

Abstract
Mammalian ras genes are thought to be critical in the regulation of cellular proliferation and differentiation and are mutated in ∼30% of all human tumors. However, N-ras and H-ras are nonessential for mouse development. To characterize the normal role of K-ras in growth and development, we have mutated it by gene targeting in the mouse. On an inbred genetic background, embryos homozygous for this mutation die between 12 and 14 days of gestation, with fetal liver defects and evidence of anemia. Thus, K-ras is the only member of the ras gene family essential for mouse embryogenesis. We have also investigated the effect of multiple mutations within the ras gene family. Most animals lacking N-ras function and heterozygous for the K-ras mutation exhibit abnormal hematopoietic development and die between days 10 and 12 of embryogenesis. Thus, partial functional overlap appears to occur within the ras gene family, but K-ras provides a unique and essential function.
Keywords: K-Ras, N-Ras, oncogene, knockout mouse, embryogenesis, hematopoiesis
During the past decade, a convergence of genetic and biochemical data from diverse systems has supported a critical role for Ras-mediated signal transduction pathways in multiple aspects of growth control and development (Khosravi and Der 1994). Although it has been known for some time that Ras proteins function as molecular switches regulated at the level of GDP/GTP binding (Bourne et al. 1990), only in the past few years have many of the components in the Ras signaling cascade been identified, cloned, and characterized through a combination of genetic studies in Drosophila melanogaster and Caenorhabditis elegans and biochemical studies using mammalian cells (Davis 1994; Daum et al. 1994; Wassarman et al. 1995; Duffy and Perrimon 1996; Wittenberg and Reed 1996). These studies have also revealed that many of the components responsible for transducing the extracellular signals are shared by many receptor tyrosine kinases and are highly conserved throughout evolution.
In mammals there are three functional ras genes located on different chromosomes that encode four highly homologous 21-kD proteins: H-Ras, N-Ras, K4A-Ras, and K4B-Ras (Barbacid 1987). K-ras is unique in that it possesses two alternative fourth coding exons, thereby allowing the synthesis of two p21 isoforms, which differ only in their carboxy-terminal residues. The first 86 amino acids of the mammalian Ras proteins, which harbor the putative effector domain, are 100% identical. In addition, two other small G proteins, R-ras and TC21, are structurally and functionally related members of this family (Hall 1994).
Despite considerable progress in elucidating the signal transduction pathways involving Ras, it is still not known what individual roles, if any, the different ras family members play. Many observations suggest that these proteins possess overlapping functions. For example, all three ras genes are expressed ubiquitously (Chesa et al. 1987; Furth et al. 1987; Leon et al. 1987). All cell types analyzed, including those that are terminally differentiated and postmitotic, have been found to express the Ras proteins. In addition, certain tumor types (e.g., thyroid) show no absolute specificity for which ras family member is mutated (Bos 1989). Finally, in yeast as well as in mice ras gene function is partially dispensable. In Saccharomyces cerevisiae, two RAS genes have been identified (Powers et al. 1984). Strains carrying mutations in both genes are inviable, whereas inactivation of either individually is compatible with growth (Kataoka et al. 1984). Importantly, mice that are homozygous null for either N-ras (Umanoff et al. 1995) or H-ras (M. Katsuki, pers. comm.) are viable and exhibit no overt abnormalities developmentally and postnatally. However, the growth inhibitory effect of dominant-negative Ras proteins in mouse cells suggests that normal proliferation is dependent on at least some Ras activity (Cai et al. 1990).
In contrast, several lines of evidence suggest the existence of unique roles for the three mammalian ras genes. For example, despite ubiquitous ras expression, levels of ras mRNA in mice appear to be regulated both temporally and spatially, with certain tissues expressing one or more members of the family preferentially (Muller et al. 1982, 1983; Leon et al. 1987); K-ras and N-ras exhibit similar expression profiles. In addition, many tumor types in humans are associated with mutation of one member of the family more frequently than the other two, suggesting a unique oncogenic role for each of these genes in specific tissues (Bos 1988, 1989). As mentioned above, K-ras is alternatively spliced at its last coding exon, resulting in two isoforms with different carboxyl termini, K-Ras4A and K-Ras4B. K-Ras4B, the predominant isoform (Capon et al. 1983; George et al. 1985), is distinct from the other three Ras proteins in the type of post-translational modifications that occur at the carboxyl terminus (Hancock et al. 1989, 1990; James et al. 1995). This difference is believed to be responsible for the specific association of K-Ras4B with the guanine nucleotide exchange factor (GEF) Smg GDS (Mizuno et al. 1991; Orita et al. 1993). Interestingly, this GEF catalyzes not only the exchange of bound GDP by GTP, but also translocates small G proteins (as shown for K-Ras4B and Rap1B) from the membrane to the cytoplasm (Kawamura et al. 1993; Nakanishi et al. 1994). This specific interaction and translocation may allow K-Ras4B to associate with a distinct subset of potential effector molecules not shared by the other Ras molecules.
Because activating ras mutations have been detected in up to 30% of all human tumors analyzed (Bos 1989; Khosravi and Der 1994), considerable research has been directed toward rational drug design aimed at selectively inhibiting Ras function in transformed cells (Hancock 1993; Gibbs et al. 1994). The efficacy of these drugs will depend, in part, on the physiological consequences of the inhibition of Ras in normal cells, including whether the loss of Ras function is compatible with cell viability. Gene targeting experiments have demonstrated that neither N-Ras nor H-Ras function are essential in the mouse. In contrast, microinjection experiments suggest that Ras activity is required for mouse embryos to develop beyond the two-cell stage (Yamauchi et al. 1994). Here, we present evidence that K-Ras has a unique and essential function in mouse embryogenesis.
Results
Disruption of the murine K-ras gene
We targeted one allele of the murine K-ras gene in mouse embryonic stem (ES) cells using the positive–negative selection method (Mansour et al. 1988). The K-ras targeting vector was constructed by replacing exon 1 sequence with the bacterial neo gene as shown in Figure 1A. Loss of exon 1 sequences should result in a nonfunctional allele of K-ras as it encodes a critical portion of the effector domain (George et al. 1985; Willumsen et al. 1986). After introduction of the K-ras targeting vector into 129/Sv D3 ES cells, the resulting G418- and gancyclovir-resistant clones were screened by Southern blot analysis using a probe located 3′ to the sequences present in the targeting vector. This probe detects an 8.1-kb StuI fragment from wild-type DNA and an additional 7.0-kb BamHI–StuI mutant-specific fragment (Fig. 1A,B). Eight of 380 ES cell clones screened had acquired the exogenous K-ras sequences by homologous recombination as detected by the 3′ probe and therefore were heterozygous for the mutation (K-ras+/−).
Figure 1.


Disruption of K-ras in ES cells. (A) The K-ras targeting vector pK-ras KO was constructed by inserting fragments from intron 0 and intron 1 of the mouse K-ras gene into the plasmid pPNT. The regions of homology consist of a 2.8-kb NotI–SalI fragment and a 5.1-kb HindIII–KpnI fragment. Both the pkg-neo and HSV-tk cassettes were positioned such that they were transcribed in the same transcriptional orientation as K-ras. (B) Southern blot analysis of BamHI plus StuI-digested genomic DNA from ES cell clones using a probe 3′ to the region of homology (3′ ext probe). Lanes 3–6 represent four independent K-ras+/− ES cells clones, as they possess both an 8.1-kb wild-type (wt) allele and a 7.0-kb mutant-specific K-ras allele. (Lane 1) The DNA is from wild-type ES cells; (lane 2) from a nonhomologous integrant; and (lanes 7,8) two independent K-ras−/− mutant ES cell clones that were obtained after exposure to increasing concentrations of G418. (C) Southern blot analysis of StuI-digested genomic DNA using a probe specific for neo. On a parallel set of samples to C the neo probe detected the expected 8.4-kb StuI fragment, as well as an additional 6.9-kb fragment in all K-ras+/− ES cell clones (lanes 3–6). As expected, no signal was detected in the wild-type ES cells (lane 1), and the lower band depicted in lane 2 represents the random integration pattern for this nonhomologous integrant. A number of different digests were performed and screened with the above probes, as well as with probes 5′ to the region of homology, within the 5′ region of homology, and spanning the PGK promoter sequence to determine the actual configuration of the mutant allele. Both the expected and actual K-ras mutant allele configurations are shown in A. This was confirmed further by PCR analysis using the primer pairs shown in A. Primer pairs 3′ neo + 5′ neo and 3′ homolog + 5′ homolog specifically amplify a 5.1- and a 1.8-kb fragment, respectively, as would be expected for this configuration. This head-to-tail integration pattern occurred either one time (lanes 3,5) or multiple times (B lanes 4,6). (D) PCR analysis of E12.5 embryos derived from a K-ras+/− intercross showing the presence of K-ras−/− embryos (lanes marked with an asterisk). The two alleles can be distinguished using primer pairs that specifically amplify a 360-bp wild-type fragment (5′ I0 + 3′ Ex1) or a 270-bp mutant specific fragment (5′ I0 + 3′ neo).
Interestingly, Southern blot analysis on the K-ras+/− clones using a probe derived from neo sequences revealed that all eight targeted clones contained an additional 6.9-kb band as well as the expected 8.4-kb fragment in a StuI (Fig. 1C). Extensive Southern blot and PCR analysis of these clones showed that the neo and 3′ fragment of K-ras carried in the targeting vector were duplicated in a head-to-tail fashion one or more times as illustrated in Figure 1A and data not shown. On the basis of the structure of the mutant allele, we could conclude that critical K-ras exon 1 sequences had been deleted in each of the heterozygous ES cell clones used for further study. This was confirmed subsequently by Southern blot analysis on homozygous mutant ES cell clones (data not shown). Further analysis using a 5′ external probe revealed that two of the eight heterozygous ES cell clones, as detected by the 3′ external probe, had undergone aberrant recombination at the 5′ side (data not shown), and these were not used in the generation of chimeras.
K-ras is an essential gene
Chimeric animals were created by injecting K-ras+/− ES cells into C57BL/6 (BL/6) blastocyst stage embryos, and these were bred to BL/6 mice to determine germ-line contribution. Three independent heterozygous ES cell clones (representing both classes of concatemeric integrations) produced chimeras that transmitted the K-ras mutant allele through the germ line as determined by Southern blot and PCR analysis of tail DNA (data not shown; Fig. 1). To establish the requirements for K-ras function during mouse development, K-ras+/− mice were mated and the genotypes of offspring determined at weaning. Genotyping of the first 83 offspring from these heterozygous intercrosses revealed no K-ras−/− animals, indicating that K-ras is an essential gene for mouse embryogenesis.
We collected and genotyped embryos at progressively earlier times in development to delineate the timing of the death of homozygous mutants. This analysis was carried out on both a mixed BL/6:129/Sv background as well as on an inbred 129/Sv background. Throughout the course of this analysis, identical results were obtained with all three independently derived lines, indicating that the composition of the concatemeric insertion did not alter the resulting phenotype. The viability plot for both genetic backgrounds is shown in Figure 2. On a mixed background, K-ras−/− embryonic viability was not affected until embryonic day 12.5 (E12.5). The number of viable K-ras−/− embryos decreased with increased gestational age, with no mutants surviving past birth. In contrast to the broad window of lethality observed on the mixed genetic background, K-ras−/− embryos died during a much more restricted developmental period when the mutation was examined on an inbred 129/Sv background. Lethality was again first evident at E12, and no K-ras−/− embryo survived beyond E14.
Figure 2.

Viability of K-ras−/− embryos. The percent recovery of viable K-ras−/− embryos (of the total viable embryos recovered) is shown as a function of increasing gestational age. All embryos were derived from K-ras+/− intercrosses; thus, 25% would be expected to be K-ras−/−. This analysis was performed on both a mixed (BL/6:129/Sv) genetic background as well as on a pure 129/Sv background. The heterozygous parents used in this analysis were the F1 progeny of chimeras bred with either pure BL/6 females (for the mixed genetic background analysis) or 129/Sv females (for the inbred genetic background analysis). On the mixed genetic background, ∼600 total implants were genotyped, ranging from 30 to 170 embryos per gestational day. On the pure 129/Sv genetic background, ∼400 total implants were genotyped. During the critical time period (E11.5–14.5), the number of embryos genotyped ranged from 30 to 130 per gestational day.
On both the mixed and inbred backgrounds, K-ras−/− embryos appeared morphologically normal and were indistinguishable from their littermates up to E10.5. Beginning at E11.5, mutant embryos could be identified morphologically based on their smaller size and delayed growth. The E12.5 K-ras−/− embryo shown in Figure 3A exemplifies the mutant phenotype; the mutant is developmentally delayed, has a less obvious superficial vasculature, and is paler than normal littermates. Moreover, the livers of mutants were very pale and reduced in size (typically two- to eightfold fewer total cells than control livers), and the embryo exhibited signs of edema, particularly in the pericardial space. These superficial features are consistent with anemia and a defect in the production or circulation of red blood cells (RBC). The developmental delay ranged from 0.5 to 3 gestational days, with those embryos surviving late in gestation (mixed background) exhibiting the most significant delay. Typically, this delay was coordinate; however, some embryos did exhibit a noncoordinate delay as shown in Figure 3B. In this K-ras−/− embryo, features such as the limbs, skin, tail, and whiskers had developed to a stage associated with E17.5–18.5, whereas the eyes did not develop beyond the equivalent of E15.5, as eye closure was not attained. Moreover, histological analysis of E15.5 or older K-ras−/− embryos on the mixed genetic background showed that a small percentage of these mutants exhibited a noncoordinate development of internal organs (data not shown).
Figure 3.
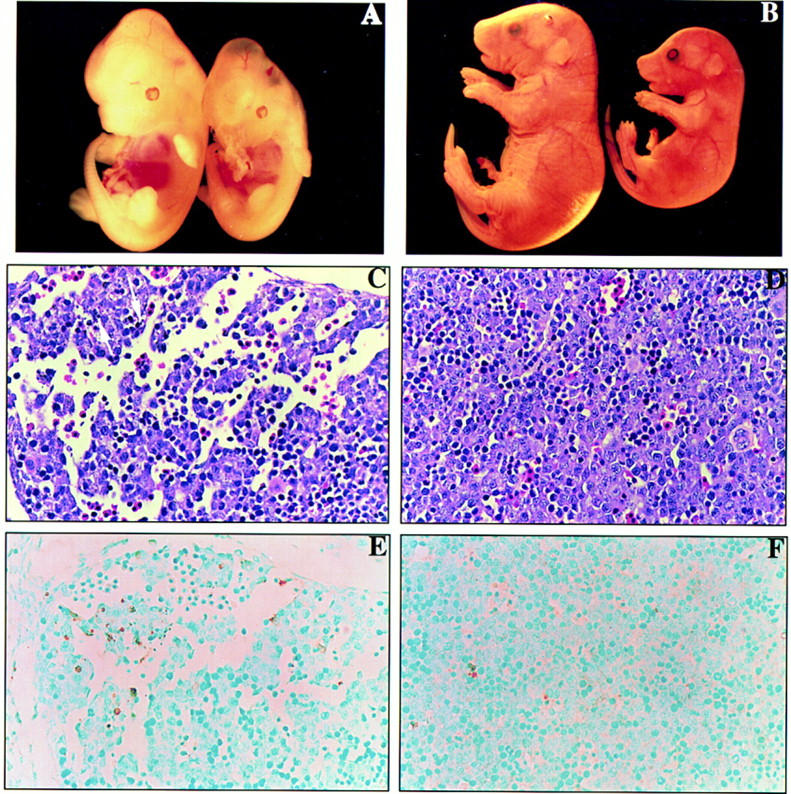
Phenotypic comparison of K-ras−/− embryos and control littermates. (A) The wild-type littermate is on the left and the K-ras−/− littermate is on the right. Note the slight developmental delay (∼0.5 gestational days) and pale coloring of the liver in the K-ras−/− embryo. (B) The control E18.5 K-ras+/− embryo is on the left and the K-ras−/− embryo is on the right. Note the marked reduction in size of the mutant relative to the control littermate as well as the noncoordinate development of the K-ras−/− embryo. The eyes have not developed beyond E15.5, whereas the limbs, tail, and skin have all advanced to at least E17.5. (C,D) Parasagittal section through an E12.5 K-ras−/− (C) and a control wild-type (D) fetal liver. Note the areas of hypocellularity in the K-ras mutant fetal liver, whereas the cells are densely packed in the control liver. Also, pyknotic nuclei (white arrows) in the distal portion of the K-ras−/− hepatic lobe are indicated. (E,F) Cell death analysis on an adjacent fetal liver section from C and D. Note the presence of significant numbers of TUNEL-positive (brown staining) cells in the K-ras−/− fetal livers (E). In more severely affected embryos, these apoptotic cells were present throughout the liver. In contrast, very few cells stain positive in the TUNEL assay from control fetal livers (F).
Fetal liver defect in K-ras−/− embryos
Morphologically, K-ras−/− exhibited a phenotype that was consistent with a defect leading to anemia. The fetal organs/tissues required at this stage in development to support a functional hematopoietic and circulatory system for the RBCs include the placenta, extraembryonic membranes (e.g., yolk sac and chorion), liver, heart, and vasculature system. Histological examination of these and other tissues was carried out on K-ras−/− embryos. Because of the more uniform expressivity of the homozygous mutant phenotype on the 129/Sv background, most of the histological and cellular analysis was performed on embryos from this genetic background. With the exception of the fetal liver (see below), no other tissues of the K-ras−/− embryos were consistently defective.
Histological examination of the livers from E12.5 to E13.5 K-ras−/− embryos indicated that they were smaller and lacked extensive cellularization compared with controls (Fig. 3C,D). In addition, cell death was observed in the livers of more severely affected mutant embryos. This cell death was first apparent in the distal portions of the hepatic lobes (Fig. 3C), but extended throughout the entire organ in the final stages of viability (data not shown). The pyknotic nuclear morphology exhibited in mutant livers is typically associated with apoptosis. To assess whether this cell death was attributable to apoptosis, we analyzed fetal liver sections by the Tdt dUTP-biotin nick-end labeling (TUNEL) assay (Morgenbesser et al. 1994). Liver sections from control embryos showed a few isolated cells that stained positive by this assay (Fig. 3F; data not shown), whereas numerous cells were TUNEL positive in K-ras−/− liver sections (Fig. 3E; data not shown). Thus, the cell death observed in K-ras-deficient embryos was attributable largely, if not exclusively, to apoptosis. Furthermore, this excessive cell death was specific to the liver as other tissues did not exhibit significantly enhanced levels of TUNEL-positive staining (data not shown).
Functional analysis of K-ras−/− hematopoiesis
At E12.5, the liver constitutes the major hematopoietic organ, predominantly for definitive erythropoiesis (Dzierzak and Medvinsky 1995). On the basis of the timing of death for the K-ras−/− embryos, it was conceivable that there may have been a specific defect in this process. Histological analysis and peripheral blood smears from E12.5 to E18.5 (mixed background embryos were used for E14.5 or older) embryos, however, established not only the presence of definitive (enucleated) red cells in the embryo but that mutants were comparable to control embryos of the same developmental stage in the percentage of enucleated versus nucleated red cells (data not shown). Therefore, erythroid cells of K-ras−/− embryos were capable of achieving end-stage differentiation within the hepatic microenvironment.
To determine the functional potential of K-ras−/− hematopoietic progenitors and stem cells, hematopoietic assays were performed both in vitro and in vivo (see Materials and Methods for details). Using fetal liver-derived hematopoietic precursors from K-ras−/− and control embryos, we examined colony formation in semisolid medium in vitro, as well as in vivo differentiation capacity after injection into lethally irradiated host animals. From both assays, we conclude that K-ras function is not required for the survival or differentiation of committed progenitors as well as the long-term repopulating hematopoietic stem cell (LTR–HSC) (data not shown), although we did note a slight reduction in in vitro colony-forming potential with the K-ras−/− cells. Together, these data suggest that the apparent anemia in K-ras−/− embryos is primarily a consequence of defects in the fetal liver microenvironment.
K-ras−/− ES cells have reduced contribution to hematopoietic compartments
Analysis of K-ras−/− embryos has shown a requirement for K-ras in the normal development of, at least, the fetal liver. However, because of the attendant lethality of the K-ras−/− embryo, it was not possible to determine what effects, if any, the absence of K-ras function would have later in gestation or in the adult animal. This issue is important, in part, given the recent efforts to develop chemotherapeutic strategies to inhibit Ras function (Hancock 1993; Gibbs et al. 1994). Therefore, K-ras+/− ES cells were subjected to enhanced concentrations of G418 to generate K-ras−/− ES cells (Mortensen et al. 1992). Of the 200 clones screened, two were found to have lost the wild-type allele (see Fig. 1B). Both K-ras−/− ES cell clones were then injected into BL/6 blastocysts to create chimeric animals. For comparison, chimeras were also generated using the parental K-ras+/− ES cell line. Only four to six K-ras−/− ES cells could be injected per blastocyst to avoid the lethality associated with K-ras homozygosity.
Contribution by the K-ras-deficient cells to chimeric animals was estimated from the extent of the agouti coat color and found to range from ∼10% to 50%. In addition, we determined the percent contribution of mutant cells to the internal tissues of 4-week-old chimeras using glucose phosphate isomerase (GPI) analysis. More than 25 tissues were examined from 8 K-ras−/− chimeras and 4 control K-ras+/− chimeras using this assay. As summarized in Figure 4, significant contribution by the K-ras−/− cells was observed in most tissues, suggesting that most cell types can develop and survive in the absence of K-ras function. However, as this assay measures the percent contribution to the whole tissue, it is possible that K-ras may be required for the proper development of certain cell types. Indeed, the homozygous mutant phenotype indicates that this must be so.
Figure 4.
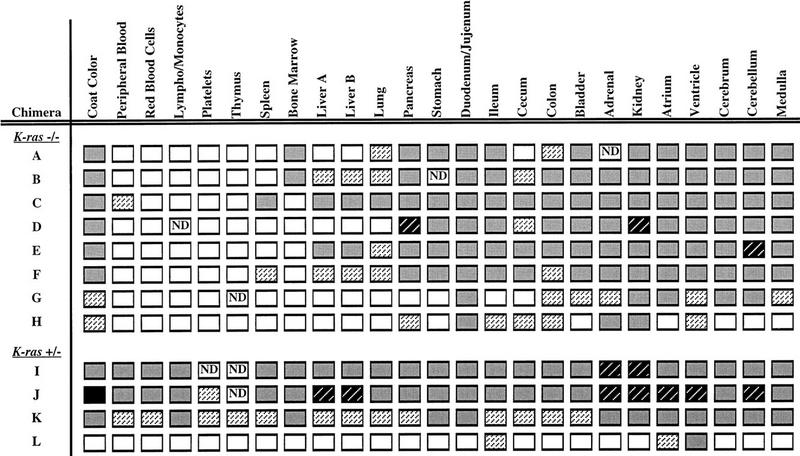
Tissue contribution analysis on K−ras+/− and K−ras−/− chimeras. The distribution of K−ras+/− and K−ras−/− ES cell derivatives to various tissues was analyzed by the GPI isoenzyme assay. The percent contribution of ES cells to each tissue was estimated and categorized as follows: 0%–10%  ; 11%–25%
; 11%–25%  ; 26%–50% ; 51%–75%
; 26%–50% ; 51%–75%  ; or 76%–100%
; or 76%–100%  . (ND) Not determined. K-ras−/− ES cells had significantly reduced contribution to multiple hematopoietic lineages and the tissues that support their production throughout embryogenesis and adulthood.
. (ND) Not determined. K-ras−/− ES cells had significantly reduced contribution to multiple hematopoietic lineages and the tissues that support their production throughout embryogenesis and adulthood.
Interestingly, a relatively low contribution by the K-ras−/− cells was observed in the lung as well as in multiple hematopoietic lineages and the tissues that support their production throughout embryogenesis and adult life (e.g., liver and spleen). Moreover, those chimeras with the highest percent contributions to the liver and spleen exhibited the lowest contribution to the hematopoietic cells of the bone marrow. In addition, in those chimeras with a relatively high contribution of K-ras-deficient cells in the bone marrow, the mutant cells were under-represented in the more differentiated cells of the blood. The low contribution to hematopoietic lineages stands in contrast with the in vitro and in vivo hematopoietic cell analyses and suggests that the lack of K-Ras function may affect hematopoietic progenitor cells directly. This discrepancy could be explained by the fact that in the chimeras, K-ras-deficient hematopoietic cells are developing in competition with wild-type cells. Thus, any subtle intrinsic defect present in the mutant cells may be accentuated. Alternatively, the reduced contribution to the blood cell compartment could be attributable to defects in the hematopoietic microenvironments of the chimeras.
No up-regulation of Ras family members in K-ras−/− embryos
Because K-ras is part of a closely related gene family, it was possible that K-ras mutation induced changes in expression of H-Ras or N-Ras, which could affect the severity of the mutant phenotypes. To determine whether N-Ras or H-Ras were up-regulated in the K-Ras mutant background, we examined the levels of these proteins in mouse embryonic fibroblasts (MEFs) derived from E13.5 embryos on both genetic backgrounds. Lysates of subconfluent cultures of MEFs were immunoprecipitated with Y13-259 antibody (which recognizes all three forms of Ras) and subsequently immunoblotted with antibodies specific for each of the three Ras proteins. As shown in Figure 5A, there was no detectable K-Ras protein in MEFs derived from K-ras−/− embryos, and neither N-Ras nor H-Ras protein levels were altered in cells lacking K-Ras. In addition, we have not detected any truncated K-Ras species expressed from the mutant allele (data not shown).
Figure 5.

N-Ras and H-Ras are not up-regulated in response to the loss of K-Ras. (A) MEFs were derived from E13.5 littermates on both genetic backgrounds and analyzed for the levels of K-Ras, N-Ras, and H-Ras proteins. Ras was immunoprecipitated from at least two independently derived clones for each genotype using the pan-Ras antibody Y13-259. Immunocomplexes were then analyzed by immunoblotting with monoclonal antibodies specific for each of the Ras proteins. (B) Tissues were prepared from E12.5 embryos derived on the mixed genetic background and analyzed as above. One immunoblot was made and probed with the same series of Ras antibodies as used in A. The order of analysis was as follows: N-Ras (F155), H-Ras (F235), K-Ras (F234), and then pan Ras (F111). The weak signal present in the K-ras−/− tissues by the K-Ras-specific antibody is attributable to background cross-reactivity to the faster migrating N-ras and H-Ras proteins. Probing with the pan Ras antibody mirrored the expression levels seen with each antibody (data not shown). Similar results were also obtained with heart and lung tissues (data not shown).
To assess whether N-Ras or H-Ras may have been up-regulated in a tissue-specific manner, extracts were prepared from E12.5 mutant and control littermate tissues (BL/6:129/Sv background) and analyzed as above. Examination of five different tissues revealed that there was no significant change in either N-Ras or H-Ras protein levels in the K-ras-deficient tissues (Fig. 5B; data not shown). It remains possible, however, that there is functional compensation for the loss of K-ras through increases in the level of GTP-bound N-Ras or H-Ras. Alternatively, other Ras-related molecules, such as R-Ras or TC21, may be up-regulated to compensate for the loss of K-Ras function.
Genetic interaction between K-ras and N-ras
In contrast to the requirement for K-ras in mouse embryogenesis described here, neither N-ras (Umanoff et al. 1995) nor H-ras (M. Katsuki, pers. comm.) are essential genes in the mouse. One possible explanation for the normal phenotype of N-ras and H-ras mutants, and for the relatively late onset phenotype in the K-ras mutants, is that different members of the family have at least partial overlapping function. To address a possible genetic interaction between K-ras and N-ras, mice carrying various combinations of the two null mutations were generated on a mixed BL/6:129/Sv genetic background. Because of the breeding considerations, the most extensive analysis was carried out on embryos that were homozygous for the N-ras mutation and heterozygous at the K-ras locus (N-ras−/−; K-ras+/−). As shown in Table 1, the majority of these embryos died during gestation. As with the K-ras−/− embryos on a mixed background, N-ras−/−; K-ras+/− mutants exhibited a variable expressivity in phenotype and a broad window of lethality. Approximately 70% of these mutants died between E10.0 and E12.0, with the remainder dying perinatally (Table 1). A few embryos were found to survive up to 2 days past birth, but subsequently were neglected by their mothers and died shortly thereafter. No apparent differences in phenotype were noted in crosses performed with parents of different mutant genotypes. These data indicate that the normal survival of N-ras-deficient embryos requires wild-type K-ras function.
Table 1.
Summary of dissection analysis for N-ras−/−; K-ras+/− embryos
| Embryonic day
|
N-ras: K-ras:
|
Genotypes of embryos from N+/−; K+/− × N−/−; K+/+
|
|||
|---|---|---|---|---|---|
| +/±/+
|
+/±/−
|
−/±/+
|
−/±/−
|
||
| 9.5–10.0 | no. viable recovered: | 11 | 14 | 16 | 12 |
| % abnormal: | 18.2d | 21.4d | 31.3d | 50.0d | |
| 10.5–11.0 | no. viable recovered: | 39 | 22 | 30 | 20 |
| % abnormal: | 5.1d | 4.6d | 3.3d | 65.0d,h | |
| 11.5–12.0 | no. viable recovered: | 13 | 18 | 12 | 4 |
| % abnormal: | 7.7d,h | 5.6d | 8.3d | 100.0d,h | |
| 12.5–13.0 | no. viable recovered: | 19 | 14 | 17 | 4 |
| % abnormal: | 5.3s | 14.3s,p | 17.6s,p | 75.0d,s,p | |
| 13.5–14.0 | no. viable recovered: | 11 | 9 | 13 | 5 |
| % abnormal: | 9.1d | 22.2d,p | 15.4d | 80.0d,p,e | |
| 14.5–15.0 | no. viable recovered: | 13 | 9 | 12 | 8 |
| % abnormal: | 7.7d | 33.3d,p,e | 16.7d,p | 87.5d,p,e,t | |
| 15.5–16.0 | no. viable recovered: | 11 | 9 | 9 | 4 |
| % abnormal: | 0.0 | 33.3d,s,p | 11.1d | 100.0d,s,p,e | |
| 16.5–17.0 | no. viable recovered: | 11 | 11 | 17 | 6 |
| % abnormal | 9.1s | 9.1s,p | 11.8s,p | 100.0d,p,e,t | |
| 17.5–18.0 | no. viable recovered: | 11 | 13 | 6 | 3 |
| % abnormal: | 0.0 | 15.4d,s | 16.7s | 33.3d | |
| 18.5–19.0 | no. viable recovered: | 11 | 10 | 9 | 4 |
| % abnormal: | 0.0 | 0.0 | 0.0 | 50.0d,t | |
Embryos resulting from an N-ras+/−; K-ras+/− × N-ras−/−; K-ras+/+ cross were dissected at various times in gestation and analyzed for their viability and gross morphological appearance and were subsequently fixed for histological purposes. Based on the parents’ genotypes, four different genotypic classes of embryos should result, each with an expected frequency of 25%. Abnormalities are denoted as follows: (d) Delayed by 0.5 day or more; (h) dilated heart and pericardial sac; (s) small for developmental stage; (p) pale and less vascularized; (e) asymmetrical development of the eye (either significantly smaller than its normal counterpart or the pigment of the eye was overgrown); (t) severe shortening of the tail.
These data suggest that N-ras and K-ras have overlapping function in one or more tissues in the developing mouse, such that mutation of both genes is necessary to uncover a phenotype in these tissues. To verify that the two genes are expressed in at least partially overlapping patterns in the mid-gestation embryo, we performed in situ hybridization as well as RT–PCR analysis. At E9.5, K-ras and N-ras exhibit broad patterns of expression (Fig. 6A–C); the genes have similar expression patterns at E11.5 as well (data not shown). Given the histological defect in the yolk sacs of these embryos (see below), we performed RT–PCR on RNA extracted from the yolk sac and demonstrated the expression of both K-ras and N-ras in this tissue as well (Fig. 6D). Although these data are not of sufficient resolution to determine expression patterns of individual cells, these results are consistent with N-ras and K-ras being expressed together in a number of cell types and are consistent with the conclusions from the genetic analysis. However, the expression data do not rule out the possibility that the double mutant phenotypes result from the combined effects of independent, subclinical defects caused by the N-ras and K-ras mutations.
Figure 6.
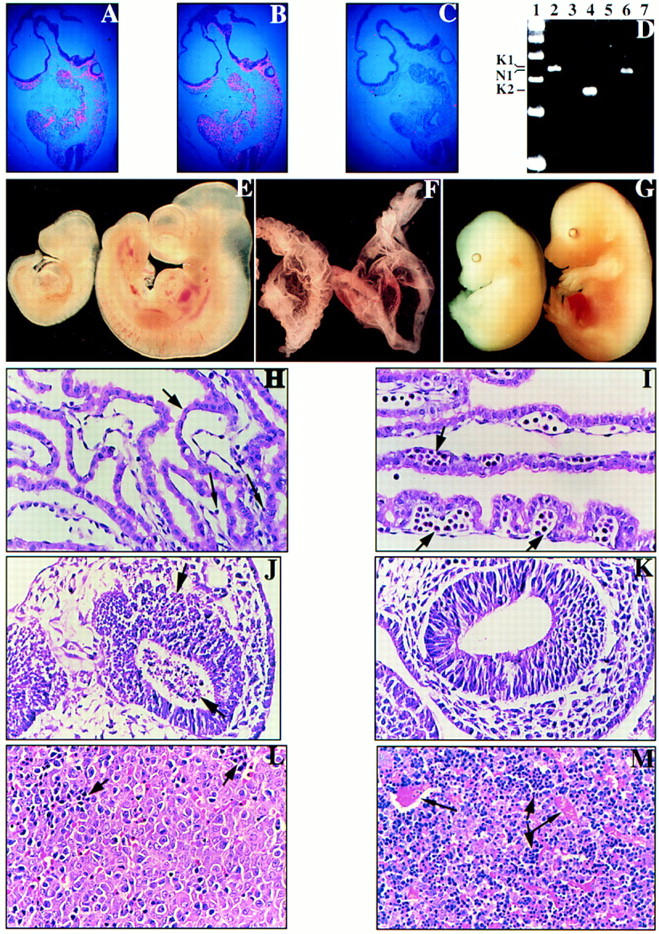
Phenotypic comparisons of N-ras−/−; K-ras+/− embryos and control littermates. In situ hybridization was performed on serial parasagittal sections of wild-type E9.5 embryos using (A) K-ras, (B) N-ras, and (C) control probes. (D) RT–PCR was performed on wild-type E9.5 yolk sacs using primer pairs specific for either K-ras [lanes 2,3 (K1); lanes 4,5 (K2)] or N-ras [lanes 6,7 (N1)] with the specific products indicated at left. RNA was omitted in the control reactions (lanes 3,5,7). A 100-bp marker is represented in lane 1. (E,F) E10.5 N-ras−/−; K-ras+/− (left) and N-ras+/−; K-ras+/− (right) embryos (E) and their respective yolk sacs (F) are shown. Note that the N-ras−/−; K-ras+/− embryo has only developed to a stage equivalent to E9.5, and is further distinguished from its normal littermate by its dilated pericardial sac and significantly reduced numbers of circulating RBCs in either the embryo or its yolk sac. The yolk sacs also take on a roughened and wrinkled appearance compared to the smooth nature of normal yolk sacs. (G) E15.5 N-ras−/−; K-ras+/− embryo (left) and a control N-ras−/−; K-ras+/+ littermate (right) are shown. The N-ras−/−; K-ras+/− embryo is developmentally delayed by 0.5 gestational days and is severely anemic and edematous. (H,I) Histological analysis of E10.5 N-ras−/−; K-ras+/− (H) and N-ras+/−; K-ras+/− (I) visceral yolk sacs. Note the strict absence of blood islands (short black arrow) and the presence of very limiting numbers of circulating primitive erythrocytes (long black arrows) in the N-ras−/−; K-ras+/− yolk sac. Otherwise, the yolk sac tissue and endothelial-lined blood vessels exhibit similar appearances in both yolk sacs. (J,K) Parasagittal sections through the forebrains of an E9.5 N-ras−/−; K-ras+/− embryo (J) and a N-ras+/+; K-ras+/− littermate control (K). Note the prevalent cell death [pyknotic nuclei (black arrows)] throughout the forebrain of the N-ras−/−; K-ras+/− embryo. This cell death extended throughout the entire embryo by E10.5. (L,M) Parasagittal sections through the fetal livers of an E15.5 N-ras−/−; K-ras+/− embryo (L) and a control N-ras−/−; K-ras+/+ littermate (M). Note the absence of blood-filled, endothelial-lined vessels (longer black arrows in M) in the N-ras−/−; K-ras+/− tissue. Moreover, the ratio of erythroblasts (dark, blue staining cells and smaller black arrows) to hepatocytes is significantly reduced in these embryos.
N-ras−/−; K-ras+/− embryos die with severe anemia
N-ras−/−; K-ras+/− embryos were indistinguishable from normal littermates at E8.5. Starting at E9.5, a higher percentage of N-ras−/−; K-ras+/− embryos were delayed by 0.5–1.0 developmental days (50% vs. ∼23% average of the other three genotypes). By E10.5, 65% of viable embryos were abnormal (Table 1). The E10.5 N-ras−/−; K-ras+/− embryo shown in Figure 6E exemplifies the phenotype observed at this stage; the mutant has arrested at ∼9.5 days of development, is markedly paler than normal littermates, and has a dilated heart and pericardial sac. Very few circulating red blood cells are seen within either the embryo proper or yolk sac (Fig. 6E,F). Furthermore, yolk sacs from these embryos exhibited a wrinkled or roughened appearance when compared to the smooth yolk sacs of littermate controls (Fig. 6F). Despite an overall growth delay in N-ras−/−; K-ras+/− embryos, many developmental processes were completed, including cardiac contraction, fusion of the allantois and chorion, rotation of the embryo, and closure of the anterior neuropore.
Histological examination of affected E10.5 N-ras−/−; K-ras+/− embryos demonstrated the near or complete absence of blood islands in their yolk sacs and significantly reduced numbers of circulating primitive erythrocytes in either the yolk sac (Fig. 6, cf. H with I) or the embryo (data not shown). However, other tissues of mesodermal origin, such as the myocardium, somites, and blood vessels, were present and appeared to develop normally. Numerous pyknotic nuclei were observed throughout the N-ras−/−; K-ras+/− embryos. Although in the yolk sac cell death appeared to be restricted to cells of mesodermal origin (data not shown), extensive cell death occurred throughout the embryo in cells derived from all three germ layers (Fig. 6, cf. J with K; data not shown). In earlier N-ras−/−; K-ras+/− embryos (E9.5), cell death was prevalent in the forebrain, and to a lesser extent along the neural axis. We cannot determine whether cell death is attributable solely to an extreme anemia and subsequent hypoxia, or whether Ras function is required for global cell survival.
A significant proportion (∼30%) of N-ras−/−; K-ras+/− embryos survived past this early developmental block. As can be seen in Table 1, almost all of these surviving embryos were abnormal and readily identifiable up until the final stages of gestation. The N-ras−/−; K-ras+/− embryo shown in Figure 6G represents some of the phenotypes observed at these later stages in development; the embryo is developmentally delayed by ∼0.5 gestational days, is small for its developmental stage, and is very anemic and edematous. Most N-ras−/−; K-ras+/− embryos were delayed by only 0.5–1.0 days in development and never exhibited the noncoordinate delay that could be observed in late stage K-ras−/− embryos. Interestingly, 22% of E13.5–18.5 N-ras−/−; K-ras+/− embryos showed an asymmetrical pattern in eye development, with only the right eye being affected, and 20% exhibited a defect in proper tail development (Table 1; data not shown). In sum, these data suggest that a critical threshold level of Ras activity must be met for development to occur normally.
Histological analysis of E15.5–16.5 N-ras−/−; K-ras+/− embryos revealed that the only defect that could be defined consistently was again in the fetal liver. As shown in Figure 6, L and M, livers from E15.5 N-ras−/−; K-ras+/− embryos have a significantly reduced ratio in the number of erythroblasts to hepatocytes as compared to normal control littermates. A high fraction of these hepatocytes were extremely vacuolated by E16.5 (data not shown). We did not observe elevated cell death in these livers, suggesting that this defect is either different than the defect in K-ras−/− fetal livers or not as severe. Moreover, there were very few circulating RBCs within the embryonic tissues (Fig. 6, cf. L and M; data not shown). Peripheral blood smears obtained from these animals confirmed that definitive erythropoiesis occurred, and that the ratio of enucleated to nucleated RBCs was appropriate for their developmental stage (data not shown). Therefore, their anemia appears to be attributable to inefficient, but apparently normal, production of definitive erythrocytes and may reflect a defect in the survival or differentiation of either the erythroblasts or a more primitive progenitor cell. As in the case of the K-ras−/− phenotype, this may be attributable to an intrinsic defect or a defect within the microenvironment to support these hematopoietic processes (see Discussion).
Other mutant combinations
In the course of these experiments, we also determined that embryos with the genotype N-ras+/−; K-ras−/− were more severely affected than K-ras−/− and N-ras−/−; K-ras+/− embryos. At E9.5 ∼40% of the N-ras+/−; K-ras−/− embryos were found dead (data not shown), whereas all of the K-ras−/− and N-ras−/−; K-ras+/− embryos examined at this developmental stage were alive (see Fig. 2, Table 1; data not shown). Also, double heterozygous mutants (N-ras+/−; K-ras+/−), although recovered at the expected frequencies throughout gestation, occasionally exhibited some of the developmental abnormalities seen more frequently in the N-ras−/−; K-ras+/− embryos (see Table 1). Finally, we have begun to examine the effects of complete lack of function of N-ras and K-ras. From double heterozygous mutant crosses, we were able to isolate double homozygous mutant (N-ras−/−; K-ras−/−) blastocyst stage embryos at approximately the expected frequency of 6%. However, when the litters were examined at E9.5, no viable double mutant embryos were recovered (data not shown). We have not determined the specific timing of this lethality.
Discussion
Analyses of mice lacking N-ras and H-ras function have demonstrated that both genes are dispensable for normal development. Here, we have shown that K-ras provides an essential function in mouse embryogenesis and have demonstrated partial functional compensation between different members of the ras gene family. On an inbred 129/Sv genetic background, K-ras−/− embryos failed in gestation between E12 and E14. Thus, although the growth and differentiation of many tissues can proceed apparently normally in the absence of K-ras function through mid-gestation, completion of embryogenesis is dependent on the activity of this gene. These mutant embryos exhibited a slight developmental growth delay starting at ∼E11, which was more pronounced in embryos late in gestation on the mixed (BL/6:129/Sv) genetic background. The lethality is likely to be attributable to hematopoietic defects, as evidenced by the overall pale color of the mutant embryos and the increased levels of cell death observed in the fetal liver (the major site of erythropoiesis in mid-gestation). However, we have shown that the K-ras function is not strictly required for the differentiation of hematopoietic progenitors in vitro or upon transplant into lethally irradiated recipients, suggesting that the apparent anemia in the mutant embryos may be caused by an abnormal hematopoietic microenvironment in the fetal liver. Interestingly, in chimeric mice composed in part of K-ras-deficient cells, the contribution of mutant cells to different hematopoietic compartments was consistently lower than to other tissues. Therefore, the absence of K-ras may cause a slight impairment in the capacity of hematopoietic cells to differentiate or survive, which is manifested when the mutant cells are in competition with wild-type cells during development and differentiation in the chimeras or in an otherwise defective fetal liver microenvironment in the homozygous mutant embryos. The phenotype of embryos mutant for both K-ras and N-ras also supports a direct role for Ras function in hematopoiesis (see below).
The requirement for K-ras in the developing mouse may reflect either a unique function of this gene not shared by H-ras or N-ras or simply the expression pattern of the different members of the gene family. Although our data and that of others (Muller et al. 1982, 1983; Leon et al. 1987), indicate broad patterns of expression of the different ras genes, it is possible that a critical cell type (e.g., within the fetal liver) expresses K-ras predominantly or exclusively. In the context of the K-ras mutation and without transcriptional up-regulation of H-ras or N-ras, such a cell would have a subthreshold level of Ras function and might fail to differentiate properly or die. In fact, upon examination of mutant MEFs in culture and various tissues in K-ras−/− embryos, we have seen no evidence for compensatory increases in the level of either N-Ras or H-Ras.
Largely on the basis of differences in post-translational modification, it has been suggested that K-Ras, and specifically the 4B isoform, may have a unique function in cell signaling. Localization of Ras proteins to the plasma membrane is critical for their biological activity (Khosravi et al. 1992; Glomset and Farnsworth 1994). Membrane association is achieved through a series of closely linked post-translational modifications that are signaled by the consensus CAAX (C, cysteine; A, aliphatic amino acid; X, any amino acid) sequence at the carboxyl terminus. In addition, N-Ras, H-Ras, and K-Ras4A are subsequently palmitoylated at upstream cysteine residues (Hancock et al. 1989). In K-Ras4B, however, these cysteine residues are substituted by a polylysine domain that serves an analogous function to that of palmitoylation (Hancock et al. 1990, 1991). This isoform has also been shown to be a substrate for geranylgeranylation as well as farnesylation in vitro, and this alternative lipid modification has been suggested to occur in vivo as well (James et al. 1995, 1996; Lerner et al. 1995). The specific association of K-Ras4B with the smg–GDP dissociation stimulator (GDS) exchange factor is believed to be mediated, at least in part, through its distinctive carboxy-terminal modifications (Mizuno et al. 1991). Thus, in certain contexts in the developing and adult animal, signal transduction may be dependent specifically on K-Ras4B. The lethality of the K-ras−/− embryos may, therefore, reflect the disruption of these pathways. This possibility could be addressed by creating a mutant allele of K-ras that blocks specifically production of the 4B isoform.
K-ras and N-ras interact genetically
Analysis of mice carrying various combinations of the K-ras and N-ras null alleles suggests strongly some form of overlapping function. For example, the majority of animals deficient for N-ras and heterozygous for K-ras (N-ras−/−; K-ras+/−) died between 10 and 12 days of gestation, with the remainder having failed later in gestation or just after birth. This result demonstrates that the normal development of N-ras-deficient mice is dependent on wild-type levels of K-ras. Similarly, the K-ras homozygous mutant phenotype was significantly worsened in embryos with only one functional N-ras allele (N-ras+/−; K-ras−/−). Therefore, in early murine development, there appears to be a critical threshold level of Ras activity, which may be achieved by various combinations of the three gene products. Beginning before organogenesis, the overall threshold appears to increase, and still later, a specific requirement for K-ras is revealed. Although we do not favor this interpretation, it is important to note that the observed compound mutant phenotypes could instead result from the additive, independent effects of the individual mutations.
Functional requirement for Ras in hematopoiesis
Various lines of evidence point to a critical role for Ras function in hematopoiesis. In addition to the apparent anemia of K-ras mutant embryos and the low contribution of K-ras−/− cells to the hematopoietic compartments of chimeric animals, we observed the absence of blood islands in the yolk sacs of the majority of N-ras−/−; K-ras+/− embryos. Before the establishment of fetal liver hematopoiesis at ∼E10.5–11.0, the earliest erythroid cells arise in yolk sac blood islands, where they produce primitive (nucleated) RBCs (Dzierzak and Medvinsky 1995; Orkin 1995). The absence of blood islands as well as the limited number of primitive erythrocytes observed in N-ras−/−; K-ras+/− yolk sacs may reflect an impairment in the ability of primitive erythrocytes or their progenitors to survive or differentiate efficiently within the yolk sac microenvironment.
Implications for oncogenesis and therapy
An estimated 30% of all human cancers carry mutations in one member of the ras family, with K-ras mutations occurring most frequently (Bos 1989; Khosravi and Der 1994). Although the predominance of K-ras mutation in certain tumor types has some correlation with the expression profile of the three ras genes in various tissues (Leon et al. 1987), our results suggest that K-Ras may have specific functions in signal transduction not shared by the other family members. Thus, mutational activation of K-Ras may result in the stimulation of a suite of signal transduction pathways common to all Ras proteins plus those that are singularly dependent on K-Ras. The further characterization of the developmental phenotype of the K-ras mutant mouse may then provide insights into the signal transduction requirements for oncogenic transformation. Moreover, the inhibition of these K-ras-specific pathways may have therapeutic value in cancer treatment.
Our results have additional implications important for the design of anticancer drugs based on the inhibition of oncogenic or normal Ras function, including the recently described farnesyltransferase inhibitors (FTIs). Our data demonstrate that inhibition of K-Ras function, at least during embryogenesis, is lethal. Moreover, analysis of the contribution of K-ras-deficient cells to various tissues in chimeric animals suggests that the gene is also important in the development or maintenance of cells at later stages of gestation or in the adult animal, including in different hematopoietic compartments and in the lung. Therefore, it is likely that drugs that inhibit Ras function nonspecifically would be highly toxic. Nevertheless, it has been reported that FTIs are well tolerated in vivo (Kohl et al. 1995). In light of our results, this lack of toxicity may be explained by the fact that K-Ras4B can be modified by geranylgeranylation as well as farnesylation (James et al. 1995, 1996; Lerner et al. 1995). In addition, K-Ras has a higher affinity for farnesyl transferase in vitro and higher concentrations of FTIs are required to suppress the transforming activity of K-Ras than H-Ras in tissue culture (Reiss et al. 1990; James et al. 1995; Lerner et al. 1995).
On the basis of the lack of phenotype in the H-ras and N-ras mutant mice, it should be possible to develop nontoxic inhibitors specific for these Ras proteins. Alternatively, it may be more sensible to screen for compounds that inhibit steps downstream of Ras in the signal transduction pathways leading to cellular transformation rather than cell growth/differentiation, should such a distinction in signaling pathways exist. Finally, our data do not address directly the requirement for K-ras in the adult, and therefore, it remains possible that inhibition of its function might be tolerated during cancer treatment. To examine what cell types in the adult are dependent on K-ras (and other ras family members), it will be necessary to develop conditional mutant alleles of these genes.
Materials and methods
Construction of targeting vector
A genomic DNA clone corresponding to K-ras was isolated from a 129/Sv genomic library using a probe derived from pHiHi3 (Ellis et al. 1981). Sequences totaling 7.9 kb surrounding exon 1 were cloned into the vector pPNT (Tybulewicz et al. 1991). Relevant restriction sites are shown in Figure 1.
Electroporation, selection of ES cell clones, and Southern blot analysis
D3 ES cells (Gossler et al. 1986) were cultured and electroporated as described by Tybulewicz et al. (1991), except that primary mouse embryonic fibroblasts were used as feeder cells. G418 (GIBCO-BRL) was used at 200 μg/ml active weight, with counterselection by gancyclovir resulting in a 13-fold reduction in the number of ES cell colonies compared with G418 selection alone. Individual clones were expanded and genomic DNAs were isolated using the method of Laird et al. (1991). DNAs were digested with BamHI plus StuI and resolved on 0.8% agarose gels. Samples were transferred onto Hybond-N (Amersham) and hybridized with the 32P-labeled probes indicated in Figure 1A using Expresshyb (Clontech). ES cell clones homozygous for the null mutation (K-ras−/−) were created by enhanced G418 selection (Mortensen et al. 1992) of the heterozygous (K-ras+/−) clones. K-ras+/− ES cell clones were plated at a density of 5 × 104 to 5 × 105 cells/p100 and 40 hr after plating the medium was supplemented with G418 at concentrations ranging from 200 μg/ml to 1.0 mg/ml active weight. Selected clones were screened by Southern blotting as shown in Figure 1. PCR analysis was also performed using Klentaq and the primer pairs indicated in Figure 1A. Primer sequences were as follows: 3′ homolog (5′-GGGATTGCAGCAATGATTTGGGGG-3′), 5′ homolog (5′-CCTGAAGATCTTACTCATCAAACTG-3′), 3′ neo (5′-AAGCTGACTCTAGAGGATCCCC-3′), 5′ neo (5′-ACGAGACTAGTGAGACGTGC-3′). PCR conditions were as recommended by the manufacturer.
Generation of chimeras
C57BL/6 blastocyst-stage embryos were injected with 10–15 K-ras+/− ES cells or 4–6 K-ras−/− ES cells and then transferred to pseudopregnant CD1 or Swiss Webster females for further development, essentially as described (Bradley 1987). Chimeric mice were mated to C57BL/6 and 129/Sv animals and agouti offspring were genotyped. Germ-line transmission of the mutant allele was detected by either Southern blot (as above) or PCR analysis of tail DNA obtained at weaning.
PCR analysis of offspring and embryos
Tail and yolk sac DNA was prepared using the method of Laird et al. (1991). All PCR reactions were performed under the following conditions: 10 mm Tris-HCl (pH 8.3), 50 mm KCl, 2 mm MgCl2, 0.001% gelatin, 0.01% NP-40, 0.01% Tween 20, 0.2 mm dNTPs, and 0.2–0.4 μm of each primer in the reaction. The annealing temperature was 60°C for both K-ras and N-ras PCR reactions and 30 rounds of amplification were performed. Primer sequences for K-ras genotyping were as follows: 5′ I0 (5′-AGGGTAGGTGTTGGGATAGC-3′), 3′ Ex1 (5′-CTCAGTCATTTTCAGCAGGC-3′), and 3′ neo (5′-ACGAGACTAGTGAGACGTGC-3′). The primer sequences for N-ras genotyping were: 5′ wild type (5′-CCCAGGATTCTTACCGAAAGC-3′), 3′ wild type (5′-CCTGTAGAGGTTAATATCTGC-3′), and 3′ mutation (5′-AATATGCGAAGTGGACCTGGG-3′). Blastocysts were isolated from superovulated females and collected into 10 μl of TE (pH 8.0). They were subsequently heated at 95°C for 5 min, treated with proteinase K (200 μg/ml) at 55°C for 90 min, and then heated at 95°C for 5 min. The sample was then split in half for K-ras and N-ras PCR analysis. PCR conditions were identical to those described above, except that 40 rounds of amplification were performed and the annealing temperature was 58°C.
Histological analysis of embryos and yolk sacs
Embryos were dissected free of decidua and uterine muscle and separated from the yolk sac. Depending on the experimental procedure, either the yolk sac or embryo was saved for genotyping by PCR. For histology, embryos (or yolk sacs) were fixed in 10% neutral buffered formalin, dehydrated in graded solutions of alcohol, embedded in paraffin, sectioned at 4–6 μm, and stained with hematoxylin and eosin (H & E).
Cell death and in situ expression analysis
Embryonic serial sections were prepared and analyzed by either TUNEL or in situ hybridization as described (Macleod et al. 1996). The K-ras and N-ras probes used in the in situ hybridization analysis have been previously described (Leon et al. 1987). The sense transcript of the K-ras probe was used as a control.
RT–PCR analysis
Poly(A)+ mRNA was made from wild-type E9.5 yolk sacs using an Oligotex direct mRNA kit (Qiagen) as per the manufacturer’s recommendations. RT–PCR was performed using an rTth reverse transcriptase RNA PCR kit (Perkin Elmer) as recommended by the manufacturer and analyzed on a 3% Metaphor (FMC) agarose gel. Primer pairs for K-ras RT–PCR analysis were as follows: 5′ K1 (5′-TGTGGATGAGTACGACC-3′) and 3′ K1 (5′-ACGGAATCCCGTAACTC-3′) yield a 338-bp specific product and 5′ K2 (5′-GTCTCTTGGATATTCTCG-3′) and 3′ K2 (5′-CCTTGCTAACTCCTGAGCC-3′) yield a 254-bp specific product. The primer pair for N-ras RT–PCR analysis was 5′ N1 (5′-AAAAGCGCCCTGACGAT-3′) and 3′ N1 (5′-CCTTGTTGGCAAGTCAC-3′) and yields a 324-bp specific product.
Hematopoietic progenitor cell analysis
E12.5 fetal livers were dissected free and collected in Iscove’s modified Dulbecco’s medium (IMDM), 2% FCS; disaggregated by passage through 23- and 26-gauge needles; counted; and plated in duplicate in α-minimal essential medium supplemented with 0.9% methylcellulose, 30% FBS, 1% BSA, 2 mm glutamine, 0.1 mm 2-mercaptoethanol, 3 U/ml of EPO, and 2% pokeweed mitogen-stimulated murine spleen cell-conditioned medium (Stem-Cell Technologies, Inc.). Colony formation was monitored at the appropriate times [colony-forming unit-erythroid (cfu-E) on days 2–3; burst-forming unit-erythroid (bfu-E) and colony-forming unit-granulocyte, macrophage (cfu-GM) on days 6–8; colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte (cfu-GEMM) on days 8–12], and subsequently colonies were picked, applied to slides, stained, and examined microscopically. Yolk sac progenitor assays were performed on E9.5 and E10.5 yolk sacs as described (Wong et al. 1986; Shivdasani et al. 1995) using the same medium as used for fetal liver colony assays. In vivo irradiation rescues were performed essentially as described (Till 1961). 129/Sv E13.5 fetal livers were isolated and disaggregated as above, counted, and injected retro-orbitally into C57BL/6 female recipient mice that had been irradiated lethally with 1200 rads of gamma irradiation. Blood samples were collected 2 weeks after injection and thereafter at 1-month intervals. Peripheral blood smears were examined and GPI analysis was performed to determine the percentage and timing of contribution by donor cells.
GPI assay
The separation and detection of GPI isoforms was performed as described (Bradley 1987). The tissues analyzed included tail, bladder, colon, cecum, small intestine, stomach, pancreas, spleen, kidney, adrenal, liver (two different lobes), atrium, ventricle, thymus, salivary gland, lung, eye, medulla oblongata, cerebrum, cerebellum, whole blood, RBC, lymphocytes and monocytes, platelets, and bone marrow. Hemoglobin assays were also performed on the RBC fractions to confirm purity and support the GPI data (Williams et al. 1994).
Preparation of embryonic tissue extracts
Embryos were collected at E12.5 in PBS and dissected free of yolk sac and placenta. Five different tissues were dissected out, frozen immediately in a dry ice/alcohol bath, and stored at −80°C until further analysis. The tissues recovered included brain, liver, heart, lung, and carcass. Yolk sacs were also retained for genotyping by PCR analysis.
Preparation of cell lysates
For MEFs, cells from three to five p100s were washed three times with cold PBS, scraped from the plates, and collected into PBS on ice. Cells were pelleted and resuspended in 1.2 ml of lysis buffer [50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5 mm MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 0.5 mm PMSF, 10 μg/ml of leupeptin, 10 μg/ml of aprotinin, and 10 μg/ml of pepstatin] using a 26-gauge needle. For E12.5 tissues, the tissues were immediately placed into 100 μl of lysis buffer and disaggregated by passage through 23- and 26-gauge needles. Extracts of equivalent genotypes were then pooled as follows: 13 for lung, heart, and liver, and 5 for brain and carcass. After this step, tissues and MEFs were treated identically. The lysates were incubated for 30 min at 4°C on a rotator, cellular debris was pelleted, and the supernatant collected. Protein concentration was determined using the Bio-Rad detergent-compatible protein assay system and equivalent amounts of protein for each of the three genotypes (+/+, +/−, and −/−) were used in immunoprecipitations.
Immunoprecipitation and Western analysis
Immunoprecipitations were performed in a final volume of 1 ml with 1–5 μg/ml of Y13-259 antibody (Santa Cruz Biotechnology, Inc.) for 2 hr to overnight at 4°C. Protein G PLUS-Agarose (Santa Cruz Biotechnology, Inc.) was added and incubated for 2 hr at 4°C. Immunoprecipitates were pelleted and washed as follows: twice with 1 ml of lysis buffer, twice with 1 ml of high salt solution [1 m NaCl, 10 mm Tris-HCl (pH 7.5), and 0.5% Triton X-100], and twice with 1 ml of KSCN wash [0.75 m KSCN, 10 mm Tris-HCl (pH 7.5), and 1% Titron X-100]. Complexes were resuspended in protein sample buffer, separated on 15% SDS-PAGE gels, and transferred onto PVDF membranes using a semidry transfer system (Owl). Western blot analysis was performed using an enhanced chemiluminescence system (Amersham) according to the manufacturer’s recommendations, with the following exceptions: blocking was done in PBS, 0.2% Tween 20, 5% nonfat dry milk; washes were done in PBS, 0.2% Tween 20; primary antibodies were incubated in blocking solution at 1 μg/ml for 2 hr at 25°C or overnight at 4°C; and the secondary anti-mouse IgG antibody conjugated to horseradish peroxidase was incubated at a 1:7000 dilution in blocking solution for 2 hr at 25°C. The primary mouse monoclonal antibodies used were K-ras (F234), N-ras (F155), H-ras (F235) (all from Santa Cruz Biotechnology, Inc.), and pan ras (Ab2 = clone F111) (Oncogene Science). Stripping of immunoblots was done by incubating the blots in 50 mm glycine (pH 2.5), 0.05% Tween 20 for 30 min at 60°C.
Acknowledgments
We wish to thank Kay Macleod, Bart Williams, Ramesh Shivdasani, and Frank McCormick for advice and assistance. L.J. wishes to dedicate her contribution to this work to Wayne Hoshor. T.J. is an Assistant Investigator at the Howard Hughes Medical Institute. This work was supported by the Searle Scholars Program/The Chicago Community Trust and a generous gift from Onyx Pharmaceuticals. L.J. was supported, in part, by a National Institutes of Health training grant to the Department of Biology at MIT. R.K. was supported by an American Cancer Society grant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tjacks@mit.edu; FAX (617) 253-9863.
References
- Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- Bos JL. The ras gene family and human carcinogenesis. Mutat Res. 1988;195:255–271. doi: 10.1016/0165-1110(88)90004-8. [DOI] [PubMed] [Google Scholar]
- ————— ras oncogenes in human cancer: A review [published erratum appears in Cancer Res 1990 Feb 15; 50 (4): 1352] Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- Bradley A. Production and analysis of Chimeric mice. In: Robertson EJ, editor. Teratocarcinomas and embryonic stem cells: A practical approach. Oxford, UK: IRL Press; 1987. pp. 113–152. [Google Scholar]
- Cai H, Szeberenyi J, Cooper GM. Effect of a dominant inhibitory Ha-ras mutation on mitogenic signal transduction in NIH 3T3 cells. Mol Cell Biol. 1990;10:5314–5323. doi: 10.1128/mcb.10.10.5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capon DJ, Seeburg PH, McGrath JP, Hayflick JS, Edman U, Levinson AD, Goeddel DV. Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature. 1983;304:507–513. doi: 10.1038/304507a0. [DOI] [PubMed] [Google Scholar]
- Chesa PG, Rettig WJ, Melamed MR, Old LJ, Niman HL. Expression of p21ras in normal and malignant human tissues: Lack of association with proliferation and malignancy. Proc Natl Acad Sci. 1987;84:3234–3238. doi: 10.1073/pnas.84.10.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Eisenmann TI, Fries HW, Troppmair J, Rapp UR. The ins and outs of Raf kinases. Trends Biochem Sci. 1994;19:474–480. doi: 10.1016/0968-0004(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Davis RJ. MAPKs: New JNK expands the group. Trends Biochem Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Duffy JB, Perrimon N. Recent advances in understanding signal transduction pathways in worms and flies. Curr Opin Cell Biol. 1996;8:231–238. doi: 10.1016/s0955-0674(96)80070-6. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, Medvinsky A. Mouse embryonic hematopoiesis. Trends Genet. 1995;11:359–366. doi: 10.1016/s0168-9525(00)89107-6. [DOI] [PubMed] [Google Scholar]
- Ellis RW, Defeo D, Shih TY, Gonda MA, Young HA, Tsuchida N, Lowy DR, Scolnick EM. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature. 1981;292:506–511. doi: 10.1038/292506a0. [DOI] [PubMed] [Google Scholar]
- Furth ME, Aldrich TH, Cordon CC. Expression of ras proto-oncogene proteins in normal human tissues. Oncogene. 1987;1:47–58. [PubMed] [Google Scholar]
- George DL, Scott AF, Trusko S, Glick B, Ford E, Dorney DJ. Structure and expression of amplified cKi-ras gene sequences in Y1 mouse adrenal tumor cells. EMBO J. 1985;4:1199–1203. doi: 10.1002/j.1460-2075.1985.tb03760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JB, Oliff A, Kohl NE. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell. 1994;77:175–178. doi: 10.1016/0092-8674(94)90308-5. [DOI] [PubMed] [Google Scholar]
- Glomset JA, Farnsworth CC. Role of protein modification reactions in programming interactions between ras-related GTPases and cell membranes. Annu Rev Cell Biol. 1994;10:181–205. doi: 10.1146/annurev.cb.10.110194.001145. [DOI] [PubMed] [Google Scholar]
- Gossler A, Doetschman T, Korn R, Serfling E, Kemler R. Transgenesis by means of blastocyst-derived embryonic stem cell lines. Proc Natl Acad Sci. 1986;83:9065–9069. doi: 10.1073/pnas.83.23.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- Hancock JF. Anti-Ras drugs come of age. Curr Biol. 1993;3:770–772. doi: 10.1016/0960-9822(93)90027-l. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991;10:4033–4039. doi: 10.1002/j.1460-2075.1991.tb04979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James GL, Goldstein JL, Brown MS. Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem. 1995;270:6221–6226. doi: 10.1074/jbc.270.11.6221. [DOI] [PubMed] [Google Scholar]
- ————— Resistance of K-rasBV12 proteins to farnesyltransferase inhibitors in Rat1 cells. Proc Natl Acad Sci. 1996;93:4454–4458. doi: 10.1073/pnas.93.9.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka T, Powers S, McGill C, Fasano O, Strathern J, Broach J, Wigler M. Genetic analysis of yeast RAS1 and RAS2 genes. Cell. 1984;37:437–445. doi: 10.1016/0092-8674(84)90374-x. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Kaibuchi K, Kishi K, Takai Y. Translocation of Ki-ras p21 between membrane and cytoplasm by smg GDS. Biochem Biophys Res Commun. 1993;190:832–841. doi: 10.1006/bbrc.1993.1124. [DOI] [PubMed] [Google Scholar]
- Khosravi FR, Der CJ. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994;13:67–89. doi: 10.1007/BF00690419. [DOI] [PubMed] [Google Scholar]
- Khosravi FR, Cox AD, Kato K, Der CJ. Protein prenylation: Key to ras function and cancer intervention? Cell Growth Differ. 1992;3:461–469. [PubMed] [Google Scholar]
- Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton K, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon J, Guerrero I, Pellicer A. Differential expression of the ras gene family in mice. Mol Cell Biol. 1987;7:1535–1540. doi: 10.1128/mcb.7.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner EC, Qian Y, Hamilton AD, Sebti SM. Disruption of oncogenic K-Ras4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J Biol Chem. 1995;270:26770–26773. doi: 10.1074/jbc.270.45.26770. [DOI] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kaibuchi K, Yamamoto T, Kawamura M, Sakoda T, Fujioka H, Matsuura Y, Takai Y. A stimulatory GDP/GTP exchange protein for smg p21 is active on the post-translationally processed form of c-Ki-ras p21 and rhoA p21. Proc Natl Acad Sci. 1991;88:6442–6446. doi: 10.1073/pnas.88.15.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens [see comments] Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, Chao S, Geisterfer LA, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller R, Slamon DJ, Tremblay JM, Cline MJ, Verma IM. Differential expression of cellular oncogenes during pre- and postnatal development of the mouse. Nature. 1982;299:640–644. doi: 10.1038/299640a0. [DOI] [PubMed] [Google Scholar]
- Muller R, Slamon DJ, Adamson ED, Tremblay JM, Muller D, Cline MJ, Verma IM. Transcription of c-onc genes c-rasKi and c-fms during mouse development. Mol Cell Biol. 1983;3:1062–1069. doi: 10.1128/mcb.3.6.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Kaibuchi K, Orita S, Ueno N, Takai Y. Different functions of Smg GDP dissociation stimulator and mammalian counterpart of yeast Cdc25. J Biol Chem. 1994;269:15085–15091. [PubMed] [Google Scholar]
- Orita S, Kaibuchi K, Kuroda S, Shimizu K, Nakanishi H, Takai Y. Comparison of kinetic properties between two mammalian ras p21 GDP/GTP exchange proteins, ras guanine nucleotide-releasing factor and smg GDP dissociation stimulation. J Biol Chem. 1993;268:25542–25546. [PubMed] [Google Scholar]
- Orkin SH. Hematopoiesis: How does it happen? Curr Opin Cell Biol. 1995;7:870–877. doi: 10.1016/0955-0674(95)80072-7. [DOI] [PubMed] [Google Scholar]
- Powers S, Kataoka T, Fasano O, Goldfarb M, Strathern J, Broach J, Wigler M. Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell. 1984;36:607–612. doi: 10.1016/0092-8674(84)90340-4. [DOI] [PubMed] [Google Scholar]
- Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell. 1990;62:81–88. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- Till JE, McCulloch EA. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Rad Res. 1961;14:213–222. [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Umanoff H, Edelmann W, Pellicer A, Kucherlapati R. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci. 1995;92:1709–1713. doi: 10.1073/pnas.92.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman DA, Therrien M, Rubin GM. The Ras signaling pathway in Drosophila. Curr Opin Genet Dev. 1995;5:44–50. doi: 10.1016/s0959-437x(95)90052-7. [DOI] [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willumsen BM, Papageorge AG, Kung HF, Bekesi E, Robins T, Johnsen M, Vass WC, Lowy DR. Mutational analysis of a ras catalytic domain. Mol Cell Biol. 1986;6:2646–2654. doi: 10.1128/mcb.6.7.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg C, Reed SI. Plugging it in: Signaling circuits and the yeast cell cycle. Curr Opin Cell Biol. 1996;8:223–230. doi: 10.1016/s0955-0674(96)80069-x. [DOI] [PubMed] [Google Scholar]
- Wong PM, Chung SW, Chui DH, Eaves CJ. Properties of the earliest clonogenic hemopoietic precursors to appear in the developing murine yolk sac. Proc Natl Acad Sci. 1986;83:3851–3854. doi: 10.1073/pnas.83.11.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi N, Kiessling AA, Cooper GM. The Ras/Raf signaling pathway is required for progression of mouse embryos through the two-cell stage. Mol Cell Biol. 1994;14:6655–6662. doi: 10.1128/mcb.14.10.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]