

Abstract
Evidence obtained in both eukaryotes and prokaryotes indicates that arbitrary contacts between DNA-bound proteins and components of the transcriptional machinery can activate transcription. Here we demonstrate that the Escherichia coli ω protein, which copurifies with RNA polymerase, can function as a transcriptional activator when linked covalently to a DNA-binding protein. We show further that ω can function as an activation target when this covalent linkage is replaced by a pair of interacting polypeptides fused to the DNA-binding protein and to ω, respectively. Our findings imply that the ω protein is associated with RNA polymerase holoenzyme in vivo, and provide support for the hypothesis that contact between a DNA-bound protein and any component of E. coli RNA polymerase can activate transcription.
Keywords: θ subunit, E. coli, RNA polymerase, transcriptional activator
Recent findings in both eukaryotes and prokaryotes indicate that arbitrary protein–protein contacts can trigger gene activation provided one of the protein partners is tethered to the DNA and the other is a component (or is tethered to a component) of RNA polymerase (RNAP) (Barberis et al. 1995; Chatterjee and Struhl 1995; Klages and Strubin 1995; Xiao et al. 1995; Apone et al. 1996; Farrell et al. 1996; Dove et al. 1997; Gaudreau et al. 1997; Gonzalez-Couto et al. 1997; Lee and Struhl 1997; for review, see Ptashne and Gann 1997). Experiments in yeast have shown further that direct fusion of a DNA-binding domain to a component of the RNAP II holoenzyme can activate transcription from a promoter bearing a recognition site for the DNA-binding domain (Barberis et al. 1995; Farrell et al. 1996; Gaudreau et al. 1997; for review, see Ptashne and Gann 1997), but analogous experiments have not been performed previously in bacteria.
RNAP in Escherichia coli consists of an enzymatic core composed of subunits α, β, and β′ in the stoichiometry α2ββ′, and one of several alternative σ factors that confer on the enzyme the ability to recognize specific promoters (Burgess 1976; Hellman and Chamberlin 1988). An additional protein, omega (θ), has been called a subunit of RNAP on the basis of its copurification with RNAP core and holoenzyme in near stoichiometric amounts (Burgess 1969). The function of θ is unknown and, unlike the other subunits, θ is not required for transcription either in vitro (Heil and Zillig 1970) or in vivo (Gentry and Burgess 1989; Gentry et al. 1991). Cells deleted for the gene encoding θ (rpoZ) have no discernible mutant phenotypes (Gentry and Burgess 1989; Gentry et al. 1991).
Many natural activators in bacteria bind the DNA near the promoters they regulate and interact directly with one or more subunits of RNAP (Busby and Ebright 1994). The best known target of these interactions is the α subunit of RNAP (Ishihama 1992; Russo and Silhavy 1992; Ebright and Busby 1995; Niu et al. 1996). Some activators, however, interact with the σ subunit (Hochschild 1994; Kuldell and Hochschild 1994; Li et al. 1994; Artsimovitch et al. 1996; Gerber and Hinton 1996), and evidence suggests that the β subunit may also serve as an activation target in at least one case (Lee and Hoover 1995). Finally the β′ subunit has been identified as the target of action of an activator that functions without binding to the DNA (Miller et al. 1997). In contrast, the θ subunit has not been implicated in activation to date.
In a previous study we fused a heterologous protein domain to the α subunit of E. coli RNAP and demonstrated that interaction between a DNA-bound protein and the heterologous protein domain tethered to α activated transcription from a test promoter (Dove et al. 1997). The magnitude of the activation correlated with the strength of the protein–protein interaction, the interaction presumably functioning to stabilize the binding of RNAP to the promoter. These findings suggest that contact between a DNA-bound protein and any subunit of RNAP could, in principle, activate transcription.
Here we show that covalent linkage of a DNA-binding protein to the θ subunit can activate transcription from a test promoter bearing a recognition site for the DNA-binding domain, and further that this covalent linkage can be replaced by a protein–protein bridge. These results support the hypothesis that any subunit of RNAP can serve as an activation target, and provide evidence that the θ protein is associated with RNAP holoenzyme in vivo and that it is accessible at the surface of the enzyme complex.
Results
The ω subunit can activate transcription from a test promoter when fused to a DNA-binding protein
To determine whether the θ subunit of RNAP can mediate transcriptional activation when tethered to the DNA upstream of a promoter, we fused the θ protein to the repressor (cI) protein of bacteriophage λ (see Fig. 1B). The λcI protein is a two-domain protein that binds DNA as a dimer; the amino-terminal domain (NTD) is the DNA-binding domain, whereas the carboxy-terminal domain (CTD) mediates dimer formation (and higher order oligomerization) (Sauer et al. 1990).
Figure 1.

(A) Basal transcription by E. coli RNA polymerase. A promoter comprised of a −10 and a −35 element is depicted bound by RNAP with subunit composition α2ββ′σθ. The θ subunit (shaded) is depicted as interacting with the β′ subunit as a monomer (see Gentry and Burgess 1993). (B) Fusion of the θ subunit of RNAP to the carboxy terminus of λcI permits interaction of a modified polymerase with a λ operator. The artificial promoter derivative plac OR2-62 is shown; it bears the λ operator OR2 centered 62 bp upstream of the transcriptional start site of the lac promoter.
We fused the entire θ protein (residues 1–90) to the carboxyl terminus of the λcI protein through a small alanine linker (see Materials and Methods). We placed the gene encoding this fusion protein downstream of an inducible promoter on a plasmid vector, thus generating plasmid pBRcI-θ. We introduced pBRcI-θ into strain KS1ΔZ, which harbors on its chromosome a lac promoter derivative (termed placOR2-62) bearing a single λ operator centered 62 bp upstream of the transcription start point. Note that λcI, which activates transcription from the λPRM promoter when bound at a site centered at position −42, cannot activate transcription from placOR2-62 (Dove et al. 1997) because the λ operator is positioned too far from the promoter. In addition, KS1ΔZ bears a deletion of the chromosomal locus encoding the θ subunit. Unlike λcI, the λcI–θ fusion protein stimulated transcription ∼70-fold, as measured by β-galactosidase assay (Fig. 2A). Primer extension analysis confirmed that the fusion protein stimulated the production of correctly initiated transcripts (Fig. 2B). A similar experiment performed with strain KS1 revealed that the λcI–θ fusion protein was unable to stimulate transcription from placOR2-62 in the presence of endogenous θ protein encoded by the chromosomal rpoZ gene (Fig. 2A).
Figure 2.

Transcriptional activation by λcI–θ fusion protein. (A) Effect of λcI–θ on transcription in vivo from plac OR2-62. KS1ΔZ cells harboring plasmids encoding either λcI–θ (▿), λcI(S45A)–θ (▾), λcI (□) or no λcI (ΔcI) (○), and KS1 cells (containing wild-type ω) (•) harboring a plasmid encoding λcI–θ, were grown in the presence of the indicated concentrations of IPTG and assayed for β-galactosidase activity. (B) Primer extension analysis of transcripts produced from placOR2-62 in the presence of λcI–θ. Total RNA was isolated from KS1ΔZ cells grown in the presence of 20 μm IPTG and harboring plasmids encoding the indicated proteins. Primer extension analysis was done by using a primer complementary to the lacZ transcript produced by the placOR2-62 promoter. Primer extension products produced by correctly initiated plac OR2-62 transcripts are indicated by +1 and excess unincorporated primer is shown.
Transcriptional activation by the λcI–ω fusion protein is dependent on its ability to bind DNA
To demonstrate that the stimulation of transcription from placOR2-62 in KS1ΔZ by λcI–θ depends on the ability of the fusion protein to bind to the λ operator of placOR2-62, we introduced a single amino acid substitution (S45A) into the λcI moiety of the fusion protein that results in a severe reduction in operator binding (Hochschild and Ptashne 1986). This mutant version of the λcI–θ fusion protein failed to stimulate transcription from placOR2-62 (Fig. 2A).
We confirmed that the λcI(S45A)–θ fusion protein is specifically defective for operator binding by measuring its binding to a consensus λ operator using an in vivo repression assay (data not shown). Western blot analysis confirmed that the λcI–θ and the λcI (S45A)–θ fusion proteins were present in the cell in comparable amounts (data not shown).
Interaction between a DNA-bound domain of Gal4 and a domain of Gal11P fused to either the α or ω subunit of RNAP results in transcriptional activation from a test promoter
We then replaced the covalent interaction between the λcI protein and the θ subunit of RNAP with a protein-protein contact. For this purpose we took advantage of a pair of protein domains originally shown to interact in yeast cells. Transcriptional activation in yeast can be triggered by an apparently fortuitous interaction between the dimerization region of the yeast transcriptional activator Gal4 and a mutant form of the Gal11 protein (Himmelfarb et al. 1990; Barberis et al. 1995), which despite its name, is a component of the RNAP II holoenzyme and is required for full transcription of many genes (Kim et al. 1994; Barberis et al. 1995; Hengartner et al. 1995). Ordinarily, in yeast, the dimerization region of Gal4 does not mediate transcriptional activation when connected to its own or another DNA-binding domain. However, in the presence of a Gal11 mutant (called Gal11P for potentiator) bearing a single amino acid substitution at position 342, the Gal4 dimerization region functions as a powerful activating region; this activation results from a specific interaction between the Gal4 dimerization region and the portion of Gal11P bearing the amino acid substitution (Barberis et al. 1995; Farrell et al. 1996).
To establish that this protein–protein interaction can also trigger gene activation in E. coli, we first tested the abilities of the relevant portions of Gal11P and Gal4 (Farrell et al. 1996) to mediate transcriptional activation when fused to the α subunit of RNAP and to the λcI protein, respectively. To do this, we proceeded as we had done previously (Dove et al. 1997), taking advantage of the domain structure of α, which initiates the assembly of RNAP by forming a dimer. The α-NTD is responsible for the assembly reaction (Hayward et al. 1991; Igarashi et al. 1991), and the α-CTD, which is connected to the α-NTD by a flexible linker region (Blatter et al. 1994; Jeon et al. 1997), can bind DNA (Ross et al. 1993; Blatter et al. 1994) and is the natural target for many transcriptional activators (Ishihama 1992; Ebright and Busby 1995). We reasoned that if we replaced the α-CTD with an appropriate domain of Gal11P, the resulting α–Gal11P chimera would display a target that could be contacted by an appropriately positioned λcI–Gal4 dimer (Fig. 3). Therefore, we created two chimeric genes, one encoding the α-NTD and linker connected to residues 263–352 of Gal11P, and the other encoding full-length λcI (residues 1–236) connected to the dimerization region of Gal4 (residues 58–97) (see Materials and Methods). We then tested the ability of the λcI–Gal4 fusion protein to activate transcription from placOR2-62 in cells containing the α–Gal11P fusion protein (as well as wild type α encoded by the chromosomal rpoA gene). Figure 4A shows that the λcI–Gal4 fusion protein activated transcription ∼45-fold in KS1 cells containing the α–Gal11P fusion protein but not in control cells containing an otherwise identical fusion protein bearing the wild type form of Gal11. Primer extension analysis confirmed that the fusion protein stimulated the production of correctly initiated transcripts (Fig. 4B). We also found that a different λcI–Gal4 fusion protein comprising only the NTD and linker region of λcI (residues 1–132) fused to Gal4(58–97) activated transcription in KS1 cells harboring the α–Gal11P fusion protein by a factor of approximately eight (data not shown).
Figure 3.

(A) Basal transcription by E. coli RNA polymerase drawn to illustrate the structure of the α subunit. A promoter comprised of a −10 and a −35 element is depicted together with the subunit composition of RNAP holoenzyme. The α subunits of RNAP consist of two independently folded domains; an NTD and a CTD connected by a flexible linker. (B) Replacement of RNAP α-CTD by Gal11P (residues 263–352) permits interaction with the Gal4 dimerization domain of a λcI–Gal4 fusion protein. The diagram depicts the test promoter placOR2-62, which bears the λ operator OR2 centered 62 bps upstream of the transcriptional start site of the lac promoter.
Figure 4.
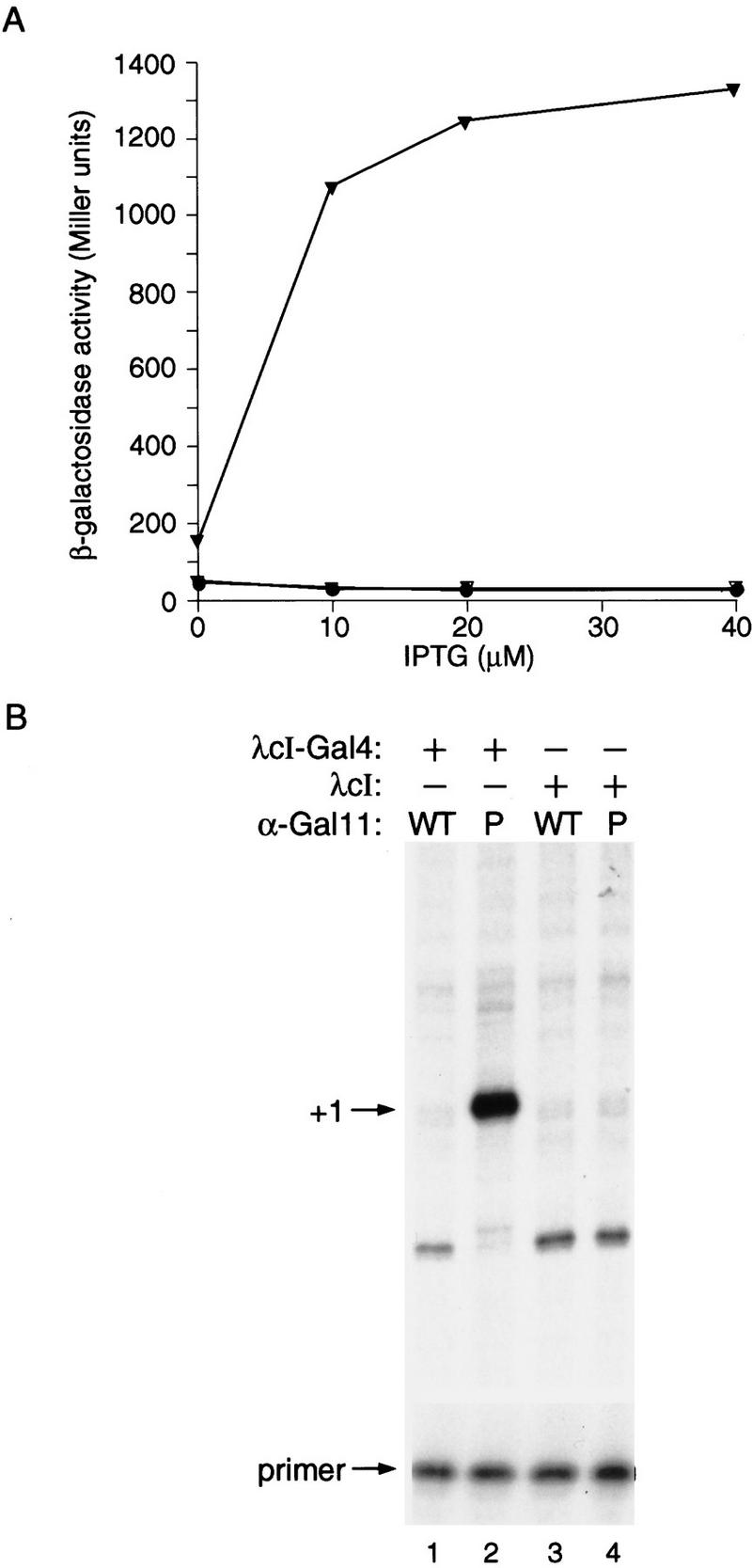
Transcriptional activation by λcI–Gal4 fusion protein in presence of α-Gal11P fusion protein. (A) Effect of λcI–Gal4 on transcription in vivo from plac OR2-62 in the presence of α-Gal11P fusion protein. KS1ΔZ cells harboring plasmids expressing the indicated proteins were grown in the presence of different concentrations of IPTG and assayed for β-galactosidase activity. (▾) λcI–Gal4 + α–Gal11P; (▿) λcI–Gal4 + α–Gal11WT; (•) λcI + α–Gal11P; (○) λcI + α–Gal11WT. (B) Primer extension analysis of transcripts produced from plac OR2-62 in the presence of λcI–Gal4 and α-Gal11P. Total RNA was isolated from KS1ΔZ cells grown in the presence of 20 μm IPTG and harboring plasmids encoding the indicated proteins. Primer extension analysis was done by using a primer complementary to the lacZ transcript produced by the plac OR2-62 promoter. Primer extension products produced by correctly initiated plac OR2-62 transcripts are indicated by +1 and excess unincorporated primer is shown.
We reasoned that if we fused the appropriate domain of Gal11P to the θ subunit of RNAP the resulting θ–Gal11P chimera would, like the α–Gal11P chimera, display a target that could be contacted by an appropriately positioned λcI–Gal4 dimer (Fig. 5A). Therefore, we constructed a chimeric gene encoding the θ subunit connected to residues 263–352 of Gal11P (see Materials and Methods). Figure 5B shows that the λcI–Gal4 fusion protein (comprising residues 1–236 of λcI fused to residues 58–97 of Gal4) activated transcription ∼20-fold in KS1ΔZ cells containing the θ–Gal11P fusion protein, but not in control cells containing the θ–Gal11WT fusion protein. Primer extension analysis confirmed that the fusion protein stimulated the production of correctly initiated transcripts (Fig. 5C). In contrast to the λcI–θ fusion protein, the λcI–Gal4 fusion protein activated transcription in KS1 cells containing both wild-type θ and the θ–Gal11P chimera (data not shown), indicating that in this case endogenous θ does not compete effectively with the θ–Gal11P chimera for binding to RNAP (see Discussion).
Figure 5.

Transcriptional activation by λcI–Gal4 fusion protein in presence of θ–Gal11P fusion protein. (A) Fusion of Gal11P (residues 263–352) to the θ subunit of RNA polymerase permits interaction with the Gal4 dimerization domain of a λcI–Gal4 fusion protein. The diagram depicts the test promoter plac OR2-62, which bears the λ operator OR2 centered 62 bp upstream of the transcriptional start site of the lac promoter. (B) Effect of λcI–Gal4 on transcription in vivo from plac OR2-62 in the presence of θ-Gal11P fusion protein. KS1ΔZ cells harboring plasmids expressing the indicated proteins were grown in the presence of different concentrations of IPTG and assayed for β-galactosidase activity. (▾) λcI–Gal4 + θ–Gal11P; (▿) λcI–Gal4 + θ–Gal11WT; (•) λcI + θ–Gal11P; (○) λcI + θ–Gal11WT. (C) Primer extension analysis of transcripts produced from plac OR2-62 in the presence of λcI–Gal4 and θ–Gal11P. Total RNA was isolated from KS1ΔZ cells grown in the presence of 50 μm IPTG and harboring plasmids encoding the indicated proteins. Primer extension analysis was done by using a primer complementary to the lacZ transcript produced by the plac OR2-62 promoter. Primer extension products produced by correctly initiated plac OR2-62 transcripts are indicated by +1 and excess unincorporated primer is shown.
Discussion
Transcriptional activation in E. coli by tethering a subunit of RNAP to DNA
We have demonstrated that covalent linkage of a DNA-binding protein (λcI) to a component of RNAP (θ) results in transcriptional activation from a promoter bearing a recognition site for the DNA-binding protein (a λ operator). This form of activation resembles the natural activation that occurs when the RNAP α subunit interacts with a DNA sequence element termed the UP element that is found upstream of the −35 hexamer of rRNA promoters (as well as some other promoters) (Ross et al. 1993; Gaal et al. 1996). As mentioned above, the α subunit has two domains, and its CTD is a sequence-specific DNA-binding domain (Blatter et al. 1994; Gaal et al. 1996). The interaction of the α-CTD with naturally occurring UP elements has been reported to increase transcription by as much as 30-fold, and this increase reflects an increase in the initial binding of RNAP to the promoter and possibly a subsequent step in the initiation process (Ross et al. 1993; Rao et al. 1994).
Experiments performed in eukaryotic cells have also shown that transcription can be activated by the direct fusion of DNA-binding domains to various components of the transcriptional machinery. For example, fusion of the E. coli LexA repressor, a sequence-specific DNA-binding protein, to the wild-type Gal11 protein creates a powerful transcriptional activator in yeast that works on promoters bearing LexA-binding sites, and this activation depends on the portion of Gal11 that mediates its association with the RNAP II holoenzyme (Himmelfarb et al. 1990; Barberis et al. 1995; see also Farrell et al. 1996; Gaudreau et al. 1997). Similarly, direct or indirect fusion of a DNA-binding domain to the yeast TATA-binding protein (TBP) creates a transcriptional activator that works on promoters bearing a recognition site for the DNA-binding domain upstream of the TATA element, and this activation depends on the ability of TBP to interact with the TATA element (Chatterjee and Struhl 1995; Klages and Strubin 1995; Xiao et al. 1995). Thus, recruitment of the polymerase II holoenzyme and TBP to promoters in yeast cells, as well as RNAP holoenzyme to bacterial promoters can be a rate-limiting step in transcription initiation in vivo.
Transcriptional activation by arbitrary protein–protein interactions
In a previous study we showed that contact between a DNA-bound protein and a heterologous protein domain fused to the α-NTD can activate transcription in E. coli (Dove et al. 1997). We have now generalized this finding by showing that an interacting pair of protein fragments that triggers gene activation in yeast also triggers gene activation in E. coli when one of the pair is fused to a DNA-binding protein and the other is fused either to the α-NTD or to the θ protein. Specifically, we fused the dimerization domain of Gal4 to the λcI protein and the relevant fragment of the Gal11P protein either to the α-NTD or to θ and demonstrated that the λcI–Gal4 fusion protein stimulated transcription from an appropriately designed test promoter in cells containing either the α–Gal11P or the θ–Gal11P fusion protein. These findings provide support for the hypothesis that contact between a DNA-bound protein and any component of RNAP can activate transcription in E. coli (Fig. 6). In particular, they indicate that the α subunit is not unique in its ability to mediate the effects of artificial activators.
Figure 6.

Any subunit of RNAP can serve as an activation target. An activator bound upstream of a promoter is depicted, and the possible interactions with any of the subunits of RNAP are illustrated with arrows. The interactions mediated by the artificial activators described herein are indicated by solid arrows; additional interactions are indicated by broken arrows. The α and σ subunits are known targets of natural DNA-bound activators (for review, see Busby and Ebright 1994), the β subunit is suggested as a target for at least one DNA-bound activator (Lee and Hoover 1995), and the β′ subunit is the target of an activator that works without binding DNA (Miller et al. 1997).
We note that the λcI–Gal4 fusion protein stimulated transcription more strongly with the α–Gal11P than with the θ-Gal11P fusion protein (45- vs. 20-fold). We do not know the reason for this difference, but we speculate that it may reflect a difference in the number of Gal11P moieties displayed by the RNAP holoenzyme in the two cases. Whereas the θ subunit is presumed to associate with RNAP as a monomer, the α subunit assembles as a dimer. Therefore, at least a fraction of the polymerase molecules assembled in the presence of chromosomally encoded wild-type α and an excess of the α–Gal11P fusion protein should display two Gal11P moieties, each of which might be able to interact with a Gal4 moiety displayed on the DNA-bound λcI dimer. It is also possible that the observed difference in activities (45-fold for the α–Gal11P fusion protein and 20-fold for the θ–Gal11P fusion protein) reflects a difference in the fraction of RNAP molecules in the cell that contain the fusion protein. Further experiments will be required to test these possibilities.
The ω protein is a subunit of RNA polymerase in vivo
Our demonstration that the θ–Gal11P fusion protein can function as an activation target provides strong evidence that the θ protein is associated with RNAP in growing cells. This demonstration taken together with the finding that the λcI–θ fusion protein is a powerful activator of transcription indicates, moreover, that θ is accessible at the surface of RNAP. Therefore, our results raise the possibility that θ might serve as a target for natural DNA-bound activators, as well.
In our experiments with the λcI–θ fusion protein (in which the amino terminus of θ is fused to the carboxyl terminus of λcI), we observed transcriptional activation only in a strain deleted for the chromosomal rpoZ gene (encoding θ). This suggests first that the λcI–θ fusion protein associates with the same surface of RNAP as native θ protein, and second, that the native θ protein associates preferentially. In contrast, the θ–Gal11P fusion protein (in which the amino terminus of θ is free) mediated transcriptional activation by the λcI–Gal4 fusion protein in both the absence and presence of chromosomally encoded θ protein. This suggests that this fusion protein competes effectively with native θ protein for association with RNAP. We suggest a possible explanation: The amino-terminal portion of the θ protein mediates its association with RNAP with the consequence that fusion of another protein at the amino terminus weakens the association.
In vitro cross-linking experiments have suggested that θ binds to the β′ subunit of RNAP (Gentry and Burgess 1993). The biological activity of our λcI–θ fusion protein should permit genetic identification of not only the residues on θ that mediate its association with RNAP, but the interacting residues on β′ (or any other subunit of RNAP) as well.
Practical implications for prokaryotic two-hybrid and one-hybrid systems
Our previous demonstration that contact between a DNA-bound protein and a protein domain fused to the α subunit of RNAP can activate transcription suggested the possibility of establishing a transcription-based two-hybrid assay for detecting protein–protein interactions in E. coli (Dove et al. 1997). Here we have demonstrated the feasibility of this approach by showing that two polypeptides known to interact in yeast and in vitro (Farrell et al. 1996) can activate transcription in E. coli when one of the pair is fused to a subunit of RNAP (either α or θ) and the other is fused to a DNA-binding protein (λcI).
Previous studies have demonstrated that heterologous protein domains that mediate dimer formation can functionally substitute for the λcI–CTD when fused to the λcI–NTD, resulting in biologically active fusion proteins that bind efficiently to λ operators (for review, see Hu 1995). In designing the λcI–Gal4 fusion protein, we sought to compare the effects of fusing the Gal4 moiety to the end of the λcI linker (at residue 132) and to the end of full-length λcI (at residue 236). Although both of the resulting fusion proteins bound to λ operators and stimulated transcription in the presence of the α–Gal11P fusion protein (Fig. 4; data not shown), the full-length λcI–Gal4 fusion protein was more active, presumably because the λcI–CTD mediates more efficient dimerization than the Gal4 moiety. The finding that a heterologous protein domain can be fused to the carboxyl terminus of λcI without interfering with λcI–CTD-mediated dimerization implies that heterologous protein domains can be tethered to the DNA through the λcI protein regardless of whether or not they have the potential to dimerize.
The utility of this strategy was confirmed by our construction of a biologically active λcI–θ fusion protein. In turn, the ability of this fusion protein to activate transcription from a promoter bearing a λ operator demonstrates the feasibility of establishing a so-called “one-hybrid” assay in E. coli to detect specific protein–DNA interactions (Li and Herskowitz 1993; Wang and Reed 1993; Inouye et al. 1994).
Mechanistic implications
Together our findings with both the α and the θ fusion proteins suggest that depending on the nature of the promoter, contact between a DNA-bound protein and any accessible surface of RNAP can activate transcription, presumably by stabilizing the binding of RNAP to the promoter. We suspect, however, that this form of activation would not work at all promoters. In particular, if the activity of a given promoter is not limited by its ability to stably bind RNAP in vivo (for example, see Morett and Buck 1989; Hidalgo and Demple 1997; for reviews, see also Kustu et al. 1991; Summers 1992; Gralla 1996; Ptashne and Gann 1997), then we hypothesize that the artificial activators we have designed would be ineffective. In this regard, it might be possible to use the λcI–θ fusion protein as a tool for classifying promoters in vivo.
Materials and methods
Media, growth conditions, and genetic techniques
Bacteria were cultured routinely in L broth or on L-agar plates (Miller 1972). Where needed, antibiotics were used at the following concentrations: carbenicillin (50 μg/ml), chloramphenicol (25 μg/ml), kanamycin (50 μg/ml), and tetracycline (20 μg/ml). Transductions were performed using generalized transducing phage P1vir as described previously (Sternberg and Maurer 1991).
Bacterial strains
E. coli strain XL1-blue (Stratagene) was used routinely as a cloning vehicle for plasmid constructions. The E. coli strain KS1 has been described previously (Dove et al. 1997) and harbors the artificial promoter derivative placOR2-62 consisting of the λ operator OR2 centered 62 bp upstream of the transcriptional start site of the lac promoter. This promoter and the linked lacZ gene are present on a λimm21 prophage. The E. coli strain KS1ΔZ was created by P1-mediated transduction of the ΔspoS3::cat mutation (an θ null allele) from CF2790 (Xiao et al. 1991) into recipient strain KS1.
DNA manipulation and oligonucleotides
Standard molecular biology techniques (Sambrook et al. 1989) were used for cloning, DNA purification, and analysis. The PCR was performed using Expand (Boehringer Mannheim) and restriction enzymes were obtained from New England Biolabs. DNA was sequenced by the dideoxy method using Sequenase (U.S. Biochemical).
Oligonucleotides used to make the different plasmids were purchased from Operon Technologies, Inc., and were as follows: OL.2 (5′-CAGTGATTCTGCATTCTGGCTTGAG-3′); OL.3 (5′-GCGGATCCTAGGTCAAAATAATCCTGTTAA-3′); OL.6 (5′-CAGACGTTTGGCGAATCAAGGCTAGAAAGACTGG-3′); OL.7 (5′-TAGCCTTGATTCGCCAAACGTCTCTTCAGG-3′); OL.13 (5′-CTGCTGTTGAGGCTCTGGTTTCTCTTCTTTCAC-3′); OL.15 (5′-GAGAAACCAGAGCCTCAACAGCAGCAAATGCAACC-3′); OL.18 (5′-AGCGGATCCTCACAAAGCTTGGATTTTTCTCAGG-3′); OL.R1 (5′-GTGCCGGTTCTACCC-3′); OL.32 (5′-TAGGATCCGGCGCGCCTAAGATCTTGCGGCCGCGCCAAACGTCTCTTCAGGCCACTG-3′); OL.39 (5′-ATATGCGGCCGCACGCGTAACTGTTCAGGACG-3′); OL.40 (5′-ATATGTCGACTTAACGACGACCTTCAGCAAT-3′); OL.41 (5′-AAAGTTCCATATGGCACGCGTAACTGTTCAGG-3′); OL.42 (5′-TATATGCGGCCGCACGACGACCTTCAGCAATAGCG-3′); OL.43 (5′-ATATGCGGCCGCACCTCAACAGCAGCAAATGCAACC-3′); OL.44 (5′-ATATGTCGACTCACAAAGCTTGGATTTTTCTCAGG-3′); OL.54 (5′-ATATATCATATGAGCACAAAAAAGAAACC-3′); OL.55 (5′-TTCTCTGGCGATTGAAGGGC-3′).
Plasmids
Plasmids used in this study are listed in Table 1. All inserts in plasmids that were generated by the PCR were subsequently sequenced to confirm that no errors had been introduced as a result of the PCR process.
Table 1.
Plasmids
| Plasmid
|
Relevant details
|
Source/Reference
|
|---|---|---|
| pACλcI | CmlR; ori–pACYC184; encodes λcI | Dove et al. (1997) |
| pACLGF2 | CmlR; ori–pACYC184; encodes λcI(1–236) + Gal4(58–97) | this work |
| pACTcλcI | TcR; ori–pACYC184; encodes λcI | this work |
| pACTcLGF2 | TcR; ori–pACYC184; encodes λcI(1–236) + Gal4(58–97) | this work |
| pBR322 | ApR cloning vector | Bolivar et al. (1977) |
| pBRα | ApR; ori–pBR322; encodes α(1–329) | Dove et al. (1997) |
| pBRα–Gal11WT | ApR; ori–pBR322; encodes α(1–248) + Gal11(263–352)WT | this work |
| pBRα–Gal11P | ApR; ori–pBR322; encodes α(1–248) + Gal11(263–352)N342V | this work |
| pBRcI–ω | ApR; ori–pBR322; encodes λcI(1–236)WT + 2 Ala + ω(1–90) | this work |
| pBRcI(S45A)–ω | ApR; ori–pBR322; encodes λcI(1–236)S45A + 2 Ala + ω(1–90) | this work |
| pBRΔcI | ori–pBR322; does not encode λcI | Whipple et al. (1994) |
| pBRω–Gal11WT | ApR; ori–pBR322; encodes ω(1–90) + 3 Ala + Gal11(263–352)WT | this work |
| pBRω–Gal11P | ApR; ori–pBR322; encodes ω(1–90) + 3 Ala + Gal11(263–352)N342V | this work |
| pE3C-1 | ApR; ori–pBR322; encodes ω(1–90) | Gentry and Burgess (1990) |
| pNS113 | encodes LexA(1–202) + Gal4(58–97) | Farrell et al. (1996) |
| pLCF3 | encodes λcI(1–132)S45A | J.K. Joung (unplubl.) |
| pLX20 | ApR; ori–pBR322; encodes λcI(1–236) | F. Whipple (unpubl.) |
| pSO23 | encodes Gal11(1–1081)WT | Gaudreau et al. (1997) |
| pSO32 | encodes Gal11(1–1081) N342V | Gaudreau et al. (1997) |
(ApR) Ampicillin resistant; (CmlR) chloramphenicol resistant; (TcR) tetracycline resistant.
Plasmid pLX20 is a derivative of pBR322 (Bolivar et al. 1977), confers ApR, and bears the cI gene under the control of the lacUV5 promoter (F. Whipple, unpubl.).
Plasmid pBRcI–θ is a derivative of pLX20, confers ApR, and encodes residues 1–236 of λcI fused to two Ala residues, which in turn are fused to residues 1–90 of the θ subunit of E. coli RNAP. The expression of the λcI–θ fusion protein is under the control of the lacUV5 promoter. The primary sequence of the λcI–θ fusion protein junction from λcI residue 235 inclusive is PheGlyAlaAlaAlaArg, where the underlined residues are the first and second, respectively, in the primary sequence of θ. Note that the initiating Met of θ was not included in the fusion protein, as it is not present in the mature protein (Gentry and Burgess 1986), and therefore, the Ala residue that follows the initiating Met is classified here as residue one. pBRcI–θ was constructed by replacing the HindIII–SalI fragment from pLX20 with two fragments of DNA. One fragment (comprising a segment of the cI gene) was a HindIII–NotI-digested PCR product that was made using primers OL.2 and OL.32 with pACλcI as template. This fragment contains a NotI site at the 3′ end of cI. The second fragment (comprising the θ gene) was a NotI–SalI-digested PCR product that was made using primers OL.39 and OL.40 with pEΔC-1 as template.
Plasmid pBRcI(S45A)–θ is identical to pBRcI–θ except that the λcI moiety of the fusion protein harbors the S45A mutation. pBRcI(S45A)–θ was constructed by replacing the NdeI–NsiI fragment from pBRcI–θ with an NdeI–NsiI-digested PCR product that was made using primers OL.54 and OL.55 with pLCF3 as template. pBRΔcI is the same as pLR1ΔcI (Whipple et al. 1994).
Plasmid pACLGF2 is a derivative of pACλcI, confers CmlR, and encodes residues 1–236 of λcI fused to residues 58–97 of Gal4 under the control of the lacUV5 promoter. pACLGF2 was made by replacing the HindIII–BstYI fragment from pACλcI with a HindIII–BamHI digested PCR product made using primers OL.2 and OL.3. The PCR product comprised a fragment of the 3′ end of the cI gene fused directly to the coding sequence of Gal4 (residues 58–97), and two PCR products (made using primers OL.2 and OL.7 with pACλcI as template and primers OL.6 and OL.3 with pNS113 as template, respectively) served as template for its generation.
Plasmid pACTcLGF2 is a derivative of pACLGF2 that confers TcR and like pACLGF2 encodes residues 1–236 of λcI fused to residues 58–97 of Gal4 under the control of the lacUV5 promoter. pACTcLGF2 was made by replacing the HindIII–EcoRI fragment from pACλcI with the EcoRI–BstYI fragment from pBR322 (encoding the TcR gene) and the appropriate BstYI–HindIII fragment from pACLGF2.
Plasmid pBRα–Gal11WT is a derivative of pBRα, confers ApR, and encodes residues 1–248 of the α subunit of E. coli RNAP fused to residues 263–352 of wild-type Gal11 under the control of tandem lpp and lacUV5 promoters. pBRα–Gal11WT was made by replacing the EcoRI–BamHI fragment from pBRα with an EcoRI–BamHI-digested PCR product made using primers OL.R1 and OL.18. The PCR product comprised a fragment of the 3′ end of the cI gene fused directly to the coding sequence of Gal11 (residues 263–352), and two PCR products (made using primers OL.R1 and OL.13 with pBRα as template and primers OL.15 and OL.18 with pSO23 as template, respectively) served as template for its generation. pBRα–Gal11P was similarly made using pSO32 instead of pSO23 as template.
Plasmid pBRθ–Gal11WT confers ApR and encodes residues 1–90 of the θ subunit of E. coli RNAP fused to three Ala residues, which in turn are fused to residues 263–352 of wild-type Gal11. The expression of the θ–Gal11WT fusion protein is under the control of the lacUV5 promoter. pBRθ–Gal11WT was made by replacing the NdeI–SalI fragment from pLX20 with two fragments of DNA. One fragment (comprising a segment of the θ gene) was an NdeI–NotI-digested PCR product that was made using primers OL.41 and OL.42 with pE3C-1 as template. This introduces an NdeI site at the start and a NotI site at the 3′ end of θ. The second fragment (comprising the Gal11WT coding sequence from residues 263–352) was a NotI–SalI-digested PCR product that was made using primers OL.43 and OL.44 with pSO23 as template. pBRθ–Gal11P was similarly made using pSO32 instead of pSO23 as template.
β-Galactosidase assays
SDS–CHCl3 permeabilized cells were assayed for β-galactosidase activity essentially as described (Miller 1972). Assays were performed at least three times in duplicate on separate occasions, with similar results. Values are the averages from one experiment and duplicate measurements differed by <10%.
Primer extension analysis
RNA isolation, primer labeling, primer extension assays, and transcriptional start site identification were as described previously (Dove et al. 1997).
Acknowledgments
We thank Mike Cashel, Luc Gaudreau, Dan Gentry, Keith Joung, Mark Ptashne, and Fred Whipple for plasmids and strains. We also thank Yan Ye Xia for excellent technical assistance. We are very grateful to Keith Joung for helpful discussions and thank Mark Ptashne, Gareth King, John Mekalanos, and Bill Forrester for comments on the manuscript. This work was supported by National Institutes of Health grant GM44025 (A.H.), by the National Science Foundation Presidential Young Investigator Award (A.H.), and by an established investigatorship from the American Heart Association (A.H.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ahochsch@warren.med.harvard.edu; FAX (617) 738-7664.
References
- Apone LM, Virbasius CA, Reese JC, Green MR. Yeast TAFII90 is required for cell-cycle progression through G2/M but not for general transcription activation. Genes & Dev. 1996;10:2368–2380. doi: 10.1101/gad.10.18.2368. [DOI] [PubMed] [Google Scholar]
- Artsimovitch I, Murakami K, Ishihama A, Howe MM. Transcription activation by the bacteriophage Mu Mor protein requires the C-terminal regions of both α and σ70 subunits of Escherichia coli RNA polymerase. J Biol Chem. 1996;271:32343–32348. doi: 10.1074/jbc.271.50.32343. [DOI] [PubMed] [Google Scholar]
- Barberis A, Pearlberg J, Simkovich N, Farrell S, Reinagel P, Bamdad C, Sigal G, Ptashne M. Contact with a component of the polymerase II holoenzyme suffices for gene activation. Cell. 1995;81:359–368. doi: 10.1016/0092-8674(95)90389-5. [DOI] [PubMed] [Google Scholar]
- Blatter EE, Ross W, Tang H, Gourse RL, Ebright RH. Domain organization of RNA polymerase α subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell. 1994;78:889–896. doi: 10.1016/s0092-8674(94)90682-3. [DOI] [PubMed] [Google Scholar]
- Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heynecker HL, Boyer HW. Construction and chracterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- Burgess RR. Separation and characterization of the subunits of ribonucleic acid polymerase. J Biol Chem. 1969;244:6168–6176. [PubMed] [Google Scholar]
- ————— . Purification and physical properties of E. coli RNA polymerase. In: Losick R, Chamberlin M, editors. RNA polymerase. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1976. pp. 69–100. [Google Scholar]
- Busby S, Ebright RH. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell. 1994;79:743–746. doi: 10.1016/0092-8674(94)90063-9. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Struhl K. Connecting a promoter-bound protein to TBP bypasses the need for a transcriptional activation domain. Nature. 1995;374:820–822. doi: 10.1038/374820a0. [DOI] [PubMed] [Google Scholar]
- Dove SL, Joung JK, Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386:627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- Ebright RH, Busby S. The E. coli RNA polymerase α subunit: Structure and function. Curr Opin Genet Dev. 1995;5:197–203. doi: 10.1016/0959-437x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- Farrell S, Simkovich N, Wu Y, Barberis A, Ptashne M. Gene activation by recruitment of the RNA polymerase II holoenzyme. Genes & Dev. 1996;10:2359–2367. doi: 10.1101/gad.10.18.2359. [DOI] [PubMed] [Google Scholar]
- Gaal T, Ross W, Blatter EE, Tang H, Jia X, Krishnan VV, Assa-Munt N, Ebright RH, Gourse RL. DNA-binding determinants of the α subunit of RNA polymerase: Novel DNA-binding domain architecture. Genes & Dev. 1996;10:16–26. doi: 10.1101/gad.10.1.16. [DOI] [PubMed] [Google Scholar]
- Gaudreau L, Schmid A, Blaschke D, Ptashne M, Horz W. RNA polymerase II holoenzyme recruitment is sufficient to remodel chromatin at the yeast PHO5 promoter. Cell. 1997;89:55–62. doi: 10.1016/s0092-8674(00)80182-8. [DOI] [PubMed] [Google Scholar]
- Gentry DR, Burgess RR. The cloning and sequence of the gene encoding the omega (θ) subunit of Escherichia coli RNA polymerase. Gene. 1986;48:33–40. doi: 10.1016/0378-1119(86)90349-5. [DOI] [PubMed] [Google Scholar]
- ————— rpoZ, encoding the omega subunit of Escherichia coli RNA polymerase, is in the same operon as spoT. J Bacteriol. 1989;171:1271–1277. doi: 10.1128/jb.171.3.1271-1277.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Overproduction and purification of the omega subunit of Escherichia coli RNA polymerase. Protein Expr Purif. 1990;1:81–86. doi: 10.1016/1046-5928(90)90050-9. [DOI] [PubMed] [Google Scholar]
- ————— Cross-linking of Escherichia coli RNA polymerase subunits: identification of β′ as the binding site of θ. Biochemistry. 1993;32:11224–11227. doi: 10.1021/bi00092a036. [DOI] [PubMed] [Google Scholar]
- Gentry D, Xiao H, Burgess R, Cashel M. The omega subunit of Escherichia coli K-12 RNA polymerase is not required for stringent RNA control in vivo. J Bacteriol. 1991;173:3901–3903. doi: 10.1128/jb.173.12.3901-3903.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber JS, Hinton DM. An N-terminal mutation in the bacteriophage T4 motA gene yields a protein that binds DNA but is defective for activation of transcription. J Bacteriol. 1996;178:6133–6139. doi: 10.1128/jb.178.21.6133-6139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Couto E, Klages N, Strubin M. Synergistic and promoter-selective activation of transcription by recruitment of transcription factors TFIID and TFIIB. Proc Natl Acad Sci. 1997;94:8036–8041. doi: 10.1073/pnas.94.15.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralla JD. Activation and repression of E. coli promoters. Curr Opin Genet Dev. 1996;6:526–530. doi: 10.1016/s0959-437x(96)80079-7. [DOI] [PubMed] [Google Scholar]
- Hayward R, Igarashi K, Ishihama A. Functional specialization within the α subunit of Escherichia coli RNA polymerase. J Mol Biol. 1991;221:23–29. doi: 10.1016/0022-2836(91)80197-3. [DOI] [PubMed] [Google Scholar]
- Heil A, Zillig W. Purification of bacterial DNA-dependent RNA-polymerase from isolated subunits as a tool for the elucidation of the role of the subunits in transcription. FEBS Lett. 1970;11:165–168. doi: 10.1016/0014-5793(70)80519-1. [DOI] [PubMed] [Google Scholar]
- Hellman JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Hengartner CJ, Thompson CM, Zhang J, Chao DM, Liao S, Koleske AJ, Okamura S, Young RA. Association of an activator with an RNA polymerase II holoenzyme. Genes & Dev. 1995;9:897–910. doi: 10.1101/gad.9.8.897. [DOI] [PubMed] [Google Scholar]
- Hidalgo E, Demple B. Spacing of promoter elements regulates the basal expression of the soxS gene and converts SoxR from a transcriptional activator into a repressor. EMBO J. 1997;16:1056–1065. doi: 10.1093/emboj/16.5.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelfarb HJ, Pearlberg J, Last DH, Ptashne M. GAL11P: A yeast mutation that potentiates the effect of weak GAL4-derived activators. Cell. 1990;63:1299–1309. doi: 10.1016/0092-8674(90)90425-e. [DOI] [PubMed] [Google Scholar]
- Hochschild A. How λ repressor talks to RNA polymerase. Curr Biol. 1994;4:440–442. doi: 10.1016/s0960-9822(00)00097-x. [DOI] [PubMed] [Google Scholar]
- Hochschild A, Ptashne M. Homologous interactions of lambda repressor and lambda Cro with the lambda operator. Cell. 1986;44:925–933. doi: 10.1016/0092-8674(86)90015-2. [DOI] [PubMed] [Google Scholar]
- Hu JC. Repressor fusions as a tool to study protein-protein interactions. Structure. 1995;3:431–433. doi: 10.1016/s0969-2126(01)00176-9. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Fujita N, Ishihama A. Identification of a subunit assembly domain in the alpha subunit of Escherichia coli RNA polymerase. J Mol Biol. 1991;218:1–6. [PubMed] [Google Scholar]
- Inouye C, Remondelli P, Karin M, Elledge S. Isolation of a cDNA encoding a metal response element binding protein using a novel expression cloning procedure: The one hybrid system. DNA Cell Biol. 1994;13:731–742. doi: 10.1089/dna.1994.13.731. [DOI] [PubMed] [Google Scholar]
- Ishihama A. Role of the RNA polymerase α subunit in transcription activation. Mol Microbiol. 1992;6:3283–3288. doi: 10.1111/j.1365-2958.1992.tb02196.x. [DOI] [PubMed] [Google Scholar]
- Jeon YH, Yamazaki T, Otomo T, Ishihama A, Kyogoku Y. Flexible linker in the RNA polymerase alpha subunit facilitates the independent motion of the C-terminal activator contact domain. J Mol Biol. 1997;267:953–962. doi: 10.1006/jmbi.1997.0902. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell. 1994;77:599–608. doi: 10.1016/0092-8674(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Klages N, Strubin M. Stimulation of RNA polymerase II transcription initiation by recruitment of TBP in vivo. Nature. 1995;374:822–823. doi: 10.1038/374822a0. [DOI] [PubMed] [Google Scholar]
- Kustu S, North AK, Weiss DS. Prokaryotic transcriptional enhancers and enhancer-binding proteins. Trends Biochem Sci. 1991;16:397–402. doi: 10.1016/0968-0004(91)90163-p. [DOI] [PubMed] [Google Scholar]
- Kuldell N, Hochschild A. Amino acid substitution in the −35 recognition motif of σ70 that result in defects in phage λ repressor-stimulated transcription. J Bacteriol. 1994;176:2991–2998. doi: 10.1128/jb.176.10.2991-2998.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Hoover TR. Protein crosslinking studies suggest that Rhizobium meliloti C4-dicarboxylic acid transport protein D, a σ54-dependent transcriptional activator, interacts with σ54 and the β subunit of RNA polymerase. Proc Natl Acad Sci. 1995;92:9702–9706. doi: 10.1073/pnas.92.21.9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Struhl K. A severely defective TATA-binding protein-TFIIB interaction does not preclude transcriptional activation in vivo. Mol Cell Biol. 1997;17:1336–1345. doi: 10.1128/mcb.17.3.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Herskowitz I. Isolation of ORC6, a component of the yeast origin recognition complex by a one-hybrid system. Science. 1993;262:1870–1874. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- Li M, Moyle H, Susskind MM. Target of the transcriptional activation function of phage λ cI protein. Science. 1994;263:75–77. doi: 10.1126/science.8272867. [DOI] [PubMed] [Google Scholar]
- Miller A, Wood D, Ebright RH, Rothman-Denes LB. RNA polymerase β′ subunit: A target of DNA binding-independent activation. Science. 1997;275:1655–1657. doi: 10.1126/science.275.5306.1655. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Morett E, Buck M. In vivo studies on the interaction of RNA polymerase-σ54 with the Klebsiella pneumoniae and Rhizobium meliloti nifH promoters. J Mol Biol. 1989;210:65–77. doi: 10.1016/0022-2836(89)90291-x. [DOI] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. Transcription activation at Class II CAP-dependent promoters: Two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- Rao L, Ross W, Appleman JA, Gaal T, Leirmo S, Schlax PJ, Record Jr MT, Gourse RL. Factor independent activation of rrnB P1. An “extended” promoter with an upstream element that dramatically increases promoter strength. J Mol Biol. 1994;235:1421–1435. doi: 10.1006/jmbi.1994.1098. [DOI] [PubMed] [Google Scholar]
- Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, Gourse RL. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science. 1993;263:1407–1413. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- Russo FD, Silhavy TJ. Alpha: The Cinderella subunit of RNA polymerase. J Biol Chem. 1992;267:14515–14518. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sauer RT, Jordan SR, Pabo CO. λ repressor: A model system for understanding protein-DNA interactions and protein stability. Adv Protein Chem. 1990;40:1–61. doi: 10.1016/s0065-3233(08)60286-7. [DOI] [PubMed] [Google Scholar]
- Sternberg NL, Maurer R. Bacteriophage-mediated generalised transduction in Escherichia coli and Salmonella typhimurium. Methods Enzymol. 1991;204:18–43. doi: 10.1016/0076-6879(91)04004-8. [DOI] [PubMed] [Google Scholar]
- Summers AO. Untwist and shout: A heavy metal-responsive transcriptional regulator. J Bacteriol. 1992;174:3097–3101. doi: 10.1128/jb.174.10.3097-3101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MM, Reed RR. Molecular cloning of the olfactory neuronal transcription factor Olf-1 by genetic selection in yeast. Nature. 1993;364:121–126. doi: 10.1038/364121a0. [DOI] [PubMed] [Google Scholar]
- Whipple FW, Kuldell NH, Cheatham LA, Hochschild A. Specificity determinants for the interaction of λ repressor and P22 repressor dimers. Genes & Dev. 1994;8:1212–1223. doi: 10.1101/gad.8.10.1212. [DOI] [PubMed] [Google Scholar]
- Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M. Residual guanosine 3′,5′-bispyrophosphate (ppGpp) synthetic activity of relA null mutants can be eliminated by spoT null mutations. J Biol Chem. 1991;266:5980–5990. [PubMed] [Google Scholar]
- Xiao H, Friesen JD, Lis JT. Recruiting TATA-binding protein to a promoter: Transcriptional activation without an upstream activator. Mol Cell Biol. 1995;15:5757–5761. doi: 10.1128/mcb.15.10.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]