

Abstract
The mammalian target of rapamycin (mTOR) kinase is present in 2 functionally distinct complexes, mTOR complex 1 (mTORC1) and complex 2 (mTORC2). Active mTORC1 mediates phosphorylation of eIF4E-binding protein (4E-BP) and p70 S6 kinase (S6K), which is important for maintaining translation. During human cytomegalovirus (HCMV) infection, cellular stress responses are activated that normally inhibit mTORC1; however, previous data show that HCMV infection circumvents stress responses and maintains mTOR kinase activity. Amino acid deprivation is a stress response that normally inhibits mTORC1 activity. Amino acids can signal to mTORC1 through the Rag proteins, which promote the colocalization of mTORC1 with its activator Rheb-GTP in a perinuclear region, thereby inducing 4E-BP and S6K phosphorylation. As expected, our results show that amino acid depletion in mock-infected cells caused loss of mTORC1 activity and loss of the perinuclear localization; however, there was no loss of activity or perinuclear localization in HCMV-infected cells where the perinuclear localization of Rheb-GTP and mTOR coincided with the perinuclear assembly compartment (AC). This suggested that HCMV infection bypasses normal Rag-dependent amino acid signaling. This was demonstrated by short hairpin RNA (shRNA) depletion of Rag proteins, which had little effect on mTORC1 activity in infected cells but inhibited activity in mock-infected cells. Our data show that HCMV maintains mTORC1 activity in an amino acid- and Rag-independent manner through the colocalization of mTOR and Rheb-GTP, which occurs in association with the formation of the AC, thus bypassing inhibition that may result from lowered amino acid levels.
INTRODUCTION
Human cytomegalovirus (HCMV) is the largest human herpesvirus, with a 230-kb double-stranded DNA genome and the potential to encode over 200 proteins (31, 32). HCMV is a slow-growing betaherpesvirus and, therefore, must maintain favorable cellular conditions for a long period. However, the numerous changes that take place in a cell following HCMV infection, including increased transcription, translation, and metabolism, will cause cellular stress. Cellular stress normally leads to the activation of stress signaling pathways to help conserve energy and resources until more favorable growth conditions return. Some outcomes of cellular stress signaling may be beneficial to a virus, such as an increase in endoplasmic reticulum chaperones (5, 22); however, other aspects, such as the inhibition of translation, will be detrimental to viral growth and survival. For example, mTOR complex 1 (mTORC1) is often inactivated by stress response pathways to inhibit cap-dependent translation in order to save energy and resources during periods of stress; while this may be beneficial for the stressed cell, the inhibition of mTORC1 and translation is deleterious to HCMV infection (10, 30). Consequently, HCMV must be able to circumvent the inhibitory effects of stress signaling so that processes such as translation are maintained during an infection. We and others have shown that HCMV has multiple mechanisms to deal with the deleterious aspects of cellular stress responses while maintaining beneficial ones (1, 5, 6, 8, 16, 21, 22, 24–26, 28–30, 44, 46). We have also previously demonstrated that both mTORC1 and mTORC2 are active during HCMV infection (24, 26) and that maintained mTOR activity is required to establish viral replication (10). From these studies, it is clear that HCMV has mechanisms to maintain mTOR kinase activity under conditions that normally induce strong inhibitory stress responses.
One of the key signaling pathways that activates mTORC1 is the phosphatidylinositol 3′-kinase-Akt-mTOR (PI3K-Akt-mTOR) pathway (1, 6) (Fig. 1). In this pathway, growth factors activate PI3K, leading to the phosphorylation and activation of Akt. Active Akt phosphorylates the tuberous sclerosis complex (TSC), which inhibits the TSC's function as a GTPase-activating protein (GAP) for the Ras homology enriched in brain (Rheb) protein, a member of the Ras family. Specifically, when active, the TSC stimulates the intrinsic GTPase activity of Rheb, converting it from Rheb-GTP to Rheb-GDP, which cannot activate mTORC1. Thus, when the TSC is inactivated by Akt phosphorylation, Rheb-GTP levels remain high, leading to the activation of mTORC1 (36). mTORC1 has multiple downstream effects, including the phosphorylation of the eIF4E binding protein (4E-BP) and p70 S6 kinase (S6K), which, when phosphorylated, promote cap-dependent translation.
Fig. 1.
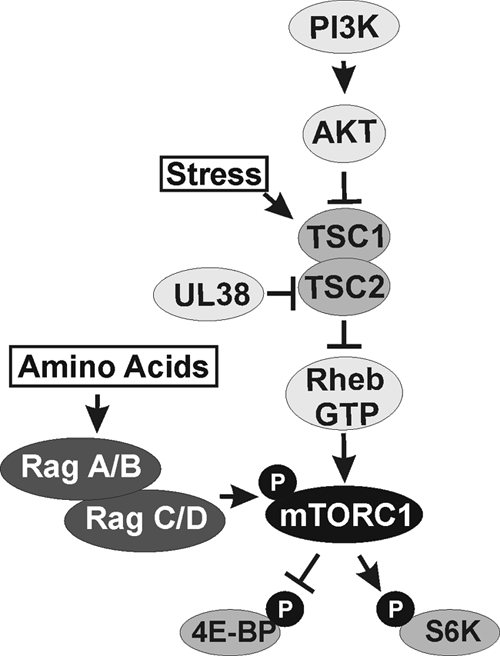
The PI3K-Akt-TSC-mTOR pathway. The general PI3K-Akt-TSC-mTOR pathway is outlined for the activation of mTOR kinase in mTORC1 and subsequent phosphorylation (P) of 4E-BP and S6K. The HCMV UL38 protein protects HCMV-infected cells from many types of cellular stress that signal through the TSC. Amino acid deprivation inhibits mTOR activity through a mechanism that does not depend on the TSC. Amino acid availability can be signaled to mTORC1 through the Rag proteins by regulating the localization of mTORC1 to a perinuclear compartment that contains its activator, Rheb-GTP (35). PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; TSC, tuberous sclerosis complex; Rheb, Ras homology enriched in brain; mTORC1, mammalian target of rapamycin complex 1; Rag, Ras-related GTP-binding proteins; 4E-BP, eIF4E binding protein; S6K, p70 S6 kinase.
Many cellular stresses, such as low energy or hypoxia, act to inhibit mTORC1 activity through a TSC-dependent manner (reviewed in reference 1). However, studies have shown that HCMV maintains mTORC1 activity in the presence of these stresses (24, 25, 29). In part, this is accomplished through an interaction between the viral UL38 protein and the TSC (29), which inactivates the TSC (Fig. 1). Thus, any stress response that intersects the pathway at the TSC, or upstream of the TSC, will be circumvented. However, some types of stress, including amino acid deprivation, inhibit mTORC1 through a TSC-independent mechanism (33, 41). Here we examine the mechanism through which HCMV maintains mTORC1 activity in the absence of amino acid signaling.
Amino acids can signal to mTORC1 through the Ras-related GTP-binding (Rag) proteins (Fig. 1) (23, 35). The 4 mammalian Rag proteins are obligatory heterodimers, with each heterodimer composed of one Rag A or B and one Rag C or D. Rags A and B are orthologs of budding yeast Gtr1p, and Rags C and D are orthologs of budding yeast Gtr2p (19, 39, 40). It has been proposed that amino acids signal to mTORC1 by stimulating the GTP charging of Rag A or B and thereby promoting the association of the Rag proteins with Raptor, the specificity factor of mTORC1. This is followed by the colocalization of mTORC1 and Rheb-GTP to a perinuclear compartment (35).
Our studies examine the importance of amino acid levels, the Rag proteins, and Rheb in regulating mTORC1 activity during an HCMV infection. Our results demonstrate that HCMV maintains mTORC1 activity in the absence of amino acids through a Rag-independent, Rheb-dependent manner. Our data suggest that HCMV accomplishes this by maintaining mTORC1 localization with its activator Rheb-GTP in a perinuclear compartment that corresponds to the HCMV cytoplasmic assembly compartment.
MATERIALS AND METHODS
Cell culture.
Life-extended human foreskin fibroblasts (HFs) (2) or the human glioblastoma-astrocytoma cell line U373-MG (U373) was cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (MEM) (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM GlutaMAX (Gibco; 35050). Serum starvation experiments were conducted using the same medium lacking FCS. HFs were used between passage 2 and 10 after thawing. For amino acid starvation experiments, cells were incubated in RPMI 1640 medium without amino acids (US Biological; R8999-04A) for 50 min. Where applicable, the medium was supplemented with MEM nonessential amino acid (100×) solution (Gibco; 11140), MEM amino acids (50×) solution (Gibco; 11130), GlutaMAX, and dialyzed FCS following the 50-min starvation. The dialyzed FCS was prepared in the lab by extensively dialyzing commercially dialyzed FCS against saline; commercially dialyzed FCS is quite variable in the extent of its depletion of amino acids and requires extensive additional dialysis.
Antibodies.
Primary antibodies used in this study include total and phospho-4E-BP1 (T37/46 and S65) and total and phospho-S6K, Rheb, Rag A/B (Cell Signaling), UL44 and pp28 (Santa Cruz Biotechnology), and β-actin (Chemicon). The antibody that recognizes the common exons 2 and 3 of the HCMV major immediate-early proteins (MIEPs) was prepared by this lab and has previously been described (17). Antibodies used for immunofluorescence include mTOR (Cell Signaling), Rheb C-19 (Santa Cruz Biotechnology), and HCMV gB (Abcam).
Virus preparation, titration, and infection.
HCMV (Towne strain) stocks were prepared and purified as previously described (24). Titers were determined using the 50% tissue culture infective dose (TCID50) method. All experiments were performed using a multiplicity of infection (MOI) of 3.
Plasmids encoding lentiviruses that express shRNAs against Rheb (TRCN0000010424), Rag A (TRCN0000047304), and Rag B (TRCN0000047308) were purchased from Open Biosystems. The lentivirus containing the luciferase shRNA, used as a control, was constructed in this lab (49). Lentiviruses were prepared in 293T cells as previously described (48). Where applicable, lentiviruses were incubated with cells for 7 h in the presence of 10 μg/ml polybrene. Cells were then maintained in serum-containing media for 2 days before serum starving.
Amino acid starvation.
HFs grown in 6-well plates were serum starved for 24 h, and mock or HCMV infection (MOI=3) in serum-free DMEM followed. At 48 h postinfection (hpi), mock-infected cells were serum stimulated for 45 min. Mock- and HCMV-infected cells were then washed 3 times with 1× phosphate-buffered saline (PBS) and incubated for 50 min in RPMI 1640 medium without amino acids (plus dialyzed serum, for the mock-infected cells only) or in the same medium supplemented with MEM nonessential amino acid solution, MEM amino acid solution, and GlutaMAX. Following the 50-min incubation, cells were harvested or were incubated for 3, 10, or 30 min in the RPMI medium supplemented with essential and nonessential amino acids and GlutaMAX.
Serum starvation.
In the experiments presented, we used a serum starvation step prior to mock or HCMV infection. We were very careful to avoid any signaling effects in infected cells that may be caused by serum. Thus, we purified the virus stocks and serum starved the cells before and during mock or HCMV infection. This procedure tells us how the virus, by itself, affects specific signaling pathways or kinases, in this case mTOR kinase. To have a positive control for active mTOR kinase, we serum stimulated mock-infected cells to activate mTOR kinase.
HCMV growth in the presence of shRNA.
HFs grown in six-well plates were infected with shRNA-expressing lentiviruses in the presence of 10 μg/ml polybrene for 7 h. Following the 7-h incubation, fresh medium was added. Two days later, cells were serum starved for 24 h followed by mock or HCMV infection at an MOI of 3. At 2 hpi, the cells were washed three times with PBS, and 2 ml of serum-free medium was added to each well. At designated time points, 1 ml of medium was removed from the well and transferred to a 15-ml conical tube. The cells in the well were scraped into the remaining 1 ml of medium, sonicated 10 times with 1-s pulses, and combined with the previously collected media. The media were then centrifuged at 4°C for 10 min at 1,800 rpm. The supernatant was transferred to a fresh tube, flash-frozen in liquid nitrogen, and stored at −80°C. Viral titers were calculated using the TCID50 method.
Time course analysis of viral infections.
HFs grown in six-well plates were serum starved for 24 h, and mock or HCMV infection at an MOI of 3 followed. At 2 hpi the cells were washed three times with PBS, and fresh serum-free DMEM was added. At various time points after infection, cells were incubated for 5 min at 4°C in cold RIPA lysis buffer (1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 0.15 M NaCl, 0.01 M sodium phosphate [pH 7.2], 2 mM EDTA, 50 mM sodium fluoride, 200 μM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 1.5 μg aprotinin/ml, 1 μg leupeptin/ml). Cells were then scraped into Eppendorf tubes, and lysates were centrifuged at 4°C for 10 min at maximum speed in a Beckman microcentrifuge. The supernatant was transferred to a fresh tube and stored at −20°C until ready for Western blot analysis.
Western analyses.
A 3× loading dye (187.5 mM Tris-HCl [pH 6.8], 6% sodium dodecyl sulfate, 30% glycerol, 0.3% bromophenol blue, 467 mM β-mercaptoethanol) was added to lysates, and the samples were boiled for 5 min. Proteins were separated by 8% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membranes. The membranes were blocked for 1 h in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and then incubated in primary antibody overnight at 4°C with the appropriate antibody diluted in 2% bovine serum albumin in TBST. Membranes were then incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (Thermo) diluted in 5% milk and visualized with ECL reagents (Roche).
Indirect immunofluorescence.
Coverslips containing U373 cells were mock- or HCMV-infected (MOI=3). At 72 hpi, cells were washed in PBS and fixed in 4% paraformaldehyde at room temperature. Cells were permeabilized in PBS containing 0.5% Triton X-100 and blocked in PBS containing 10% human serum (blocking buffer). Primary and secondary antibodies (Alexa Fluor 594 and 647 [Invitrogen] and donkey anti-goat IgG-FITC [Santa Cruz Biotechnology]) were diluted in blocking buffer. Coverslips were washed in PBS, rinsed in H2O, and mounted on slides using Vectashield mounting medium containing 4′6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Slides were examined using a Nikon Eclipse E600 (40×) or a Leica AF6500 (63×) microscope, and pictures were taken using a Hamamatsu camera. LAS-AF 3D deconvolution software was used to deconvolute a maximum projection z-axis stack of 64 sections.
RESULTS
mTOR kinase activity is maintained following amino acid starvation of HCMV-infected cells.
In our experiments, we analyze mTOR kinase activity by measuring the levels of phosphorylation of 4E-BP1 and S6K. The reason that the virus maintains phosphorylation of 4E-BP is to maintain cap-dependent translation (6). Specifically, phosphorylated 4E-BP cannot bind eukaryotic initiation factor 4E (eIF4E); thus, it cannot function to inhibit cap-dependent translation. Mammalian 4E-BP1 has been reported to be phosphorylated on multiple sites: Thr37, Thr46, Ser65, Thr70, Ser83, Ser101, and Ser112 (14, 18, 45). Thr37 and Thr46 are coordinately phosphorylated by mTOR kinase (as part of mTORC1), priming the hierarchical phosphorylation of Thr70, followed by that of Ser65, which ultimately results in 4E-BP1 release from eukaryotic initiation factor 4E (eIF4E), thus allowing cap-dependent translation (15). It is not clear what kinase(s) phosphorylates Ser65 and Thr70. The phosphorylation of Ser83 is not required for 4E-BP1 release from eIF4E (9). Reports suggest an importance for Ser101 and Ser112 phosphorylation in 4E-BP1 function, but mechanisms are not established (45). In our experiments, we analyze 4E-BP phosphorylation in two ways; first, we utilize an antibody that recognizes total 4E-BP1. This not only indicates the total amount of 4E-BP1, but also indicates the phosphorylated forms which migrate more slowly in SDS-PAGE (as many as 3 to 5 bands can be distinguished). This provides a good general measure of phosphorylation since events that cause hypophosphorylation result in a decrease in the number of bands, and these hypophosphorylated forms migrate faster in SDS-PAGE. Therefore, increased migration indicates loss of phosphorylation and correlates with loss of mTOR kinase activity. The second means of analyzing mTOR phosphorylation uses phospho-specific antibodies that detect 4E-BP phosphorylated on residues Thr37/Thr46 (the mTOR sites) or Ser65, which is described above. For the detection of S6K, we use an antibody that detects total S6K and one that detects phosphorylation at the mTOR site T389.
Amino acid starvation inhibits mTORC1 activity in uninfected cells through a TSC-independent mechanism (Fig. 1). To determine whether mTOR activity was inhibited in HCMV-infected cells in the absence of amino acid signaling, HFs or U373 cells were serum starved for 24 h followed by mock or HCMV infection (MOI=3). After 48 h the mock-infected cells were serum stimulated for 45 min. Mock- and HCMV-infected cells were subsequently washed 3 times with PBS, and a 50-min incubation in RPMI without amino acids or the same medium supplemented with essential and nonessential amino acids and GlutaMAX followed; the medium added to the mock-infected cells also contained dialyzed serum. After a 50-min incubation in RPMI, cells were harvested or cells that were amino acid starved were incubated in RPMI supplemented with essential and nonessential amino acids and GlutaMAX (and dialyzed serum for the mock-infected cells) for an additional 3, 10, or 30 min. Cells were then harvested, and lysates analyzed by Western analysis using antibodies that recognize total or phospho-4E-BP1 or S6K.
Figure 2 shows the results of Western blot analysis examining the phosphorylation of 4E-BP1 and S6K in HFs (Fig. 2A) or U373 cells (Fig. 2B). Phosphorylation of 4E-BP1 and S6K was significantly lowered by the 50-min amino acid starvation in both HFs and U373 cells that had been mock infected (Fig. 2A and B, compare lanes 1 and 2). This is demonstrated using a phospho-specific S6K antibody and antibodies specific for total 4E-BP1 or 4E-BP1 phosphorylated on amino acids threonine 37/46 or serine 65. Upon restoration of amino acids to the starved, mock-infected cells, 4E-BP1 and S6K phosphorylation was incrementally regained by between 3 and 30 min (Fig. 2A and B, lanes 3, 4, and 5). For HF cells the 4E-BP1 and S6K phosphorylation at 30-min post-amino acid restoration was substantial, nearing the level seen in cells maintained in amino acid-containing medium (Fig. 2A, compare lanes 1 and 5). In U373 cells, the levels of 4E-BP1 and S6K phosphorylation at 30 min post-amino acid restoration were comparable to those observed with the cells maintained in amino acid-containing medium (Fig. 2B, compare lanes 1 and 5). Conversely, mTOR activity, as measured by 4E-BP1 and S6K phosphorylation, was maintained in HCMV-infected cells following 50 min of amino acid starvation (Fig. 2A and B, compare lanes 6 and 7) and remained active under all conditions tested (Fig. 2A and B, lanes 6 to 10) in both cell types. These data show that HCMV-infected cells are protected from the inhibition of mTOR activity normally induced by amino acid depletion.
Fig. 2.

mTORC1 activity is maintained in the absence of amino acid signaling. Western blot analysis of mock- or HCMV-infected HFs (A) or U373s (B) following incubation in complete medium (lanes 1 and 6) or in medium lacking amino acids (lanes 2 and 7) for 50 min. Following 50 min of amino acid starvation, amino acids were restored for 3, 10, or 30 min before extraction (lanes 3 to 5 and 8 to 10). All samples were harvested at 48 hpi and analyzed for expression of total or phospho-S6K, total or phospho-4E-BP1, and actin. N/A, not applicable.
HCMV can establish infection, maintain mTOR activity, and grow in cells depleted of Rag A and B.
A major mechanism for signaling amino acid availability to mTORC1 involves the Rag proteins, a family of Ras-related small GTP-binding proteins (Fig. 1) (23, 27, 35, 39). There are 4 mammalian Rag proteins: Rag A, B, C, and D. These proteins are obligate heterodimers; each heterodimer contains one Rag A or B and one Rag C or D (40). Rag proteins do not directly stimulate the kinase activity of mTORC1 but are thought to bind to Raptor and promote the intracellular localization of mTORC1 to a perinuclear compartment that also contains its activator Rheb-GTP (35). Thus, we asked whether HCMV is regulating the Rag proteins in order to maintain mTORC1 activity under conditions of amino acid deprivation. U373 cells were used for the following experiments because knockdown of the Rag proteins was more effective in U373 cells than in HFs. U373 cells were infected with lentiviral vectors expressing shRNAs specific for luciferase (control) or the combination of Rag A and B; since Rag A and B obligatorily dimerize with Rag C or D, depletion of Rag A and depletion of Rag B together result in the inability to form functional dimers. A total of 48 h after the addition of luciferase- or Rag-specific shRNAs, the cells were serum starved for 24 h, and mock or HCMV infection (MOI=3) followed. At 48 hpi, the cells were harvested, and lysates were subjected to Western analysis to monitor the expression of phosphorylated S6K, total 4E-BP1, or 4E-BP1 specifically phosphorylated on amino acid residues threonine 37/46 or serine 65, Rag A/B and the early viral protein UL44. The results in Fig. 3 show that the depletion of the Rag proteins was effective and caused a substantial loss of S6K and 4E-BP1 phosphorylation in mock-infected cells; however, in HCMV-infected cells, the loss of S6K and 4E-BP1 phosphorylation was much less. In the case of 4E-BP1, the most significant effect of Rag A and B depletion is a decrease in the amount of total 4E-BP1 rather than a significant loss in the ability to phosphorylate 4E-BP1. These results suggest that the HCMV-mediated activation of mTOR activity is substantially independent of the Rag proteins and suggests that HCMV mediates an mTOR-activating mechanism downstream of, or independent of, the Rag proteins; this virally induced mechanism is also unaffected by amino acid availability.
Fig. 3.

mTOR kinase remains active following depletion of the Rag proteins in HCMV-infected cells. Western analysis of mock- or HCMV-infected cells treated with shRNA specific for luciferase (Luc) or Rags A and B (Rag). Cells were harvested 48 h after mock or HCMV infection (48 hpi), and expression of Rags A and B, phospho-S6K, and total- and phospho-4E-BP1 and expression of the viral early protein UL44 and actin were examined.
At the time of HCMV infection, the cells expressing the Rag shRNAs were already substantially depleted of Rag A and B, yet an infection was established, as indicated by the accumulation of the HCMV early protein UL44 (Fig. 3). Nonetheless, we also examined the effect of Rag knockdown on HCMV growth. A total of 48 h after the addition of luciferase- or Rag-specific shRNA, U373 cells were serum starved for 24 h, and HCMV infection (MOI=3) followed. The cells were harvested for progeny viruses at 0 (mock), 72, or 96 hpi to determine the viral titer using the TCID50 method (Fig. 4 A), or for Western analysis (Fig. 4B). The viral titration data show that viral growth in Rag A- and B-depleted cells lags behind that of normal cells by no more than one log at each time point. The Western analysis shows that the Rag A and B depletion was very successful, and in agreement with the data shown in Fig. 3, there was a lowering of total 4E-BP in the Rag-depleted cells, but phosphorylation was maintained in the infected cells. Overall, the data show that an HCMV infection can be established and proceed in Rag A/B-depleted cells; albeit, the lack of Rag proteins from the beginning of infections does appear to slow the accumulation of progeny virions. This may be due, at least in part, to the reduced accumulation of late proteins in Rag-depleted cells; this is suggested by the decreased amounts of pp28 in Rag-depleted cells at 72 and 96 hpi in comparison to control-infected cells (Fig. 4B). The results again show that HCMV infection can proceed and maintain mTOR activity utilizing mechanisms that are, in large part, independent of the Rag proteins and amino acid signaling.
Fig. 4.

HCMV growth is moderately inhibited following knockdown of the Rag proteins. An HCMV infection time course (0, 72, and 96 hpi) analysis was performed with U373 cells treated with luciferase- (Luc) or Rag A and B (Rag)-specific shRNA. (A) Viral titer was determined at 72 and 96 hpi for cells treated with luciferase-specific (black bars) or Rag-specific (gray bars) shRNAs. (B) Western blot analysis of corresponding protein samples shows effective knockdown of the Rag proteins at these time points; mock is equivalent to that at the zero time point of analysis described for panel A.
HCMV-infected cells require Rheb expression for viral growth and mTORC1 activity.
Rheb (Ras homology enriched in brain) is a small GTPase in the Ras family which was first identified in a screen examining genes that are induced in brain neurons by synaptic activity; it is ubiquitously expressed but particularly abundant in muscle and brain (47). As discussed in the introduction, Rheb-GTP activates mTORC1 under normal conditions (Fig. 1). Although it has been assumed that Rheb is required for an HCMV infection, it has never been tested. Since the Rag proteins largely can be eliminated from an HCMV infection, it is possible that HCMV can induce an alternative mTORC1 activation mechanism independent of Rheb. However, in contrast to the Rag proteins, our data show that HCMV cannot establish an infection or grow in Rheb-depleted cells (Fig. 5). The Western analysis shown in Fig. 5A demonstrates that the levels of Rheb are substantially increased during an infection time course, suggesting that Rheb may play an important role during infection.
Fig. 5.

HCMV infection fails to activate mTOR kinase in Rheb-depleted cells. (A) Western analysis of samples from a viral infection time course analysis show that levels of Rheb are increased during HCMV infection of U373 cells. (B) Western analysis of mock- or HCMV-infected cells treated with shRNA specific for luciferase (L) or Rheb (R). U373 cells were harvested at 48 hpi, and expression of Rheb, phosphorylated S6K, and 4E-BP1 and expression of the viral early and late proteins UL44 and pp28 were examined. (C) An HCMV infection time course analysis was performed with U373 cells treated with luciferase (L)- or Rheb (R)-specific shRNA. Viral titer was determined at 72 and 96 hpi for cells treated with a luciferase-specific (black bars) or Rheb-specific (gray bars) shRNA.
To examine whether HCMV-infected cells maintain mTORC1 activity following Rheb depletion, U373 cells were infected for 48 h with lentivirus vectors which express either a control luciferase shRNA or a Rheb-specific shRNA and then serum starved for 24 h, and mock or HCMV infection (MOI=3) followed. Cells were collected at 48 hpi, and lysates were separated by SDS-PAGE and subjected to Western analysis examining the levels of total and phospho-4E-BP1 and S6K as well as several viral proteins. The results (Fig. 5B) show that Rheb depletion was very efficient, resulting in a dramatic loss of S6K and 4E-BP1 phosphorylation in both mock- and HCMV-infected cells. This correlated with a substantial decrease in the expression of the early and late viral proteins UL44 and pp28. Correspondingly, infectious progeny virion levels at 72 and 96 hpi show no viral growth in Rheb-depleted cells (Fig. 5C). These data suggest that Rheb depletion in serum-starved cells reduces HCMV replication approximately 1,000-fold.
Perinuclear localization of mTOR and Rheb during HCMV infection corresponds with the cytoplasmic assembly compartment.
Previously published data demonstrate that mTOR localizes to the endomembrane system of cells, including the endoplasmic reticulum, Golgi apparatus, and endosomes (13). The results of studies of mTOR localization in the presence and absence of amino acids (35) demonstrated that mTOR localizes to tiny puncta throughout the cytoplasm in amino acid-starved HEK-293T cells, whereas mTOR localizes to a perinuclear region of the cell and/or to large vesicular structures in cells which had been starved and then restimulated with amino acids for as little as 3 min. We used immunofluorescence microscopy to examine whether this mTOR localization pattern also occurred in uninfected U373 cells. Figure 6 shows that uninfected U373 cells in complete medium (+AA) maintained mTOR localization to perinuclear regions. However, amino acid starvation for 50 min caused mTOR to become dispersed in the cytoplasm in tiny puncta. Upon readdition of amino acids to the starved cells (Fig. 6, −AA +AA), mTOR returned to the perinuclear location within 30 min of amino acid addition, corresponding to the return of mTOR activity in amino acid-starved cells in which amino acids had been restored (Fig. 2). Thus, mTOR localization in response to amino acids in U373 cells is the same as that reported for HEK-293T cells (35).
Fig. 6.

Amino acid (AA) regulation of mTOR localization in normal U373 cells. U373 cells were maintained in complete medium (+AA), medium lacking amino acids for 50 min (−AA), or in medium lacking amino acids for 50 min and subjected to the restoration of amino acids for 30 min (−AA +AA). Cells were stained for mTOR (green), and the nuclei were visualized with DAPI (blue). Under complete-medium conditions, 70% of the cells showed definable mTOR-containing perinuclear regions; this dropped to less than 10% when the amino acids were removed and returned to 70 to 80% when the amino acids were restored.
We next determined the localization of mTOR in HCMV-infected U373 cells under the same amino acid starvation and restoration conditions. Figure 7A shows that mTOR is quite concentrated in a perinuclear structure in infected cells in complete medium (+AA). Unlike uninfected cells, mTOR is maintained in this perinuclear structure during amino acid starvation (Fig. 7A, −AA), suggesting that HCMV has induced a mechanism that maintains mTORC1 in its activated state during amino acid depletion.
Fig. 7.

mTOR and Rheb localize to the perinuclear viral assembly compartment during HCMV infection. (A) At 72 hpi, HCMV-infected U373 cells were maintained in complete medium (+AA), medium lacking amino acids for 50 min (−AA), or in medium lacking amino acids for 50 min and subjected to the restoration of amino acids for 30 min (−AA +AA). Cells were stained for mTOR (green), HCMV gB (red), and DAPI (blue). Colocalized staining of mTOR and gB was observed in 80% of the cell. (B) Colocalization of mTOR and Rheb in the viral assembly compartment. At 72 hpi, HCMV-infected U373 cells were stained for mTOR (red), Rheb (green), and DAPI (blue).
Interestingly, the perinuclear structure is very similar to the HCMV-induced cytoplasmic assembly compartment (AC) (4, 12, 37). To determine whether mTOR was localized to the AC, we stained for HCMV glycoprotein B (gB), a known component and marker of the AC. As shown in Fig. 7A, gB and mTOR colocalize, indicating that mTOR is maintained in the AC. These data suggest that during HCMV infection, mTOR can be maintained in an active state in its perinuclear location due to its inclusion or association with the AC. This can explain why mTOR activity is maintained in the absence of amino acids and substantially maintained when the Rag proteins are depleted. These findings suggest that mTOR's activator Rheb may also be in the AC. Figure 7B shows that there is a close association of mTOR and Rheb in the AC. These data also show the mTOR-Rheb association in the perinuclear region of mock-infected cells maintained in complete medium. These data suggest that HCMV maintains mTORC1 activity in the absence of amino acids by maintaining mTOR and Rheb localization in the viral AC.
DISCUSSION
Our previous data and the data of others (reviewed in reference 1 and 6) show that HCMV is a master at manipulating cell signaling and stress response signaling to maintain cellular activity when it would normally be inhibited. The present data expand this to include amino acid stress. Our data demonstrate that HCMV-infected cells, but not mock-infected cells, are able to maintain mTORC1 activity during periods of amino acid depletion (Fig. 2). To elucidate the mechanism used to maintain mTORC1 activity during amino acid deprivation, cells were depleted of the Rag proteins (Fig. 3), which have been demonstrated to signal amino acid availability to mTORC1 (23, 35). HCMV-infected cells, but not mock-infected cells, substantially maintained mTORC1 activity in the absence of the Rag proteins. Since the Rag proteins are involved in regulating the localization of mTORC1 to a perinuclear compartment with its activator Rheb-GTP, our results suggested that HCMV has a mechanism to maintain mTORC1 and Rheb-GTP in the active, perinuclear location in an amino acid- and Rag-independent manner. The significance of Rheb-GTP in maintaining mTORC1 activity in HCMV-infected cells was indicated by the inability of HCMV to complete an infection in Rheb-depleted cells (Fig. 5).
Thus, our results show that HCMV is able to maintain mTORC1 activity during amino acid depletion and that the mechanism used is dependent on the colocalization of Rheb-GTP and mTOR in a perinuclear location which is maintained under amino acid starvation and Rag depletion conditions. Interestingly, the perinuclear localization of Rheb and mTOR corresponds to the cytoplasmic viral assembly compartment (AC) (Fig. 7), an HCMV-specific perinuclear structure that is important for HCMV virion assembly and egress. The AC is found nestled against the concave surface of the enlarged, kidney-shaped nucleus characteristic of HCMV infection. It is centered on a microtubule organizing center (MTOC), which is necessary for AC integrity (37, 38). A three-dimensional model of the AC proposes that it is a compartment composed of organelle-specific vesicles which form nested cylinders that make up ordered layers (12). Each layer is thought to contain a specific set of tegument proteins which are transferred to the developing virions as nucleocapsids move successively from outside to the center of the AC. The ordered layers of the AC consist of reorganized components of the secretory apparatus and early-endosomal, trans-Golgi, and Golgi components (12, 38). The endoplasmic reticulum (ER) is found at the periphery of the AC, as shown by antibodies that detect the KDEL ER localization signal; however, the ER chaperone glucose-regulated protein 78 (GRP78; also known as BiP) is relocalized from the ER to the AC (4). Defining the exact origin of this compartment has been complicated by the observation of specific organellar markers in and around the compartment, while other markers of the same organelle are not detected (12). This suggests that the virus recruits and relocalizes specific cellular factors to form the AC. In addition to the cellular markers, many viral proteins are present in the AC. These include both tegument proteins (pp28, pp65, and pp150) and glycoproteins (gB, gO, gH, gL, gM, gN, and gp65) (37, 42). It is proposed that as the nucleocapsid progresses through the rings of the AC, it acquires its tegument layer and final envelope before egressing from the cell (12).
The derivation of the AC by reorganization of the secretory apparatus correlates with reports that mTOR localizes to the endomembrane system of cells, including the ER, Golgi apparatus, and endosomes (13). Thus, mTOR is localized to components similar to those that make up the AC. Recent studies have suggested that amino acids promote the localization of mTORC1, in a Rag-dependent manner, to the lysosomal surface, where the Rag proteins are localized regardless of the presence of amino acids (34). Immunofluorescence studies of the AC suggest that lysosomal markers may not be a feature of the AC; specifically, antibodies to lysosome-associated membrane protein 1 (LAMP1), a lysosomal marker, have been reported to visualize a fine-granular distribution that had no particular relationship with the AC (11). Thus, it is possible that HCMV uncouples mTORC1 localization from Rag protein function in order to diminish the lysosomal localization of mTORC1. At present, the mechanism for localizing mTOR and Rheb to the perinuclear compartment in normal cells is not known; additionally, the means for the formation of the AC largely are not understood, but an involvement of dynein has been established (3, 20).
It will be interesting to determine whether HCMV commandeers the mTORC1/Rheb perinuclear localization mechanisms to form the AC or whether the perinuclear localization of mTORC1 and Rheb-GTP is a scaffold for AC formation. Either mechanism could result in sequestration of mTORC1 and Rheb-GTP such that mTORC1 would remain active and, potentially, out of contact with the Rag proteins. The sequestration of mTOR and Rheb-GTP also explains the rapamycin resistance of mTOR kinase that develops during the course of an HCMV infection (24). Rapamycin functions to inhibit mTOR kinase through an interaction with a small protein FKBP-12; it is possible that this complex cannot permeate the AC; thus, once mTORC1 and Rheb-GTP are sequestered in the AC, mTOR kinase would be resistant to rapamycin.
It is intriguing to speculate that the mTOR/Rheb-GTP sequestration in the AC may allow mTORC1 to function as a kinase for new substrates, such as viral tegument or structural proteins in the AC, thereby facilitating assembly and egress. Equally interesting is the possibility that the AC localization of mTORC1-Rheb GTP may set up a condition for localized protein synthesis within or adjacent to the AC. The precedence for the translation of viral mRNAs in a discrete compartment has been well established for poxviruses (43) and African swine fever virus (7).
ACKNOWLEDGMENTS
We thank all the members of the Alwine lab for support and critical evaluation of the experiments and data and Eric Witze for his assistance with the Leica AF 6500 microscope.
A.J.C. was supported by Training in Tumor Virology Grant T32 CA115299-04 awarded to Erle Robertson. This work was supported by NIH Grant R01 CA157679-01 awarded to J.C.A.
Footnotes
Published ahead of print on 6 July 2011.
REFERENCES
- 1. Alwine J. C. 2008. Modulation of host cell stress responses by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 325:263–279 [DOI] [PubMed] [Google Scholar]
- 2. Bresnahan W. A., Hultman G. E., Shenk T. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchkovich N. J., Maguire T. G., Alwine J. C. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J. Virol. 84:7005–7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchkovich N. J., Maguire T. G., Paton A. W., Paton J. C., Alwine J. C. 2009. The endoplasmid reticulum chaperone BiP/GRP78 is important in the structure and function of the HCMV assembly compartment. J. Virol. 83:11421–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchkovich N. J., Maguire T. G., Paton A. W., Paton J. C., Alwine J. C. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78 which is required for virion assembly. J. Virol. 82:31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buchkovich N. J., Yu Y., Zampieri C. A., Alwine J. C. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signaling pathway. Nat. Rev. Microbiol. 6:266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castello A., et al. 2009. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 5:e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Child S. J., Hakki M., De Niro K. L., Geballe A. P. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78:197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi K. M., McMahon L. P., Lawrence J. C. 2003. Two motifs in the translational repressor PHAS-I required for efficient phosphorylation by mammalian target of rapamycin and for recognition by raptor. J. Biol. Chem. 278:19667–19673 [DOI] [PubMed] [Google Scholar]
- 10. Clippinger A. J., Maguire T. G., Alwine J. C. 2011. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J. Virol. 85:3930–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das S., Pellett P. E. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 85:5864–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das S., Vasanji A., Pellett P. E. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drenan R. M., Liu X., Bertram P. G., Zheng X. F. 2004. FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J. Biol. Chem. 279:772–778 [DOI] [PubMed] [Google Scholar]
- 14. Fadden P., Haystead T. A., Lawrence J. C. 1997. Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J. Biol. Chem. 272:10240–10247 [DOI] [PubMed] [Google Scholar]
- 15. Gingras A. C., et al. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hakki M., Marshall E. E., De Niro K. L., Geballe A. P. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 80:11817–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harel N. Y., Alwine J. C. 1998. Phosphorylation of the human cytomegalovirus 86-kilodalton immediate early protein IE2. J. Virol. 72:5481–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heesom K. J., Avison M. B., Diggle T. A., Denton R. M. 1998. Insulin-stimulated kinase from rat fat cells that phosphorylates initiation factor 4E-binding protein 1 on the rapamycin-insensitive site. Biochem. J. 336.:39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirose E., Nakashima N., Sekiguchi T., Nishimoto T. 1998. RagA is a functional homologue of S. cerevisiae Gtr1p involved in the Ran/Gsp1-GTPase pathway. J. Cell Sci. 111:11–21 [DOI] [PubMed] [Google Scholar]
- 20. Indran S. V., Ballwstas M. E., Britt W. J. 2010. Bicaudal D1-dependent trafficking of HCMV tegument protein pp150 in virus-infected cells. J. Virol. 84:3162–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isler J. A., Maguire T. Goldberg, Alwine J. C. 2005. Production of infectious human cytomegalovirus virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J. Virol. 79:15338–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Isler J. A., Skalet A. H., Alwine J. C. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim E., Goraksha-Hicks P., Li L., Neufeld T., Guan K.-L. 2008. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10:935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kudchodkar S., Yu Y., Maguire T., Alwine J. C. 2004. Human cytomegalovirus infection induces rapamycin insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 78:11030–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kudchodkar S. B., Del Prete G. Q., Maguire T. G., Alwine J. C. 2007. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J. Virol. 81:3649–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kudchodkar S. B., Yu Y., Maguire T. G., Alwine J. C. 2006. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc. Natl. Acad. Sci. U. S. A. 103:14182–14187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Memmott R. M., Dennis P. A. 2009. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 21:656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mohr I. 2006. Phosphorylation and dephosphorylation events that regulate viral mRNA translation. Virus Res. 119:89–99 [DOI] [PubMed] [Google Scholar]
- 29. Moorman N. J., et al. 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moorman N. J., Shenk T. 2010. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 84:5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murphy E., Rigoutsos I., Shibuya T., Shenk T. E. 2003. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. U. S. A. 100:13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murphy E., et al. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nobukuni T., Kozma S. C., Thomas G. 2007. hvps34, an ancient player, enters a growing game: mTOR Complex1/S6K1 signaling. Curr. Opin. Cell Biol. 19:135–141 [DOI] [PubMed] [Google Scholar]
- 34. Sancak Y., et al. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sancak Y., et al. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sancak Y., et al. 2007. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25:903–915 [DOI] [PubMed] [Google Scholar]
- 37. Sanchez V., Greis K. D., Sztul E., Britt W. J. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanchez V., Sztul E., Britt W. J. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-golgi-intermediate compartment. J. Virol. 74:3842–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schurmann A., Brauers A., Massmann S., Becker W., Joost H. G. 1995. Cloning of a novel family of mammalian GTP-binding proteins (RagA, RagBs, RagB1) with remote similarity to the Ras-related GTPases. J. Biol. Chem. 270:28982–28988 [DOI] [PubMed] [Google Scholar]
- 40. Sekiguchi T., Hirose E., Nakashima N., Li M., Nishimoto T. 2001. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins Rag A and Rag B. J. Biol. Chem. 276:7246–7257 [DOI] [PubMed] [Google Scholar]
- 41. Smith E. M., Finn S. G., Tee A. R., Browne G. J., Proud C. G. 2005. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J. Biol. Chem. 280:18717–18727 [DOI] [PubMed] [Google Scholar]
- 42. Theiler R. N., Compton T. 2002. Distinct glycoprotein O complexes arise in a post-Golgi compartment of cytomegalovirus-infected cells. J. Virol. 76:2890–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walsh D., et al. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 28:2648–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walsh D., Perez C., Notary J., Mohr I. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X., Li W., Parra J. L., Beugnet A., Proud C. G. 2003. The C terminus of initiation factor 4E-binding protein 1 contains multiple regulatory features that influence its function and phosphorylation. Mol. Cell. Biol. 23:1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xuan B., Qian Z., Torigoi E., Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 83:3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamagata K., et al. 1994. Rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 269:16333–16339 [PubMed] [Google Scholar]
- 48. Yu Y., Alwine J. C. 2008. Interaction between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J. Virol. 82:4521–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu Y., Maguire T. G., Alwine J. C. 2011. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 85:1573–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]