

Abstract
Although Epstein-Barr virus (EBV) usually establishes an asymptomatic lifelong infection, it is also implicated in the development of germinal center (GC) B-cell-derived malignancies, including Hodgkin's lymphoma (HL). Following primary infection, EBV remains latent in the memory B-cell population, where host-driven methylation of viral DNA contributes to the repression of viral gene expression. However, it is still unclear how EBV harnesses the cell's methylation machinery in B cells, how this contributes to viral persistence, and what impact this has on the methylation of cellular genes. We show that EBV infection of GC B cells is followed by upregulation of the DNA methyltransferase DNMT3A and downregulation of DNMT3B and DNMT1. We show that the EBV latent membrane protein 1 (LMP1) oncogene downregulates DNMT1 and that DNMT3A binds to the viral promoter Wp. Genome-wide promoter arrays performed with these cells showed that EBV-associated methylation changes in cellular genes were not randomly distributed across the genome but clustered at chromosomal locations, consistent with an instructive pattern of methylation, and were in part determined by promoter CpG content. Both DNMT3B and DNMT1 were downregulated and DNMT3A was upregulated in HL cell lines, recapitulating the pattern of expression observed following EBV infection of GC B cells. We also found, by using gene expression profiling, that genes differentially expressed following EBV infection of GC B cells were significantly enriched for those reported to be differentially expressed in HL. These observations suggest that EBV-infected GC B cells are a useful model for studying virus-associated changes contributing to the pathogenesis of HL.
INTRODUCTION
DNA methylation is a common epigenetic modification which contributes to the regulation of gene expression in mammalian cells. However, both hypomethylation-associated oncogene activation and hypermethylation-associated tumor suppressor gene (TSG) silencing can also contribute to cancer initiation and progression. DNA methylation is catalyzed by three DNA methyltransferases (DNMT). Whereas DNMT1 has a preference for hemimethylated DNA and is involved in the maintenance of methylation, DNMT3A and DNMT3B function as de novo methyltransferases. Increased expression of one or more of the DNMT has been reported at a number of sites of cancer and has been shown to be an adverse prognostic factor (1, 23, 39).
Oncogenic viruses are known to modulate the expression of the DNMT. For example, the hepatitis B virus X protein upregulates DNMT1 and DNMT3A but downregulates DNMT3B, the hepatitis C virus core protein upregulates DNMT1 and DNMT3B, human papillomavirus E7 and adenovirus E1A upregulate DNMT1, and Kaposi's sarcoma-associated herpesvirus LANA protein upregulates DNMT3A (2, 6, 18, 24, 30, 31, 40). The major Epstein-Barr virus (EBV) oncogene, the latent membrane protein 1 (LMP1) gene, upregulates DNMT1, DNMT3A, and DNMT3B in nasopharyngeal carcinoma cell lines and has been shown to induce methylation of the tumor suppressor genes, the RARB and CDH13 genes, in these lines (30, 35). LMP2A, another EBV latent gene, has been shown to upregulate DNMT1 and to induce methylation of the tumor suppressor gene, the PTEN gene, in gastric cancer cell lines (7, 13). However, whether EBV modulates the expression of the DNMT in primary B cells has not been reported. This is an important consideration, because EBV has also been implicated in the development of germinal center (GC) B-cell-derived malignancies, including Hodgkin's lymphoma (HL), Burkitt's lymphoma, and posttransplant lymphoproliferative disorder.
EBV infection of resting primary human B cells induces their indefinite proliferation in vitro. Expansion of these B cells gives rise to stable lymphoblastoid cell lines (LCL). The first viral promoter to be activated during in vitro transformation is Wp, which initiates the transcription of the EBV nuclear antigens (EBNA). Thereafter, levels of Wp-initiated transcripts decline and Cp becomes the dominant EBNA promoter in most established LCL. This switch from Wp to Cp usage is important, because it leads to the broadening of virus antigen expression to include all six EBNA and the upregulation of the latent membrane proteins, all of which are probably critical to the virus' strategy for establishing persistence in vivo (14). Although the silencing of Wp is known to be associated with the methylation of its promoter, the DNMT responsible for this has yet to be identified.
In this study, we have focused on HL. We investigate how EBV and its latent genes modulate the expression of the DNMT in GC B cells, the presumptive progenitors of HL. We measure the effect of virus-induced alterations in the expression of these proteins on the methylation of viral and cellular genes before investigating factors which might determine the distribution of methylation changes in cellular genes.
MATERIALS AND METHODS
Isolation and infection of tonsillar GC B cells.
Tonsillar tissue was obtained from the Children's Hospital Birmingham following informed consent (reference number for ethical approval, 06/Q2702/50). Mononuclear cells were isolated by Ficoll-Isopaque centrifugation and CD10+ GC B cells isolated by magnetic separation on LS columns (Miltenyi Biotec Ltd.) using α-CD10-phycoerythrin (PE) (eBioscience) and α-PE microbeads (Miltenyi Biotec Ltd.). Wild-type 2089 EBV particles were produced from 293 cells carrying a recombinant B95.8 EBV genome (kindly provided by Claire Shannon-Lowe), and virus copy number was measured using a BALF5 quantitative PCR (qPCR) assay (32). GC B cells (2 × 106) were infected overnight on a fibroblast feeder layer with wild-type 2089 EBV at a multiplicity of infection of 50.
Maintenance of cell lines.
GC B-cell-derived LCL, HL cell lines (L591, L428, L540, L1236, KMH2), Rael-Burkitt's lymphoma cells (Qp-using cell line), X50-7 LCL (Wp-using cell line), and 11W LCL (an LCL carrying recombinant EBV with 11BamHI W repeats) (14) were maintained at 37°C in RPMI 1640 medium (Sigma) supplemented with 10% (vol/vol) fetal calf serum (FCS) and 1% (vol/vol) penicillin-streptomycin (Gibco).
Quantitative reverse transcriptase PCR.
Total RNA was extracted using the RNeasy minikit (Qiagen). For the detection of EBV transcripts, cDNA synthesis and qPCR assays were performed as previously described (4). For the detection of human transcripts, cDNA was generated using the Superscript III first-strand synthesis system (Invitrogen) with a random primer (Promega). qPCR assays were prepared in a final volume of 25 μl which contained 1 μl cDNA, TaqMan universal PCR master mix (Applied Biosystems), B2M housekeeping assay (Applied Biosystems), and TaqMan assay for the DNMT1 Hs00154749_m1, DNMT3A Hs00171876_m1, DNMT3B Hs01027166_m1, UHRF1 Hs00273589_m1, and GADD45A Mm00432802_m1 target genes (Applied Biosystems). qPCR assays were performed in triplicate using an ABI Prism 7700 sequence detection system (Applied Biosystems). The 2−ΔΔCT method was used to quantify expression relative to the housekeeping control.
Western blotting.
Cells (1 × 107) were lysed in 100 μl radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium vanadate, and protease inhibitor cocktail [Roche]). Denatured protein was run on a sodium dodecyl sulfate-polyacrylamide gel before being transferred to a BioTrace NT membrane (VWR International) and then incubated overnight with primary antibody diluted in 5% (wt/vol) milk. Antibodies used were DNMT1 mouse monoclonal antibody (Abcam) at a 1:500 dilution, DNMT3A goat polyclonal antibody (Santa Cruz) at a 1:100 dilution, and DNMT3B rabbit monoclonal antibody (Abcam) at a 1:200 dilution. MCM-7 (Sigma-Aldrich) at a 1:2,000 dilution was used as a loading control. After being washed with Tris-buffered saline–Tween 20 (TBS-T), blots were incubated for 1 h with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (DakoCytomation). Proteins were visualized using the enhanced chemiluminescence (ECL) technique (Amersham).
Gene expression array analysis.
Ten micrograms of fragmented cRNA was hybridized to HGU133Plus2 microarrays. Microarray chips were analyzed using the GCOS software from Affymetrix, Inc. Probe-level quantile normalization and robust multiarray analysis were performed using the Affymetrix package from the Bioconductor (http://www.bioconductor.org) project. The transcriptional profile of EBV-infected GC B cells was compared to that of GC B cells. Differentially expressed genes were identified using significance analysis of microarrays (SAM) with a Q value threshold of 5% and no fold change threshold.
Methylation microarray experiments.
Genomic DNA was isolated from GC B cells and LCL using phenol-chloroform extraction and ethanol precipitation. Methylated DNA was immunoprecipitated from 5 μg of sonicated genomic DNA using 10 μg 5-methyl cytosine antibody (Eurogentec) as previously described (37). Immunoprecipitated DNA was assayed by SYBR green qPCR (Qiagen) using GAPDH as the unmethylated control and XIST as the methylated control. Immunoprecipitated DNA was amplified according to the Affymetrix protocol, and 7.5 μg of GC B cell and LCL DNA was hybridized to promoter tiling arrays (Affymetrix). The raw array data from each of the biological replicates were quantile normalized. Following normalization, the default settings provided on the Tilemap software program were used when assigning methylation change (15). In brief, the moving average (MA) method was used to analyze normalized data from the methylation arrays. The window size was set to 11 probes, and the maximum gap allowed between each set of 11 probes was set to 300 bp. The peaks were merged if the size of the gap between two peaks was less than that of the maximal gap allowed and the number of probes that failed to pass the cutoff between the two peaks was less than 6. Peaks were discarded if peak length was less than 100 bp or did not contain at least 5 continuous probes passing the cutoff. Left tail analysis was used to calculate the false discovery rate (FDR). All probe sequences were mapped to the genome assembly (Hg18). Genomic regions were visualized using the Affymetrix Integrated Human Genome (IGB) browser.
Bisulfite modification and pyrosequencing.
Genomic DNA (500 ng) was bisulfite converted using the EZ DNA methylation kit (Zymo Research). When validating methylation changes predicted on the array, pyrosequencing primers were designed within the region of predicted change, using Biotage PSQ primer design software. Biotinylated, nonbiotinylated, and sequencing primers are listed in Table S1 in the supplemental material. The PCR was performed in a total volume of 50 μl using 25 μl HotStart Taq master mix (Thermo Scientific), 5 pmol biotinylated primer, 10 pmol nonbiotinylated primer, and 10 μl bisulfite-modified DNA. The pyrosequencing reactions were performed on a Pyromark ID system (Biotage) and analyzed using Pyro Q-CpG software (Biotage).
Chromatin immunoprecipitation analysis (X-ChIP).
Cells were fixed in 1% formaldehyde (Fisher) prior to incubation in lysis buffer (10 mM EDTA, 50 mM Tris-HCl [pH 8], 1% [wt/vol] SDS, 5 mM Na butyrate, 0.01 mM phenylmethylsulfonyl fluoride [PMSF; Sigma], protease inhibitor cocktail [Roche]). DNA fragments were found to be between 200 and 1,000 bp following sonication. For each immunoprecipitation (IP), 100 μl protein G Dynabeads (Invitrogen) was incubated overnight with 2.4 μg of antibody in 0.5 ml RIPA buffer. Antibodies used were DNMT1 mouse monoclonal antibody (Abcam; catalog no. ab13537), DNMT3A goat polyclonal antibody (Santa Cruz; catalog no. sc-10231), DNMT3B rabbit polyclonal antibody (Abcam; catalog no. ab2851), and rabbit IgG antibody (Santa Cruz; catalog no. sc-2027). Antibody-bound beads were isolated using a magnet and incubated with 10 μg sonicated chromatin. Isolated immune complexes were resuspended in elution buffer (20 mM Tris-HCl [pH 7.5], 5 mM EDTA, 5 mM sodium butyrate, 50 mM NaCl, 1% SDS, 50 mM proteinase K) and incubated overnight at 68°C. Input and IP DNA were isolated using the ChIP DNA clean and concentrator kit (Zymo Research). SYBR green qPCR was performed using primers listed in Table S2 in the supplemental material. The extent of DNA binding was calculated as a percentage of input.
RESULTS
EBV and LMP1 modulate the expression of the DNA methyltransferases.
We first performed genome-wide transcriptional profiling for EBV-infected CD10+ GC B cells, the presumptive progenitors of HL. Six weeks following infection, these cells were shown to be polyclonal in nature and to express the typical latency III pattern of viral genes (see Fig. S1 and S2 in the supplemental material). Analysis of these arrays revealed downregulation of DNMT1 and DNMT3B and upregulation of DNMT3A (see File S1 in the supplemental material). We next confirmed these changes, at both the mRNA and protein levels, using three newly established LCL derived from GC B cells taken from different donors (Fig. 1). Using RNA collected from earlier time points, we found that DNMT1 and DNMT3B were downregulated within 48 and 24 h of infection, respectively. DNMT3A expression was first raised 72 h after EBV infection and continued to increase gradually thereafter (Fig. 2). We next investigated whether the LMP1, LMP2A, and EBNA1 viral oncogenes modulated the expression of one or more of the DNMT when transfected into CD10+ GC B cells (36). We found no evidence to suggest that LMP1 alone regulates the expression of DNMT3A or DNMT3B in GC B cells or that the expression of the DNMT is modulated by LMP2A or EBNA1 alone (data not shown). However, we did find that DNMT1 was downregulated at the transcriptional level by LMP1 in these cells (Fig. 3).
Fig. 1.

DNMT1, DNMT3B, and DNMT3A expression in GC B cells and EBV-infected GC B cells. (A) Quantitative reverse transcriptase PCR (qRT-PCR) showing DNMT expression in LCL relative to uninfected GC B cells. Gray bars represent expression in GC B cells isolated from three different patients (1, 2, 3), and black bars represent expression in each of the corresponding EBV-infected GC B cells. The GC B cells with the highest expression of the gene in question served as the reference sample. Assays were carried out in triplicate, and results are presented as 2−ΔΔCT values. (B) Western blots showing DNMT expression in GC B cells and in corresponding EBV-infected GC B cells; MCM7 was used as a loading control.
Fig. 2.

Kinetics of DNMT1, DNMT3B, and DNMT3A expression in LCL derived from GC B cells. qRT-PCR showing changes in DNMT expression in LCL compared to that in GC B cells. Assays were carried out in triplicate, and results are presented as 2−ΔΔCT values. Experiments were performed with the three LCL, and representative results for one LCL are shown.
Fig. 3.
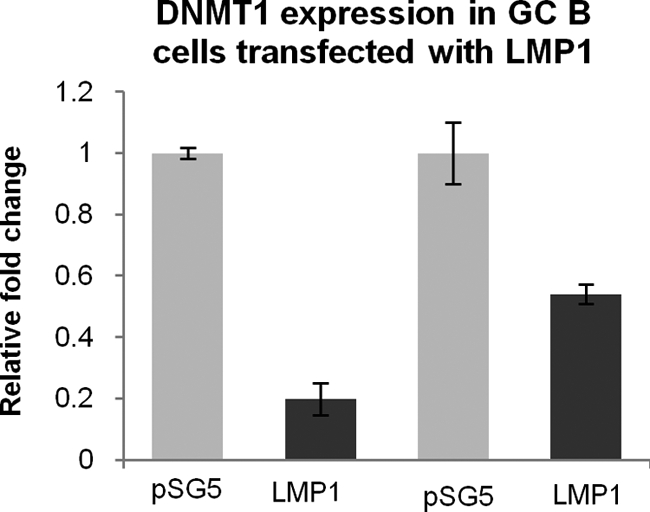
LMP1 downregulates DNMT1 in GC B cells. qRT-PCR showing downregulation of LMP1 following transfection of GC B cells with LMP1. Assays were performed in triplicate, and the results are presented as 2−ΔΔCT values compared to the vector control, pSG5.
Binding of DNA methyltransferases to the methylated Wp promoter.
EBV-induced deregulation of the DNMT may be followed by a change in the methylation status of both viral and cellular genes. We first consider the impact of changes in expression of the DNMT on the methylation status of the Wp viral promoter. This is the first viral promoter to be activated following EBV infection and is known to be silenced shortly thereafter by methylation (14). We first established the methylation status of the Wp promoter in our GC B-cell-derived LCL using pyrosequencing primers covering those regions which bind the YY1 and BSAP transcription factors (Fig. 4 A). At each CpG site, the proportions of viral genomes found to contain methylated forms were similar across each of the LCL (Fig. 4B). Binding of the DNMT to Wp and Cp promoters was then assessed using X-ChIP. In all three LCL, the binding of DNMT3A to regions within Wp was significantly greater than DNMT3A binding either to Cp or to controls (IgG and no antibody). Neither DNMT1 nor DNMT3B was found to bind to Wp (Fig. 5). These analyses were extended to include two LCL (11W LCL and X50-7) and a BL cell line (Rael-BL) in which the methylation status of the Wp promoter had previously been defined using bisulfite genomic sequencing (14). We confirmed, using pyrosequencing, that the Wp promoter is heavily methylated in both Rael-BL and 11W LCL but only partially methylated in X50-7 (see Table S3 in the supplemental material). In both Rael-BL and 11W LCL, DNMT3A binding to Wp was found to be greater than that observed in controls. In contrast, in X50-7, an LCL with a transcriptionally active Wp promoter, there was only minimal binding of DNMT3A and DNMT3B (Fig. 6). In none of these cell lines was there evidence of DNMT binding to Cp.
Fig. 4.

Methylation status of Wp in three GC B-cell-derived LCL. (A) Schematic showing the region of Wp analyzed by pyrosequencing. Black lollipops represent CpG sites, and boxes denote transcription factor binding sites. (B) Percentage methylation at CpG sites in each LCL.
Fig. 5.

Binding of DNMT3A to Wp. (A) Schematic showing regions of Wp analyzed by X-ChIP. Numbers 1 to 9 represent locations of X-ChIP primers. (B) DNMT1, DNMT3B, or DNMT3A binding to nine regions of the Wp and Cp promoters was measured using X-ChIP. Cp provided a negative control, and Sat2, which has previously been shown to bind DNMT3A and DNMT3B (12), provided a positive control. Nonspecific binding was controlled for by including IgG and no-antibody reactions. Each assay was carried out in duplicate, and results are shown as a percentage of input. Experiments were performed with three GC B-cell-derived LCL, and representative results for one are shown.
Fig. 6.

Binding of DNMT1, DNMT3B, and DNMT3A to Wp in 11W LCL, Rael-BL, and X50-7 cell lines. (A) qPCR results showing binding of DNMT3A to regions 5, 6, and 7 of Wp in 11W LCL. (B) Binding of DNMT3A to region 5 in Rael-BL. (C) The absence of DNMT binding in X50-7. Although all regions of Wp were examined, only results for regions 5, 6, and 7 are reported, as the remaining regions showed no evidence of binding (data not shown). Nonspecific binding was controlled for by including IgG and no-antibody reactions. Each assay was carried out in duplicate, and results are shown as a percentage of input. Experiments were performed at least twice, and representative results for one are shown.
Promoter methylation arrays predict widespread changes in cellular genes following EBV infection of GC B cells.
To explore the impact on the cellular epigenome of EBV-induced changes in the expression of the DNMT, genome-wide methylation profiling was performed with DNA harvested from three GC B-cell-derived LCL 6 weeks following infection. Of 19,885 genes listed on the promoter array, 831 were found to be significantly more methylated in the three LCL following infection, and 914 to be less methylated than uninfected GC B cells (see File S2 in the supplemental material). Twelve genes reported to be differentially expressed in microdissected Hodgkin and Reed-Sternberg (HRS) cells, HL cell lines, or both were selected for further validation of their change in methylation status (5, 36). For 11 of these genes, the change in methylation status predicted on the array was confirmed using pyrosequencing (Table 1). Whereas the methylation array predicted an increase in methylation for the remaining gene, the ID2 gene, pyrosequencing revealed both an increase and a decrease in methylation at different CpG sites within this region.
Table 1.
Confirmation of array predictions: summary of pyrosequencing resultsa
| Gene | Cell line | % of genes predicted by array to be hypo- or hypermethylated following EBV infection |
% avg change in methylation status across all CpGs examined | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CpG 1 | CpG 2 | CpG 3 | CpG 4 | CpG 5 | CpG 6 | CpG 7 | |||
| Hypomethylated | |||||||||
| CSMD1 | GC B | 98 | 89 | 87 | 83 | −12.5 | |||
| LCL | 94 | 80 | 70 | 63 | |||||
| SPRY2 | GC B | 14 | 12 | 12 | 16 | 13 | 16 | −11 | |
| LCL | 2 | 0 | 2 | 7 | 0 | 6 | |||
| MAGEA3 | GC B | 18 | 24 | 37 | 39 | −9.75 | |||
| LCL | 10 | 18 | 29 | 22 | |||||
| PRDM1 | GC B | 31 | 29 | 30 | 24 | 26 | −18 | ||
| LCL | 8 | 7 | 14 | 12 | 9 | ||||
| ICMT | GC B | 44 | 49 | 48 | 88 | 54 | −44 | ||
| LCL | 11 | 13 | 6 | 26 | 7 | ||||
| FGFR2 | GC B | 74 | 65 | 72 | 76 | 100 | 86 | 51 | −13.8 |
| LCL | 23 | 62 | 64 | 56 | 94 | 78 | 50 | ||
| GRB10 | GC B | 48 | 91 | 100 | 100 | 80 | 74 | −8.6 | |
| LCL | 20 | 73 | 99 | 100 | 85 | 65 | |||
| Hypermethylated | |||||||||
| ELL3 | GC B | 97 | 62 | 100 | 73 | 8 | |||
| LCL | 97 | 94 | 100 | 75 | |||||
| TCL6 | GC B | 31 | 68 | 56 | 100 | 21 | 23.2 | ||
| LCL | 57 | 95 | 88 | 100 | 52 | ||||
| RBM5 | GC B | 4 | 0 | 4 | 6 | 4 | 2.6 | ||
| LCL | 11 | 6 | 4 | 6 | 4 | ||||
| SMAD4 | GC B | 62 | 63 | 50 | 89 | 100 | 6 | ||
| LCL | 75 | 70 | 57 | 92 | 100 | ||||
| ID2 | GC B | 69 | 69 | 64 | 69 | 61 | 31 | 0.7 | |
| LCL | 56 | 90 | 87 | 56 | 52 | 26 | |||
The direction of the methylation change predicted on the array is recorded for each candidate gene along with the percentage of methylation at each CpG site in both LCL and GC B cells. The average percentage of methylation change is also shown.
Methylation changes are not randomly distributed across the genome.
To determine whether methylation changes in the EBV-infected GC B cells were randomly distributed across the genome, we first investigated whether a gene with increased or decreased methylation following infection was more likely to be found immediately adjacent to one which was concordantly changed. To do this, genes on the promoter array were first ordered by chromosomal location. Excluded from subsequent analyses were 1,596 genes with a transcription start site which fell between the transcription start and end site of the gene which it immediately preceded. Of the remaining 18,389 genes with nonoverlapping transcriptional start sites listed on the promoter array, 751 were found to have increased methylation following EBV infection, and 834 had decreased methylation. Of those genes with increased methylation, 177 (23.6%) were immediately adjacent to another hypermethylated gene, and of those with decreased methylation, 162 (19.4%) were immediately adjacent to another hypomethylated gene. For both hypermethylated and hypomethylated genes, the probability of observing the number of adjacent genes with a concordant change in methylation status, or one greater than this number, is less than 1 in 100,000. We next explored whether methylation changes clustered at certain chromosomal locations. Each of the 19,885 genes listed on the promoter array was assigned one of 806 cytobands. A cytoband was considered a hypermethylation hot spot when it included significantly (P < 0.001) more genes with increased methylation following EBV infection than would have been predicted from the overall frequency of hypermethylated genes on the array, and it was considered a hypomethylation hot spot when it was similarly enriched for hypomethylated genes. Seventeen hypermethylation hot spots and 31 hypomethylation hot spots were identified (see Table S4 in the supplemental material).
CpG content of cellular genes predict frequency and direction of methylation change.
To investigate possible determinants of EBV-associated methylation changes in cellular genes, we first explored how the incidence of methylation events varied with the CpG content of the promoter region using a classification system defined by Weber et al. in 2007 and since adopted by a number of other investigators (26, 28). Compared to genes with a low CpG content, the risk of increased methylation following EBV infection increased with increasing CpG content, with the highest risk observed in those genes with a high CpG content (odds ratio [OR] = 2.11; 95% confidence interval [CI] of 1.7 to 2.6; P < 0.0001). However, compared to genes with a low CpG content, the risk of a decrease in methylation following EBV infection decreased with increasing CpG content, with the lowest risk observed for those genes with the highest CpG content (OR = 0.36; 95% CI of 0.3 to 0.4; P < 0.0001) (Table 2).
Table 2.
Risk of decreased or increased methylation in EBV-infected GC B cells in relation to CpG contenta
| Methylation in EBV-infected GC B cells | CpG content | No. of genes with and without methylation status change in EBV-infected GC B cells |
Odds ratio | 95% CI | P value | |
|---|---|---|---|---|---|---|
| With | Without | |||||
| Increased | Low | 102 | 3,737 | 1 (reference) | ||
| Intermediate | 58 | 1,824 | 1.17 | 0.8 to 1.6 | 0.3597 | |
| High | 465 | 8,058 | 2.11 | 1.7 to 2.6 | <0.0001 | |
| Decreased | Low | 294 | 3,737 | 1 (reference) | ||
| Intermediate | 118 | 1,824 | 0.82 | 0.66 to 1 | 0.082 | |
| High | 230 | 8,058 | 0.36 | 0.3 to 0.4 | <0.0001 | |
Shown is the risk of decreased or increased methylation as defined in reference 37a. Excluded from this analysis were 13 genes that were both hypermethylated and hypomethylated in different regions.
Ontological profile of genes with decreased methylation following EBV infection of GC B cells.
Whereas ontological profiling of genes with increased methylation following EBV infection revealed no significant associations, those with decreased methylation following EBV infection of GC B cells were significantly enriched for a number of biological processes, listed in Table 3. As with all ontological classifications, one gene can be assigned to more than one biological process. For example, the 533 genes with decreased methylation reported in Table 3 correspond to 209 unique genes, of which 73 (35%) were involved in G protein-mediated signaling. Thus, it would seem that the EBV-associated hypomethylation of G protein-mediated signaling genes can explain many of the ontological associations observed for this data set (Table 3). We also found that 54 cancer testis antigens (CTA) identified from a database compiled by the Ludwig Institute (http://www.cancerimmunity.org/CTdatabase/) and supplemented by a search of the NCBI database (n = 232 genes) were also hypomethylated following EBV infection of GC B cells (OR = 6.6; 95% CI of 4.8 to 9.1; P < 0.0001) (see Table S5 in the supplemental material). Some of these CTA, for example, the SSX genes and others from the same genomic location (CT45A1), have been shown to be overexpressed in HL (8).
Table 3.
Biological processes significantly enriched (P < 0.001) among genes with reduced methylation following EBV infection of GC B cellsa
| Biological process | No. of genes with no change in methylation status | No. of genes with decreased methylation (n = 533) | No. of genes with decreased methylation and involved in G protein-mediated signaling | Expected no. of genes | P value (Bonferroni correction applied) |
|---|---|---|---|---|---|
| Chemosensory perception | 149 | 38 | 36 | 7.19 | 4.76E−14 |
| Olfaction | 142 | 37 | 36 | 6.85 | 8.31E−14 |
| Signal transduction | 2739 | 204 | 73 | 132.15 | 1.34E−09 |
| G protein-mediated signaling | 653 | 73 | 73 | 31.51 | 1.09E−08 |
| Sensory perception | 389 | 49 | 39 | 18.77 | 7.49E−08 |
| Cell surface receptor-mediated signal transduction | 1312 | 113 | 73 | 63.3 | 2.47E−07 |
| Cell adhesion-mediated signaling | 310 | 39 | 0 | 14.96 | 2.16E−05 |
PANTHER classification system was used to perform ontological profiling.
The GC B-cell-derived LCL is a useful model for studying the contribution of EBV to the pathogenesis of Hodgkin's lymphoma.
The GC B-cell-derived LCL model appears to be a useful model for studying the contribution of early EBV-associated changes to the pathogenesis of HL. Gene expression profiling performed 6 weeks following infection revealed upregulation of 6,766 genes and downregulation of 7,196 genes compared to that of uninfected GC B cells (FDR < 0.05; no fold change cutoff). Genes previously reported to be differentially expressed in HL cell lines, in microdissected HRS cells, and in LMP1-transfected GC B cells compared with GC B cells are significantly enriched for genes differentially expressed following EBV infection of GC B cells (5, 36) (Table 4). Furthermore, compared with normal GC B cells, DNMT1 and DNTM3B were found to be downregulated and DNMT3A upregulated at the protein level in HL cell lines. This pattern of expression is broadly consistent with the transcriptional changes observed in these HL cell lines and recapitulates those observed following EBV infection of GC B cells (Fig. 7). Two other components of the cells epigenetic machinery, UHRF1, which recruits DNMT1 to hemimethylated replication foci, and GADD45A, a DNA demethylase, were also found to be concordantly regulated in HL cell lines and in GC B cells infected with EBV (Fig. 8) (3, 22, 25).
Table 4.
Genes previously reported to be differentially expressed in HL cell lines, in microdissected HRS cells, and in LMP1-transfected GC B cells compared with GC B cells are significantly enriched for genes differentially expressed following EBV infection of GC B cells (FDR < 0.05; no fold-change cutoff)
| Increased or decreased expression of genes in LCL | Array comparison | No. of genes with increased or decreased expression: |
% of genes concordantly increased or decreased | P value (binomial test) | |
|---|---|---|---|---|---|
| On array | On array and in EBV-infected GC B cells | ||||
| Increased (n = 6,766; 33%) | HL cell lines vs GC B cells | 1,724 | 1,313 | 76 | P < 0.000001 |
| HRS cells vs centrocytes | 6,569 | 2,558 | 39 | P < 0.000001 | |
| HRS cells vs centroblasts | 4,304 | 1,856 | 43 | P < 0.000001 | |
| LMP1-GC B vs GC B cells | 849 | 556 | 65 | P < 0.000001 | |
| Decreased (n = 7,196; 35%) | HL cell lines vs GC B cells | 2,207 | 1,764 | 80 | P < 0.000001 |
| HRS cells vs centrocytes | 2,292 | 1,375 | 60 | P < 0.000001 | |
| HRS cells vs centroblasts | 3,607 | 2,017 | 56 | P < 0.000001 | |
| LMP1-GC B vs GC B cells | 1,347 | 860 | 64 | P < 0.000001 | |
Fig. 7.

DNMT1, DNMT3B, and DNMT3A expression in five HL cell lines. (A) qRT-PCR showing DNMT expression in HL cell lines (black) compared with that of GC B cells (gray). Assays were performed in triplicate, and results are presented as 2−ΔΔCT values. (B) Western blot showing DNMT expression in HL cell lines compared to that in GC B cells. MCM7 was used as a loading control.
Fig. 8.

UHRF1 and GADD45A expression in GC B cells infected with EBV and in HL cell lines. qRT-PCR showing UHRF1 and GADD45A expression in GC B cells infected with EBV (dark gray) and HL cell lines (black) compared to that in GC B cells (light gray). Assays were performed in triplicate, and results are presented as 2−ΔΔCT values.
DISCUSSION
We have shown that whereas EBV downregulates the expression of the DNA methyltransferases DNMT1 and DNMT3B in GC B cells, it upregulates that of DNMT3A; these changes are seen shortly following the onset of EBV infection. This pattern of expression recapitulates that seen in HL cell lines and is different from that associated with EBV infection of epithelial cells. These findings are also consistent with gene expression profiling performed with microdissected HRS cells, which showed a downregulation of DNMT1 and DNMT3B and an upregulation of DNMT3A (5). We also show that LMP1, the major EBV oncogene expressed in HL, is responsible for the downregulation of DNMT1.
Although we have shown deregulation of DNMT3A and DNMT3B in HL cell lines, we were unable to reproduce this pattern of deregulation following transfection of GC B cells with viral genes usually expressed in HL. However, in these experiments, these genes were transfected singly into GC B cells, and we cannot exclude the possibility that the deregulation of DNMT3A and DNMT3B in HL may be dependent on achieving levels of expression not attained in our transfection experiments or that deregulation may be dependent on the cooperative activity of two or more of these viral genes. Such cooperation between EBV latent genes has been shown to modulate the behavior of B cells in other systems (38).
Our observations also show how EBV-induced changes in the expression of the DNMT might contribute to the flexibility of latent promoter usage critical to the virus's strategy for persistence in vivo (14). It has been known for some time that the Wp promoter is silenced by DNA methylation in GC-derived tumors (33). We were able to reveal a potential role for EBV-induced upregulation of DNMT3A in maintaining viral persistence by demonstrating that the expression of Wp begins to decline when that of DNMT3A first increases and also that DNMT3A binds to Wp in the critical BSAP region in a GC B-cell-derived LCL (see Fig. S1 in the supplemental material).
EBV-induced changes in the expression of the DNMT in GC B cells were associated with widespread changes in the methylation status of cellular genes. These changes were not randomly distributed across the genome but appeared to cluster at certain chromosomal locations. Concordant methylation of adjacent CpG island gene promoters has also been reported for a number of gene clusters in cancer, and recent genome-wide analyses have identified large chromosomal regions containing several CpG islands which are often methylated and transcriptionally repressed in cancer (9). Although these observations suggest that coordinated epigenetic control over large regions is common in cancer, this phenomenon has not previously been reported following the infection of primary cells with an oncogenic virus (10, 34). Why particular genes become more methylated and others less methylated following EBV infection of GC B cells has yet to be determined. However, we were able to reveal a relationship between a change in methylation status and promoter CpG content. Whereas promoters with a high CpG content were significantly more likely to have increased methylation following EBV infection, those with a low CpG content were more likely to become less methylated. Similar associations have been reported in melanoma, prostate cancer, and in hematological malignancies. In these tumors, genes with a high CpG content are more likely to be hypermethylated, whereas those with a low CpG content are more likely to be hypomethylated (11, 17, 20, 27). Not surprisingly, this association with CpG content also appears to explain in part the ontological profile of those genes in which methylation changes were observed. For example, we have shown that genes with decreased methylation following EBV infection are significantly enriched for those involved in G protein-mediated signaling. Genes involved in G protein-mediated signaling are in turn significantly enriched for those with a low CpG content (P = 3.98E−132). Thus, it would appear that in both cancer and in primary GC B cells infected with EBV, an instructive pattern of de novo methylation change is determined in part by cis-acting susceptibility factors, such as promoter CpG content. Other determinants of CpG methylation in cancer under investigation, for example, local sequence context, may yet be found to determine the pattern of oncogenic virus-induced methylation changes (21).
We found only one example (TP73) of a TSG which is hypermethylated following EBV infection and which is reported to be epigenetically silenced in HL. However, as there are only 22 TSG reported to be silenced by methylation in HL, and no instance of a gene is reported in the literature to be hypomethylated in this lymphoma, a final judgment on the relevance of EBV-associated methylation changes in GC B cells must be postponed until we have the results of a comparable methylation array performed on microdissected HRS cells. However, our observations offer some clues as to how EBV-induced deregulation of the DNMT might contribute to the pathogenesis of HL. EBV is known to drive the differentiation of B cells toward a post-GC stage, and we have shown that its major oncoprotein, LMP1, reprograms GC B cells toward an HRS-like phenotype by hijacking the B-cell transcriptional program and subverting normal B-cell differentiation (16, 36). Here, we show that LMP1 downregulates DNMT1 in GC B cells. This is of interest because DNMT1 has recently been shown to have an essential role in maintaining the progenitor state of constantly replenishing somatic tissue. DNMT1 depletion is followed by the exit of epidermal cells from the progenitor compartment and their premature differentiation (29). This transition is associated with the downregulation of UHRF1, a component of the cell's DNA methylation machinery, and with the upregulation of GADD45, a putative DNA demethylase. Similar transcriptional changes are seen during normal B-cell differentiation, when DNMT1 and UHRF1 are downregulated and GADD45A is upregulated in both plasma cells and in memory cells compared with results for centrocytes (5). We have confirmed not only the transcriptional downregulation of DNMT1 but also the downregulation of UHRF1 and the upregulation of GADD45A in both EBV-infected GC B cells and in HL cell lines compared with that in GC B cells (Fig. 8). DNMT1 and UHRF1 have also been found to be downregulated and GADD45A to be upregulated on gene expression profiling of microdissected HRS cells compared with that of centrocytes (5). Given that the balance between renewal of progenitor cells and differentiation appears to be controlled by a dynamic antagonism between regulators of DNA methylation, it is tempting to speculate that EBV and its latent genes contribute to the pathogenesis of HL by disrupting the expression of those epigenetic regulators which control this balance.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Frederike von Bonin for performing the immunoglobulin heavy-chain rearrangement PCR.
This work was funded by the Leukaemia and Lymphoma Research Fund (J. A. and M. V.).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 13 July 2007.
REFERENCES
- 1. Amara K., et al. 2010. DNA methyltransferase DNMT3b protein overexpression as a prognostic factor in patients with diffuse large B-cell lymphomas. Cancer Sci. 101:1722–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arora P., Kim E. O., Jung J. K., Jang K. L. 2008. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 261:244–252 [DOI] [PubMed] [Google Scholar]
- 3. Barreto G., et al. 2007. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445:671–675 [DOI] [PubMed] [Google Scholar]
- 4. Bell A. I., et al. 2006. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 87:2885–2890 [DOI] [PubMed] [Google Scholar]
- 5. Brune V., et al. 2008. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 205:2251–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgers W. A., et al. 2007. Viral oncoproteins target the DNA methyltransferases. Oncogene 26:1650–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang M. S., et al. 2006. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin. Cancer Res. 12:2995–3002 [DOI] [PubMed] [Google Scholar]
- 8. Chen Y. T., et al. 2010. Expression of cancer testis antigen CT45 in classical Hodgkin lymphoma and other B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 107:3093–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coolen M. W., et al. 2010. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat. Cell Biol. 12:235–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davidsson J., et al. 2009. The DNA methylome of pediatric acute lymphoblastic leukemia. Hum. Mol. Genet. 18:4054–4065 [DOI] [PubMed] [Google Scholar]
- 11. Gal-Yam E. N., et al. 2008. Frequent switching of polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. U. S. A. 105:12979–12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geiman T. M., et al. 2004. Isolation and characterization of a novel DNA methyltransferase complex linking DNMT3B with components of the mitotic chromosome condensation machinery. Nucleic Acids Res. 32:2716–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hino R., et al. 2009. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 69:2766–2774 [DOI] [PubMed] [Google Scholar]
- 14. Hutchings I. A., et al. 2006. Methylation status of the Epstein-Barr virus (EBV) BamHI W latent cycle promoter and promoter activity: analysis with novel EBV-positive Burkitt and lymphoblastoid cell lines. J. Virol. 80:10700–10711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ji H., Wong W. H. 2005. TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics 21:3629–3636 [DOI] [PubMed] [Google Scholar]
- 16. Klein U., et al. 2006. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 7:773–782 [DOI] [PubMed] [Google Scholar]
- 17. Koga Y., et al. 2009. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 19:1462–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laurson J., Khan S., Chung R., Cross K., Raj K. 2010. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 31:918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Linke B., et al. 1997. Automated high resolution PCR fragment analysis for identification of clonally rearranged immunoglobulin heavy chain genes. Leukemia 11:1055–1062 [DOI] [PubMed] [Google Scholar]
- 20. Martin-Subero J. I., et al. 2009. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One 4:e6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCabe M. T., Lee E. K., Vertino P. M. 2009. A multifactorial signature of DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res. 69:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meilinger D., et al. 2009. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 10:1259–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oh B. K., et al. 2007. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 20:65–73 [PubMed] [Google Scholar]
- 24. Park I. Y., et al. 2007. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology 132:1476–1494 [DOI] [PubMed] [Google Scholar]
- 25. Rai K., et al. 2008. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135:1201–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rauch T. A., Wu X., Zhong X., Riggs A. D., Pfeifer G. P. 2009. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. U. S. A. 106:671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richter J., et al. 2009. Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer 9:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruike Y., Imanaka Y., Sato F., Shimizu K., Tsujimoto G. 2010. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 11:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sen G. L., Reuter J. A., Webster D. E., Zhu L., Khavari P. A. 2010. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 463:563–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seo S. Y., Kim E. O., Jang K. L. 2008. Epstein-Barr virus latent membrane protein 1 suppresses the growth-inhibitory effect of retinoic acid by inhibiting retinoic acid receptor-beta2 expression via DNA methylation. Cancer Lett. 270:66–76 [DOI] [PubMed] [Google Scholar]
- 31. Shamay M., Krithivas A., Zhang J., Hayward S. D. 2006. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. U. S. A. 103:14554–14559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shannon-Lowe C., et al. 2005. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 86:3009–3019 [DOI] [PubMed] [Google Scholar]
- 33. Tao Q., Young L. S., Woodman C. B., Murray P. G. 2006. Epstein-Barr virus (EBV) and its associated human cancers—genetics, epigenetics, pathobiology and novel therapeutics. Front. Biosci. 11:2672–2713 [DOI] [PubMed] [Google Scholar]
- 34. Taylor K. H., et al. 2007. Large-scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res. 67:2617–2625 [DOI] [PubMed] [Google Scholar]
- 35. Tsai C. N., Tsai C. L., Tse K. P., Chang H. Y., Chang Y. S. 2002. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. U. S. A. 99:10084–10089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vockerodt M., et al. 2008. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin's Reed-Sternberg-like phenotype. J. Pathol. 216:83–92 [DOI] [PubMed] [Google Scholar]
- 37. Weber M., et al. 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 37:853–862 [DOI] [PubMed] [Google Scholar]
- 37a. Weber M., et al. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39:457–466 [DOI] [PubMed] [Google Scholar]
- 38. White R. E., et al. 2010. Extensive cooperation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS One 5:e13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xing J., et al. 2008. Expression of methylation-related genes is associated with overall survival in patients with non-small cell lung cancer. Br. J. Cancer 98:1716–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zheng D. L., et al. 2009. Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J. Hepatol. 50:377–387 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.