used at 30 μg/ml for P. fluorescens and at 10 μg/ml for E. coli. Ampicillin (Ap) was used at 50 μg/ml for culturing E. coli cells harboring pET vectors. Tetracycline (Tet) was used at 45 μg/ml when growing P. fluorescens on plates, 15 μg/ml in liquid, and 10 μg/ml for E. coli. For complementation and overexpression studies, arabinose was used to induce expression from pMQ72 at the concentrations noted in the text. P. fluorescens was grown in K10T-1 medium for assaying biofilm formation, LapA localization, and protein abundance. This medium, used in prior studies as a phosphate-rich condition, contains 50 mM Tris-HCl (pH 7.4), 0.2% (wt/vol) Bacto tryptone, 0.15% (vol/vol) glycerol, 0.6 mM MgSO4, and 1 mM K2HPO4 and was prepared as described previously (25).
Transposon mutagenesis and transposon mutation mapping.
The pUT plasmid harboring miniTn5-KmlacZ1 was used to generate transposon insertions in the chromosome of P. fluorescens Pf0-1. Transposon insertions were mapped using arbitrarily primed PCR. Mutagenesis and mapping were performed exactly as described previously (25).
Microarray experiment.
Details concerning the microarray experiment described here can be found in the supplemental material.
Biofilm formation assay.
An aliquot (1.5 μl) of an overnight culture grown in LB was transferred into 100 μl of K10T-1 medium in a 96-well plate (353911; BD Falcon) and grown statically for 6 h at 30°C. After incubation, the cells were discarded and material attached to the wells was stained with a 0.1% solution of crystal violet (CV) in water. Staining, imaging, and quantification of attached biomasses were performed as described previously (24).
Swimming motility assay.
For swimming motility assays, K10T-1 plates containing 0.35% agar were prepared by adding each filter-sterilized component of K10T-1 medium to an autoclaved, molten agar solution. After solidifying for 3 h, plates were inoculated with overnight cultures using sterile, 2- to 200-μl micropipette tips. For each point of inoculation, a fresh tip was dipped into 100 μl of culture in a 96-well plate and then plunged into the swim agar to the bottom of the petri plate. After incubation at 30°C for ∼40 h, plates were photographed. Each plate contained at least two replicates of the WT in addition to mutant strains; the swim area was calculated using ImageJ software (http://rsbweb.nih.gov/ij/) by encircling each swim zone with an oval selection and measuring its area.
Construction of single-crossover knockouts.
To construct the single-crossover knockouts, ∼500-bp PCR fragments of the N-terminal portion of each gene were amplified and cloned into the pKO3 vector (Tetr) after digestion with appropriate restriction enzymes (noted in primer sequences; all primers are listed in Table S2 in the supplemental material). Each pKO3 plasmid was transformed into E. coli S17-1 for conjugation with P. fluorescens Pf0-1, and conjugation was performed at 30°C for ∼5 h on an LB plate without antibiotics as described previously (25). P. fluorescens transconjugates were selected on plates containing 45 μg/ml Tet and 30 μg/ml chloramphenicol (Cm) to eliminate E. coli and subsequently passaged in 15 μg/ml Tet for liquid culture and 30 μg/ml Tet on solid plates. The proper location of each insertion was confirmed by PCR using a primer that anneals to the vector backbone (pKO3 verify) and one targeting the chromosomal DNA outside the initial amplicon (XXXX verify [where XXXX represents the designated gene name]).
Constructs for clean deletion, complementation, and overexpression of DGCs.
Saccharomyces cerevisiae strain InvSc1 (Invitrogen) was used to construct plasmids for clean deletions and complementation and overexpression studies through in vivo homologous recombination as described previously (46). All deletion mutants were constructed using the same basic strategy: ∼1-kbp PCR fragments were amplified from upstream and downstream of the gene to be deleted. The fragments were cloned adjacent to each other in the allelic exchange vector pMQ30. Deletion constructs were introduced into P. fluorescens by conjugation, and transconjugates were selected for on 30 μg/ml Gm and 30 μg/ml Cm plates. Gm-resistant colonies were subcultured in the absence of antibiotic and then subjected to selection on 5% sucrose. The deletion of the target genes was confirmed by PCR across the area of recombination and subsequent sequencing of the PCR product.
Complementation and overexpression studies utilized the pMQ72 plasmid, which contains the araC gene and a PBAD promoter directed toward the cloned insert. This plasmid does not include a ribosomal binding site (RBS). Target genes were amplified by PCR using primers that included the native RBS. The basal expression of different genes from pMQ72 varies due to the use of each gene's native RBS. For GcbA, a 6× histidine (6×His) tag was added to the N-terminal end, while a C-terminal HA tag was utilized for GcbC and C-terminal 6×His tags were used for GcbB and WspR. Site-directed mutations to GcbA were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) by following the manufacturer's instructions. Site-directed mutations to all other constructs were made using the following strategy. Two fragments were amplified by PCR from the WT complementation plasmid template using a primer on the vector backbone (pMQ72 Fwd or pMQ72 Rev) and a primer divergent from (and excluding) the codons for the two acidic residues in the GGDEF signature motif. Divergent primers included additional bases encoding replacement alanine codons as well as sequence to facilitate recombination in yeast. The transformation of yeast with pMQ72 and both PCR fragments yielded a recombined plasmid identical to the parent except for the desired mutations. The successful construction of each plasmid and each chromosomal alteration was confirmed by sequencing.
Construction of pET expression vectors, expression and purification of enzymes.
GcbA and variants were cloned from pMQ72 into pET3a by PCR amplification, restriction digestion, and ligation. Two primers, pET-Pfl01_0623 fwd and P729, were used to produce the gene fragment possessing N-terminal NdeI and C-terminal BamHI sites. After digestion of the PCR product and pET3a with NdeI and BamHI, these pieces were ligated with T4 DNA ligase (New England BioLabs, Beverly, MA).
The GcbA proteins with the N-terminal 6×His tag were expressed in E. coli BL21(DE3). Six hundred μl of overnight culture was inoculated into 300 ml of LB medium in a 2-liter flask, which was cultured at 37°C with gentle shaking (100 rpm). After 4.5 h the temperature was set to 28°C, and 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to the culture to induce protein expression. Cultivation was continued for an additional 20 h, and then the cells were harvested by centrifugation. The cells were stored at −80°C until use.
The frozen E. coli cells were resuspended in a binding buffer (75 mM Tris-HCl, pH 7.8, 250 mM NaCl, 25 mM KCl, 10 mM MgCl2, 20% glycerol, 1 mM dithiothreitol [DTT], and 20 mM imidazole) containing a protease inhibitor cocktail (Roche) and then sonicated on ice. After centrifugation at 19,000 rpm for 30 min, the supernatant was separated on a 5-ml-bed-volume HisTrap FF column (GE Healthcare, Piscataway, NJ). The column was extensively washed with the binding buffer. The absorbed proteins were eluted with the buffer containing 200 mM imidazole. Protein was collected and dialyzed against the DGC reaction buffer, which is described below. The purity of the protein was checked by SDS-PAGE using 7% polyacrylamide gels stained with Coomassie brilliant blue (CBB).
Hybrid DGC proteins were constructed by fusing GGDEF domains from GcbB (D637 to the C terminus) or GcbC (D342 to the C terminus) to the N terminus of GcbA after residue R377. This was accomplished by amplifying the appropriate gene fragments by PCR with primers containing nonannealing bases to direct homologous recombination with the adjacent fragment and pMQ72 via in vivo recombination. The growth regimen (timing of induction, etc.) and subsequent purification procedures (described above for GcbA) were employed for the hybrid DGC constructs, except that arabinose was used as the inducer.
Diguanylate cyclase assay.
In vitro diguanylate cyclase assays were performed as described by Paul et al. (38) with slight modifications. Glycerol and DTT were added to the reaction mixture to avoid protein aggregation. Each purified protein (200 μg/3.1 nmol) was mixed with 1.66 nmol of [α32P]GTP (3,000 Ci/mmol) to initiate the reaction in a total volume of 50 μl. Reactions were stopped by the addition of 5 μl 0.5 M EDTA. The reaction buffer contained 75 mM Tris-HCl, pH 7.8, 250 mM NaCl, 25 mM KCl, 10 mM MgCl2, 20% glycerol, and 1 mM DTT. Nucleotide reactants and products were separated by thin-layer chromatography (TLC) as described previously (24) on cellulose PEI TLC plates (Selecto Scientific, Suwanee, GA) with a running buffer of 1:1.5 saturated NH4SO4 and 1.5 M KH2PO4 (pH 3.6). Plates were exposed to a phosphor storage screen and then read on a Storm 860 scanner (Molecular Devices, Sunnyvale, CA). The effect of acetyl phosphate on the enzymatic activity was assayed by addition to a final concentration of 10 mM in the reaction buffer.
Measurement of c-di-GMP levels.
Nucleotide extraction from P. fluorescens cultures was performed as previously reported (26), with modifications. Briefly, an overnight, LB-grown culture was subcultured 1:75 into K10T-1 medium and grown at 230 rpm for 6 h. Cells were harvested by centrifugation at 4°C for 5 min at 8,000 × g. Pellets were quickly resuspended in a small amount of supernatant, transferred to a 1.5-ml microcentrifuge tube, and then pelleted again. After completely removing the supernatant, pellets were resuspended in 250 μl of extraction buffer (acetonitrile-methanol-water [40:40:20] plus 0.1 N formic acid) and incubated at −20°C for 24 h. Cell debris was pelleted for 5 min at 4°C, and resulting supernatant was adjusted to pH ∼7.5 by adding 15% (NH4)2HCO3. Nucleotide extractions were dried under nitrogen gas at room temperature and then resuspended in 10 mM tributylamine plus 15 mM acetic acid in water-methanol (97:3). Resuspended samples were analyzed via an Acquity ultraperformance liquid chromatography (UPLC) system coupled to a Quattro Premier XE mass spectrometer (Waters Corporation, Milford, MA). The mass spectrometry (MS) parameters were the following: capillary voltage, 3.5 kV; cone voltage, 50 V; collision energy, 34 V; source temperature, 120°C; and desolvation temperature, 350°C. UPLC separation was achieved using an Acquity UPLC BEH C18 column (particle size, 1.7 μm; 2.1 by 50 mm; Waters Corporation, Milford, MA). Solvent A was 10 mM tributamine plus 15 mM acetic acid in water-methanol (97:3), and solvent B was methanol. The gradient was the following: time 0, 1% solvent B; 1 min, 1% solvent B; 2.5 min, 20% solvent B; 4 min, 20% solvent B; 7 min, 65% solvent B; 7.5 min, 95% solvent B; 9 min, 95% solvent B; 9.01 min, 1% solvent B; 10 min, 1% solvent B. The running time for each sample was 10 min. Other liquid chromatography parameters were the following: column temperature, 50°C; and solvent flow rate, 300 μl/min. Each sample was compared to a standard curve of c-di-GMP resuspended in water to quantify the amount of nucleotide.
Statistical methods.
All statistical tests were performed in Excel (Microsoft Corp., Redmond, WA). Unless noted otherwise in the text, the test employed was a two-tailed, Student's t test assuming equal variance.
Microarray accession number.
Our microarray data and design information were deposited in the GEO database under accession number GSE30681.
RESULTS
The genome of P. fluorescens Pf0-1 encodes 5 EAL, 21 GGDEF, and 17 tandem GGDEF/EAL domain proteins (47). Despite ample evidence that c-di-GMP regulates biofilm formation by this organism, to date no diguanylate cyclases have been characterized in this strain. To identify DGCs that promote biofilm formation by P. fluorescens Pf0-1, we undertook a transposon mutagenesis screen for biofilm-deficient mutants. Approximately 14,600 transposon insertion mutants were screened using a static-growth, microtiter dish assay. As with our previous studies of P. fluorescens Pf0-1, we utilized K10T-1 medium, a dilute medium with 0.2% tryptone, as the primary source of carbon and nitrogen (see Materials and Methods for more information). We focused on this growth condition because our previous work showed that c-di-GMP signaling through the effector protein LapD is required for biofilm formation in this medium (33), and multiple lines of evidence in previously published studies suggest that robust changes in biofilm formation result from the modulation of c-di-GMP levels under these conditions (24, 26, 29, 31).
A total of 145 mutants, about 1% of mutants tested, showed a measurable reduction in biofilm formation. The location of each transposon mutation yielding a reduced biofilm phenotype was identified by arbitrarily primed PCR and sequencing (see Table S3 in the supplemental material). Mutations were found in genes with a wide variety of predicted functions, with mutations occurring most frequently in genes involved in flagellar synthesis and modification or LapA production and regulation. About a third of the mutations were in one of the lap genes and already have been characterized for their roles in adhesion and biofilm formation (lapA, lapB, lapC, lapD, and lapE) (13, 14). Other genes found in the screen which have established roles in biofilm regulation include pstC (24) and sadB (22). No putative exopolysaccharide synthesis loci were identified, which is consistent with previous observations that P. fluorescens Pf0-1 does not contain homologues of exopolysaccharide synthesis genes common to other pseudomonads (32). When mutants with a more subtle reduction in biofilm formation were characterized, one was found to contain a transposon insertion in a putative diguanylate cyclase gene, Pfl01_0623.
Putative DGC, GcbA, promotes biofilm formation.
The strain bearing a transposon insertion in Pfl01_0623 showed a 25% reduction in attached biomass in the static biofilm formation assay (Fig. 1 A). To confirm the necessity of Pfl01_0623 for the effect observed, an unmarked deletion of the gene was constructed in a WT genetic background. The resulting strain showed a reduction in biofilm biomass relative to that of the WT that was comparable to that exhibited by the transposon mutant (Fig. 1A). Given the reduced biofilm phenotype and the predicted function of Pfl01_0623, we named the gene gcbA (for di-guanylate cyclase promoting biofilm) and the deletion mutant ΔgcbA.
Fig. 1.

Genetic characterization of gcbA. (A) A quantitative biofilm assay comparing the WT, Pfl01_0623 transposon mutant, and unmarked gcbA deletion strains. Data presented are the mean absorbances of dissolved crystal violet-stained biofilm material ± standard deviations (SD) (n = 12). Representative images are shown above the graph. (B) A schematic of the GcbA protein showing the amino acid numbers and predicted domains. The lowercase “rec” indicates a degenerate REC domain. (C) A quantitative biofilm assay examining the complementation of the ΔgcbA mutant biofilm phenotype. pMQ72 is the empty complementation vector; 0.2% arabinose was added to the medium. (D) A representative image of a swimming motility assay in 0.35% agar assessing the complementation of the ΔgcbA mutant. (E) Quantitative measurements of swim zone areas are presented for eight replicates of the assay shown in panel D. Data presented are mean percentages of WT swim areas on the same plate ± standard errors (SE) (n = 8).
The gcbA gene encodes a protein with three predicted domains: rec, REC, and GGDEF (Fig. 1B). The first receiver domain is too degenerate to match the reference in the Conserved Domain Database (CDD; http://ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) but was predicted by the simple modular architecture research tool (E = 0.15; http://smart.embl-heidelberg.de). The second receiver domain appears to be well conserved (E = 1 × 10−27 according to the CDD) and contains an aspartic acid residue at the expected phosphorylation site, D299. The predicted GGDEF domain of gcbA also is well conserved (E = 3 × 10−48 according to the CDD) and has all residues known to be required for DGC activity in other GGDEF domain proteins. A BLAST search identified genes homologous to gcbA in other strains of P. fluorescens and other Pseudomonas species, including P. syringae, P. putida, and P. aeruginosa, each with more than 70% sequence identity. No cognate histidine kinase was found in proximity to gcbA or its homologues.
A genetic complementation analysis was utilized to test the sufficiency of gcbA for the observed effects on biofilm formation and to test which residues of the GcbA protein are required for function in vivo. A WT copy of gcbA was placed under the control of the PBAD promoter on a multicopy plasmid, pMQ72, and amended with an N-terminal 6×His tag. When introduced into the ΔgcbA strain, this construct restored WT biofilm formation, with the addition of 0.2% arabinose to the assay medium (Fig. 1C). The empty vector did not restore a WT biofilm phenotype to ΔgcbA.
To test if the putative phosphorylation site, D299, or the GGDEF motif is necessary for GcbA function, we introduced mutations into the complementation construct, generating alleles with D299A or GGDEF-to-GGAAF mutations. The resulting constructs could not restore biofilm formation to the ΔgcbA strain (Fig. 1C). These results suggest that GcbA functions as a DGC to promote biofilm formation through the synthesis of c-di-GMP and imply that phosphorylation at residue D299 is important for this activity.
Despite significant effort, we were unable to detect the expression of the His-tagged GcbA proteins, WT, or mutants in P. fluorescens by Western blotting. Even attempts to concentrate the WT or mutant proteins by applying large volumes of cell lysate to nickel resin failed to yield detectable protein. In contrast, all three GcbA proteins were stable and soluble when expressed in E. coli, as discussed below. Thus, while mutant proteins did not show reduced stability in vitro, it is possible that the D299A and GGAAF mutant alleles do not complement the ΔgcbA strain because they are unstable in vivo.
gcbA inhibits swimming motility of P. fluorescens.
One way c-di-GMP promotes biofilm formation by P. fluorescens Pf0-1 is by supporting the localization and maintenance of the LapA adhesin on the cell surface via the c-di-GMP receptor LapD (33). We assessed the effect of the ΔgcbA mutation on LapA localization to test the hypothesis that c-di-GMP generated by GcbA promotes biofilm formation through this pathway. Interestingly, the deletion of gcbA had only a minor effect on cell surface LapA levels (this phenotype is presented and discussed below).
We next tested the hypothesis that c-di-GMP generated by GcbA regulates another output: flagellar motility. In many bacteria, c-di-GMP is known to inhibit flagellar motility, and these effects often affect biofilm formation (52). We found that the ΔgcbA mutant swam through a larger area than the WT strain when grown in 0.35% agar plates for 40 h (Fig. 1D). In contrast, the complemented strain, ΔgcbA pGcbA, covered an area comparable to that of the WT strain. This indicates that gcbA inhibits swimming motility under these conditions. Neither the GGAAF nor the D229A allele of gcbA could complement the ΔgcbA mutant in this assay, suggesting that functional GGDEF and REC domains are required for GcbA's effects on swimming. A quantitative analysis of these swimming phenotypes is presented in Fig. 1E.
Investigating connections between GcbA and the regulation of biofilm formation by inorganic phosphate.
Biofilm formation by P. fluorescens Pf0-1 is inhibited by phosphate starvation via the induction of the Pho regulon (24). This response includes the Pho-induced expression of RapA, a phosphodiesterase which depletes cellular c-di-GMP under phosphate-limiting conditions (24). Our prior work indicated that RapA accounts for most, but not all, of the observed Pho-dependent reduction of biofilm formation. Thus, we were intrigued to discover that gcbA is adjacent to ctpL (Pfl01_0622), a Pho-regulated gene which was shown to act in chemotaxis toward inorganic phosphate in P. aeruginosa (53) (see Fig. S1A in the supplemental material for a diagram of the locus). To investigate connections between this locus and the Pho-dependent regulation of biofilm formation, we sought to confirm the Pho regulation of ctpL in P. fluorescens Pf0-1, test if gcbA expression also is affected by the Pho system, and assess the effect of mutations in ctpL and gcbA on the inhibition of biofilm formation by phosphate limitation.
A microarray experiment was performed comparing gene expression in the WT (Pho-off) to that of the pst mutant (Pho-on) strains in high-phosphate medium. The expression of ctpL was induced >7-fold in the Pho-on condition, while gcbA expression was slightly reduced (see Tables S4 and S5 in the supplemental material). Only rapA, of all of the genes encoding GGDEF and EAL domain proteins, showed differential regulation of ≥2-fold in the microarray data set (see Table S5). Despite evidence for Pho regulation at the ctpL locus, the mutation of neither gcbA nor ctpL had any effect on the inhibition of biofilm formation by phosphate limitation (see Fig. S1B).
The GcbA protein exhibits DGC activity stimulated by phosphorylation.
The genetic characterization of gcbA suggested that it acts as a DGC. To test if the GcbA protein possesses DGC activity, constructs were made in the conventional pET expression system for WT, D299A, and GGAAF proteins, each amended with an N-terminal 6×His tag. We expressed and purified proteins from E. coli and obtained yields of ≥60 mg of pure protein/liter of culture for each protein. The purity of each protein was more than 90% as judged by Coomassie-stained polyacrylamide gels (data not shown). No differences in protein purity or stability (such as aggregation) were observed between the WT and mutant proteins during purification.
To test for DGC activity, WT or mutant GcbA was incubated with [α32P]GTP at room temperature for 16 h. These assay conditions were chosen so that even weak in vitro DGC activity could be detected. PleD* was used as a positive control, as previously published (38). Reactions were stopped by the addition of EDTA to 50 mM and resolved by thin-layer chromatography. As shown in Fig. 2 A, both the WT and D299A proteins converted GTP to c-di-GMP, while the GGAAF mutant protein did not. These data show that GcbA can act as a DGC, and that the GGDEF motif is required for this activity while D299 is not.
Fig. 2.
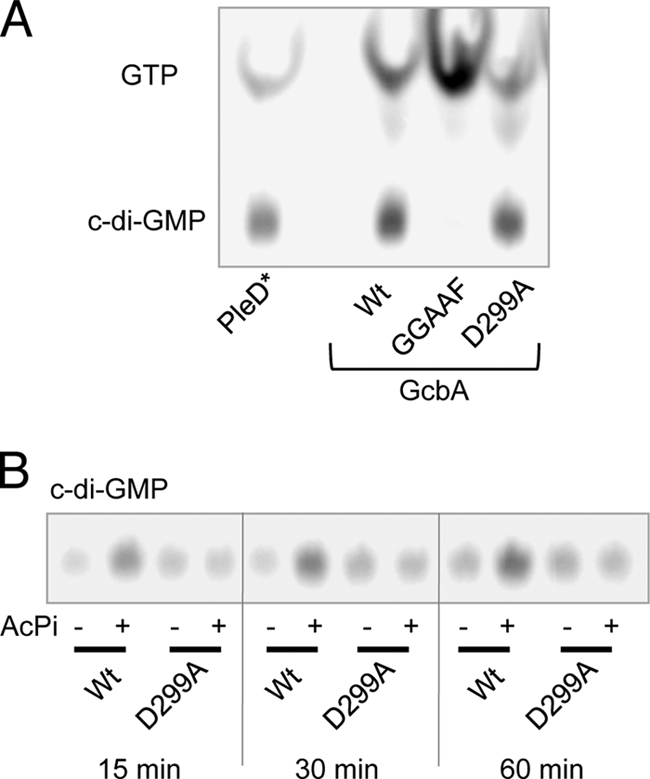
Enzymatic characterization of GcbA. (A) In vitro DGC assays comparing GcbA and mutant variants to the control DGC, PleD*, are shown resolved on a thin-layer chromatography plate. Compounds are labeled on the left; the image was generated by a phosphor screen scanner. (B) Assay comparing the effect of acetylphosphate (AcPi) treatment on the DGC activity of WT GcbA and the D299A mutant.
To resolve the role of D299 in regulating GcbA activity, we treated samples of the WT and D299A proteins with acetyl phosphate, which can function to phosphorylate and activate REC domain proteins in vitro (18). Using acetyl phosphate-treated and untreated samples, we assayed the DGC activity of WT and D299A proteins during shorter reaction times. We found that acetyl phosphate-treated WT GcbA had elevated DGC activity compared to that of the untreated control (Fig. 2B). In contrast, the acetyl phosphate treatment of the D299A protein had no effect on its DGC activity, which was similar to that of untreated WT protein (Fig. 2B). This experiment suggests that the phosphorylation of GcbA stimulates its DGC activity, and residue D299, in the REC domain proximal to the GGDEF domain, is the likely site of phosphorylation. This stands in contrast to PleD, which has a REC-REC-GGDEF domain structure but is activated by the phosphorylation of the first REC domain (37). Therefore, the activation mechanisms of GcbA and PleD may be different even though they share structural similarities.
Identification of additional GGDEF domain proteins that promote biofilm formation.
For P. fluorescens Pf0-1, mutations that block c-di-GMP binding to LapD completely abolish biofilm formation (33), implying that c-di-GMP is required for biofilm formation. Given the partial loss of biofilm formation exhibited by the gcbA mutant, we predicted that additional DGCs act to promote biofilm formation by this organism. The genome of P. fluorescens Pf0-1 encodes 37 putative GGDEF domain proteins in addition to GcbA. Two of these proteins were characterized in our laboratory, LapD (Pfl01_0131) and RapA (Pfl01_1678), and do not possess DGC activity (24, 33). Six other putative GGDEF domain proteins show signature motif sequences that deviate from what is known to be required for DGC activity, and these were not investigated (Pfl01_ 460, Pfl01_2295, Pfl01_4487, Pfl01_4552, Pfl01_5581, and Pfl01_5643), leaving 29 candidate DGCs in addition to GcbA (Table 1).
Table 1.
Putative diguanylate cyclases investigated in this study
| Locus tag | Name or homologuea | DGC motifb | PDE motif | Other domain(s)c | Biofilm phenotyped | Mutatione |
|---|---|---|---|---|---|---|
| Pfl01_0050 | GGEEF | No change | Insertion | |||
| Pfl01_0190 | GGEEF | PAS 3 | No change | Insertion | ||
| Pfl01_0192 | GGDEF | EAL | TM, PAS | Increased | Insertion | |
| Pfl01_0623 | GcbA | GGDEF | REC | Decreased | Deletion, Tn | |
| Pfl01_0692 | AwsR/YfiN (19, 21) | GGDEF | TM, HAMP | No change | Insertion | |
| Pfl01_1058 | WspR (12, 20) | GGEEF | REC | Decreased | Insertion | |
| Pfl01_1252 | SwsR (21) | GGDEF | EAL | TM, MHYT | No change | Insertion |
| Pfl01_1323 | GGDEF | PAS 3, GAF | No change | Insertion | ||
| Pfl01_1336 | GGEEF | TM | No change | Insertion | ||
| Pfl01_1789 | GcbB | GGEEF | TM, CHASE, PAS | Decreased | Deletion | |
| Pfl01_1887 | GGDEF | EAL | PAS, PAS 3, PAS 4 | Increased | Insertion | |
| Pfl01_2049 | Similar to AdrA (16) | GGDEF | MASE2, TM | No change | Insertion | |
| Pfl01_2170 | GGDEF | 7 TM | No change | Insertion | ||
| Pfl01_2176 | GGDEF | GAF | No change | Insertion | ||
| Pfl01_2297 | GGEEF | TM, CACHE_1 | No change | Insertion | ||
| Pfl01_2525 | Similar to MucR (11) | GGDEF | EAL | TM, MHYT | No change | Insertion |
| Pfl01_2709 | GGDEF | EAL | TM, CHASE4, PAS | Increased | Insertion | |
| Pfl01_3508 | GGDEF | TM, PAS 3 | No change | Insertion | ||
| Pfl01_3550 | GGDEF | TM, HAMP | No change | Insertion | ||
| Pfl01_3800 | GGDEF | TM | No change | Insertion | ||
| Pfl01_4084 | GGEEF | No change | Deletion | |||
| Pfl01_4086 | RbdA (1) | GGDEF | EIL | TM, PAS | Increased | Insertion |
| Pfl01_4307 | GGDEF | MASE2, TM | No change | Insertion | ||
| Pfl01_4451 | Similar to SadC (22) | GGDEF | 7 TM | No change | Insertion | |
| Pfl01_4551 | GGDEF | aaq | PAS, PAC, REC | No change | Insertion | |
| Pfl01_4666 | GcbC | GGEEF | TM, CACHE_1 | Decreased | Deletion | |
| Pfl01_4876 | MwsR/MorA (4, 21) | GGDEF | EAL | TM, PAS | Increased | Insertion |
| Pfl01_5150 | GGDEF | EAL | SBP bac3, TM, PAS | No change | Insertion | |
| Pfl01_5168 | GGEEF | 7 TM | No change | Insertion | ||
| Pfl01_5255 | GGDEF | EAL | REC, PAS | No change | Insertion |
Names for DGCs characterized in this study, or homologues (≥65% amino acid identity) that have been characterized in other organisms, are given with references indicated. “Similar to” indicates <65% amino acid identity but similar domain structures.
Amino acid sequences at the signature GGDEF and EAL motifs are given. Lowercase letters indicate degenerate sequence, and boldface letters indicate phenotypic evidence suggesting the domain is enzymatically active in P. fluorescens Pf0-1.
Other predicted domains are indicated using Pfam symbols (http://pfam.sanger.ac.uk/).
Biofilm phenotypes are given relative to that of WT P. fluorescens Pf0-1 in K10T-1 medium.
The type of mutant(s) tested for each locus is given: Tn, Tn5 insertion; insertion, single-crossover insertion of pKO3; deletion, unmarked deletion of the entire gene.
To investigate their potential roles in biofilm formation, each gene encoding a candidate DGC was inactivated by single-crossover disruption, and the resulting mutant was compared to the WT in a static biofilm assay. A single-crossover mutation in gcbA was utilized for comparison. Most of the mutants exhibited biofilm formation levels comparable to that of the WT strain (Fig. 3 A). Mutations in five genes showed statistically significant increases in biofilm formation compared to that of the WT (P < 0.01): Pfl01_0192, Pfl01_1887, Pfl01_2709, Pfl01_4086, and Pfl01_4876. Mutations in four genes caused significant (P < 0.01) reductions in biofilm formation compared to that of the WT in a two-tailed Student's t test assuming equal variance (for details on statistical methods, see Materials and Methods): the strain carrying a single-crossover mutation in gcbA, Pfl01_0623KO, showed a 27% reduction (P = 7.5 × 10−8); Pfl01_1058KO showed an 11% reduction (P = 6.8 × 10−3); Pfl01_1789KO showed a 27% reduction (P = 8.5 × 10−7); and Pfl01_4666KO showed a 15% reduction (P = 1.8 × 10−3).
Fig. 3.

Effect of mutations in putative DGCs on biofilm formation. (A) A quantitative biofilm assay analyzing the WT and single-crossover insertion mutants in gcbA and 29 putative DGCs; data are mean percentages of the WT ± SD (n = 8). Asterisks indicate a statistically significant decrease in biofilm relative to that of the WT (P < 0.01 in a two-tailed Student's t test assuming equal variance). (B) A quantitative biofilm assay comparing the WT and strains with deletion mutations of Pfl01_0623 (ΔgcbA), Pfl01_1789 (ΔgcbB), Pfl01_4666 (ΔgcbC), and Pfl01_1058 (ΔwspR).
From these results we hypothesized that, in addition to gcbA, genes Pfl01_1058, Pfl01_1789, and Pfl01_4666 also encode DGCs that promote biofilm formation. Unmarked deletions of the entire open reading frames of these three new candidate genes were constructed in the WT background. A biofilm assay on the resulting mutant strains confirmed the necessity of Pfl01_1058, Pfl01_1789, and Pfl01_4666 for achieving WT levels of biofilm formation (Fig. 3B).
Following the nomenclature introduced for GcbA, Pfl01_1789 and Pfl01_4666 were dubbed gcbB and gcbC, respectively. Pfl01_1058 encodes a homologue of the well-characterized DGC WspR. WspR can regulate pellicle formation by P. fluorescens SBW25 and biofilm formation by P. aeruginosa PAO1 through the synthesis of c-di-GMP (12, 20). Pfl01_1058 shares 90 and 75% amino acid identity with the SBW25 and PAO1 WspR proteins, respectively, and amino acid sequence alignments performed by De et al. suggest the complete conservation of important structural and functional features in Pfl01_1058 (8). We refer to Pfl01_1058 as wspR.
Complementation analysis of wspR.
To investigate the role of WspR in biofilm formation by P. fluorescens Pf0-1, we conducted a complementation analysis. We constructed a C-terminally 6×His-tagged allele of wspR and also a variant of this allele containing the GGAAF mutation in the signature motif. The introduction of pWspR-6H into the ΔwspR strain restored biofilm formation to WT levels, while pWspR-GGAAF did not (Fig. 4 A). Both WT and mutant WspR proteins had comparable levels of expression in vivo (Fig. 4B). These data support the conclusion that WspR acts as a DGC to promote biofilm formation by P. fluorescens Pf0-1. Consistent with the small reduction in biofilm formation by the ΔwspR mutant, it had limited phenotypic effects on biofilm-associated phenotypes such as LapA localization and swimming motility under the conditions tested (see below).
Fig. 4.

Complementation analysis of wspR. (A) A quantitative biofilm assay examining the complementation of the ΔwspR mutant phenotype; 0.1% arabinose was added to the medium. (B) Western blot comparing the abundance of WspR-6H and WspR-GGAAF proteins in vivo. Samples were probed for the 6×His epitope on each protein, and cells were cultured in K10T-1 with 0.1% arabinose.
Complementation analysis of gcbB.
The deletion of the gcbB gene caused a decrease in biofilm formation on par with that of the ΔgcbA mutation (Fig. 3B). The gcbB gene is predicted to encode a membrane-localized DGC with an extracellular CHASE domain and intracellular PAS domains (Fig. 5 A). A BLASTP search with the GcbB sequence returns 21 homologous proteins (≥70% similarity across the whole sequence) in strains of P. fluorescens, P. putida, and P. syringae. We tested the ability of WT GcbB bearing a C-terminal 6×His tag and a GGAAF mutant allele to complement the ΔgcbB mutation. We first confirmed that both WT and GGAAF proteins were expressed in vivo, where the GGAAF protein was slightly more abundant than the WT (Fig. 5B).
Fig. 5.

Complementation analysis of gcbB. (A) A schematic of the GcbB protein showing the amino acid numbers (above) and predicted domains in boxes. TM, transmembrane. (B) Western blot comparing the abundance of GcbB-6H and GcbB-GGAAF proteins in vivo. Samples were probed for the 6×His epitope on each protein, and cells were cultured in K10T-1 with 0.01% arabinose. (C) A quantitative biofilm assay examining the complementation of the ΔgcbB mutant phenotype by pGcbB-6H and pGcbB-GGAAF at three concentrations of inducer (ara, arabinose). (D) A quantitative immunoblot assay probing whole cells for the surface expression of LapA. Pixel densities of blot images were calculated using ImageJ, and the mean density for the WT was set to 100. Data shown are means ± SE (n = 7). Representative images are shown above the graph.
We next assessed the ability of these constructs to restore biofilm formation to ΔgcbB. For this experiment we utilized the arabinose-inducible PBAD promoter on the pMQ72-based complementation constructs, which can increase expression when arabinose is added (46). Without the addition of arabinose, pGcbB-6H provided a partial restoration of biofilm formation, but with the addition of 0.01% arabinose to the medium, biofilm formation by ΔgbcB pGcbB-6H was comparable to that of the WT strain (Fig. 5C). This stands in contrast to the empty vector, pMQ72, and the pGcbB-GGAAF construct, which could not complement the ΔgcbB mutant even with the addition of arabinose. When the highest concentration of arabinose was added to the medium, 0.1%, the ΔgcbB pGcbB-6H strain formed more biofilm than the WT, while the vector and pGcbB-GGAAF construct still had no effect on biofilm formation by ΔgcbB (Fig. 5C).
In previous studies of P. fluorescens Pf0-1, we have observed a close correspondence between the amount of cell surface LapA adhesin and the level of biofilm formation in a static microtiter dish assay (24, 26, 33). Biofilm formation by Pf0-1 relies on the adhesin LapA, whose localization to and maintenance on the outer membrane is tightly controlled by c-di-GMP via the LapDG effector system (31). The cell surface LapA localization assay consists of drying washed cells onto a nitrocellulose membrane and immunoblotting with an antibody directed toward the HA epitope on the LapA protein. This assay detects only extracellular LapA that is tightly associated with the cell surface, as shown previously (33).
We examined the impact of gcbB on cell surface LapA localization and found that the ΔgcbB strain carrying the empty vector exhibited a reduction in cell surface LapA compared to that of the WT (Fig. 5D). The pGcbB-6H construct could restore WT levels of cell surface LapA to ΔgcbB, while the pGcbB-GGAAF construct could not (Fig. 5D). These data are consistent with GcbB acting as a DGC in vivo and promoting biofilm formation through the cell surface localization of LapA.
Complementation analysis of the DGC gcbC.
gcbC encodes a protein predicted to contain two transmembrane domains, a periplasmic CACHE_1 superfamily domain and a cytoplasmic GGDEF domain (Fig. 6 A). A BLASTP search identified 20 proteins likely to be homologues of GcbC (>65% amino acid similarity over the whole sequence) encoded in genomes of environmental pseudomonads as well as other bacteria. We performed a complementation analysis to determine if gcbC was sufficient to account for the ΔgcbC mutant phenotype and assess if the GGDEF motif is important for GcbC function.
Fig. 6.

Complementation analysis of gcbC. (A) A schematic of the GcbC protein showing the amino acid numbers and predicted domains in boxes. TM, transmembrane. (B) Western blot comparing the abundance of GcbC-HA and GcbC-GGAAF proteins in vivo. Samples were probed for HA on each protein, and cells cultured in K10T-1 without arabinose. (C) A quantitative biofilm assay examining the complementation of the ΔgcbC mutant phenotype by pGcbC-HA and pGcbC-GGAAF with and without inducer (ara, arabinose). (D) A quantitative immunoblot assay probing whole cells for the surface expression of LapA. Data shown are means ± SE (n = 7). Representative images are shown above the graph. (E) A representative image of a swimming motility assay in 0.35% agar, assessing the complementation of the ΔgcbC mutant. (F) Quantitative measurements of swim zone areas are presented for eight replicates of the assay shown in panel E. Data presented are mean percentages of WT swim area on the same plate ± SE (n = 8).
We built expression constructs for WT and GGAAF mutant alleles of gcbC, incorporating a C-terminal HA tag to track protein abundance in vivo. In the ΔgcbC strain, the GcbC-HA and GcbC-GGAAF proteins were expressed at comparable levels, with the GcbC-GGAAF protein consistently showing slightly higher abundance (Fig. 6B). The addition of pGcbC-HA to ΔgcbC led to an increase in biofilm above WT levels (Fig. 6C). When arabinose was added to induce a higher level of expression of GcbC-HA, biofilm formation was increased to nearly twice the level of the WT. To our surprise, the addition of pGcbC-GGAAF to ΔgcbC restored biofilm formation to nearly WT levels despite the mutation to the GGDEF domain (Fig. 6C). However, the overexpression of GcbC-GGAAF with arabinose showed no stimulatory effect on biofilm formation.
We examined the effect of the ΔgcbC mutation on LapA localization and swimming motility and assessed the complementation of these phenotypes by wild-type and mutant gcbC expression constructs. The ΔgcbC mutant carrying an empty vector exhibited a substantial decrease in LapA expression on the cell surface (Fig. 6D) and increased swimming motility compared to that of the WT strain (Fig. 6E). Similarly to the results for complementation in the biofilm assay, the ΔgcbC pGcbC-HA strain showed a gain of function: increased LapA-HA localization and reduced motility compared to those of the WT (Fig. 6D to F). While pGcbC-GGAAF had shown some ability to promote biofilm formation (Fig. 6C), its effect on the ΔgcbC mutant's LapA localization and swimming phenotypes was less pronounced, showing a small but measurable increase above the level for the empty vector control (Fig. 6D to F). The complementation data set is consistent with GcbC acting as a DGC to promote biofilm formation; GcbC's DGC activity likely is required to support the surface localization of LapA and also may inhibit motility. The basis for the partial complementation of the biofilm phenotype of ΔgcbC by the GcbC-GGAAF mutant construct is unclear at this time. The complementation phenotype observed for GcbC-HA was identical to that of an untagged gcbC allele in pMQ72 (data not shown).
Assessing DGC activity of GcbB and GcbC.
We took several approaches to investigate whether or not GcbB or GcbC possesses DGC activity. The first was to purify full-length versions of each protein by nickel affinity chromatography and assess their activity in vitro. The addition of a C-terminal 6×His tag to full-length GcbC resulted in a loss of function and stability in vivo (data not shown), so this construct was not pursued further. Full-length, 6×His-tagged GcbB was stably expressed in E. coli and could be purified in 1% Thesit or Triton X-100. These preparations did not exhibit DGC activity in vitro (data not shown).
As an alternative strategy, we constructed hybrid proteins in which the GGDEF domains from either GcbB or GcbC were fused to the N terminus of GcbA, replacing its native GGDEF domain. A similar strategy was successfully employed by our laboratory in the past, fusing the N terminus of PleD to GGDEF domains from other membrane proteins (22, 23). Site-directed GGAAF mutant forms of these constructs also were made.
The hybrid proteins were expressed and purified in the same manner as that for the GcbA protein. While we were able to purify hybrid proteins containing the GGDEF domain of GcbB, they did not show DGC activity in vitro (data not shown). In contrast, the GcbC-Hyb protein was able to produce c-di-GMP in vitro, while the GGAAF mutant form was not (Fig. 7 A). This indicates that the GGDEF domain of GcbC is enzymatically active and supports the idea that GcbC acts as a DGC in vivo.
Fig. 7.

In vitro and in vivo assessments of DGC activity. (A) In vitro DGC assays comparing GcbC-Hyb and GcbC-Hyb-GGAAF mutant variants to GcbA as a positive control. An image of a thin-layer chromatography plate generated by a phosphor screen scanner is shown with compounds labeled on the left. (B) Quantitative measurements of cellular c-di-GMP from the Δ4DGC strain expressing the indicated plasmids. Formic acid-extracted c-di-GMP was measured by LC/MS and normalized to mg of dry weight of bacteria after extraction.
To provide further evidence that GcbB and GcbC act as DGCs in vivo, we measured the impact of each protein and its GGAAF mutant on cellular c-di-GMP levels by acid extraction followed by LC/MS (23). This experiment was performed using the Δ4DGC strain lacking four DGCs (gcbA, gcbB, gcbC, and wspR) to enable robust changes in cellular c-di-GMP to be observed (the Δ4DGC mutant phenotype is discussed below). The expression of each construct was induced using the concentration of arabinose that had previously resulted in WT complementation phenotypes in the single-mutant strains: 0.01% arabinose for pGcbB constructs and no arabinose for pGcbC constructs. The expression of GcbB-6H boosted cellular c-di-GMP 1.6-fold compared to that of GcbB-GGAAF (Fig. 7B; P = 0.001 in one-tailed Student's t tests; n = 3). Likewise, the expression of GcbC-HA in the Δ4DGC strain caused a 2.2-fold increase in cellular c-di-GMP compared to that of GcbC-GGAAF in the same strain (Fig. 7B; P = 0.0002 in one-tailed Student's t tests; n = 3). There were no significant changes in c-di-GMP levels due to the expression of either GcbB-GGAAF or GcbC-GGAAF compared to that of a vector-only control strain (Fig. 7B; P = 0.35 and P = 0.19, respectively). Taken together, these data reinforce the conclusion that GcbB and GcbC act as DGCs in vivo.
Combinatorial effects of DGC mutations on biofilm formation.
An intriguing question in c-di-GMP research is the following: to what extent do different c-di-GMP signaling proteins form distinct pathways in the same cell? As an initial step toward addressing this question, we undertook a combinatorial mutant analysis of the four DGCs identified here. Our goal was to assess whether the effects of combined mutations were additive across all phenotypes assessed or if one or more DGC had a disproportionate effect on a particular phenotype.
Beginning with strains bearing an unmarked deletion in each DGC, we constructed each combination of double- and triple-deletion mutants, as well as a quadruple-deletion mutant lacking all four DGC genes. The full panel of deletion mutants was tested for biofilm formation and compared to the WT (Fig. 8 A). The biofilm reductions that resulted from single deletions of gcbA, gcbB, and gcbC were roughly additive when these mutations were combined in double and triple mutants.
Fig. 8.

Combinatorial analysis of DGC mutants. (A) A quantitative biofilm assay comparing each single-, double-, triple-, and quadruple-mutant combination for the DGC genes indicated (means ± SD; n = 8). (B) A quantitative immunoblot assay probing whole cells for the surface expression of LapA by the indicated strains. Data shown are means ± SE (n = 7). Representative images are shown above the graph. (C) Quantitative swimming motility assay for the strains indicated. Data presented are mean percentages of WT swim area on the same plate ± SE (n = 8). Representative images of each mutant and adjacent WT control are shown above the corresponding bar in the graph.
Interestingly, mutations to wspR did not yield additive phenotypes in double and triple mutants. Some double mutants that include ΔwspR showed a reduction compared to that of their single-mutant parent strain (ΔgcbA ΔwspR or ΔgcbC ΔwspR), but these reductions were less than an additive relationship would predict (Fig. 8A). The ΔgcbB ΔwspR double mutant showed no additional reduction in biofilm compared to the level for the ΔgcbB single mutant. Furthermore, each triple mutant incorporating ΔwspR showed no additional reduction in biofilm formation relative to that of the wspR+ parent strain (Fig. 8A). On the other hand, the quadruple deletion mutant showed a measurable reduction in biofilm relative to that of the wspR+ triple mutant. Taken together, these data show that most DGC mutations affect biofilm formation in an additive manner, while the effect of deleting wspR is apparent only in single- and quadruple-mutant backgrounds.
Effect of DGC gene deletions on LapA localization.
We analyzed the effects of each DGC deletion mutation, and some combination mutations, on LapA localization to the cell surface to assess how they affect this c-di-GMP-regulated output. Compared to the WT, each DGC mutant showed some reduction in cell surface LapA (Fig. 8B), while levels of the LapA protein did not differ in whole-cell lysates of any of the strains analyzed (data not shown). The ΔwspR and ΔgcbA mutations resulted in mild reductions in LapA that were not statistically significant (P = 0.57 and P = 0.41, respectively). The ΔgcbC and ΔgcbB mutations caused more severe reductions, 38 and 48%, respectively, that were significant (P < 0.005).
We included two double mutants and the Δ4DGC mutant in the analysis. Our rationale was to combine the two mutations with the smallest effect on LapA, ΔgcbA ΔwspR, and those with the largest effect, ΔgcbB ΔgcbC. In combination, the mild effects of the ΔgcbA and ΔwspR mutations were additive, resulting in a modest but significant reduction compared the level for the WT (P = 0.002). The more pronounced effects of the ΔgcbB and ΔgcbC mutations also were additive, with the ΔgcbB ΔgcbC mutant showing a 65% reduction relative to WT effects (Fig. 8B). This defect was comparable to that exhibited by the Δ4DGC mutant, a 72% reduction, and the difference between the Δ4DGC mutant and ΔgcbB ΔgcbC was not statistically significant (P = 0.15).
The LapA localization results are interesting, because the degree of reduction in cell surface LapA exhibited by each single mutant does not strictly follow its effect on biofilm formation. This observation is consistent with DGCs GcbA and WspR affecting biofilm formation through outputs other than LapA. Furthermore, the deletion of gcbB and gcbC seems to be sufficient to explain the bulk of the c-di-GMP-dependent localization of LapA to the cell surface.
Effect of DGC gene deletions on swimming motility.
As described above, single deletions of either gcbA or gcbC caused an increase in swimming motility compared to that of the WT (Fig. 1D and 6E). We evaluated all single mutants, select double mutants, and the Δ4DGC mutant for effects on swimming through semisolid agar (Fig. 8C). In this assay, the ΔwspR and ΔgcbB mutations were found to have minimal effects on motility, with 4 and 19% increases, respectively, in swim area compared to the level for the WT. The ΔgcbC and ΔgcbA mutations caused more substantial increases in swim area: 46 and 81%, respectively. For all mutants except ΔwspR, swim areas proved to be statistically different from that of the WT (P ≤ 0.01).
Double mutants were analyzed for swimming motility by combining mutations that have the smallest or greatest effect on the phenotype of interest, similarly to the rationale for analyzing LapA localization. Both the ΔgcbB ΔwspR and ΔgcbA ΔgcbC strains showed additive phenotypes in the swimming motility assay compared to their single-mutant counterparts. The ΔgcbA ΔgcbC mutant showed a 120% increase in swim area compared to that of the WT, which is comparable to the 100% increase exhibited by the Δ4DGC mutant. The swim zone areas exhibited by these two strains, ΔgcbA ΔgcbC and Δ4DGC, were not statistically different (P = 0.15), suggesting that gcbA and gcbC are sufficient to account for the regulation of swimming motility by DGCs under the tested conditions.
DISCUSSION
In this study, we sought to determine which DGCs promote biofilm formation by P. fluorescens Pf0-1. We found that, out of 30 predicted DGCs, mutations in only 4 caused significant reductions in biofilm formation under the conditions tested. The genetic and biochemical evidence presented here is consistent with the conclusion that these proteins function as DGCs in vivo. Mutations in five genes caused increases in biofilm formation above the WT level, suggesting that they encode tandem GGDEF/EAL domain proteins with c-di-GMP phosphodiesterase activity.
Of the DGCs characterized here, gcbB and gcbC are predominantly responsible for promoting LapA localization to the cell surface, a critical step for the transition to stable surface attachment and subsequent biofilm formation (13, 14, 33). We also found that the swimming motility of P. fluorescens Pf0-1 is inhibited by DGCs, mainly gcbA and gcbC. These data raise the possibility that biofilm formation by this organism is affected by changes in motility, in addition to adherence via LapA. Further experimentation is required to determine whether gcbA and gcbC affect motility and biofilm formation independently or if the modulation of motility contributes to their role(s) in biofilm formation. The possibility that motility regulation does affect biofilm formation by P. fluorescens Pf0-1 is consistent with a growing consensus that biofilm initiation results from the combined consequences of a reduction motility and increase in adhesion (34). Dissecting the mechanistic basis for the observed specificity in the outputs of gcbA and gcbB (motility and LapA localization, respectively) may help solve a broader puzzle in c-di-GMP research: how are independent c-di-GMP signals segregated and/or integrated in the cell?
We chose to focus our study on biofilm formation in one growth medium, K10T-1. Further investigation will be required to identify which parameters of this growth condition serve to activate the DGCs characterized here. Surveying additional growth conditions may reveal roles for other GGDEF domain proteins in biofilm formation and consequently facilitate the identification of specific signals for additional putative DGCs.
While it was not the focus of this study, it is interesting that more PDEs than DGCs may regulate biofilm formation under the conditions tested. Four predicted DGC mutants had significant phenotypes, while five mutants in genes encoding predicted, tandem GGDEF/EAL proteins showed significant increases in biofilm formation. An additional 11 candidate PDEs were not examined here, leaving open the possibility that yet more PDEs could affect biofilm formation by P. fluorescens Pf0-1. This observation suggests that keeping c-di-GMP production in check through robust PDE activities plays an important part in biofilm regulation by this organism, although additional work will be required to test this hypothesis. Rainey and colleagues have made an analogous finding while examining the mutational activation of the wrinkly spreader phenotype of P. fluorescens SBW25. Their work highlights that the negative regulation of c-di-GMP production is common, and the inactivation of negative regulators of DGC activity has dramatic phenotypic consequences (21).
c-di-GMP promotes adherence and inhibits motility in an array of different bacterial species. The commonality of this observation is in contrast to the overwhelming diversity of c-di-GMP signaling pathways and proteins employed within and among bacterial species. In some groups, particularly the gammaproteobacteria (45), it seems that each species or even each strain has a unique complement of dozens of c-di-GMP signaling proteins. For example, Pseudomonas fluorescens Pf0-1 contains 43 proteins with either GGDEF or EAL or both domains. Of these, 12 are not shared among the three completed P. fluorescens genomes (including gcbC), and just 10 have close homologues in four or more Pseudomonas species (unpublished observations).
Both wspR and gcbA belong to the latter group, having homologues in most Pseudomonas species. The hyperactivation of WspR through mutations to inhibitory components of the Wsp chemosensory system causes the potent stimulation of biofilm or pellicle formation in multiple pseudomonads (3, 12) and the inhibition of motility in P. aeruginosa PAO1 (6). However, the inactivation of wspR alone showed subtle or no effect on WT phenotypes in the same studies. One recent study of P. fluorescens F113 reports increased swimming motility in wspR mutants compared to that of the WT (30). Our data indicate that wspR makes a small but measurable contribution to biofilm formation by P. fluorescens Pf0-1 but has no effect on swimming and a limited impact on LapA localization. The identification of a growth condition or environmental signal that activates the Wsp pathway and increases its impact on biofilm formation by Pf0-1 above that observed in our test conditions could provide an opportunity to further clarify its role in this strain.
Much less is known about homologues of gcbA than those of wspR. One study utilizing P. aeruginosa PAO1 shows a >40% reduction in biofilm formation for a mutant with a transposon insertion in PA4843 (51), a gcbA homologue. However, a gross survey of c-di-GMP signaling genes in P. aeruginosa PA14 reported similar levels of biofilm formation for a PA4843 mutant and the WT (17). Likewise, other exhaustive surveys of P. aeruginosa PA14 mutants with reductions in biofilm formation failed to identify PA4843 (28, 36). Any potential role for gcbA homologues in regulating the flagellar motility of other bacteria has not been reported.
If any conclusion can be drawn from comparing the phenotypes of mutants in the same gene across different Pseudomonas strains and species, it is that one should not assume that conserved c-di-GMP signaling components serve an identical function in different organisms. While conserved components may function similarly in different systems, these components are likely to be pieced together differently within each organism's unique c-di-GMP signaling repertoire. Finding commonalities in the function and organization of analogous pathways may be the key to understanding the underlying logic of these signaling networks. By generating a detailed knowledge of each signaling module and the role it plays in an individual organism's c-di-GMP network, we can build a broader understanding of how bacteria integrate signals to regulate biofilm formation. This study represents a first step toward discerning the architecture of c-di-GMP signaling in P. fluorescens Pf0-1, a versatile model organism in which attachment to surfaces is governed by a well-defined c-di-GMP effector system (31).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Science Foundation grant MCB-9984521 (G.A.O.), a National Institutes of Health predoctoral fellowship (T32 GM08704), and the John H. Copenhaver, Jr., and William H. Thomas fellowship (P.D.N.). A program for young researchers to study abroad from The Graduate University for Advanced Studies (Sokendai) supported S.Y.
We thank Urs Jenal for sharing plasmids and the Michigan State University Mass Spectrometry Facility for the quantitative analysis of c-di-GMP.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 15 July 2011.
REFERENCES
- 1. An S., Wu J., Zhang L. H. 2010. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-Di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl. Environ. Microbiol. 76:8160–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banin E., Vasil M. L., Greenberg E. P. 2005. Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 102:11076–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bantinaki E., et al. 2007. Adaptive divergence in experimental populations of Pseudomonas fluorescens. III. Mutational origins of wrinkly spreader diversity. Genetics 176:441–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choy W. K., Zhou L., Syn C. K., Zhang L. H., Swarup S. 2004. MorA defines a new class of regulators affecting flagellar development and biofilm formation in diverse Pseudomonas species. J. Bacteriol. 186:7221–7228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Christen M., Christen B., Folcher M., Schauerte A., Jenal U. 2005. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J. Biol. Chem. 280:30829–30837 [DOI] [PubMed] [Google Scholar]
- 6. D'Argenio D. A., Calfee M. W., Rainey P. B., Pesci E. C. 2002. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J. Bacteriol. 184:6481–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davey M. E., O'Toole A. G. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64:847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De N., et al. 2008. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galperin M. Y., Nikolskaya A. N., Koonin E. V. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11–21 [DOI] [PubMed] [Google Scholar]
- 10. Haas D., Defago G. 2005. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3:307–319 [DOI] [PubMed] [Google Scholar]
- 11. Hay I. D., Remminghorst U., Rehm B. H. 2009. MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 75:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hickman J. W., Tifrea D. F., Harwood C. S. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. U. S. A. 102:14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hinsa S. M., Espinosa-Urgel M., Ramos J. L., O'Toole G. A. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol. Microbiol. 49:905–918 [DOI] [PubMed] [Google Scholar]
- 14. Hinsa S. M., O'Toole G. A. 2006. Biofilm formation by Pseudomonas fluorescens WCS365: a role for LapD. Microbiology 152:1375–1383 [DOI] [PubMed] [Google Scholar]
- 15. Jackson D. W., et al. 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J. Bacteriol. 184:290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kader A., Simm R., Gerstel U., Morr M., Romling U. 2006. Hierarchical involvement of various GGDEF domain proteins in rdar morphotype development of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 60:602–616 [DOI] [PubMed] [Google Scholar]
- 17. Kulasakara H., et al. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. U. S. A. 103:2839–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lukat G. S., McCleary W. R., Stock A. M., Stock J. B. 1992. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. U. S. A. 89:718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malone J. G., et al. 2010. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog. 6:e1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Malone J. G., et al. 2007. The structure-function relationship of WspR, a Pseudomonas fluorescens response regulator with a GGDEF output domain. Microbiology 153:980–994 [DOI] [PubMed] [Google Scholar]
- 21. McDonald M. J., Gehrig S. M., Meintjes P. L., Zhang X. X., Rainey P. B. 2009. Adaptive divergence in experimental populations of Pseudomonas fluorescens. IV. Genetic constraints guide evolutionary trajectories in a parallel adaptive radiation. Genetics 183:1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merritt J. H., Brothers K. M., Kuchma S. L., O'Toole G. A. 2007. SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J. Bacteriol. 189:8154–8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Merritt J. H., et al. 2010. Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. mBio 1:e00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monds R. D., Newell P. D., Gross R. H., O'Toole G. A. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 63:656–679 [DOI] [PubMed] [Google Scholar]
- 25. Monds R. D., Newell P. D., Schwartzman J. A., O'Toole G. A. 2006. Conservation of the Pho regulon in Pseudomonas fluorescens Pf0-1. Appl. Environ. Microbiol. 72:1910–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monds R. D., et al. 2010. Di-adenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways. J. Bacteriol. 192:3011–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Monds R. D., O'Toole G. A. 2009. The developmental model of microbial biofilms: ten years of a paradigm up for review. Trends Microbiol. 17:73–87 [DOI] [PubMed] [Google Scholar]
- 28. Müsken M., Di Fiore S., Dotsch A., Fischer R., Haussler S. 2010. Genetic determinants of Pseudomonas aeruginosa biofilm establishment. Microbiology 156:431–441 [DOI] [PubMed] [Google Scholar]
- 29. Navarro M. V., et al. 2011. Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol. 9:e1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Navazo A., et al. 2009. Three independent signalling pathways repress motility in Pseudomonas fluorescens F113. Microb. Biotechnol. 2:489–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Newell P. D., Boyd C. D., Sondermann H., O'Toole G. A. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol. 9:e1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Newell P. D., O'Toole G. A. 2010. Environmental control of cyclic di-GMP signaling in Pseudomonas fluorescens: from signal to output, p. 282–290 In Wolfe A. J., Visick K. L. (ed.), The second messenger cyclic Di-GMP. ASM Press, Washington DC [Google Scholar]
- 33. Newell P. D., Monds R. D., O'Toole G. A. 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. U. S. A. 106:3461–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Toole G. A. 2008. How Pseudomonas aeruginosa regulates surface behaviors. Microbe 3:65–71 [Google Scholar]
- 35. O'Toole G. A., Gibbs K. A., Hager P. W., Phibbs P. V., Jr., Kolter R. 2000. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 182:425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Toole G. A., Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304 [DOI] [PubMed] [Google Scholar]
- 37. Paul R., et al. 2007. Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. J. Biol. Chem. 282:29170–29177 [DOI] [PubMed] [Google Scholar]
- 38. Paul R., et al. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 18:715–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pratt J. T., McDonough E., Camilli A. 2009. PhoB regulates motility, biofilms, and cyclic di-GMP in Vibrio cholerae. J. Bacteriol. 191:6632–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rao F., et al. 2009. The functional role of a conserved loop in EAL domain-based cyclic di-GMP-specific phosphodiesterase. J. Bacteriol. 191:4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryan R. P., et al. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U. S. A. 103:6712–6717 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Ryjenkov D. A., Tarutina M., Moskvin O. V., Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187:1792–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sawai H., et al. 2010. Molecular oxygen regulates the enzymatic activity of a heme-containing diguanylate cyclase (HemDGC) for the synthesis of cyclic di-GMP. Biochim. Biophys. Acta 1804:166–172 [DOI] [PubMed] [Google Scholar]
- 44. Schmidt A. J., Ryjenkov D. A., Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 187:4774–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seshasayee A. S., Fraser G. M., Luscombe N. M. 2010. Comparative genomics of cyclic-di-GMP signalling in bacteria: post-translational regulation and catalytic activity. Nucleic Acids Res. 38:5970–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shanks R. M., Caiazza N. C., Hinsa S. M., Toutain C. M., O'Toole G. A. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Silby M. W., et al. 2009. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 10:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stanley N. R., Lazazzera B. A. 2004. Environmental signals and regulatory pathways that influence biofilm formation. Mol. Microbiol. 52:917–924 [DOI] [PubMed] [Google Scholar]
- 49. Tarutina M., Ryjenkov D. A., Gomelsky M. 2006. An unorthodox bacteriophytochrome from Rhodobacter sphaeroides involved in turnover of the second messenger c-di-GMP. J. Biol. Chem. 281:34751–34758 [DOI] [PubMed] [Google Scholar]
- 50. Tuckerman J. R., et al. 2009. An oxygen-sensing diguanylate cyclase and phosphodiesterase couple for c-di-GMP control. Biochemistry 48:9764–9774 [DOI] [PubMed] [Google Scholar]
- 51. Wagner V. E., Li L. L., Isabella V. M., Iglewski B. H. 2007. Analysis of the hierarchy of quorum-sensing regulation in Pseudomonas aeruginosa. Anal. Bioanal. Chem. 387:469–479 [DOI] [PubMed] [Google Scholar]
- 52. Wolfe A. J., Visick K. L. 2008. Get the message out: cyclic-Di-GMP regulates multiple levels of flagellum-based motility. J. Bacteriol. 190:463–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu H., et al. 2000. Identification and characterization of two chemotactic transducers for inorganic phosphate in Pseudomonas aeruginosa. J. Bacteriol. 182:3400–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.