

Abstract
DNA replication and recombination generate intertwined DNA intermediates that must be decatenated for chromosome segregation to occur. We showed recently that topoisomerase IV (topo IV) is the only important decatenase of DNA replication intermediates in bacteria. Earlier results, however, indicated that DNA gyrase has the primary role in unlinking the catenated products of site-specific recombination. To address this discordance, we constructed a set of isogenic strains that enabled us to inhibit selectively with the quinolone norfloxacin topo IV, gyrase, both enzymes, or neither enzyme in vivo. We obtained identical results for the decatenation of the products of two different site-specific recombination enzymes, phage λ integrase and transposon Tn3 resolvase. Norfloxacin blocked decatenation in wild-type strains, but had no effect in strains with drug-resistance mutations in both gyrase and topo IV. When topo IV alone was inhibited, decatenation was almost completely blocked. If gyrase alone were inhibited, most of the catenanes were unlinked. We showed that topo IV is the primary decatenase in vivo and that this function is dependent on the level of DNA supercoiling. We conclude that the role of gyrase in decatenation is to introduce negative supercoils into DNA, which makes better substrates for topo IV. We also discovered that topo IV has an unexpectedly strong DNA relaxation activity that, together with gyrase and topo I, is able to set the supercoiling levels in Escherichia coli.
Keywords: DNA supercoiling, quinolones, λ integrase, Tn3 resolvase, topoisomerase I
Catenated DNA dimers are important intermediates of two major biological processes, DNA replication and recombination. Topoisomerases decatenate these intermediates, allowing segregation of daughter chromosomes at cell division (for review, see Bjornsti 1991; Osheroff et al. 1991; Ullsperger et al. 1995; Wang 1996). In Escherichia coli four topoisomerases have been identified (for review, see Wang 1991; Ullsperger et al. 1995). These enzymes alter DNA topology by passing intact DNA through either transient single-stranded breaks (type-1 topoisomerase) or double-stranded breaks (type-2 topoisomerase) in DNA. The type-2 enzymes, topoisomerase II (topo II; DNA gyrase) and topoisomerase IV (topo IV), are essential for cell viability (Kreuzer and Cozzarelli 1979; Orr et al. 1979; Filutowicz and Jonczyk 1983; Kato et al. 1988; Schmid 1990). The genes for the two topo IV subunits are parC, which is homologous to gyrA of gyrase, and parE, which is homologous to gyrB of gyrase (Kato et al. 1990; Luttinger et al. 1991; Springer and Schmid 1993).
Topo I, encoded by the topA gene, and topo III, encoded by the topB gene, are the type-1 enzymes (Sternglanz et al. 1981; Trucksis and Depew 1981; DiNardo et al. 1982; Dean et al. 1983; Srivenugopal et al. 1984). Topo III is dispensable for cell viability, but may have important biological roles (DiGate and Marians 1988, 1989; Gangloff et al. 1994). topA deletions are tolerated only in the presence of compensatory mutations in other genes that influence the DNA supercoiling level (DiNardo et al. 1982; Raji et al. 1985). Decatenation of intact duplex DNA rings can be carried out only by a type-2 topoisomerase. However, DNA replication produces catenated intermediates with gaps and nicks that could be unlinked by a type-1 topoisomerase. Topo III does this efficiently in vitro (Dean et al. 1983; Hiasa and Marians 1994).
We show that topo IV is, by far, the major decatenase of DNA replication products in E. coli and Salmonella typhimurium (Adams et al. 1992b; Khodursky et al. 1995; Zechiedrich and Cozzarelli 1995). Previous results from our laboratory, however, indicated that DNA gyrase, instead, was the principal decatenase of the products of site-specific recombination (Bliska and Cozzarelli 1987; Bliska et al. 1991; Adams et al. 1992a). To reconcile these results, several models have been suggested: The two topoisomerases might be compartmentalized within the cell; topo IV might be limited to action at the replication fork, whereas gyrase is free to unlink catenanes arising elsewhere; or the structures of the catenated products of replication and recombination may differ (Wang 1991; Adams et al. 1992b; de Boer 1993; Watt and Hickson 1994; Luttinger 1995; Ullsperger et al. 1995; Zechiedrich and Cozzarelli 1995).
The conclusion that gyrase was the principal decatenase was based chiefly on the observation that the quinolone norfloxacin blocked decatenation of the products of λ integrase (Int) recombination in a wild-type strain, but not in a drug-resistant gyrase mutant (Bliska and Cozzarelli 1987). Resolvase-generated catenanes also accumulated upon treatment of a wild-type strain with norfloxacin (Bliska et al. 1991; Adams et al. 1992a). At the time the recombination experiments were performed, however, topo IV had not yet been discovered. Furthermore, until recently, the only known target in bacteria of the quinolone antibiotics, such as norfloxacin and ciprofloxacin, was DNA gyrase (for review, see Drlica 1984). With the discovery and purification of topo IV, it was found that topo IV is also a target of the quinolones in vitro (Kato et al. 1992; Peng and Marians 1993). We showed that topo IV was a drug target in an E. coli strain containing a quinolone-resistant mutant gyrase (Khodursky et al. 1995). Mutations in parC could impart drug resistance to topo IV in vitro and in vivo (Khodursky et al. 1995). These results have been confirmed (Chen et al. 1996; Heisig 1996; Kumagai et al. 1996; Yamagiahi et al. 1996) and other studies had already indicated that topo IV can be a primary drug target in other bacteria (Belland et al. 1994; Ferrero et al. 1994). In addition, a clinical isolate of E. coli that is hyper-resistant to the quinolone antibiotics (Heisig and Wiedemann 1991) contains a mutation in topo IV that confers an extremely high level of drug resistance (Khodursky et al. 1995; Heisig 1996). Hence, topo IV is an important clinical target of quinolones. Therefore, we needed to test the role of topo IV in recombination.
We constructed an isogenic set of strains that enabled us to inhibit topo IV, gyrase, both enzymes, or neither enzyme rapidly and selectively with quinolones in vivo. These strains allowed us to address directly the roles of topo IV and gyrase in decatenation of the products of site-specific recombination by λ Int or Tn3 resolvase. We show that topo IV decatenates the products of recombination just as it unlinks DNA replication intermediates. Our reinterpretation of the earlier recombination experiments in wild-type strains is that the amount of norfloxacin used inhibited gyrase completely and topo IV only partially. The residual topo IV was insufficient to decatenate the recombination catenanes as they were no longer supercoiled because of DNA relaxation by topo I in the absence of gyrase activity. In the drug-resistant gyrase mutant, the partial activity of topo IV was sufficient to decatenate the negatively supercoiled catenanes. The role of gyrase in decatenation is to maintain negative supercoiling in the substrate DNA, which makes it a superior substrate for topo IV.
In the process of delineating the roles of the topoisomerases in recombination, we made the surprising discovery that topo IV has a strong DNA relaxation activity in vivo that, with gyrase and topo I, sets the cellular DNA supercoiling level. It was thought previously that only gyrase and topo I carried out this essential function (for review, see Wang, 1991).
Results
Effect of topo IV and gyrase on Int recombination in vivo
Our first series of experiments employed the Int recombination reaction. This site-specific recombinase reacts with (−) supercoiled plasmids containing directly repeated DNA-binding sites to form multiply linked catenanes (Spengler et al. 1985). In vivo, these catenanes are quickly unlinked by type-2 topoisomerases into free circles (Bliska and Cozzarelli 1987). The reaction protocol we used to determine which enzyme unlinks the Int products is schematized in Figure 1. The strains were lysogens for a mutant phage λ, which contains the Int gene under control of the temperature-sensitive repressor, cI857 (Bliska and Cozzarelli 1987). The strains harbored the Int recombination substrate pJB3.5d (Bliska and Cozzarelli 1987). Int expression was initiated by shifting the cultures to 43°C for 10 min to inactivate the repressor. Norfloxacin was added (or not) and the cultures were shifted back to 30°C to activate the thermolabile wild-type Int protein (Guarneros and Echols 1973). Thus, recombination of the plasmid substrate and topoisomerase inhibition were initiated at the same time (time 0). Cultures were incubated at 30°C for up to 60 min. If the topoisomerase responsible for decatenation is drug inhibited, then the recombined plasmid DNA would be trapped as catenanes.
Figure 1.

Experimental system for measuring the unlinking of catenanes produced by λ Int recombination. Cells were grown at 30°C to a density of 70 Klett units and shifted to 43°C where Int is expressed but is inactive. Upon downshift to 30°C, Int recombines the substrate plasmid pJB3.5d to make multiply interlinked catenanes. A type-2 topoisomerase unlinks the catenanes to release free 0.6- and 2.9-kb circles.
Before we consider the fraction of products that is catenated, it is important to note that norfloxacin affects the frequency of recombination. The inhibition of topoisomerases not only blocks decatenation, but also influences the level of cellular DNA supercoiling, which, in turn, modulates recombination (Nash 1990). Because supercoiling affects transcription [indeed, even of the topoisomerases themselves (Menzel and Gellert 1983)], we limited transcription of Int to the time before the addition of norfloxacin to insure that the resultant changes in DNA supercoiling have no effect on Int synthesis, only on the ability of Int to recombine (Fig. 1).
We measured the effect of norfloxacin on the extent of recombination in each of six test strains, LZ33–LZ38 (see Table 1). For convenience, we refer to these strains by the topo IV and gyrase genotypes. Plasmids were isolated at various times throughout the recombination reactions, nicked with DNase I, and analyzed by high resolution gel electrophoresis and Southern blotting. The amount of label in the recombinant products as a percentage of the total label in all plasmid DNA for each strain is shown in Figure 2. In this and two subsequent figures, the results are presented as bar graphs. The three panels on the left are gyrA+ strains and the three on the right are strains with gyrAL83, which renders gyrase highly norfloxacin resistant. In the top row, parC is wild type; in the middle row, parC is resistant to norfloxacin to an intermediate degree (parCL80); and in the bottom row, parC is highly resistant to norfloxacin (parCK84). For each strain, we show the amount of recombination in the presence of 0, 30, 60, 90, or 120 μm norfloxacin. For each drug concentration, data are shown for 0, 20, 30, 45, and 60 min after drug addition and shift down to 30°C. The drug concentrations were chosen based upon the Ki values that we determined for the inhibition of topo IV or gyrase in vitro and in vivo (Table 2A; see Materials and Methods). From these Ki values, we constructed Table 2B showing qualitatively the inhibition of wild-type or mutant gyrase and topo IV at the concentrations of drug used in this work.
Table 1.
Bacterial strains
| Strain
|
Genotype
|
Constructiona or Reference
|
|---|---|---|
| CAG12183 | zei-3143::Tn10kan | Singer et al. (1989) |
| C600 | F- thr-1 leu-6 thi-1 lacY1 supE44 tonA21 | Kato et al. (1988) |
| KL16 | Hfr thi | Yoshida et al. (1988) |
| LZ1 | Hfr thi gyrAL83zei-723::Tn10 | Zechiedrich and Cozzarelli (1995) |
| LZ2 | Hfr thi zei-723::Tn10 | Zechiedrich and Cozzarelli (1995) |
| LZ21 | C600 except gyrAL83 zei-723::Tn10 parCL80, kanR | Khodursky et al. (1995) |
| LZ22 | C600 except zei- 723::Tn10 parCL80, kanR | Khodursky et al. (1995) |
| LZ23 | C600 except gyrAL83 zei-723::Tn10 kanR | Khodursky et al. (1995) |
| LZ24 | C600 except zei-723::Tn10 kanR | Khodursky et al. (1995) |
| LZ27 | LZ21 except TetS | fusaric acidR |
| LZ28 | LZ22 except TetS | fusaric acidR |
| LZ29 | LZ23 except TetS | fusaric acidR |
| LZ30 | LZ24 except TetS | fusaric acidR |
| LZ31 | W3101λ except gyrAL83zei-723::Tn10 | P1(LZ1) × W3101λ ,TetR |
| LZ32 | W3101λ except zei-723::Tn10 | P1(LZ2) × W3101λ, TetR |
| LZ33 | LZ31 except parCL80, kanR | P1(LZ22) × LZ31, KanR |
| LZ34 | LZ32 except parCL80, kanR | P1(LZ22) × LZ32, KanR |
| LZ35 | LZ31 except kanR | P1(LZ24) × LZ31, KanR |
| LZ36 | LZ32 except kanR | P1(LZ24) × LZ32, KanR |
| LZ37 | LZ31 except parCK84, kanR | P1(1644) × LZ31, KanR |
| LZ38 | LZ32 except parCK84, kanR | P1(1644) × LZ32, KanR |
| LZ49 | KL16 except zei-3143::Tn10kan | P1(CAG12183) × KL16, KanR |
| LZ50 | N51 except zei-3143::Tn10kan | P1(CAG12183) × N51, KanR |
| LZ53 | RS2λ except zei-3143::Tn10kan | P1(LZ49) × RS2λ, KanR |
| LZ54 | RS2λ except gyrAL83zei-3134::Tn10kan | P1(LZ50) × RS2λ, KanR |
| N51 | Hfr thi gyrAL83 | Yoshida et al. (1988) |
| RS2λ | pyrF287 nirA trpR72 iclR7 gal25 rpsL195 top10 λ (P80 xis1 cI857) | Bliska and Cozzarelli (1987) |
| W3101λ | F-λ (P80 red114 xis-1 cI857) | Bliska and Cozzarelli (1987) |
| 1643 | C600 except gyrAL83zei-723::Tn10 parCK84, kanR | Khodursky et al. (1995) |
| 1644 | C600 except zei-723::Tn10 parCK84, kanR | Khodursky et al. (1995) |
Constructions are shown as chemical used for selection or as donor x recipient, selection. Abbreviations: (R) Resistant; (S) sensitive; (Tet) tetracycline; (Kan) kanamycin; (P1) source of transducing phage lysate.
Figure 2.
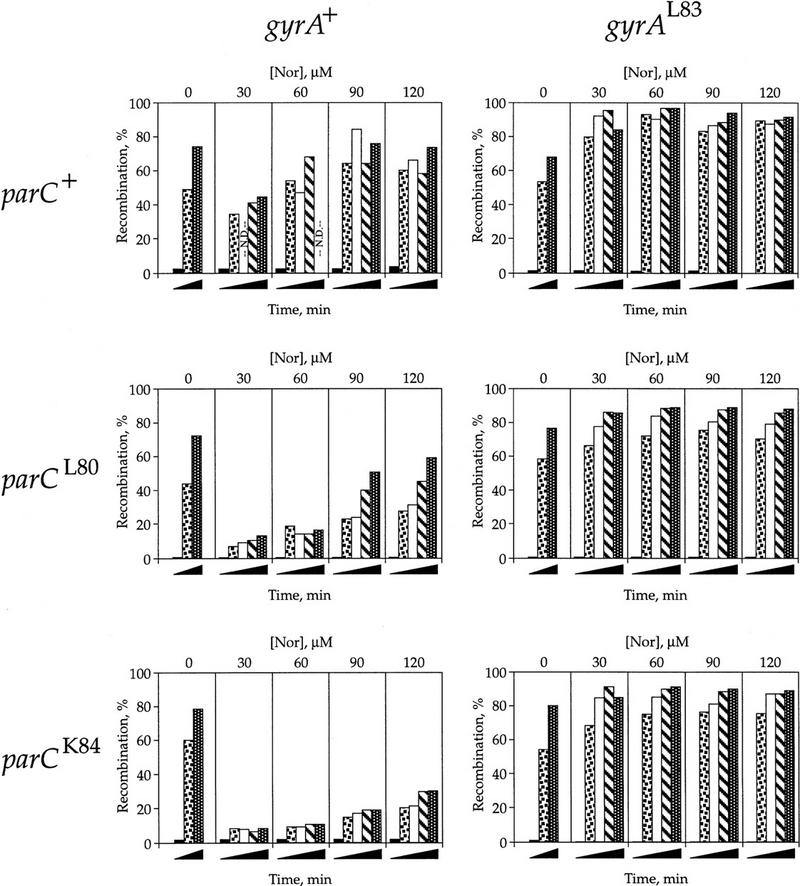
Effect of norfloxacin on Int recombination. The experiment was as described in the legend of Fig. 1. Plasmid DNA was isolated, subjected to electrophoresis on high resolution agarose gels, Southern blotted, and quantified by PhosphorImager analysis. The total amount of plasmid DNA label was normalized to 100%. The percentage of the total recombined plasmid is shown for various time points and drug concentrations [Nor] in each of the six strains. Time after shift down to 30°C is represented as black wedges. Samples were analyzed at the following times after the addition of drug: 0 min (▪), 20 min (squares in box), 30 min (white box), 45 (hatched box), and 60 min (dotted). The graphs are presented in a matrix array depending upon the states of the topo IV and gyrase alleles. (parC+ gyrA+) LZ36; (parC+ gyrAL83) LZ35; (parCL80 gyrA+) LZ34; (parCL80 gyrAL83) LZ33; (parCK84 gyrA+) LZ38; (parCK84 gyrAL83) LZ37. (N.D.) Not determined. Parts of these experiments were repeated three times. The exact values varied somewhat, but the relative values were always the same. For example, the 30 μm drug, 20 min point for the double wild-type strain was done three times. The amount of recombination averaged 30% ± 7% and the amount of catenation averaged 75% and varied <4%. The results here and in Figs. 3 and 5 are from one extensive experiment performed all on the same day.
Table 2.
Effects of norfloxacin on topo IV and DNA gyrase
| A. Ki values for gyrase and topo IV | ||||
|---|---|---|---|---|
| Enzyme
|
In vitro Ki (μm)
|
In vivo Ki (μm)
|
||
| gyrasea | topo IVb | gyrasea | topo IVb | |
| Wild type | 0.9 | 7.6 | 6 | 15 |
| ParCL80 | — | 88 | — | >90 |
| ParCK84 | — | 245 | — | >120 |
| GyrAL83 | 34.5 | — | 180 | — |
| B. Inhibition of gyrase and topo IVc | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain
|
30 μm
|
60 μm
|
90 μm
|
120 μm
|
|||||
| gyrA | parC | gyrase | topo IV | gyrase | topo IV | gyrase | topo IV | gyrase | topo IV |
| wt | wt | − | +/− | − | − | − | − | − | − |
| wt | L80 | − | + | − | + | − | +/− | − | +/− |
| wt | K84 | − | + | − | + | − | + | − | + |
| L83 | wt | + | +/− | + | + | + | − | + | − |
| L83 | L80 | + | + | + | + | + | +/− | + | +/− |
| L83 | K80 | + | + | + | + | + | + | + | + |
Measure by DNA supercoiling
Measured by decatenation
The concentration of norfloxacin is shown; −, +/−, and + correspond to complete, partial, and no inhibition, respectively, of the enzyme in the strain composed of the gyrA and parC alleles indicated in the first two columns. (wt) Wild type.
In the absence of norfloxacin, the extent and rate of recombination were the same for each strain [Fig. 2, (Nor) = 0], showing that the topoisomerase mutants have the same Int expression and activity as the wild-type strain. When gyrase was drug-resistant and thus able to maintain negative supercoiling, recombination occurred at all drug concentrations tested (Fig. 2, right panels). The drug resistance imparted by this gyrA allele exceeded 180 μm norfloxacin (Table 2; data not shown).
In contrast, when gyrase was wild type, and thus inhibited by norfloxacin, recombination was clearly reduced by even the lowest drug concentration used, 30 μm (Fig. 2, cf. left and right panels in each row). The surprising result was that the inhibition of recombination by norfloxacin was greatly affected by the drug resistance of topo IV (Fig. 2, left panels). When parC was wild type, recombination was decreased, but still robust in the gyrA+ strain, compared with the isogenic gyrAL83 strain. Recombination was reduced with drug in the parCL80 strain and even further reduced in the presence of the more resistant parCK84 allele.
The most direct explanation for these results is that topo IV lowers the (−) supercoiling density of the substrate plasmids. The Int reaction studied here requires (−) supercoiled DNA (Nash 1990) and uninhibited topo IV relaxes the plasmid DNA and makes it an inferior substrate for Int. Accordingly, DNA relaxation by topo IV is almost completely blocked by norfloxacin in wild-type parC strains, partially blocked in the strain carrying the intermediate drug-resistant parCL80 allele, and is barely affected in the parCK84 strain when gyrase is wild type. The dependence of recombination on drug concentration is best explained by the inhibition of topo IV DNA relaxation activity at the highest norfloxacin doses. Thus, without the topoisomerases to alter it, the DNA remains negatively supercoiled and highly recombinogenic.
Previously, the equilibrium balance of (−) supercoiling has been thought to be controlled in a simple fashion by the opposing action of gyrase and topo I (Gellert 1981; Pruss and Drlica 1982; Wang 1985; Liu and Wang 1987). DNA gyrase pumps (−) supercoils into DNA, whereas topo I selectively removes (−) supercoils. These functions are influenced by tracking processes such as transcription, which separates supercoiled DNA into (+) and (−) domains, and also depend upon DNA-anchoring sites that maintain these domains (Liu and Wang 1987; Cook et al. 1992; Lynch and Wang 1993). Previously, no role for topo IV in DNA supercoiling was observed (Wang 1991; Adams et al. 1992b; Khodursky et al. 1995; Zechiedrich and Cozzarelli 1995). However, topo IV is known to relax DNA in vitro (Kato et al. 1992) and in vivo when overexpressed (Free and Dorman 1994; McNairn et al. 1995).
Effect of topo IV and gyrase on DNA supercoiling levels in vivo
To test directly whether normal levels of topo IV have an effect on DNA supercoiling in vivo, we examined a portion of the DNA samples described in Figure 2 (before they were nicked) on agarose gels containing various concentrations of chloroquine. Chloroquine untwists circular DNA in a controlled manner and thereby allows the electrophoretic separation of DNA topoisomers and the measurement of supercoiling density (σ) (Keller 1975). Gels were blotted and the results were analyzed with a PhosphorImager.
The results are presented as a histogram similar to Figure 2 except the ordinate shows σ (Fig. 3). Completely relaxed DNA has a σ value of 0. In the absence of drug, the σ for plasmid pJB3.5d in all strains was −0.075 (leftmost set in each panel). This shows that the topoisomerases in the mutant strains are functioning equally in the absence of drug. When gyrA was drug-resistant, norfloxacin had little effect on σ (Fig. 3, right panels). When gyrase was wild type, however, the supercoiling level was reduced to a degree dependent upon the parC allele (Fig. 3, left panels). The more drug-resistant the parC allele was, the greater the observed DNA relaxation. Therefore, the decrease in recombination seen in Figure 2 is caused by the simultaneous inhibition of (−) supercoiling by DNA gyrase and the rapid DNA relaxation by drug-resistant topo IV (and topo I). Further, in the wild-type strain, topo IV is inhibited by norfloxacin and only topo I relaxes the plasmid DNA (Fig. 2, top left). Topo I alone, then, must not be able to relax DNA beyond σ ∼ −0.05 in vivo. Topo IV, on the other hand, can relax the DNA to near completion (σ ∼ −0.012) in the drug-resistant parC strains.
Figure 3.
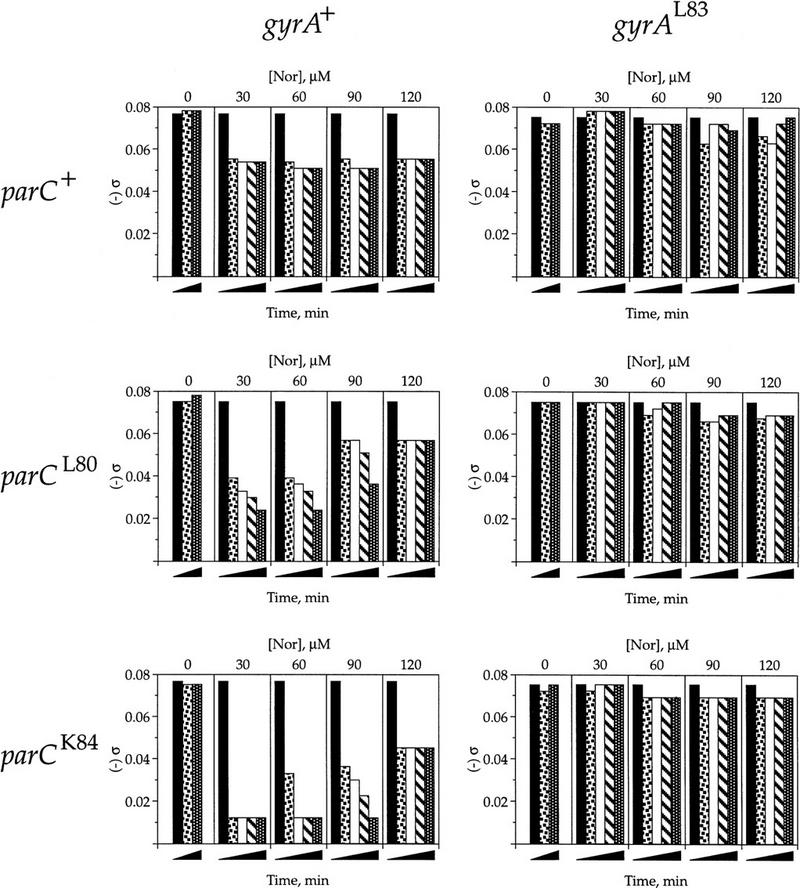
Effect of norfloxacin on plasmid DNA supercoiling levels. Supercoiled plasmid DNA from the experiment described in Fig. 2 was separated and visualized on agarose gels containing 0, 2, 4, or 8 μg/ml chloroquine and compared with plasmids of known σ. Shown are the σ values as quantified by the band counting method (Keller 1975). A ς of zero was taken as the midpoint of the topoisomer distribution after plasmid DNA relaxation in vitro with calf thymus topo I. The time points and symbols are the same as in Fig. 2.
To illustrate the effect of (−) supercoiling on Int recombination, we plotted the percentage of recombination as a function of σ for all the data in Figures 2 and 3 (Fig. 4). The transition from a baseline level of recombination to maximal recombination is surprisingly sharp and the midpoint lies around a σ of −0.05. This supercoiling level required for recombination is near measured wild-type σ levels for various plasmids and the chromosome (Sinden et al. 1980; for review, see Wang 1984). Clearly, λ Int requires highly (−) supercoiled DNA in vivo and this DNA is modulated by topo IV relaxation.
Figure 4.

Effect of DNA supercoiling (σ) on Int recombination. The amount of recombination as a function of σ was assessed by plotting all the data from Figs. 2 and 3. The line is drawn for ease of visualization.
Topo IV decatenates products of Int recombination in vivo
Even when σ was lowest (when gyrase was inhibited and topo IV was not), recombination could be detected. Thus, decatenation of the recombination products could be measured. The high-resolution gels used for Figure 2 allow separation of the catenanes from both unreacted Int substrate plasmids and unlinked products. From these data, we measured the amount of recombination (catenanes plus free circles) for each time point and normalized it to 100%. Of that, the amount of recombined plasmid DNA that was catenated was plotted for each strain (Fig. 5). The histogram panels are as in Figures 2 and 3. In the absence of drug, no appreciable amount of catenanes was seen at any time point. Nearly all of the plasmids that were recombined in the parC+ gyrA+ strain remained as catenanes when norfloxacin was present, even 60 min after recombination was initiated (Fig. 5, upper left). At the other extreme, the double drug-resistant mutants, parCL80 gyrAL83 or parCK84 gyrAL83, showed very few catenanes with drug, about 10% (middle and lower right panels). These data establish that topo IV and gyrase are the only relevant decatenases of the products of Int recombination in E. coli.
Figure 5.

Effect of norfloxacin on decatenation of Int recombination products. The amount of recombined DNA from Fig. 2 was further broken down into catenanes and free circles (decatenated). The percentage of the total recombined plasmid that was catenated is shown.
If gyrase were solely responsible for decatenation (as previously thought), then a gyrA+ strain with drug-resistant topo IV should have the same level of catenation as the parC+ gyrA+ strain. Instead, the strains with drug-resistant topo IV, parCL80 gyrA+ (Fig. 5, middle left) and parCK84 gyrA+ (bottom left) have substantially lower levels of catenanes than the strain with drug-resistant gyrase (parC+ gyrAL83). At higher drug concentrations, first the parCL80 and then the parCK84 drug-resistance is partially overcome, as indicated by the increase in catenanes at 90 μm and 120 μm norfloxacin, respectively. These results are expected if topo IV, not gyrase, is the primary decatenase of recombination products.
Gyrase plays some role in decatenation of recombination products because there is roughly a 30% difference in the levels of catenation between the parC+ gyrA+ and parC+ gyrAL83 strains (Fig. 5, cf. top left and right panels). Gyrase may directly decatenate this fraction of catenanes. Alternatively, because topo IV unlinks supercoiled catenanes four-fold better than it unlinks relaxed catenanes in vitro (Ullsperger and Cozzarelli 1996), it is possible that the role played by gyrase is to boost the decatenation activity of residual uninhibited topo IV by supercoiling the plasmids. As a result, when supercoiling is normal (in gyrAL83 strains), residual topo IV decatenates more efficiently than it does when the DNA is relaxed.
To visualize better the relationship between supercoiling (σ) and decatenation, we plotted this dependence for every data point from Figure 5 for parC+ strains, in which catenanes are preserved by drug addition. As with recombination (Fig. 4), σ affects strikingly the amount of decatenation (Fig. 6). There is an abrupt decrease in the fraction of catenanes over the relatively small supercoiling density range (−0.05 to −0.075).
Figure 6.

Effect of DNA supercoiling (σ) on decatenation. The correlation between decatenation and σ is shown for every parC+ data point in Fig. 5.
We carried out the following experiment to test whether the role of DNA gyrase is to introduce negative supercoils into the DNA, which is then decatenated by topo IV exclusively. We reasoned that if this hypothesis is correct, then if we could block DNA relaxation, the inhibition of gyrase would not result in a loss of supercoiling. The consequence would be that recombination, supercoiling, and catenation should now be the same in the gyrA+ and gyrAL83 strains after the treatment with norfloxacin. Although we knew from the data in Figures 2 and 3 that topo IV was able to relax DNA in the presence of topo I, we expected the role of topo I to predominate because it has been considered to be the primary (−) supercoil relaxation enzyme in the cell (for review, see Wang 1991). We moved our drug-resistant and wild-type parC and gyrA alleles into a strain RS2λ, which contains a mutation in topo I, top10, which reduces topo I activity 100-fold (DiNardo et al. 1982).
The experiment proved to be more complex than we had anticipated, but for an interesting reason. After inhibition of gyrase by drug, topo IV alone was able to relax the DNA to near completion in the topo I mutant (data not shown). Although this result confounded our original intent, we obtained useful information. In these experiments, topo IV was wild-type and topo I was mutant. In the presence of drug, there was a fourfold faster initial rate of recombination in the gyrAL83 strain than in the gyrA+ control (Fig. 7A). In addition, recombination reached 95% in the strain with drug-resistant gyrase, but never exceeded 50% in the wild-type strain. Even without topo I activity to relax the DNA, the level of DNA supercoiling was dependent upon gyrase activity. The value of σ after the addition of drug was −0.053 in the strain with wild-type gyrase, but reached the very high level of −0.093 in the gyrAL83 strain. Therefore, through its effect on σ, gyrase has an effect on recombination.
Figure 7.
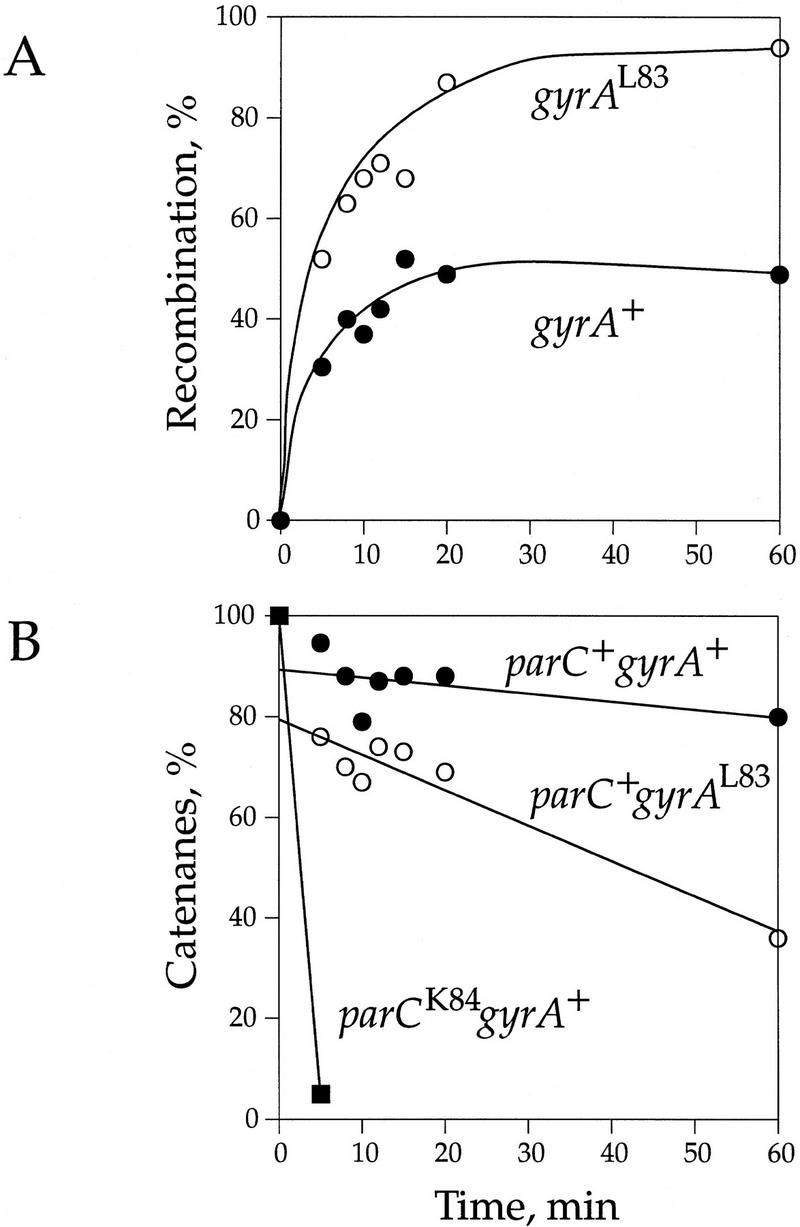
Kinetics of recombination and decatenation. (A) Rate of recombination in top10 parC+ gyrA+ (LZ53; •) and top10 parC+ gyrAL83 (LZ54; ○) strains. The experiment was as outlined in Figs. 1 and 2. The drug concentration was 90 μm. (B) Amount of catenation in top10 parC+ gyrA+ (•) and top10 parC+ gyrAL83 (○) strains. Catenane turnover in a parCK84 gyrAL83 strain (LZ37; ▪) is shown for comparison. From the relative slopes, catenane unlinking when gyrase is active and topo IV is inhibited (○) is 3.5-fold faster than when both enzymes are inhibited (•) and 25-fold faster than when topo IV is uninhibited (▪).
Our chief interest is in the portion of recombined DNA that was catenated (Fig. 7B). The majority (70%–90%) of the DNA remained catenated after drug treatment, but as in Figure 5, there was ∼20% difference in catenation depending on whether gyrase was drug resistant. This difference is small compared with the differences in recombination and σ, however. With time, the catenanes are unlinked slightly in both strains. As revealed from the slopes of the least squares fit lines of the kinetic data, the unlinking activity when gyrase is drug resistant is ∼3.5-fold greater than the unlinking activity when gyrase is wild type. For comparison, we show the rate of catenane turnover when topo IV is active and gyrase is not in a parCK84 gyrA+ strain. This rate is ∼25-fold faster than when gyrase alone is active. Therefore, the gyrase contribution to the rate of decatenation is far less than that of topo IV. Overall, these results are the same as those for the wild-type topo I strains and indicate that there are two rates of decatenation—an efficient unlinking by uninhibited topo IV and a rate ∼25-fold slower, which we believe is carried out by incompletely inhibited topo IV. Residual topo IV is boosted by the high level of negative supercoiling in the gyrAL83 strain.
Topo IV decatenates the products of resolvase recombination in vivo
The most common plasmid catenane type in E. coli is the singly linked catenane (Wasserman and Cozzarelli 1986). A singly linked (two-noded) catenane can arise from DNA replication, type-2 topoisomerization, or recombination (Wasserman et al. 1988). The two-noded catenane is structurally unique among catenanes in that there is no strain in the DNA introduced by the link (Wasserman and Cozzarelli 1986). Therefore, there is not the same enthalpic drive to decatenation as with multiply interlinked catenanes. Because of this and because of the complexity of the results with Int, an independent evaluation of the roles of topoisomerases in unlinking recombination catenanes was desirable. Therefore, we tested whether topo IV also played the key role in the unlinking of the two-noded catenanes formed by Tn3 resolvase.
As with Int, resolvase expression was under the λ phage pL promoter controlled by the temperature-sensitive cI857 repressor (Bliska et al. 1991). There were two differences between the Int and resolvase systems (Fig. 8). First, because resolvase is active at 43°C, the entire experiment was conducted at 43°C and some recombination takes place before the addition of the topoisomerase inhibitor (140 sec later). Second, the resolvase gene was in a multicopy plasmid instead of a single λ chromosomal insertion and was, therefore, expressed at a much higher level (Bliska 1990). Because of the high expression levels and the recombination before topoisomerase inactivation, the time scale was much faster for resolvase than for Int. The substrate used, pRR51, contains directly repeated resolvase-binding sites and is recombined into two-noded catenanes, which are unlinked by topoisomerases in vivo (Bliska et al. 1991).
Figure 8.

Experimental system for measuring decatenation of the products of resolvase recombination. The cells were grown at 30°C and then shifted to 43°C to induce resolvase expression. The enzyme recombines the two directly repeated sites on the substrate plasmid DNA and the product is a singly linked (two-noded) catenane. The sizes of the rings were 2.75 and 3.2 kb. A type-2 topoisomerase unlinks the catenanes to generate free circles. Drug (norfloxacin) was added after recombination but before most unlinking.
We focused on the gyrAL83 strains because in gyrA+ strains there was minimal recombination as the plasmid substrate was relaxed at even the lowest drug levels examined and resolvase absolutely requires (−) supercoiling (Reed 1981; Krasnow and Cozzarelli 1983; Bliska et al. 1991; Benjamin et al. 1996). In the gyrAL83 strains, the plasmid DNA remained fully (−) supercoiled in up to 200 μm norfloxacin (data not shown). In the absence of drug, the rate of resolvase recombination and subsequent decatenation was identical in all strains (data not shown). With increasing concentrations of norfloxacin, increasing amounts of recombination products were trapped as catenanes when topo IV alone was inhibited (parC+ gyrAL83) (Fig. 9). At 30 μm, there was only a slight block of decatenation. At 60 μm, half of the recombined plasmid was catenated. In contrast, when topo IV and gyrase were both resistant to drug (parCL80 gyrAL83), there was a rapid unlinking of the catenanes into free circles even with norfloxacin as high as 300 μm. Although we could make no reliable quantitation, we observed catenanes in the parC+ gyrA+ strain and not in the parCL80 gyrA+ strain (data not shown). We conclude that topo IV is also the primary decatenase of singly linked recombination products.
Figure 9.

Effect of topo IV on decatenation of resolvase recombination products. The fraction of plasmid DNA that was catenated after 10 min at 43°C is shown as a function of norfloxacin (Nor) concentration. Two strains were assayed: parC+ gyrAL83 (LZ29; •) and parCL80 gyrAL83 (LZ27; ▪).
Discussion
By use of isogenic strains with various combinations of alleles encoding wild-type and drug-resistant gyrase and topo IV, we have determined the division of labor of the type-2 topoisomerases in the cell. We find that topo IV, not gyrase, is the enzyme that unlinks the catenated intermediates of two different recombinases. Our conclusion that topo IV is the primary decatenase of recombination intermediates differs from the earlier conclusion that DNA gyrase was responsible (Bliska and Cozzarelli 1987; Bliska et al. 1991). Our data agree with the earlier results, but because they are far more extensive, lead to the revised interpretation. These results, combined with our previous work with DNA replication intermediates (Zechiedrich and Cozzarelli 1995), allow us to conclude that topo IV is the primary enzyme for all decatenation in the cell.
The role of gyrase in decatenation
Although topo IV is the major unlinking enzyme, gyrase does have an effect on decatenation. This is apparent from the reduction in decatenation after inhibition of gyrase with drug (Figs. 5 and 7). There are two ways that gyrase could promote decatenation. First, gyrase might directly unlink catenanes, but at a rate 25-fold lower than that of topo IV (see Fig. 7). This contribution is similar to what we previously estimated for DNA gyrase decatenation of replication intermediates, which was roughly 100-fold less than the contribution of topo IV (Zechiedrich and Cozzarelli 1995). The weak activity of gyrase was not sufficient to prevent catenation of replicated plasmids and a partition defect phenotype of conditionally lethal topo IV mutants (Kato et al. 1988; Schmid 1990).
The second possibility is that gyrase negatively supercoils the catenanes, which makes them superior substrates for topo IV decatenation. There are three arguments in favor of this possibility. (1) Purified topo IV unlinks supercoiled catenanes at least fourfold faster than relaxed catenanes in vitro (Ullsperger and Cozzarelli 1996) and at equilibrium supercoiling favors plasmid decatenation by more than two orders of magnitude (Rybenkov et al. 1997). The reduction in decatenation after norfloxacin addition to the gyrA+ strain compared with the resistant mutant in all experiments correlated with σ value; (−) supercoiling promotes decatenation (Fig. 6). (2) If drug-resistant gyrase were directly decatenating some fraction of the catenanes, then the inhibition of topo IV should not alter that fraction. Instead, decatenation is far better when topo IV is drug resistant (Fig. 5). If anything, the presence of a drug-resistant topo IV should impair any direct decatenation by gyrase because topo IV relaxes the plasmid DNA and gyrase was also found to unlink supercoiled substrates better than relaxed substrates in vitro (Ullsperger and Cozzarelli 1996). (3) The turnover of the catenanes is blocked in the parC+ gyrAL83 strain at the highest drug concentrations used (Fig. 5) even though gyrase is still fully functional [as evidenced by the complete recombination (Fig. 2) and unaltered supercoiling (Fig. 3) at the same drug concentrations]. Uninhibited gyrase should be able to continue to decatenate if it is responsible directly for decatenation.
If supercoiling by gyrase is inhibited, the activity of the drug-resistant topo IV on the relaxed DNA is still sufficient to completely unlink the catenanes. This is shown by the complete decatenation in the parCK84 gyrA+ strain in 90 μm or less norfloxacin. Topo IV is only barely able to keep up with decatenation, however, because even the partial inhibition of topo IV in the parCL80 gyrA+ strain causes an increase in the proportion of catenanes.
We conclude that the role of gyrase in decatenation is to introduce negative supercoils into plasmid DNA, which makes a superior substrate for topo IV.
The role of topo IV in DNA supercoiling
It has been long thought that only two topoisomerases, topo I and DNA gyrase, counteracted each other to maintain the level of DNA supercoiling in bacteria (for review, see Wang 1991). Topo I removes (−) supercoils and DNA gyrase introduces (−) supercoils. Here, we show that topo IV has a potent relaxing activity in vivo that almost completely removes (−) supercoils after the inhibition of gyrase (Fig. 3). The extent of DNA relaxation by topo IV (σ = −0.012; Fig. 3, bottom left) exceeds the extent of relaxation by topo I (σ = −0.055; Fig. 3, top left). The relaxation activity of topo IV was missed before because the drugs used to inhibit gyrase also inhibited topo IV, the same reason that the role of topo IV in decatenation of recombination intermediates was missed. Only when we used a drug-resistant topo IV were the new activities revealed. These results call for a re-examination of previous research on DNA supercoiling in bacteria and we have addressed the relative importance of the topoisomerases in DNA supercoiling in a separate study (E.L. Zechiedrich, A.B. Khodursky, and N.R. Cozzarelli, in prep.).
Our findings reveal a complete division of labor between topo IV and DNA gyrase. Through the same DNA strand-passage mechanism, these homologous topoisomerases perform radically different functions in the cell. Topo IV decatenates replication and recombination intermediates as well as relaxes DNA supercoils. DNA gyrase introduces (−) supercoils in DNA. We speculate that, when examined more closely, perhaps other enzymes that are considered to be redundant may also have distinct roles in vivo.
Materials and methods
Chemicals and reagents
[α-32P]dCTP (250 mCi/mmole; 1 Ci/ml) was from NEN. The supercoiled DNA and 1 kilobase ladders were from GIBCO BRL. Proteinase K was from Boehringer-Mannheim. Norfloxacin, fusaric acid, chlorotetracycline, chloroquine, RNaseA, and DNAse I were from Sigma. Multiprime DNA-labeling kit was from Amersham and Nytran (MSI) transfer membrane from Fisher.
Bacterial strains and plasmids
The bacterial strains used in this study are listed in Table 1. Four isogenic sets of strains containing all possible combinations of drug-resistant and wild-type alleles of gyrA and parC were constructed. Two sets were based upon the parental strain C600 that we used in earlier work (Khodursky et al. 1995; Zechiedrich and Cozzarelli 1995). The set of strains used to measure Ki values in vivo were LZ21–24, 1643, and 1644. For the resolvase experiments, the set was LZ27–30. The strains used for the Int experiments were derived from the parental lysogen strain W3101λ (Bliska and Cozzarelli 1987; Adams et al. 1992a) and were designated LZ33–38. The topo I mutant strains were LZ53 and LZ54. In the text, for simplicity, we refer to these strains mostly by their parC and gyrA allelic states. The Int substrate plasmid was pJB3.5d (Bliska and Cozzarelli 1987). The resolvase expression vector, pJBREScI (Bliska et al. 1991), and the resolvase substrate, pRR51 (Reed 1981), have been described previously.
Strain construction
The construction of the tet-linked gyrA+ and gyrAL83 (Zechiedrich and Cozzarelli 1995) and kan-linked parC+, parCL80, and parCK84 (Khodursky et al. 1995) strains were described previously. Because the resolvase substrate plasmid pRR51 conferred tetracycline resistance, we used the method of Bochner et al. (1980) to isolate tets versions of these strains except that the final concentration of fusaric acid was increased to 24 μg/ml and chlorotetracycline to 100 μg/ml. For the Int experiments, the tet-linked gyrA or kan-linked parC alleles were transduced with P1vira (Miller 1972) into W3101λ. The strains containing mutant topo I were made by transferring the kan marker from CAG12183 into strains N51 and KL16 (Yoshida et al. 1988; Singer et al. 1989). Then the gyrAL83 or gyrA+ gene with its linked kan marker was transduced into the lysogen RS2λ (Bliska and Cozzarelli 1987). The presence of the lysogen was verified by temperature sensitivity at 42°C. The presence of the topo I mutation was verified by the increased plasmid supercoiling.
Experimental rationale
To repeat the earlier experiments as closely as possible, we used the same recombinase expression systems and substrates for Int and resolvase (Bliska and Cozzarelli 1987; Bliska et al. 1991; Adams et al. 1992a). We looked at two different recombinases not only to test the generality of our results, but also to assay the decatenation of two different types of catenanes—singly linked and multiply linked. Because the sensitivity of DNA gyrase and topo IV to norfloxacin in vivo varies only by about twofold (Table 2; Khodursky et al. 1995), we had to use drug-resistant forms of the enzymes to distinguish them from each other. These drug-resistant enzymes have Ki values one to two orders of magnitude greater than their wild-type counterparts (Table 2A). By use of drug-resistant and wild-type alleles, we could inhibit one enzyme without affecting the other (Table 2B).
Inhibition of topo IV and gyrase with norfloxacin
To determine the drug sensitivity of the catalytic activity of the enzymes in vivo, we measured the amount of drug required to inhibit by 50% the supercoiling activity of gyrase and the decatenation activity of topo IV. Of course, quinolone antibiotics are not merely inhibitors of the topoisomerases. They act by the so-called poisoning mechanism whereby the cleaved DNA intermediate in the topoisomerase reaction is stabilized (Kreuzer and Cozzarelli 1979; Tewey et al. 1984). We do not include such covalent enzyme–DNA complexes in our analyses. However, they represent <5% of the plasmids (data not shown). Therefore, the change in plasmid topology levels that we analyze result from the removal of the topoisomerase being tested. Steady-state supercoiling of plasmid pBR322 DNA was chosen to be the measure of gyrase activity. The Ki value for gyrase was defined as the minimal concentration of antibiotic that caused a twofold decrease in the amount of the topoisomer closest to the mean value.
Accumulation of DNA replication catenanes was used to determine the extent of topo IV inhibition by norfloxacin. The Ki value for topo IV was defined as the minimal drug concentration at which half of the newly replicated DNA was catenated after a 0.5-min pulse (see Khodursky et al. 1995; Zechiedrich and Cozzarelli 1995). Measurement of the inhibition of topo IV was possible only in the presence of the drug-resistant gyrA because with wild-type gyrase, DNA replication was inhibited at drug concentrations much lower than those that inhibited topo IV (Khodursky et al. 1995). The Ki values in vitro and in vivo are summarized in Table 2A. We were unable to determine the amount of drug needed to overcome the resistance of parCK84 in vivo because the resistance of the gyrAL83 allele was overcome first. For the same reason, we could only estimate the Ki value for parCL80. We used the Ki values in vitro and in vivo (Table 2A) to assess the range of drug concentrations that would be active against the mutant enzymes in our experiments (Table 2B). For 30 μm or higher norfloxacin, wild-type gyrase was inhibited. The (−) supercoiling activity of drug-resistant gyrase in vivo was not inhibited until >180 μm norfloxacin. ParC+ was mostly inhibited at 30 μm and completely blocked at higher concentrations. ParCL80 was unaffected at 60 μm and was inhibited at 90 μm or higher. ParCK84 was slightly inhibited at 120 μm, but was not at the lower drug concentrations.
Int recombination
The assay used for Int recombination (Bliska and Cozzarelli 1987; Adams et al. 1992a) is schematized in Figure 1. Lysogens harboring the plasmid pJB3.5d were grown at 30°C in LB with 50 μg/ml of ampicillin. At a cell density of 70 Klett units, cultures were shifted to 43°C to inactivate the temperature-sensitive λ repressor (cI) and induce Int expression. After 10 min with shaking, cultures were returned to 30°C because Int is not active at 43°C. Norfloxacin was added to inhibit the parC+ and/or gyrA+-containing cells when the cultures were shifted to 30°C (Bliska and Cozzarelli 1987). Samples (1.9 ml) were removed at various times during the incubation, immersed in liquid nitrogen, stored at −80°C, and thawed on ice (Zechiedrich and Cozzarelli 1995). Plasmid DNA was isolated by the alkaline lysis procedure (Sambrook et al. 1989), treated with RNase A, and divided into two parts. One part was nicked with DNase I and separated by high resolution electrophoresis on agarose gels (Sundin and Varshavsky 1981). The remaining portion of supercoiled plasmids was analyzed by electrophoresis through 1.2% agarose gels (TAE) with 0, 2, 4, or 8 μg/ml of chloroquine. DNA in the gels was transferred to Nytran membranes and probed with labeled nick-translated pJB3.5d. The Southern blots were exposed to Kodak XAR film or PhosphorImager (Molecular Dynamics) cassettes for various times (15 min to 18 hr). The amount of radioactivity in each band was determined by PhosphorImager analysis.
For the catenane turnover shown in Figure 7 for a parCK84 gyrA+ strain, we assumed that all unlinked circles came from catenanes (100%), as we never observed more than a few percent of catenanes in this strain.
Resolvase recombination
The assay used for resolvase recombination was as described (Bliska et al. 1991) and is depicted in Figure 8. Cells harboring pJBREScI and pRR51 were grown at 30°C to a density of 70 Klett units in LB with 30 μg/ml of chloramphenicol, 50 μg/ml of ampicillin, and 15 μg/ml of tetracycline. The cells were shifted to 43°C (time = 0) to induce resolvase expression. Cultures were shaken for up to 10 min at 43°C. To block decatenation of resolvase recombination products, norfloxacin was added 140 sec after the upshift. This time for drug addition gave a maximal ratio of catenanes to free circles (data not shown). Aliquots (1.9 ml) were removed before upshift and at various times after shift to 43°C. Plasmids were isolated and analyzed as described above.
Acknowledgments
We thank E.L. Beall, D.B. Roth, and members of the laboratory for critical reading of the manuscript and C.A. Gross and S. Nakamura for strains. This work was supported by National Institutes of Health grant GM31657 and National Institute on Environmental Health Sciences grant ESO1896. E.L.Z. is a Special Fellow of the Leukemia Society of America.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL cozzlab@mendel.berkeley.edu; FAX (510) 643-1079.
References
- Adams DE, Bliska JB, Cozzarelli NR. Cre-lox recombination in E. coli cells: Mechanistic differences from the in vitro reaction. J Mol Biol. 1992a;226:661–673. doi: 10.1016/0022-2836(92)90623-r. [DOI] [PubMed] [Google Scholar]
- Adams DE, Shekhtman EM, Zechiedrich EL, Schmid MB, Cozzarelli NR. The role of topoisomerase IV in partitioning bacterial replicons and the structure of catenated intermediates in DNA replication. Cell. 1992b;71:277–288. doi: 10.1016/0092-8674(92)90356-h. [DOI] [PubMed] [Google Scholar]
- Belland RJ, Morrison SG, Ison C, Huang WM. Neisseria gonorrhoeae acquires mutations in analogous regions of gyrA and parC in fluoroquinolone-resistant isolates. Mol Microbiol. 1994;14:371–380. doi: 10.1111/j.1365-2958.1994.tb01297.x. [DOI] [PubMed] [Google Scholar]
- Benjamin KR, Abola AP, Kanaar R, Cozzarelli NR. Contributions of supercoiling to Tn3 resolvase and phage mu gin site-specific recombination. J Mol Biol. 1996;256:50–65. doi: 10.1006/jmbi.1996.0067. [DOI] [PubMed] [Google Scholar]
- Bjornsti M-A. DNA topoisomerases. Curr Opin Struct Biol. 1991;1:99–103. [Google Scholar]
- Bliska, J.B. 1990. “Studies on site-specific recombination, DNA structure and DNA metabolism in vivo” Ph.D. Thesis, University of California, Berkeley, CA.
- Bliska JB, Cozzarelli NR. Use of site-specific recombination as a probe of DNA structure and metabolism in vivo. J Mol Biol. 1987;194:205–218. doi: 10.1016/0022-2836(87)90369-x. [DOI] [PubMed] [Google Scholar]
- Bliska JB, Benjamin HW, Cozzarelli NR. Mechanism of Tn3 resolvase recombination in vivo. J Biol Chem. 1991;266:2041–2047. [PubMed] [Google Scholar]
- Bochner BR, Huang HC, Schieven GL, Ames BN. Positive selection for loss of tetracycline resistance. J Bacteriol. 1980;143:926–933. doi: 10.1128/jb.143.2.926-933.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Malik M, Snyder M, Drlica K. DNA gyrase and topoisomerase IV on the bacterial chromosome: Quinolone-induced DNA cleavage. J Mol Biol. 1996;258:627–637. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- Cook DN, Ma D, Pon NG, Hearst JE. Dynamics of DNA supercoiling by transcription in E. coli. Proc Natl Acad Sci. 1992;89:10603–10607. doi: 10.1073/pnas.89.22.10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer PA. Chromosome segregation and cytokinesis in bacteria. Curr Opin Cell Biol. 1993;5:232–237. doi: 10.1016/0955-0674(93)90108-3. [DOI] [PubMed] [Google Scholar]
- Dean F, Krasnow MA, Otter R, Matzuk MM, Spengler SJ, Cozzarelli NR. Escherichia coli type-1 topoisomerases: Identification, mechanism, and role in recombination. Cold Spring Harbor Symp Quant Biol. 1983;47:769–777. doi: 10.1101/sqb.1983.047.01.088. [DOI] [PubMed] [Google Scholar]
- DiGate RJ, Marians KJ. Identification of a potent decatenating enzyme from Escherichia coli. J Biol Chem. 1988;263:13366–13373. [PubMed] [Google Scholar]
- ————— Molecular cloning and DNA sequence analysis of Escherichia coli topB, the gene encoding topoisomerase III. J Biol Chem. 1989;264:17924–17930. [PubMed] [Google Scholar]
- DiNardo S, Voelkel KA, Sternglanz R, Reynolds AE, Wright A. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell. 1982;31:43–51. doi: 10.1016/0092-8674(82)90403-2. [DOI] [PubMed] [Google Scholar]
- Drlica K. Biology of bacterial deoxyribonucleic acid topoisomerases. Microbiol Rev. 1984;48:273–289. doi: 10.1128/mr.48.4.273-289.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero L, Cameron B, Manse B, Lagneaux D, Crouzet J, Famechon A, Blanche F. Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: A primary target of fluoroquinolones. Mol Microbiol. 1994;13:641–653. doi: 10.1111/j.1365-2958.1994.tb00458.x. [DOI] [PubMed] [Google Scholar]
- Filutowicz M, Jonczyk P. The gyrB gene product functions in both initiation and chain polymerization of Escherichia coli chromosome replication: Suppression of the initiation deficiency in gyrB-ts mutants by a class of rpoB mutations. Mol & Gen Genet. 1983;191:282–287. doi: 10.1007/BF00334827. [DOI] [PubMed] [Google Scholar]
- Free A, Dorman CJ. Escherichia coli tyrT gene transcription is sensitive to DNA supercoiling in its native chromosomal context: Effect of DNA topoisomerase IV overexpression on tyrT promoter function. Mol Microbiol. 1994;14:151–161. doi: 10.1111/j.1365-2958.1994.tb01275.x. [DOI] [PubMed] [Google Scholar]
- Gangloff S, McDonald J, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M. DNA topoisomerases. Annu Rev Biochem. 1981;50:879–910. doi: 10.1146/annurev.bi.50.070181.004311. [DOI] [PubMed] [Google Scholar]
- Guarneros G, Echols H. Thermal asymmetry of site-specific recombination by bacteriophage lambda. Virology. 1973;52:30–38. doi: 10.1016/0042-6822(73)90395-4. [DOI] [PubMed] [Google Scholar]
- Heisig P. Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother. 1996;40:879–885. doi: 10.1128/aac.40.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisig P, Wiedemann B. Use of a broad host-range gyrA plasmid for genetic characterization of fluoroquinolone-resistant gram-negative bacteria. Antimicrob Agents Chemother. 1991;35:2031–2036. doi: 10.1128/aac.35.10.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa H, Marians KJ. Topoisomerase III, but not topoisomerase I, can support nascent chain elongation during theta-type DNA replication. J Biol Chem. 1994;269:32655–32659. [PubMed] [Google Scholar]
- Kato J-i, Nishimura Y, Yamada M, Suzuki H, Hirota Y. Gene organization in the region containing a new gene involved in chromosome partition in Escherichia coli. J Bacteriol. 1988;170:3967–3977. doi: 10.1128/jb.170.9.3967-3977.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J-i, Nishimura Y, Imamura R, Niki H, Hiraga S, Susuki H. New topoisomerase essential for chromosome segregation in Escherichia coli. Cell. 1990;63:393–404. doi: 10.1016/0092-8674(90)90172-b. [DOI] [PubMed] [Google Scholar]
- Kato J-i, Suzuki H, Ikeda H. Purification and characterization of DNA topoisomerase IV in Escherichia coli. J Biol Chem. 1992;267:25676–25684. [PubMed] [Google Scholar]
- Keller W. Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc Natl Acad Sci. 1975;72:4876–4880. doi: 10.1073/pnas.72.12.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodursky AB, Zechiedrich EL, Cozzarelli NR. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc Natl Acad Sci. 1995;92:11801–11805. doi: 10.1073/pnas.92.25.11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnow MA, Cozzarelli NR. Site-specific relaxation and recombination by the Tn3 resolvase: Recognition of the DNA path between oriented res sites. Cell. 1983;32:1313–1324. doi: 10.1016/0092-8674(83)90312-4. [DOI] [PubMed] [Google Scholar]
- Kreuzer KN, Cozzarelli NR. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: Effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J Bacteriol. 1979;140:424–435. doi: 10.1128/jb.140.2.424-435.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y, Kato J-i, Hoshino K, Akasaka T, Sato K, Ikeda H. Quinolone-resistant mutants of Escherichia coli DNA topoisomerase IV parC gene. Antimicrob Agents Chemother. 1996;40:710–714. doi: 10.1128/aac.40.3.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttinger A. The twisted “life” of DNA in the cell: Bacterial topoisomerases. Mol Microbiol. 1995;15:601–606. doi: 10.1111/j.1365-2958.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]
- Luttinger AL, Springer AL, Schmid MB. A cluster of genes that affects nucleoid segregation in Salmonella typhimurium. New Biol. 1991;3:687–697. [PubMed] [Google Scholar]
- Lynch AS, Wang JC. Anchoring of DNA to the bacterial cytoplasmic membrane through contranscriptional synthesis of polypeptides encoding membrane proteins or proteins for export: A mechanism of plasmid hypernegative supercoiling in mutants deficient in DNA topoisomerase I. J Bacteriol. 1993;175:1645–1655. doi: 10.1128/jb.175.6.1645-1655.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNairn E, Ni Bhriain N, Dorman CJ. Overexpression of the Shigella flexneri genes coding for DNA topoisomerase IV compensates for loss of DNA topoisomerase I: Effect on virulence gene expression. Mol Microbiol. 1995;15:507–517. doi: 10.1111/j.1365-2958.1995.tb02264.x. [DOI] [PubMed] [Google Scholar]
- Menzel R, Gellert M. Regulation of the genes for E. coli DNA gyrase: Homeostatic control of DNA supercoiling. Cell. 1983;34:105–113. doi: 10.1016/0092-8674(83)90140-x. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Nash HA. Bending and supercoiling of DNA at the attachment site of bacteriophage lambda. Trends Biochem Sci. 1990;15:222–227. doi: 10.1016/0968-0004(90)90034-9. [DOI] [PubMed] [Google Scholar]
- Orr E, Fairweather NF, Holland IB, Pritchard RH. Isolation and characterization of a strain carrying a conditional lethal mutation in the cou gene of Escherichia coli K-12. Mol & Gen Genet. 1979;177:103–112. doi: 10.1007/BF00267259. [DOI] [PubMed] [Google Scholar]
- Osheroff N, Zechiedrich EL, Gale KC. Catalytic function of DNA topoisomerase II. BioEssays. 1991;13:269–273. doi: 10.1002/bies.950130603. [DOI] [PubMed] [Google Scholar]
- Peng H, Marians KJ. Escherichia coli topoisomerase IV; purification, characterization, subunit structure, and subunit interactions. J Biol Chem. 1993;268:24481–24490. [PubMed] [Google Scholar]
- Pruss GJ, Drlica K. Escherichia coli DNA topoisomerase I mutants: Increased supercoiling is corrected by mutations near gyrase genes. Cell. 1982;31:35–42. doi: 10.1016/0092-8674(82)90402-0. [DOI] [PubMed] [Google Scholar]
- Raji A, Zabel DJ, Laufer CS, Depew RE. Genetic analysis of mutations that compensate for loss of Escherichia coli DNA topoisomerase I. J Bacteriol. 1985;162:1173–1179. doi: 10.1128/jb.162.3.1173-1179.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed RR. Transposon-mediated site-specific recombination: A defined in vitro system. Cell. 1981;25:713–719. doi: 10.1016/0092-8674(81)90178-1. [DOI] [PubMed] [Google Scholar]
- Rybenkov VV, Vologodskii AV, Cozzarelli NR. The effect of ionic conditions on the conformations of supercoiled DNA. II. Equilibrium catenation. J Mol Biol. 1997;267:312–323. doi: 10.1006/jmbi.1996.0877. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schmid MB. A locus affecting nucleoid segregation in Salmonella typhimurium. J Bacteriol. 1990;172:5416–5424. doi: 10.1128/jb.172.9.5416-5424.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden RR, Carlson JO, Pettijohn DE. Torsional tension in the DNA double helix measured with trimethylpsoralen in living E.coli cells: Analogous measurements in insect and human cells. Cell. 1980;21:773–783. doi: 10.1016/0092-8674(80)90440-7. [DOI] [PubMed] [Google Scholar]
- Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erikson JW, Gross CA. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev. 1989;53:1–24. doi: 10.1128/mr.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler SJ, Stasiak A, Cozzarelli NR. The stereostructure of knots and catenanes produced by phage λ integrative recombination: Implications for mechanism and DNA structure. Cell. 1985;42:325–334. doi: 10.1016/s0092-8674(85)80128-8. [DOI] [PubMed] [Google Scholar]
- Springer AL, Schmid MB. Molecular characterization of the Salmonella typhimurium parE gene. Nucleic Acids Res. 1993;21:1805–1809. doi: 10.1093/nar/21.8.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivenugopal KS, Lockshon D, Morris DR. Escherichia coli DNA topoisomerase III: Purification and characterization of a new type I enzyme. Biochemistry. 1984;23:1899–1906. doi: 10.1021/bi00304a002. [DOI] [PubMed] [Google Scholar]
- Sternglanz R, DiNardo S, Voelkel KA, Nishimura Y, Hirota Y, Becherer K, Zumstein L, Wang JC. Mutations in the gene coding for Escherichia coli DNA topoisomerase I affect transcription and transposition. Proc Natl Acad Sci. 1981;78:2747–2751. doi: 10.1073/pnas.78.5.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin O, Varshavsky A. Arrest of segregation leads to accumulation of highly intertwined catenated dimers: Dissection of final stages of SV40 DNA replication. Cell. 1981;25:659–669. doi: 10.1016/0092-8674(81)90173-2. [DOI] [PubMed] [Google Scholar]
- Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- Trucksis M, Depew RE. Identification and localization of a gene that specifies production of Escherichia coli DNA topoisomerase I. Proc Natl Acad Sci. 1981;78:2164–2168. doi: 10.1073/pnas.78.4.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullsperger C, Cozzarelli NR. Contrasting enzymatic activities of topoisomerase IV and DNA gyrase from Escherichia coli. J Biol Chem. 1996;271:31549–31555. doi: 10.1074/jbc.271.49.31549. [DOI] [PubMed] [Google Scholar]
- Ullsperger CJ, Vologodskii AA, Cozzarelli NR. Unlinking of DNA by topoisomerases during DNA replication. In: Lilley DMJ, Eckstein F, editors. Nucleic acids and molecular biology. Berlin, Germany: Springer-Verlag; 1995. pp. 115–142. [Google Scholar]
- Wang, J.C. 1984. DNA supercoiling and its effects on the structure of DNA. J. Cell. Sci. [Suppl.] 1: 21–29. [DOI] [PubMed]
- ————— DNA topoisomerases. Annu Rev Biochem. 1985;54:665–697. doi: 10.1146/annurev.bi.54.070185.003313. [DOI] [PubMed] [Google Scholar]
- ————— DNA topoisomerases: Why so many? J Biol Chem. 1991;266:6659–6662. [PubMed] [Google Scholar]
- ————— DNA topoisomerases. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- Wasserman SA, Cozzarelli NR. Biochemical topology: Applications to DNA recombination and replication. Science. 1986;232:951–960. doi: 10.1126/science.3010458. [DOI] [PubMed] [Google Scholar]
- Wasserman SA, White JH, Cozzarelli NR. The helical repeat of double-stranded DNA varies as a function of catenation and supercoiling. Nature. 1988;334:448–450. doi: 10.1038/334448a0. [DOI] [PubMed] [Google Scholar]
- Watt PM, Hickson ID. Structure and function of type II DNA topoisomerases. Biochem J. 1994;303:681–695. doi: 10.1042/bj3030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi J-I, Kojima T, Oyamada Y, Fujimoto K, Hattori H, Nakamura S, Inoue M. Alterations in the DNA topoisomerase IV grlA gene responsible for quinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1996;40:1157–1163. doi: 10.1128/aac.40.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kojima T, Yamagishi J-i, Nakamura S. Quinolone-resistant mutations of the gyrA gene of Escherichia coli. Mol & Gen Genet. 1988;211:1–7. doi: 10.1007/BF00338386. [DOI] [PubMed] [Google Scholar]
- Zechiedrich EL, Cozzarelli NR. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes & Dev. 1995;9:2859–2869. doi: 10.1101/gad.9.22.2859. [DOI] [PubMed] [Google Scholar]