

Abstract
Strong evidence has indicated that protein phosphatase 2A (PP2A) is a tumor suppressor, but a mouse model for testing the tumor suppressor activity was missing. The most abundant forms of trimeric PP2A holoenzyme consist of the scaffolding Aα subunit, one of several regulatory B subunits, and the catalytic Cα subunit. Aα mutations were discovered in a variety of human carcinomas. All carcinoma-associated mutant Aα subunits are defective in binding the B or B and C subunits. Here we describe two knock-in mice expressing cancer-associated Aα point mutants defective in binding B′ subunits, one knockout mouse expressing truncated Aα defective in B and C subunit binding, and a floxed mouse for generating conditional Aα knockouts. We found that the cancer-associated Aα mutations increased the incidence of cancer by 50 to 60% in lungs of FVB mice treated with benzopyrene, demonstrating that PP2A acts as a tumor suppressor. We show that the effect of Aα mutation on cancer incidence is dependent on the tumor suppressor p53. The finding that the Aα mutation E64D, which was detected in a human lung carcinoma, increases the lung cancer incidence in mice suggests that this mutation also played a role in the development of the carcinoma in which it was discovered.
INTRODUCTION
Protein phosphatase 2A (PP2A) plays a role in many fundamental cellular processes, such as signal transduction, DNA repair, transcription, translation, and growth control (15, 24, 26, 43, 77). The basis for this multifunctionality of PP2A rests on the large number of subunits that control its phosphatase activity, substrate specificity, and subcellular localization. The trimeric holoenzyme is composed of a catalytic C subunit, a scaffolding A subunit, and one of many regulatory B subunits. The dimeric core enzyme consists of one A and one C subunit. Both forms coexist in cultured cells and in tissue (5, 30, 42). The A subunit exists as two isoforms, Aα and Aβ, which are 87% identical. The catalytic C subunit also exists as two isoforms, Cα and Cβ, which are 96% identical. The B subunits fall into four families designated B, B′, B", and B‴. The B or PR55 family has four members, the B′ family (also designated B56 or PR61) consists of five isoforms and additional splice variants, and the B" or PR72 family has four members, including splice variants. B, B′, and B" are largely unrelated by sequence except for two common regions involved in binding to the A subunit (34). The B‴ family has two members, striatin and S/G2 nuclear autoantigen (40). The combination of all subunits could give rise to over 70 different holoenzymes. In addition, the ability of PP2A to associate with approximately 150 other proteins further increases its regulatory potential (15, 24, 26, 42). Figure 1 shows a schematic diagram of the B′-containing holoenzyme whose structure has been revealed by biochemical analysis (53, 56) and X-ray crystallography (9, 19, 44, 78, 79).
Fig. 1.

Diagram of PP2A holoenzyme containing B′γ. The B′γ subunit binds to intrarepeat loops 2 to 8, and the Cα subunit binds to intrarepeat loops 11 to 15. Amino acid E64 of Aα binds to L309 of the C-terminal tail of Cα. E64 mutants are found in human lung and mammary carcinomas and are defective in B′ binding. R183 of Aα interacts with E214 of B′. R183 mutants were found in ovary carcinomas and are predicted to be defective in B′ binding.
The first indication that PP2A might be a tumor suppressor came from the finding that okadaic acid is both a tumor promoter and an inhibitor of PP2A (18, 25, 43, 69, 77). Subsequently, it was discovered that the simian virus 40 (SV40) small tumor antigen, SV40-ST, polyomavirus small tumor antigen (Py-ST), and Py middle tumor antigen (Py-MT) associate with PP2A by replacing B subunits (49, 57, 73). This association is essential for the transforming activity of the viral tumor antigens, suggesting a link between transformation and inhibition or modification of PP2A activity (3, 45, 49, 57, 73). Evidence that PP2A plays a role in human cancer came from the discovery that Aα and Aβ are mutated in a variety of human malignancies, including cancer of the breast, lung, colon, skin, and ovaries (6, 27, 70, 74). Importantly, all Aα and most Aβ cancer-associated mutant proteins are defective in binding B or both B and C subunits (7, 54, 55). Two Aα point mutants, E64G discovered in a breast carcinoma and E64D in a lung carcinoma, are strongly defective in binding B′γ subunits, whereas binding of Bα and B" is normal. These findings raised the questions of whether loss of binding B′γ to Aα causes loss of tumor suppressor activity and whether B′γ itself or the B′γ holoenzyme is a tumor suppressor (7, 55). Strong support for the idea that the B′γ holoenzyme has tumor suppressor activity comes from the finding that suppression of B′γ in human embryonic kidney cells promotes growth in agar and tumor formation in mice. In addition, it was shown that B′γ is not expressed in various human lung cancer cell lines and that overexpression of B′γ in these cells partially reverses their carcinogenicity (8). Furthermore, a reduced amount of B′γ was found in a human melanoma (14), and a truncated form of B′γ was discovered in B16 mouse melanoma cells, which have an enhanced metastatic potential compared to cells with normal B′γ (23). Thus, the presumed tumor suppressor function of the B′γ holoenzyme might be destroyed either by mutations of the Aα subunit rendering it unable to bind B′γ or by mutating B′γ.
An important discovery was that SV40-ST is required for the transformation of primary human cells in combination with SV40 large T antigen, H-ras, and telomerase (20, 80). The transforming function of SV40-ST depends on its ability to replace B′γ from the holoenzyme. Thus, it is likely that B′γ holoenzyme is a form of PP2A with tumor suppressor activity. This activity is obliterated when SV40-ST replaces B′γ on the A subunit. Conversely, overexpression of B′γ reverts SV40-ST-dependent transformation (7, 8). It has also been reported that a 50% reduction of Aα expression in HEK293 cells leads to a reduction of B′γ holoenzyme levels and increased tumor formation in immunodeficient mice (7). We reported a 10-fold reduced expression of Aα in 43% of primary human gliomas, suggesting that PP2A might be a tumor suppressor in glial cells (11).
Further evidence that PP2A is a tumor suppressor comes from the discovery that the B′α holoenzyme plays a key role in Myc degradation (2) and from the identification of an inhibitor, CIP2A, which binds to PP2A and Myc, thus preventing PP2A from dephosphorylating Myc at serine 62, a prerequisite for Myc degradation (28). Another PP2A inhibitor, SET, induced by the BCR/ABL kinase, causes marked reduction of PP2A activity in cells from chronic myelogenous leukemia patients. Importantly, restoring PP2A activity suppresses BCR/ABL activity by inducing its degradation (46, 50). Recently, evidence was provided that the Aβ-holoenzyme can also function as a tumor suppressor (58).
Mouse models are powerful tools for studying the functions of tumor suppressor proteins and their ability to counteract oncogenes (35). Based on the Aα mutations discovered in human carcinomas (6), we generated the following mouse strains: E64D/+, expressing the E64D mutation found in human lung carcinoma; E64G/+, expressing the E64G mutation found in a breast carcinoma; F5-6/+, harboring a floxed Aα allele for conditional knockout; Δ5-6/+, expressing the Aα knockout allele in the germ line. In the present report, we describe the construction and properties of these new strains. We also demonstrate that PP2A is a tumor suppressor and that the Aα mutations that were discovered in human lung and breast carcinomas cause loss of PP2A tumor suppressor activity.
MATERIALS AND METHODS
Gene targeting.
To introduce the point mutation E64D into the Aα gene (accession number ENSMUST00000007708), a targeting construct was assembled using plasmid pFloxΔtk, generously provided by Steven Hedrick (10). pFloxΔtk contains a loxP-flanked neomycin selection cassette plus a third loxP site. Using genomic DNA of R1 embryonic stem (ES) cells of strain 129, a BamHI-flanked DNA fragment was synthesized by high-fidelity PCR (PfuUltra; Stratagene) introducing an NheI site into exon 3 (Fig. 2 A and 3) and reaching about 200 bp into intron 3. This fragment was cloned into the BamHI site of pFloxΔtk. Then, a XhoI site was placed 5′ of the 5′ homology arm reaching from before exon 2 into exon 3, introducing the E64D mutation and NheI site shown in Fig. 2A and 3. Cloning this XhoI-NheI fragment into pFloxΔtk replaced the third loxP site. Finally, an XhoI site was placed 5′ of the 3′ homology arm encompassing most of intron 3, reaching into exon 9, and ending on a new SfiI site. This XhoI-SfiI fragment was cloned into the SalI and SfiI sites of pFloxΔtk, thereby destroying both the SalI and XhoI sites. The finished targeting construct was sequenced, linearized with XhoI (cutting the site 5′ of the 5′ arm, shown to the right of the vector backbone in Fig. 2A), and electroporated into R1 ES cells (strain 129) by the Transgenic Mouse Core facility at UCSD. By screening 252 neomycin-resistant colonies with primers A> and <D (Fig. 2A and B and 3), 46 clones were found to be positive (18% targeting efficiency). Clones considered for injection into blastocysts were validated in two ways. To ensure intact loxP sites, DNA surrounding the 5′ and 3′ loxP sites was amplified and sequenced. To ensure that the targeted allele expressed the intended point mutation without additional deviations from the wild-type Aα sequence, E64D mRNA was sequenced as described in the legend to Fig. 3. Chimeras were obtained from two ES cell clones. They were crossed with 129S6/SvEvTac mice, and about 1/2 of the offspring were positive with primers A> and <D, demonstrating germ line transmission of the E64Dneo targeted allele as well as viability of heterozygous mice. The neo cassette was removed by breeding with a Cre deleter mouse, prion-Cre in strain 129S6/SvEvTac, generously provided by Anthony Wynshaw-Boris (59).
Fig. 2.

Generation of E64D mice. (A) The Aα allele spans nearly 20 kb from exon 1 to exon 15 (first row, scale). The region targeted reaches from before exon 2 into exon 9 (black horizontal line, second row). Amino acid E64 of wild-type (WT) Aα is encoded in exon 3 (second row). The targeting construct (i) replaces codon E64 with codon D64, (ii) introduces an NheI site due to silent mutations, and (iii) places a neomycin (neo) cassette flanked by loxP sites (open triangles) into intron 3 (third row). ES cells were screened for the E64Dneo allele by using forward primer A (A>) and reverse primer D (<D) (fourth row). A> binds 5′ of the targeted region, while <D binds specifically to D64/NheI but not WT E64 (fourth row). E64D mice were identified with primers D> and <A (fifth row). D> binds to D64/NheI but not WT E64, while <A binds past neo. (B) PCR screening with primer pair A> and <D yields a product of 3,625 bp from the E64Dneo (Dneo) and E64D (D) alleles. (C) Primer pair D> and <A distinguishes E64Dneo (2,950 bp) from E64D mice (1,050 bp).
Fig. 3.

Correct sequence of E64D and E64G mRNAs. RNA from ES cell clones that yielded E64D and E64G mice was reverse transcribed and amplified by PCR with primers based on the reference sequence for mouse Aα mRNA, NM_016891.3 (top line, wild type [WT]). Nucleotide (nt) numbers are given in the right column. Only relevant sequence stretches are shown. The forward primer (nt 24 to 44) binds 5′ of the ATG start codon (nt 53 to 55), the reverse primer (nt 2047 to 2026) 3′ of the TGA stop codon (nt 1820 to 1822). The PCR products were cloned, and clones sensitive to NheI (GCTAGC at nt 250 to 255) were sequenced. The obtained sequences showed codon 64 as GAC for E64D and as GGC for E64G (nt 242 to 244). Stars indicate identity. Silent substitutions (nt 247 to 259) 3′ of E64D and E64G permitted design of primers that distinguish WT (primer <E), E64D (<D), and E64G (<G). The sequence of E64D mRNA from liver and kidney of E64D mice was also correct.
The targeting construct for the E64G mouse was derived from the E64D construct by site-directed mutagenesis (QuikChange II XL using PfuUltra; Stratagene), changing codon GAC for D to codon GGC for G (Fig. 3). Instead of D-specific primers <D and D>, shown in Fig. 2A and 3, corresponding G-specific primers <G and G> were used for screening (Fig. 3 and Table 1).
Table 1.
Primers used for screening targeted and transgenic mice
| Primera | Sequence | Target (nt) |
|---|---|---|
| A> | TCTCAAGTGTTCTCTGTAGTAGTGGGG | 5522–5548b |
| <E | CAAGGCCAAGAGGACC | 9147–9132b |
| <D | GAGAGCTAGCAATACG | 9147–9132 of E64Dc |
| <G | GAGCTAGCAATACGC | 9147–9131 of E64Gc |
| D> | GATGAAGATGACGTATTGCTAGCTCTC | 9121–9147 of E64Dc |
| G> | GATGAAGATGGCGTATTGCTAGCTCTC | 9121–9147 of E64Gc |
| <A | GGCAAGGCTATTGCATTGCTC | 10072–10052b |
| U> | GGGTTCATGGGTAAGACAAGC | 6633–6653b |
| <P | TGCTATACGAAGTTATGGCGC | 5′ of the 5′ loxP site |
| P> | CAGCACCAGGATATCAGGATCCG | 5′ of the 5′ loxP site |
| <I | TGCAGGGCCTTGTACTGTACTTATGAG | 12702–12676,b intron 6 |
| 6> | AGGACAAGTCTTGGCGTG | 11558–11575,b exon 6 |
| Cre> | CCAGGCGTTTTCTGAGCATACC | 102–123 of the Cre CDSd |
| <Cre | GGCAACACCATTTTTTCTGACCC | 744–722 of the Cre CDSd |
| p53> | TGGAGGAGTCACAGTCGGATATCAG | 11–35 of the p53 CDSe |
| <p53 | CCTTCTTGGTCTTCAGGTAGCTGG | 1111–1088 of the p53 CDSe |
>, forward primer; <, reverse primer.
Nucleotide range, counted from the start codon of the Aα gene (ENSMUST00000007708).
See also Fig. 3.
Based on the Cre coding sequence (accession number DQ023272). The PCR product was 642 bp.
Based on the p53 coding sequence (accession number ENSMUST00000108658). Note that the CCSP-dnp53 transgene contains only part of the p53 coding region (amino acids 1 to 14 and 302 to 390), resulting in a PCR product of only 239 bp.
To generate the conditional Aα knockout mouse, a targeting construct was designed that surrounded exons 5 and 6 with loxP sites (F5-6). The construct is based on pSniper_FL and was assembled by Ozgene by first inserting a fragment containing the 5′ loxP site and exons 5 and 6 upstream of an FRT-flanked neo cassette followed by the 3′ loxP site, then introducing the 5′ arm containing exons 3 and 4, and finally adding the 3′ arm spanning exons 7 to 11 (Fig. 4). One F5-6neo chimera was shipped to us and crossed with a Flpe deleter mouse (catalog number 003946; The Jackson Laboratory) (16). Pups were screened for the absence of the neo cassette by using primers P> and <I. Aα-floxed pups, F5-6, were then bred with the prion-Cre deleter mouse and yielded Aα knockout mice, Δ5-6.
Fig. 4.

Generation of Aα-floxed (F5-6) and Aα knockout (Δ5-6) mice. (A) The region targeted reaches from before exon 3 past exon 11 (black horizontal line, second row, Aα wild type [WT]). The targeting construct contains (i) a loxP site (open triangle) 5′ of exon 5, (ii) a neo cassette flanked by FRT sites (black triangles) 3′ of exon 6, and (iii) a second loxP site 3′ of neo (third row). F5-6neo ES cells and mice were identified by Southern blotting of EcoRV-digested DNA (Eco) with the 3′ probe (black box) (second and fourth rows). F5-6 and Δ5-6 mice were identified by PCR using primer P>, binding to the 5′ loxP site, and primer <I, binding to intron 6 past neo (fifth and sixth rows). (B) Southern blotting of EcoRV-digested DNA with the 3′ probe identifies WT (24,000 bp) and F5-6neo (Fneo) alleles (8,200 bp). (C) PCR screening with primer pair P> and <I identifies F5-6 (F) (1,836 bp), Δ5-6 (Δ) (1,032 bp), and Δ5-6/F5-6 (1,032 and 1,836 bp) mice. (D) F5-6 and Δ5-6 mice were routinely screened with primer U> binding 5′ of the targeted region and primer <P binding to the 5′ loxP site (4,507 bp).
All mice were backcrossed to FVB/NCrl (catalog number 207; Charles River Laboratories), screened for the absence of the Cre transgene, then further backcrossed, and currently are at N11 or above. All mouse work was performed in accordance with federal and local standards and was approved by the IACUC at UCSD.
PCR.
PfuUltra was used to synthesize targeting constructs and fragments for sequencing, and PfuUltra or PfuTurbo (Stratagene) was used for screenings. Screening reaction mixtures of 50 μl contained the 10× buffer supplied, a 200 μM concentration of each deoxynucleoside triphosphate, 300 nM each primer, 1.5 U of Pfu, and 60 ng tail DNA. Tail DNA was isolated using Omega Bio-Tek's tissue DNA kit. We ran one cycle at 94°C for 3 min, 39 cycles consisting of 94°C for 30 s, 56°C for 30 s, and 72°C for 8 min, followed by a final cycle at 72°C for 24 min. Repetitive sequences in the Aα gene were identified with the software tool CENSOR (girinst.org/censor) (29) and avoided as targets for primers. Primers for screening are shown in Table 1.
Embryo and yolk sac preparation.
A Δ5-6/+ female was set up with a Δ5-6/+ male in the evening, the male was removed the next morning, and females with plugs (embryonic day 0.5 [E0.5]) were used to harvest uterine horns 10 days later (E10.5). The uteri were washed in ice-cold phosphate-buffered saline (PBS), and implantation sites were individually dissected under a stereomicroscope. Embryos were removed and rinsed in PBS. When harvesting yolk sacs, the section attached to maternal tissue was cut off. Yolk sacs were rigorously rinsed in PBS to remove any maternal contaminants. Yolk sacs or embryos were placed into 200 μl of buffer TL with proteinase OB of Omega Bio-Tek's tissue DNA kit, frozen on dry ice, and stored at −20°C until further processing according to Omega's protocol for tail DNA. Due to their small mass, yolk sacs yielded DNA at low concentrations (4 to 20 ng/μl). Nonetheless, 60 ng was used per PCR screening reaction mixture (see Fig. 6, below).
Fig. 6.
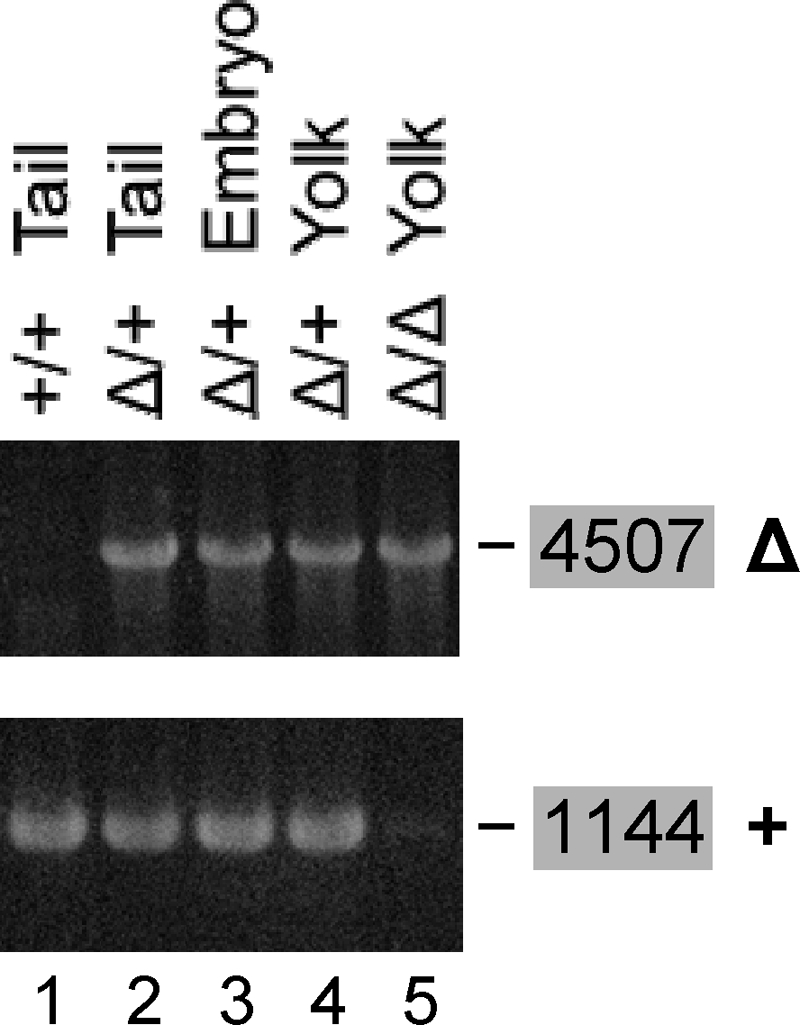
Lethality of the Δ5-6/Δ5-6 genotype. Inbreeding of Δ5-6/+ mice yielded +/+ and Δ5-6/+ (Δ/+) offspring (lanes 1 and 2) but no Δ5-6/Δ5-6 pups. Δ5-6/+ inbreeding yielded Δ5-6/+ embryos and corresponding yolk sacs at E10.5 (lanes 3 and 4) but no Δ5-6/Δ5-6 embryos. Several unusually small implantation sites contained no embryos but contained Δ5-6/Δ5-6 (Δ/Δ) yolk sacs at E10.5 (lane 5). The Δ5-6 allele was identified with primer pair U> and <P (4,507 bp). Since <P has no target in the + allele, the +/+ genotype yields no product (lane 1). The + allele was identified with the pair 6> and <I (1,144 bp). Since 6> has no target in the Δ5-6 allele, the Δ5-6/Δ5-6 genotype yields no product (lane 5).
Tamoxifen-inducible Cre mouse.
Hayashi and McMahon generated a transgenic mouse (CreER) ubiquitously expressing Cre fused to a mutant estrogen receptor (ER) that binds tamoxifen (TM) but not endogenous estrogen (21). CreER is restricted to the cytoplasm until application of TM leads to nuclear translocation, enabling recombination to be carried out by Cre, which converts the F5-6 allele into the Δ5-6 allele. The CreER mouse was obtained from The Jackson Laboratory (catalog number 004682). TM (catalog number T5648; Sigma-Aldrich) was dissolved in corn oil to 25 g/liter and stored as 1-ml aliquots in liquid N2. TM was injected intraperitoneally once per day on five consecutive days. Wild-type mice survived 0.1 mg TM per g of body weight, barely survived 0.2 mg/g, and succumbed to 0.3 mg/g. Therefore, 0.1 mg TM/g of body weight was chosen for the experiments, about 1/2 the maximum dose reported by Hayashi and McMahon.
Organ lysis and Western blotting.
Mice were sacrificed using CO2. Organs were removed, rinsed in PBS, and placed into ice-cold PBS until all organs were harvested. Approximately 40 mg was cut off, weighed, placed into a 5-ml round-bottom tube, and stored on dry ice until all organs were weighed. Per mg of organ, 50 μl of TCEP-SB was added (25 mM TCEP-HCl [catalog number 20490; Pierce], 2% SDS, 5% glycerol, 50 mM Tris [pH 6.9]). TCEP-SB permits an easy protein assay (see below), but samples turn acidic upon long-term storage or repeated freeze-thawing. The organs were homogenized at 8,000 rpm for 1 min by using a tissuemizer, Ultra-Turrax T25, with a rotor-stator generator of 8 mm outer diameter. Aliquots (1,200 μl) of the homogenates were pipetted into microcentrifuge tubes and placed onto dry ice until frozen, then heated to 95°C for 4 min. Insolubles were spun out at 14,000 × g and 25°C for 5 min, then 805 μl of the supernatants was transferred into new tubes and 5 μl was diluted in 1,000 μl water for absorbance determination at 200 nm (A200). Bovine serum albumin (BSA) amounts of 2 to 32 μg in 5 μl TCEP-SB and 1,000 μl water were used to generate standard curves. All samples were adjusted to 1.5 g/liter, 3 μl saturated BPB was added per 100-μl sample, and 10 to 20 μg was loaded onto SDS-PAGE gels. Proteins were transferred onto polyvinylidene difluoride membranes (Immobilon-P; Millipore). Membranes were blocked in TBST (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.2% Tween 20) containing 3% BSA (BP1600; Fisher). Primary antibodies were diluted in TBST with 3% BSA and 0.03% sodium azide. After use, they were stored at 4°C and reused numerous times. The blocking solution was used again with horseradish peroxidase-labeled secondary antibodies and then discarded. The membranes were developed by enhanced chemiluminescence using SuperSignal West Pico (Pierce).
The following antibodies were used: rat monoclonal antibody 6F9, recognizing human and mouse Aα but not Aβ (30); anti-B′δ (A301-100A; Bethyl); anti-B′α (A300-967A; Bethyl); rabbit anti-Bα (30); anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 14C10 (2118; Cell Signaling); anti-β-actin (A5441; Sigma).
Quantitation of Western blot bands was achieved by loading dilution series of samples. Bands in question were compared to bands of the dilution series by using appropriate film exposures. A dilution series was made for each antibody.
Organ extraction and immunoprecipitation.
For organ extraction, approximately 30 mg of organ was cut off, weighed, frozen in liquid N2, transferred into an ice-cold mortar, and ground with an ice-cold pestle. Homogenization buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol [DTT], 1× HALT protease inhibitor cocktail, 1× HALT phosphatase inhibitor cocktail, 1 μM okadaic acid) was added (30 μl per mg of organ). DTT was added fresh from a 1 M stock stored in liquid N2, and the HALT cocktails were added fresh from 100× stocks stored at 4°C (catalog numbers 87785 and 78420; Pierce). Okadaic acid (catalog number O-5857; LC Labs) was added fresh from a 1 M stock stored in liquid N2. After additional grinding, the homogenate was pipetted into a microcentrifuge tube and placed on ice. A 50-μl aliquot was centrifuged at 14,000 × g and 4°C for 3 min, and 10 μl of the supernatant was mixed with 2,000 μl Coomassie Plus reagent (catalog number 23236; Pierce) before the A595 was determined. The homogenates of different genotypes were adjusted to the same protein concentration, 2.5 g/liter for +/+ and E64D/E64D or +/+ and E64G/E64G lung and 1.0 g/liter for +/+ and E64D/E64D or +/+ and E64G/E64G cortex. Aliquots of 600 μl were extracted by adding Triton X-100 to 0.5% from a 10% stock and incubating on ice for 15 min. Following centrifugation at 14,000 × g and 4°C for 3 min, supernatants were transferred into fresh tubes. Aliquots of 60 μl were added to antibody beads prepared beforehand as follows.
GammaBind Plus Sepharose beads (catalog number 17-0886-02; Pharmacia) were coupled to monoclonal antibody 6F9 as described previously (60). Beads were washed using homogenization buffer without inhibitors but with Triton X-100, then resuspended in homogenization buffer with inhibitors and Triton X-100, aliquoted to 30 μl of settled beads per tube, and drained. After addition of 60 μl extract mentioned above for a total of 90 μl, immunoprecipitations proceeded for 2 h at 4°C while rotating the tubes slowly along their axes (rotator model 151; Scientific Industries). Subsequently, the beads were pelleted at 300 × g and 4°C for 1 min. Supernatant (45 μl) was removed, mixed with 15 μl 4× DTT-SB, and heated to 95°C for 4 min. The beads were then washed with homogenization buffer containing 0.1× inhibitors without Triton X-100, drained, and suspended in 30 μl of 2× plus 60 μl of 1× DTT-SB for a total of 120 μl, which was heated to 95°C for 4 min. Aliquots of 10 μl were loaded onto SDS-PAGE gels. With the indicated volumes, equal loading volumes of depleted supernatant and immunoprecipitate represent the same fraction of the corresponding homogenate, so that band intensities on the Western blot are quantitatively comparable.
Tumor induction, detection, and evaluation.
PP2A mutant mice were backcrossed to strain FVB, because FVB mice have a predisposition for lung cancer (20% incidence at 18 months of age) (36, 71). When used for the cancer studies, they were at the N7 generation (99.2% FVB). Transgenic FVB mice expressing a dominant negative mutant of p53 (dnp53) under the control of the lung-specific promoter for the Clara cell-specific protein (CCSP) were generously provided by Kam Meng Tchou-Wong (71). One E64D/+; dnp53 male was mated 2 to 3 times with 15 Δ5-6/+ females in order to obtain approximately 25 pups of each resulting genotype. At 5 weeks of age, all pups received one intraperitoneal injection of 0.1 mg benzo[a]pyrene (BP; catalog number B1760; Sigma) per g of body weight (71, 75). One g of BP was dissolved (suspended) in 50 ml tricaprylin (catalog number T9216; Sigma) to 0.02 mg/μl at 37°C with gentle shaking over several hours and then spun once at 2,000 × g for 3 min. The supernatant was mixed, split into aliquots of 800 to 1,000 μl, and stored in liquid N2. Each aliquot was used only once to inject a litter, e.g., 100 μl per 20-g mouse, and only aliquots of the same BP preparation were used for a particular study group of mice. We harvested when 40 to 50% of wild-type mice had lung tumors, in order to allow for a doubling of cancer incidence in the mutant mice. For test mice, the appropriate time point was determined to be 10 months after BP application for mice without dnp53 and 6 months after BP for mice with dnp53. Necropsies rarely revealed cancers other than lung cancer. Lungs were inflated with 10% formalin and inspected under a stereomicroscope for surface tumors. The smallest tumors detected were 0.1 mm in diameter, and the largest were 10 mm. Tumor diameters were determined by using a scale in the eye piece with 100 distance markings, calibrated with a ruler to 4 mm. Tumors were processed for paraffin embedding by the UCSD Histology Core. Sections of 4 μm were stained with hematoxylin and eosin for evaluation by Nissi Varki.
Statistics.
The significance of the weight loss of mice injected with tamoxifen was assessed using the 1-tailed paired t test (Excel). To evaluate Mendelian ratios, the chi-square test for comparing observed frequencies with expected frequencies was applied (Graphpad). To compare the increase in cancer incidence in mutant strains versus wild-type mice, the 1-tailed chi-square test for analyzing 2 × 2 contingency tables was used (Graphpad). The average number of tumors per tumor-bearing mouse was calculated after outliers were excluded (Graphpad). Higher tumor numbers in mutant strains versus wild-type mice were evaluated with the 1-tailed 2-sample t test for unequal variances (Excel).
RESULTS
Generation of E64D and E64G mice.
To generate mice expressing cancer-associated mutations of the Aα subunit of PP2A, we employed gene targeting by homologous recombination in ES cells. E64D point mutant mice were constructed as shown in Fig. 2. To generate E64G mice, a single nucleotide of the E64D targeting vector was exchanged by site-directed mutagenesis to obtain the E64G vector (cf. Fig. 3). Otherwise, E64G mice were made the same way as E64D mice.
Since Western blotting cannot distinguish wild-type Aα from E64D and E64G mutant proteins, we sequenced mRNA to prove that E64D and E64G mice express the intended mutation. RNA was isolated from E64Dneo/+ and E64Gneo/+ ES cells and from liver and kidney of E64D/+ mice. The RNA was reverse transcribed, cDNA was amplified, and PCR products were cloned. E64D and E64G clones were distinguished from wild-type clones by cleaving the NheI site that was introduced near D64 and G64 (Fig. 2A and 3). Of 40 clones derived from E64Dneo/+ ES cells, 23 were cut by NheI (data not shown), demonstrating that E64D and wild-type mRNA were expressed at similar levels in heterozygous ES cells, i.e., that the neo cassette did not generate a hypomorphic E64Dneo allele. Similar results were obtained for E64Gneo/+ ES cells. Not unexpectedly, the E64D allele without neo was expressed as efficiently as the wild-type allele in E64D/+ liver and kidney (data not shown). Complete sequencing of several E64D and E64G clones verified the intended mutations as well as the absence of unintended coding mutations (Fig. 3).
Generation of Aα-floxed and knockout mice.
A single-nucleotide insertion was found in the Aα gene of a human breast carcinoma, generating a frame shift and a stop codon (6). The protein encoded by the mutated gene consists of amino acids 1 to 170 of wild-type Aα followed by 6 amino acids (PEPVLR-stop) encoded by an alternate reading frame. We called this mutant Δ171-589. Based on previous studies, this truncated protein binds neither B nor C subunits and is functionally equivalent to an Aα knockout (55). Since the mutation lies near the 5′ end of exon 5, a nearly identical knockout mutation can be constructed and made conditional by floxing exons 5 and 6, F5-6. In the presence of Cre, the floxed exons are deleted, and exon 4 is joined with exon 7 out of frame, which generates knockout allele Δ5-6. The knockout allele encodes deletion mutant Δ168-589 which consists of amino acids 1 to 167 of wild-type Aα followed by 21 amino acids (HSRKQWGLRSPRQIWCLPSRT-stop) encoded by the alternate reading frame. The Aα-floxed and knockout mice were constructed as shown in Fig. 4.
Viability of mutant mice.
All heterozygous mice, E64D/+, E64G/+, F5-6/+, and Δ5-6/+, appeared healthy and were fertile, and the females nursed their pups. To check whether homozygous point mutants are viable, E64D/+ and E64G/+ mice were inbred and yielded E64D/E64D and E64G/E64G pups which grew up normally (Fig. 5 A and B). When crossing E64D/+ and E64G/+ with Δ5-6/+ mice, Δ5-6/E64D and Δ5-6/E64G pups were born and grew up normally (Fig. 5C and D). Subsequently, E64D/E64D, E64G/E64G, Δ5-6/E64D, and Δ5-6/E64G mice were inbred. All proved fertile, and the females nursed their pups.
Fig. 5.

Viability of E64D/E64D, E64G/E64G, Δ5-6/E64D, and Δ5-6/E64G mice. (A) Inbreeding of E64D/+ mice yields +/+, E64D/+, and E64D/E64D progeny. Tail DNA was screened for the E64D (D) allele with primer pair A> and <D (3,625 bp) and for the wild-type (WT) allele with primer pair A> and <E (3,625 bp) (cf. Fig. 2 and 3). +/+ mice showed the + band only, E64D/+ mice showed the D and the + band, and E64D/E64D mice showed the D band only. Lane 4 shows that A> and <D are positive on E64G/E64G DNA, since <D matches both the E64D and E64G alleles (Fig. 3). (B) Inbreeding of E64G/+ mice yields +/+, E64G/+, and E64G/E64G progeny. The E64G (G) allele was identified with primer pair A> and <G (3,623 bp), and the WT allele was identified with pair A> and <E (3,625 bp). Lane 4 shows that A> and <G are negative on E64D/E64D DNA, since <G is specific for the E64G allele (Fig. 3). (C) Crossing E64D/+ with Δ5-6/+ mice yields all four genotypes expected. The E64D allele was identified with the pair A> and <D, and the Δ5-6 (Δ) allele was identified with the pair U> and <P (4,507 bp) (Fig. 4). (D) Crossing E64G/+ with Δ5-6/+ mice yields all four genotypes expected. The E64G allele was identified with the pair A> and <G, and the Δ5-6 allele was identified with the pair U> and <P.
Requirement of Aα for embryonic development.
When inbreeding Δ5-6/+ mice, no Δ5-6/Δ5-6 pups were obtained among over 100 offspring, demonstrating lethality of Δ5-6/Δ5-6 embryos. To investigate at what stage lethality occurs, embryos were harvested at E10.5. Among 41 embryos recovered, we detected 13 +/+ and 28 Δ5-6/+, but no Δ5-6/Δ5-6 embryos, implying that development is blocked before E10.5. In addition to the 41 appropriately sized implantation sites, we noticed 9 smaller ones (one-half to two-thirds of normal size) not containing embryos but yolk sacs, of which one was +/+, three Δ5-6/+, and five Δ5-6/Δ5-6 (Fig. 6). The presence of Δ5-6/Δ5-6 yolk sacs suggests that Δ5-6/Δ5-6 embryos developed at least until implantation, which occurs around E5. Yolk sacs are formed around E7.5 (51). The persistence of Δ5-6/Δ5-6 yolk sacs suggests either that yolk sac cells can live without Aα, that maternal Aα is still present at E10.5, or that Aβ, which is 87% identical to Aα, can replace Aα in yolk sac cells but not in embryos.
Requirement of Aα in adult mice.
To test whether Aα is required in adult mice, we generated animals containing one Δ5-6 allele, one F5-6 allele, and a transgene (CreER) expressing Cre fused to a mutant ER that binds TM (21). Upon application of TM, CreER translocates to the nucleus, where Cre converts the F5-6 allele into the Δ5-6 allele. Four Δ5-6/F5-6; CreER mice died on day 6 after the first TM injection (day 1), while all nine Δ5-6/+; CreER control mice survived. This demonstrated that Aα is continuously required in adult animals. Figure 7 A shows the relative average body weights of the mice. Both genotypes displayed an initial drop in weight by approximately 5%, from which the controls recovered while the floxed mice continued to lose weight to a maximum of 19% by the time of death (day 6). They also displayed a hunched back and had difficulty walking. The initial drop in body weight was probably due to TM toxicity (see Materials and Methods), while the continued loss of weight in the floxed mice was likely the result of Aα loss. Western blotting of several organs showed, indeed, a drop in Aα levels to 30% in pancreas, 70% in heart, 80% in lung, and 80% in cortex (Fig. 7B). These reductions are based on comparing a Δ5-6/F5-6; CreER mouse with a Δ/+; CreER mouse. Since Δ5-6/+ mice contain only approximately half as much Aα as +/+ mice (except for brain), the Aα levels in the floxed mouse compared to a +/+ mouse dropped in pancreas to 15%, in heart to 35%, and in lung to 40%. Similar reductions were seen in spleen, liver, and kidney (data not shown). Despite the drops in Aα, histopathology did not provide hints for the cause of death.
Fig. 7.

Lethality of knockout of Aα in adult mice. Mice were generated containing one Δ5-6 (Δ) knockout allele, one F5-6 (F) floxed allele, and a transgene (CreERTM) expressing Cre ubiquitously and inducible by TM. (A) Nine control mice, Δ5-6/+; CreERTM, and four floxed littermates, Δ5-6/F5-6; CreERTM, ages 2 to 4 months, were injected with TM on days 1 through 5 and weighed daily. All four Δ5-6/F5-6; CreERTM mice died on day 6, while the nine control mice survived. The error bars represent the 95% confidence intervals. The weight losses of the floxed mice were significant, with P values of 0.02 (day 1 versus 2), 0.0002 (day 4 versus 5), and 0.006 (day 5 versus 6). (B) One control and one floxed mouse were treated as described for panel A and sacrificed on day 6, at which time the floxed mouse had a hunched back and difficulty walking. The weights of both mice were similar to the averages shown in panel A. Several organs were Western blotted for Aα (6F9). GAPDH was used as a loading control.
Reduced Aα levels in Δ5-6/+ organs, except brain.
To check whether Aα levels are reduced in Δ5-6/+ mice due to expression of only one allele, Western blotting was performed. As shown in Fig. 8, Δ5-6/+ mice contained approximately 50% Aα in mammary gland, liver, kidney, heart, and lung compared to +/+ controls. Similar results were obtained with salivary gland, spleen, pancreas, adrenal gland, thymus, and skeletal muscle (data not shown). This confirms the successful knockout of one Aα allele in Δ5-6/+ mice. Surprisingly, Aα levels were not reduced in cerebellum, cortex, and brain stem (brain stem data not shown) of Δ5-6/+ mice. Thus, mouse brain has a mechanism for maintaining Aα at high levels when the gene dosage is reduced. Our finding that mouse brain has 2- to 5-fold more Aα than other organs (Fig. 8 and 9) is consistent with reports for rat brain (68).
Fig. 8.

Reduced Aα levels in Δ5-6/+ mice. Various organs of Δ5-6/+ (Δ/+) mice and +/+ control littermates were Western blotted for Aα using 6F9 antibody. All organs except brain showed approximately 50% Aα in the heterozygous knockout mice compared to +/+ controls. GAPDH and β-actin were used as loading controls. MG, mammary gland; Cereb, cerebellum. The results are representative of three experiments.
Fig. 9.
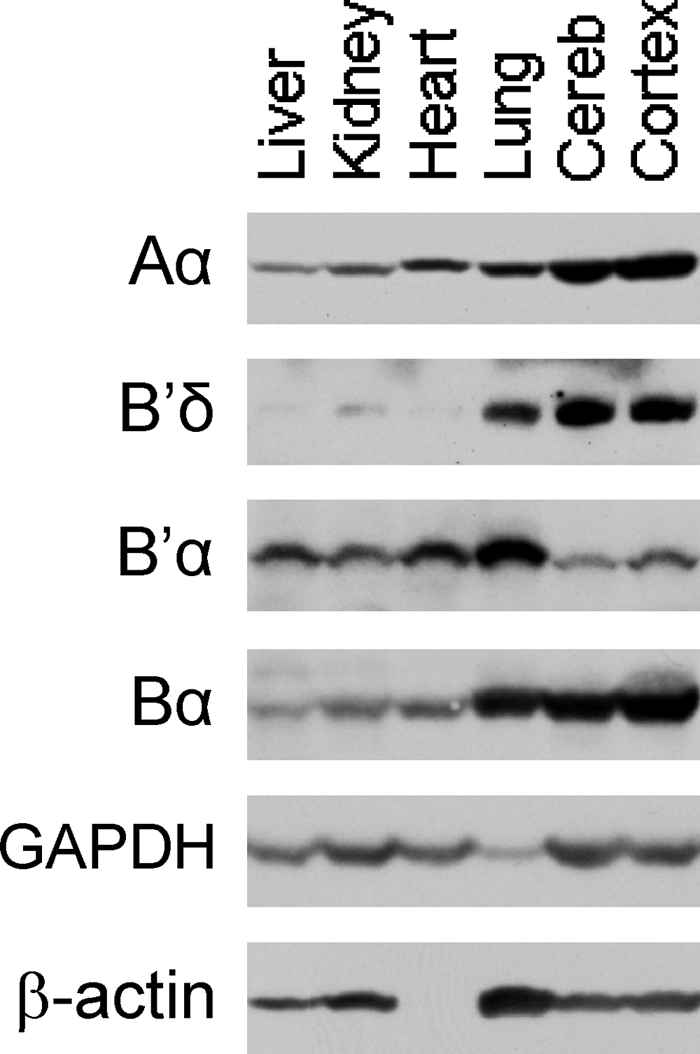
PP2A subunit levels in wild-type mouse organs. Various organs were Western blotted for Aα, B′δ, B′α, and Bα. GAPDH and β-actin were used as loading controls. Cereb, cerebellum. The same results were obtained in two separate experiments.
Organ-specific expression of B′α and B′δ.
To check whether E64D and E64G are defective in binding B′ in mice, we first determined which organs express high levels of B′α and B′δ. These two isoforms were chosen because specific antibodies were available. As demonstrated in Fig. 9, B′α was highest in lung, followed by liver, kidney, heart, cerebellum, and cortex. B′δ was highest in cortex and cerebellum followed by lung. Based on these findings, we selected lung and cortex for coimmunoprecipitation experiments (see below). Consistent with our data, high levels of B′δ mRNA were found by Northern blotting in mouse and rabbit brain (12, 38, 47).
Reduced binding of B′α and B′δ to E64D and E64G.
To investigate whether E64D and E64G are defective in binding B′ subunits in mice, as expected from in vitro and tissue culture data, we carried out immunoprecipitations with organ extracts from E64D/E64D and E64G/E64G mice. We chose the lung, which is rich in B′α and B′δ, and cortex, which is rich in B′δ (Fig. 9). Monoclonal antibody 6F9 was used, which recognizes the N terminus of Aα and precipitates all holoenzymes, including B′ holoenzymes (30). The antibody, coupled to Sepharose beads, was used in excess and precipitated nearly all Aα from lung and cortex extracts of either +/+, E64D/E64D, or E64G/E64G mice. Figure 10 A, row a, lanes 2 and 10 [abbreviated Fig. 10A(a)2 and 10] shows that after depletion only 5% of Aα remained in lung supernatant from wild-type animals. The same result was obtained with lung extracts from E64D/E64D mice [Fig. 10A(a)6] and E64G/E64G mice [Fig. 10A(a)14]. As expected, all depleted wild-type and mutant Aα (95%) was found in the immunoprecipitates [Fig. 10A(a)4, 8, 12, and 16]. Nearly identical results were obtained with cortex from +/+, E64D/E64D, and E64G/E64G mice [Fig. 10B(a)].
Fig. 10.

Reduced binding of B′δ and B′α to E64D and E64G. Extracts of lung (A) and cortex (B) of +/+, E64D/E64D (D/D), and E64G/E64G (G/G) mice were used for immunodepletion of Aα using 6F9. Both the depleted supernatants (Sup) and the immunoprecipitates (IP) were Western blotted for Aα, B′δ, B′α (A), and Bα (B). Controls (Con) are immunodepletions for which we used beads without antibody. The first rows of percentages (below the blots) compare subunit amounts between supernatants and IP from the same organ. The second rows of percentages (in bold italics) compare input amounts of subunits between wild-type and mutant organs. GAPDH was used as a loading control. Coimmunoprecipitation of B′α and B′δ with E64G from extracts of lung, cortex, and liver (liver not shown) was carried out twice in independent experiments using different mice. Coimmunoprecipitation of B′α and B′δ with E64D from extracts of lung, cortex, and kidney (kidney not shown) was carried out once. In addition, quantitatively similar binding defects of B′α and B′δ to E64D were observed when we used extracts from +/+ and E64D/E64D mouse embryo fibroblasts from embryonic day 13.5 (data not shown).
Most importantly, immunoprecipitation clearly demonstrated that the E64D and E64G mutant proteins are defective in binding B′ subunits. In +/+ lung extract precipitated with 6F9, 30% of B′δ remained in the supernatant [Fig. 10A(b)2], and all codepleted B′δ (70%) appeared in the precipitate [Fig. 10A(b)4]. On the other hand, with E64D/E64D extract, 60% of B′δ remained in the supernatant [Fig. 10A(b)6], and only 40% was precipitated [Fig. 10A(b)8]. In comparison to E64D, E64G was more defective. As much as 80% of B′δ was not codepleted from E64G/E64G lung extract [Fig. 10A(b)14], and only 20% coprecipitated [Fig. 10A(b)16]. Very similar results were obtained with cortex extracts, in which 30% of B′δ remained in the +/+ supernatant [Fig. 10B(b)2], 50% in the E64D/E64D supernatant [Fig. 10B(b)6], and 75% in the E64G/E64G supernatant [Fig. 10B(b)14]. Accordingly, 70% of B′δ coprecipitated with Aα [Fig. 10B(b)4], 50% with E64D [Fig. 10B(b)8], and only 25% with E64G [Fig. 10B(b)16]. These data are consistent with previous reports by us and others that in vitro and in tissue culture cell extracts E64G is more defective than E64D in B′ binding (7, 55, 84).
Furthermore, we found that B′α is a weak binder of wild-type Aα and a very weak or nonbinder of E64D and E64G. A large fraction, 60%, was not codepleted from +/+ lung supernatant [Fig. 10A(c)2], and of the 40% removed, only one-half of that ended up in the final precipitate (20%) [Fig. 10A(c)4]. Presumably, some B′α got lost during the washing of the precipitate. In E64D/E64D and E64G/E64G lung extracts, 95% of B′α remained undepleted [Fig. 10A(c)6 and 14], and no precipitation was observed [Fig. 10A(c)8 and 16]. Figure 10 also shows that there was much less B′α in E64G/E64G lung (10%) [Fig. 10A(c)13] than in E64D/E64D (70%) [Fig. 10A(c)5] and +/+ lung (100%) [Fig. 10A(c)1 and 9] (lower row of percentages). Presumably, this was due to degradation of B′α in E64D/E64D and E64G/E64G lung. This view is consistent with reports in the literature demonstrating that the expression levels of PP2A subunits are strictly coordinated, such that knocking down expression of one subunit type (e.g., the A subunit) leads to ablation of the other types (B and C) (33, 63, 67, 72). For the same reason, we assumed that there was less B′δ in E64G/E64G (30%) [Fig. 10A(b)13] and in E64D/E64D lung (60%) [Fig. 10A(b)5] than in +/+ lung (100%) [Fig. 10A(b)1] (lower row of percentages). Moreover, there was only 40% E64G in E64G/E64G lung [Fig. 10A(a)13] compared to 100% Aα in +/+ lung [Fig. 10A(a)9], although the E64G allele is transcribed as efficiently as the wild-type allele (see above). It is possible that 60% of E64G was degraded because B′ subunits do not bind to it and there are not enough B and B" subunits to compensate for B′. Thus, E64G/E64G mice are viable with only 40% Aα in the lung. By comparison, knockdown of Aα levels to less than one-third of the wild-type level causes cell death (7, 33, 63, 67).
We have not tested whether E64D and E64G in mouse tissues are as defective in binding B′γ as they are in binding B′δ and B′α. However, there is evidence that this is likely the case, because the central Aα binding region in all B′ subunit isoforms is highly conserved (34), suggesting that Aα mutations that affect binding of one B′ isoform will affect all isoforms (this does not exclude that the binding affinities may differ between B′ isoforms). In addition, it has been shown in tissue culture that E64D and E64G are defective in B′α, B′γ, and B′δ binding (7). This argues that E64D and E64G are also defective in the mouse, since all binding properties of Aα mutants that have been observed in vitro or in tissue culture are qualitatively and quantitatively similar in mouse organs.
To further confirm that E64D and E64G are specifically defective in B′ subunit binding, we tested their ability to bind Bα in extracts of cortex. As shown in Fig. 10, Bα codepleted equally well with Aα, E64D, and E64G. In each case, 50% remained in the lysate [Fig. 10B(c)2, 6, 10, and 14], and 10 to 20% coprecipitated [Fig. 10B(c)4, 8, 12, and 16]. These data are consistent with previous in vitro and tissue culture data and further confirm that E64D and E64G are specifically defective in B′ binding.
Increased lung cancer incidence in E64D/+, Δ5-6/+, and Δ5-6/E64D mice.
We examined whether the E64D mutation that was found in a human lung carcinoma promoted lung carcinogenesis in E64D knock-in mice. Since the authors who discovered E64D did not report the state of the second Aα allele (6), we tested E64D in the heterozygous state as well as in combination with the Δ5-6 knockout allele. To this end, E64D/+ and Δ5-6/+ mice were crossed, yielding +/+, E64D/+, Δ5-6/+, and Δ5-6/E64D mice for the cancer study. Since FVB mice have a predisposition for lung cancer (20% incidence at 18 months of age) (36, 71), we reasoned that if the E64D mutation abrogates the tumor suppressor activity of PP2A, it would preferentially promote carcinogenesis in the lung rather than in other organs. To investigate whether PP2A suppresses carcinoma formation induced by benzopyrene, a powerful carcinogen in cigarette smoke, all mice were treated with benzopyrene at 5 weeks of age.
As shown in Fig. 11 (black columns), the cancer incidence in E64D/+, Δ5-6/+, and Δ5-6/E64D mice was 72%, 77%, and 75%, respectively, representing a 50 to 60% increase over wild-type mice (for which the incidence was 48%). These data represent the first demonstration that PP2A acts as a tumor suppressor in mice. They also indicate that PP2A suppresses lung cancer induced by benzopyrene (Discussion).
Fig. 11.

Increased lung cancer risk in Aα mutant mice. E64D/+;dnp53 and Δ5-6/+ mice were crossed to obtain the eight genotypes indicated. dnp53 is a dominant-negative fragment of p53. All mice received BP at 5 weeks of age. Mice without dnp53 were harvested 10 months later (black columns), and mice with dnp53 were harvested 6 months after BP application (grey columns). The table on the bottom shows the total number of mice, the number of mice with tumors, lung tumor incidences, and the average numbers of tumors (Tu) per tumor-bearing mouse. The higher tumor numbers in Δ/D (2.5) and Δ/D;dnp53 (2.8) have P values of 0.05 and 0.01, respectively, compared to the wild type. The P values above columns 2 to 4 are for the incidences in the mutant mice compared to the wild-type mice. D, E64D; Δ, Δ5-6. Note that neither tumor numbers nor sizes were comparable between mice with and without dnp53, since they were harvested at different times.
Tumor suppressor activity of PP2A depends on p53.
Loss of the p53 tumor suppressor function occurs in over 50% of human lung carcinomas, either by direct mutation of the p53 gene (22) or indirectly through alteration of pathways that control p53. In contrast, loss of p53 tumor suppressor activity is rare in genetically or chemically induced murine lung carcinomas (37). Nevertheless, mice have been a useful model for investigating the role of p53 in lung carcinogenesis (71, 75, 76, 82). We asked whether loss of PP2A tumor suppressor activity enhances the oncogenic effect of p53 inactivation. To answer this question, we used the system developed by Tchou-Wong et al. (71), who demonstrated that elimination of p53 tumor suppressor activity by expression of the dominant negative p53 mutant, dnp53 (61), doubles the lung cancer incidence in FVB mice. We generated wild-type, E64D/+, Δ5-6/+, and Δ5-6/E64D mice expressing dnp53 and treated with benzopyrene at 5 weeks of age. The mice were harvested sooner (after 6 months) than those without dnp53, because dnp53 accelerates carcinoma formation. Surprisingly, the lung cancer incidence was not affected in PP2A mutant mice expressing dnp53. As shown in Fig. 11 (gray columns), the cancer incidence in E64D/+, Δ5-6/+, and Δ5-6/E64D mice expressing dnp53 was 29%, 33%, and 33%, respectively, compared to 40% in the control mice. We conclude that the tumor suppressor function of PP2A depends on the presence of functional p53.
Tumor number, size, and type.
Wild-type, E64D/+, and Δ5-6/+ mice had approximately two tumors per lung without dnp53 and one with dnp53 (Fig. 11, data table). Individual mice of these three genotypes rarely had more than 3 tumors. In contrast, several Δ5-6/E64D mice had 4, 5, or 6 tumors, resulting in the higher averages of 2.5 and 2.8, as shown in the table in Fig. 11. The average tumor size was approximately 1 mm for all genotypes. Some tumors were larger, up to a maximum of 10 mm. We observed both adenomas and adenocarcinomas in small tumors. Larger tumors were primarily adenocarcinomas. Examples of one adenoma and one adenocarcinoma are shown in Fig. 12.
Fig. 12.

Lung adenomas and adenocarcinomas in Aα mutant mice. (A and C) An adenoma at low magnification (40×; A) appears as a small parenchymal nodule (n →). At high magnification (400×; C), there is a focus of neoplastic cells, each with abundant cytoplasm and uniform monomorphic, small round nuclei. (B and D) An adenocarcinoma presented on gross evaluation as a large nodule that protruded above the pleural surface. Low magnification (40×; B) showed that the ill-defined capsule (c →) was eroded and that small foci of neoplastic cells infiltrated (i →) into the surrounding parenchyma. At high magnification (400×; D), the tumor mass shows anaplastic pleomorphic cells with tubular (t →) differentiation within solid (s →) areas. The majority of neoplastic cells contains large pleomorphic nuclei with an altered nuclear-to-cytoplasmic ratio, prominent nucleoli (o →), and occasional mitotic (m →) figures, all indicative of malignancy.
DISCUSSION
To generate a mouse model for studying the tumor suppressor function of PP2A, we constructed two strains with point mutations in the Aα subunit. One strain contains the mutation E64D, which was discovered in a lung carcinoma, and the other strain has the mutation E64G, which was found in a mammary carcinoma (6). Both strains show reduced binding of B′α and B′δ in lung and brain tissue as expected based on previous in vitro binding assays and immunoprecipitations of extracts from tissue culture cells (7, 55, 84). We were surprised that E64D/E64D and E64G/E64G mice were viable and showed no signs of disease. One reason could be that the E64D and E64G proteins still bind enough B′ subunits for survival. Another possibility is that large amounts of B′ holoenzymes are only required when animals are subjected to stress, such as by radiation, exposure to carcinogens, or infection. We also generated a strain in which exons 5 and 6 of the Aα gene are deleted (Δ5-6). This strain expresses an N-terminal fragment of Aα that binds neither C nor B subunits, equivalent to a knockout and almost identical to a mutation that was found in breast cancer. It is noteworthy that the heterozygous Δ5-6/+ knockout mice were healthy, although they contained 50% less Aα subunit in all organs tested except brain. Apparently, PP2A is available in large excess. It is even more remarkable that Δ5-6/E64G mice are viable, even though they have no wild-type Aα at all.
The main conclusion from our data is that PP2A acts like a tumor suppressor in the lungs of mice, and likely in other organs. This activity manifests itself in a 50 to 60% higher cancer incidence in the lung of benzopyrene-treated mice with mutations in the Aα gene (genotypes E64D/+, Δ5-6/+, and Δ5-6/E64D) than in benzopyrene-treated wild-type mice. It is remarkable that the increase in lung cancer incidence is very similar in E64D/+, Δ5-6/+, and Δ5-6/E64D mice, although their potential in generating holoenzymes was strikingly different both quantitatively and qualitatively. E64D/+ mice are partially defective in the formation of B′ holoenzyme, while the generation of all B and B" holoenzymes should be unaffected. Δ5-6/+ mice are expected to have reduced amounts of all holoenzymes due to the loss of one allele, while Δ5-6/E64D mice are hampered by the loss of one allele and mutation of the other. Since E64D/+ mice showed the same increase in cancer incidence as the other two strains, even though they had the least damage in their capacity of holoenzyme formation, we conclude that this damage, i.e., the partial reduction of B′ holoenzymes, is sufficient to increase the cancer incidence. Therefore, PP2A does not fall into the group of classic tumor suppressors, such as Rb, that become dysfunctional only when both alleles are inactivated, but instead belongs to the type of tumor suppressors, such as PTEN, that fail to protect from carcinogenesis when one allele is dysfunctional (4, 31). Haploinsufficiency of Aα has been demonstrated before in tissue culture cells by using shRNA (7). Taken together, our results indicate that only B′ holoenzymes are tumor suppressors, as suggested previously (8, 55), whereas B and B" holoenzymes are not. Whether all possible forms of B′ holoenzymes function as tumor suppressors is an open question, but both B′γ and B′α holoenzymes have been implicated (2, 8).
Our results strongly suggest that PP2A is a tumor suppressor in humans and that the E64D mutation contributed to the development of a lung carcinoma in the individual in which it was discovered. They also suggest that PP2A provides substantial protection against lung cancer in smokers, since the tumor incidence in benzopyrene-treated wild-type FVB mice was approximately 30% lower than in PP2A mutant mice. By comparison, p53 tumor suppressor activity in benzopyrene-treated mice causes a 50% reduction in lung cancer incidence compared to mice with inactive p53 (71).
Unexpectedly, the increased lung cancer incidence in E64D/+, Δ5-6/+, and Δ5-6/E64D mice depends on p53, since it does not occur if the p53 tumor suppressor activity has been destroyed by overexpressing a dominant negative fragment of p53. Since mutations of the p53 gene are detected in over 50% of human lung cancers (22), PP2A may only function in the remaining cases with intact p53. The question arises whether PP2A directly dephosphorylates p53, which is phosphorylated on numerous serine and threonine residues. However, since phosphorylation of most sites leads to p53 activation or stabilization (13), dephosphorylation by PP2A would inactivate or destabilize p53, which is incompatible with a PP2A tumor suppressor function. However, one site whose dephosphorylation by PP2A results in activation of human p53 is threonine 55. This dephosphorylation is carried out by the B′γ holoenzyme which forms a complex with p53, leading to transactivation of the cell cycle inhibitor p21 (32). Importantly, a small region in B′γ that interacts with p53 has been identified. Furthermore, a B′γ point mutation was discovered in a human lung cancer, which is located in this p53 binding region and renders the B′γ holoenzyme inactive in T55 dephosphorylation and p53 transcriptional activation (62). While these findings strongly support a role of B′γ as tumor suppressor in the human lung, it is puzzling that T55 appears to be human specific with no equivalent site in mouse (32).
PP2A could also act indirectly by dephosphorylating p53-regulating proteins. An attractive possibility is that it attenuates the phosphatidylinositol 3-kinase (PI3K)–Akt pathway, which is frequently upregulated in cancer. In this pathway, Akt is activated by phosphorylation at T308 and S473. Activated Akt then phosphorylates and stabilizes MDM2, resulting in p53 degradation. Importantly, PP2A has been shown to inactivate Akt by dephosphorylating both T308 and S473, leading to reduced MDM2 phosphorylation and increased degradation while p53 accumulates (17, 85). Thus, PP2A could function as a tumor suppressor through p53 upregulation in the PI3K-Akt pathway. Additional evidence that PP2A controls the phosphorylation state of Akt has come from studies with SV40-ST and Py-ST, which inhibit T308 and S473 dephosphorylation (1, 3, 81, 83). Furthermore, suppression of B′γ with short hairpin RNA leads to increased phosphorylation of S473, implicating B′γ holoenzyme in Akt dephosphorylation (7). Our finding that the tumor suppressor function of PP2A depends on the presence of active p53 is consistent with the idea that both PP2A and p53 exert their tumor suppressor activities in the same pathway, in contrast to human tissue culture cells, in which p53 and PP2A appear to act independently and inactivation of both is essential for transformation by oncogenic Ras (20). Our data are consistent with the idea that one or more B′ holoenzymes dephosphorylate Akt, since they are the only holoenzymes that are reduced in E64D/+ mice.
Another pathway that involves PP2A in the control of p53 is the Arf-p53 signaling pathway, in which the tumor suppressor Arf (ADP-ribosylation factor), in response to oncogenes, inhibits MDM2 and thus prevents p53 degradation (48). That PP2A has a positive effect on p53 in this pathway was demonstrated by the finding that inhibition of PP2A by Py-ST prevents Arf-mediated activation of p53 (41). It remains to be tested whether expression of E64D, E64G, or knockout of one allele in lung carcinomas causes downregulation of Arf and p53. It is important to note that Arf expression is frequently reduced in human lung adenocarcinomas.
K-ras mutations occur in 25% of human lung adenocarcinomas and in 90% of spontaneous and chemically induced mouse lung tumors, mainly adenocarcinomas. Furthermore, it has been shown that the K-ras codon 12 mutation is the most frequent mutation observed in benzopyrene-induced carcinomas (39). Therefore, it is likely that the majority of benzopyrene-treated mice in our study had codon 12 mutations in the K-ras gene. This raises the question whether the Ras–mitogen-activated protein kinase–extracellular signal-regulated kinase pathway, which is inhibited by PP2A (64), is upregulated in E64D/+, Δ5-6/+, and Δ5-6/E64D mutant mice. Our mouse model offers a possibility to investigate this and similar questions related to the role of PP2A in human cancer, including analyses of the status of the wild-type allele in tumors from heterozygous PP2A mutant mice to confirm the haploinsufficiency of Aα. Another question is whether the p53, Arf, and K-ras genes are mutated.
Of great interest is the recent discovery that in two ovarian clear cell carcinomas, arginine 183 of Aα is mutated to glycine in one case and to tryptophan in the other. In a third ovarian cancer, arginine 182 is mutated to tryptophan (27). We previously demonstrated that two artificial R183 mutations, R183A and R183E, render the Aα subunit highly defective in binding Bα, B′γ, and B"/PR72 (52). They were constructed because, based on our model of the A subunit (56), they appeared to be located in intrarepeat loop 5 (Fig. 1), which we suspected to be involved in B subunit binding (53). It seems likely that both ovarian cancer-associated mutant proteins R183G and R183W are also defective in B, B′, and B" binding, resulting in loss or reduction of PP2A tumor suppressor activity. Importantly, Xu et al. demonstrated by X-ray crystallography of the B′γ holoenzyme that R183, which is highly conserved in evolution, forms hydrogen bonds with E214 in B′γ (Fig. 1) (79). With regard to R182W, we previously constructed R182A and R182E mutants, and we found that both had reduced affinity for B, B′, and B" (52). It seems likely that R182 mutations act similarly to R183 mutations. Taken together, the available data suggest that PP2A is a tumor suppressor in ovaries.
The new mouse strains could be used for investigating the role of PP2A in fundamental cellular processes unrelated to growth control and cancer. For example, the finding that the levels of Aα (Fig. 5) and Cα (data not shown) are highest in brain indicates that PP2A plays an important role in this organ. Additional support for this idea comes from our analysis of Δ5-6/+ mice. In 11 organs from these mice, Aα levels were approximately 50% lower than in corresponding organs from wild-type animals, as expected due to the reduced gene dosage. However, in Δ5-6/+ cortex, cerebellum, and brain stem, the Aα levels were as high as in +/+ controls. Apparently, brain does not tolerate a reduction of Aα, i.e., of PP2A activity, without functional impairment. To avoid damage, it must have developed mechanisms for sensing low Aα levels and for reconstituting wild-type levels. It is important to find out which brain substrates require continuously high levels of PP2A. Of particular interest is the abundant microtubule-associated Tau protein, which is phosphorylated at multiple sites (65, 66). When hyperphosphorylated, Tau dissociates from microtubules, leading to formation of neurofibrillary tangles, a process that takes place in Alzheimer's disease. The Bα holoenzyme is the major phosphatase that protects Tau from hyperphosphorylation (65). Therefore, Δ5-6/+ mice might be a useful model system for studying the formation and prevention of tangles. In addition, a Cre-inducible homozygous knockout of Aα, in combination with a mouse model for Alzheimer's disease, could provide new insight into the disease process.
ACKNOWLEDGMENTS
We thank Kam-Meng Tchou-Wong for the dnp53 mouse, Robert Oshima for many helpful discussions, Steven Hedrick for plasmid pFloxΔtk, Ella Kothari for her excellent service in the UCSD Transgenic Mouse Core facility, and Anthony Wynshaw-Boris for the prion-Cre deleter mouse. We thank Nissi Varki for her thorough histological examination of mouse tumors. We thank Wolfgang Deppert for insightful ideas about p53 and Arf.
This work was supported by U.S. Public Health Service grant CA118015 and the Tobacco-Related Disease Research Program (grant 14RT-0065) to G.W.
Footnotes
Published ahead of print on 26 July 2011.
REFERENCES
- 1. Andrabi S., Gjoerup O. V., Kean J. A., Roberts T. M., Schaffhausen B. 2007. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 104:19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arnold H. K., Sears R. C. 2008. A tumor suppressor role for PP2A-B56α through negative regulation of c-Myc and other key oncoproteins. Cancer Metastasis Rev. 27:147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arroyo J. D., Hahn W. C. 2005. Involvement of PP2A in viral and cellular transformation. Oncogene 24:7746–7755 [DOI] [PubMed] [Google Scholar]
- 4. Berger A. H., Pandolfi P. P. 2011. Haplo-insufficiency: a driving force in cancer. J. Pathol. 223:137–146 [DOI] [PubMed] [Google Scholar]
- 5. Brewis N., et al. 2000. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am. J. Physiol. Heart Circ. Physiol. 279:H1307–H1318 [DOI] [PubMed] [Google Scholar]
- 6. Calin G. A., et al. 2000. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene 19:1191–1195 [DOI] [PubMed] [Google Scholar]
- 7. Chen W., Arroyo J. D., Timmons J. C., Possemato R., Hahn W. C. 2005. Cancer-associated PP2A Aα subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 65:8183–8192 [DOI] [PubMed] [Google Scholar]
- 8. Chen W., et al. 2004. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5:127–136 [DOI] [PubMed] [Google Scholar]
- 9. Cho U. S., Xu W. 2007. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445:53–57 [DOI] [PubMed] [Google Scholar]
- 10. Chui D., et al. 1997. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell 90:157–167 [DOI] [PubMed] [Google Scholar]
- 11. Colella S., et al. 2001. Reduced expression of the Aα subunit of protein phosphatase 2A in human gliomas in the absence of mutations in the Aα and Aα subunit genes. Int. J. Cancer 93:798–804 [DOI] [PubMed] [Google Scholar]
- 12. Csortos C., Zolnierowicz S., Bako E., Durbin S. D., DePaoli-Roach A. A. 1996. High complexity in the expression of the B′ subunit of protein phosphatase 2A0. Evidence for the existence of at least seven novel isoforms. J. Biol. Chem. 271:2578–2588 [DOI] [PubMed] [Google Scholar]
- 13. Dai C., Gu W. 2010. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deichmann M., Polychronidis M., Wacker J., Thome M., Naher H. 2001. The protein phosphatase 2A subunit Bγ gene is identified to be differentially expressed in malignant melanomas by subtractive suppression hybridization. Melanoma Res. 11:577–585 [DOI] [PubMed] [Google Scholar]
- 15. Eichhorn P. J., Creyghton M. P., Bernards R. 2009. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795:1–15 [DOI] [PubMed] [Google Scholar]
- 16. Farley F. W., Soriano P., Steffen L. S., Dymecki S. M. 2000. Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28:106–110 [PubMed] [Google Scholar]
- 17. Feng J., et al. 2004. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 279:35510–35517 [DOI] [PubMed] [Google Scholar]
- 18. Fujiki H., Suganuma M. 1993. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: the okadaic acid class of compounds. Adv. Cancer Res. 61:143–194 [DOI] [PubMed] [Google Scholar]
- 19. Groves M. R., Hanlon N., Turowski P., Hemmings B. A., Barford D. 1999. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 96:99–110 [DOI] [PubMed] [Google Scholar]
- 20. Hahn W. C., et al. 2002. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 22:2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hayashi S., McMahon A. P. 2002. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244:305–318 [DOI] [PubMed] [Google Scholar]
- 22. Hernandez-Boussard T. M., Hainaut P. 1998. A specific spectrum of p53 mutations in lung cancer from smokers: review of mutations compiled in the IARC p53 database. Environ. Health Perspect. 106:385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ito A., et al. 2000. A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. EMBO J. 19:562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Janssens V., Goris J. 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353:417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Janssens V., Goris J., Van Hoof C. 2005. PP2A: the expected tumor suppressor. Curr. Opin. Genet. Dev. 15:34–41 [DOI] [PubMed] [Google Scholar]
- 26. Janssens V., Longin S., Goris J. 2008. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem. Sci. 33:113–121 [DOI] [PubMed] [Google Scholar]
- 27. Jones S., et al. 2010. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330:228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Junttila M. R., et al. 2007. CIP2A inhibits PP2A in human malignancies. Cell 130:51–62 [DOI] [PubMed] [Google Scholar]
- 29. Jurka J., et al. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110:462–467 [DOI] [PubMed] [Google Scholar]
- 30. Kremmer E., Ohst K., Kiefer J., Brewis N., Walter G. 1997. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol. Cell. Biol. 17:1692–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwabi-Addo B., et al. 2001. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl. Acad. Sci. U. S. A. 98:11563–11568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H. H., Cai X., Shouse G. P., Piluso L. G., Liu X. 2007. A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 26:402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li X., Scuderi A., Letsou A., Virshup D. M. 2002. B56-associated protein phosphatase 2A is required for survival and protects from apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 22:3674–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li X., Virshup D. M. 2002. Two conserved domains in regulatory B subunits mediate binding to the A subunit of protein phosphatase 2A. Eur. J. Biochem. 269:546–552 [DOI] [PubMed] [Google Scholar]
- 35. Lozano G. 2010. Mouse models of p53 functions. Cold Spring Harb. Perspect. Biol. 2:a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mahler J. F., Stokes W., Mann P. C., Takaoka M., Maronpot R. R. 1996. Spontaneous lesions in aging FVB/N mice. Toxicol. Pathol. 24:710–716 [DOI] [PubMed] [Google Scholar]
- 37. Malkinson A. M. 1998. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp. Lung Res. 24:541–555 [DOI] [PubMed] [Google Scholar]
- 38. Martens E., et al. 2004. Genomic organisation, chromosomal localisation tissue distribution and developmental regulation of the PR61/B′ regulatory subunits of protein phosphatase 2A in mice. J. Mol. Biol. 336:971–986 [DOI] [PubMed] [Google Scholar]
- 39. Mass M. J., et al. 1993. Ki-ras oncogene mutations in tumors and DNA adducts formed by benz[j]aceanthrylene and benzo[a]pyrene in the lungs of strain A/J mice. Mol. Carcinog. 8:186–192 [DOI] [PubMed] [Google Scholar]
- 40. Moreno C. S., et al. 2000. WD40 repeat proteins striatin and S/G(2) nuclear autoantigen are members of a novel family of calmodulin-binding proteins that associate with protein phosphatase 2A. J. Biol. Chem. 275:5257–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moule M. G., Collins C. H., McCormick F., Fried M. 2004. Role for PP2A in ARF signaling to p53. Proc. Natl. Acad. Sci. U. S. A. 101:14063–14066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mumby M. 2001. A new role for protein methylation: switching partners at the phosphatase ball. Sci. STKE 2001:pe1. [DOI] [PubMed] [Google Scholar]
- 43. Mumby M. 2007. PP2A: unveiling a reluctant tumor suppressor. Cell 130:21–24 [DOI] [PubMed] [Google Scholar]
- 44. Mumby M. 2007. The 3D structure of protein phosphatase 2A: new insights into a ubiquitous regulator of cell signaling. ACS Chem. Biol. 2:99–103 [DOI] [PubMed] [Google Scholar]
- 45. Mumby M. C., Walter G. 1991. Protein phosphatases and DNA tumor viruses: transformation through the back door? Cell Regul. 2:589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neviani P., et al. 2005. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 8:355–368 [DOI] [PubMed] [Google Scholar]
- 47. Ortega-Lazaro J. C., del Mazo J. 2003. Expression of the B56δ subunit of protein phosphatase 2A and Mea1 in mouse spermatogenesis. Identification of a new B56γ subunit (B56γ4) specifically expressed in testis. Cytogenet. Genome Res. 103:345–351 [DOI] [PubMed] [Google Scholar]
- 48. Ozenne P., Eymin B., Brambilla E., Gazzeri S. 2010. The ARF tumor suppressor: structure, functions and status in cancer. Int. J. Cancer 127:2239–2247 [DOI] [PubMed] [Google Scholar]
- 49. Pallas D. C., et al. 1990. Polyoma small and middle T antigens and SV40 small T antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176 [DOI] [PubMed] [Google Scholar]
- 50. Perrotti D., Neviani P. 2008. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 27:159–168 [DOI] [PubMed] [Google Scholar]
- 51. Rossant J., Tam P. P. L. 2002. Mouse development: patterning, morphogenesis, and organogenesis. Academic Press, San Diego, CA [Google Scholar]
- 52. Ruediger R., Fields K., Walter G. 1999. Binding specificity of protein phosphatase 2A core enzyme for regulatory B subunits and T antigens. J. Virol. 73:839–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ruediger R., Hentz M., Fait J., Mumby M., Walter G. 1994. Molecular model of the A subunit of protein phosphatase 2A: interaction with other subunits and tumor antigens. J. Virol. 68:123–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruediger R., Pham H. T., Walter G. 2001. Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the A beta subunit gene. Oncogene 20:1892–1899 [DOI] [PubMed] [Google Scholar]
- 55. Ruediger R., Pham H. T., Walter G. 2001. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene 20:10–15 [DOI] [PubMed] [Google Scholar]
- 56. Ruediger R., et al. 1992. Identification of binding sites on the regulatory A subunit of protein phosphatase 2A for the catalytic C subunit and for tumor antigens of simian virus 40 and polyomavirus. Mol. Cell. Biol. 12:4872–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rundell K., Major E. O., Lampert M. 1981. Association of cellular 56,000- and 32,000-molecular-weight protein with BK virus and polyoma virus T-antigens. J. Virol. 37:1090–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sablina A. A., et al. 2007. The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell 129:969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scheel J. R., et al. 2003. An inbred 129SvEv GFPCre transgenic mouse that deletes loxP-flanked genes in all tissues. Nucleic Acids Res. 31:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schneider C., Newman R. A., Sutherland D. R., Asser U., Greaves M. F. 1982. A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem. 257:10766–10769 [PubMed] [Google Scholar]
- 61. Shaulian E., Zauberman A., Ginsberg D., Oren M. 1992. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol. Cell. Biol. 12:5581–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shouse G. P., Nobumori Y., Liu X. 2010. A B56γ mutation in lung cancer disrupts the p53-dependent tumor-suppressor function of protein phosphatase 2A. Oncogene 29:3933–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Silverstein A. M., Barrow C. A., Davis A. J., Mumby M. C. 2002. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. U. S. A. 99:4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sontag E., et al. 1993. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75:887–897 [DOI] [PubMed] [Google Scholar]
- 65. Sontag E., et al. 2004. Altered expression levels of the protein phosphatase 2A ABαC enzyme are associated with Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 63:287–301 [DOI] [PubMed] [Google Scholar]
- 66. Sontag E., Nunbhakdi-Craig V., Lee G., Bloom G. S., Mumby M. C. 1996. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 17:1201–1207 [DOI] [PubMed] [Google Scholar]
- 67. Strack S., Cribbs J. T., Gomez L. 2004. Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J. Biol. Chem. 279:47732–47739 [DOI] [PubMed] [Google Scholar]
- 68. Strack S., Zaucha J. A., Ebner F. F., Colbran R. J., Wadzinski B. E. 1998. Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J. Comp. Neurol. 392:515–527 [PubMed] [Google Scholar]
- 69. Suganuma M., et al. 1988. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc. Natl. Acad. Sci. U. S. A. 85:1768–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Takagi Y., et al. 2000. Alterations of the PPP2R1B gene located at 11q23 in human colorectal cancers. Gut 47:268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tchou-Wong K. M., et al. 2002. Lung-specific expression of dominant-negative mutant p53 in transgenic mice increases spontaneous and benzo(a)pyrene-induced lung cancer. Am. J. Respir. Cell Mol. Biol. 27:186–193 [DOI] [PubMed] [Google Scholar]
- 72. Tsao C. C., et al. 2007. Mitochondrial protein phosphatase 2A regulates cell death induced by simulated ischemia in kidney NRK-52E cells. Cell Cycle 6:2377–2385 [DOI] [PubMed] [Google Scholar]
- 73. Walter G., Ruediger R., Slaughter C., Mumby M. 1990. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc. Natl. Acad. Sci. U. S. A. 87:2521–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang S. S., et al. 1998. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 282:284–287 [DOI] [PubMed] [Google Scholar]
- 75. Wang Y., Zhang Z., Kastens E., Lubet R. A., You M. 2003. Mice with alterations in both p53 and Ink4a/Arf display a striking increase in lung tumor multiplicity and progression: differential chemopreventive effect of budesonide in wild-type and mutant A/J mice. Cancer Res. 63:4389–4395 [PubMed] [Google Scholar]
- 76. Wang Y., Zhang Z., Lubet R., You M. 2005. Tobacco smoke-induced lung tumorigenesis in mutant A/J mice with alterations in K-ras, p53, or Ink4a/Arf. Oncogene 24:3042–3049 [DOI] [PubMed] [Google Scholar]
- 77. Westermarck J., Hahn W. C. 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14:152–160 [DOI] [PubMed] [Google Scholar]
- 78. Xing Y., et al. 2006. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 127:341–353 [DOI] [PubMed] [Google Scholar]
- 79. Xu Y., et al. 2006. Structure of the protein phosphatase 2A holoenzyme. Cell 127:1239–1251 [DOI] [PubMed] [Google Scholar]
- 80. Yu J., Boyapati A., Rundell K. 2001. Critical role for SV40 small-t antigen in human cell transformation. Virology 290:192–198 [DOI] [PubMed] [Google Scholar]
- 81. Yuan H., Veldman T., Rundell K., Schlegel R. 2002. Simian virus 40 small tumor antigen activates AKT and telomerase and induces anchorage-independent growth of human epithelial cells. J. Virol. 76:10685–10691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Z., et al. 2000. A germ-line p53 mutation accelerates pulmonary tumorigenesis: p53-independent efficacy of chemopreventive agents green tea or dexamethasone/myo-inositol and chemotherapeutic agents taxol or adriamycin. Cancer Res. 60:901–907 [PubMed] [Google Scholar]
- 83. Zhao J. J., et al. 2003. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3:483–495 [DOI] [PubMed] [Google Scholar]
- 84. Zhou J., Pham H. T., Ruediger R., Walter G. 2003. Characterization of the Aα and Aβ subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem. J. 369:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhuravleva E., Tschopp O., Hemmings B. 2010. Role of PKB/Akt in liver diseases, p. 243–259 In Dufour J.-F., Clavien P.-A., (ed.), Signaling pathways in liver diseases. Springer-Verlag, Berlin, Germany [Google Scholar]