

Abstract
Oncolytic viruses have been tested against many carcinomas of ectodermal and endodermal origin; however, sarcomas, arising from mesoderm, have received relatively little attention. Using 13 human sarcomas representing seven tumor types, we assessed the efficiency of infection, cytolysis, and replication of green fluorescent protein (GFP)-expressing vesicular stomatitis virus (VSV) and its oncolytically enhanced mutant VSV-rp30a. Both viruses efficiently infected and killed 12 of 13 sarcomas. VSV-rp30a showed a faster rate of infection and replication. In vitro and in vivo, VSV was selective for sarcomas compared with normal mesoderm. A single intravenous injection of VSV-rp30a selectively infected all subcutaneous human sarcomas tested in mice and arrested the growth of tumors that otherwise grew 11-fold. In contrast to other sarcomas, synovial sarcoma SW982 demonstrated remarkable resistance, even to high titers of virus (multiplicity of infection [MOI] of 100). We found no dysfunction in VSV binding or internalization. SW982 also resisted infection by human cytomegalovirus and Sindbis virus, suggesting a virus resistance mechanism based on an altered antiviral state. Quantitative reverse transcriptase (qRT)-PCR analysis revealed a heightened basal expression of interferon-stimulated genes (ISGs). Pretreatment, but not cotreatment, with interferon attenuators valproate, Jak1 inhibitor, or vaccinia virus B18R protein rendered SW982 highly susceptible, and this correlated with downregulation of ISG expression. Jak1 inhibitor pretreatment also enhanced susceptibility in moderately VSV-resistant liposarcoma and bladder carcinoma. Overall, we find that the potential efficacy of VSV as an oncolytic agent extends to nonhematologic mesodermal tumors and that unusually strong resistance to VSV oncolysis can be overcome with interferon attenuators.
INTRODUCTION
Soft tissue sarcomas and osteosarcomas are relatively rare in adults but represent approximately 15% of pediatric malignancies and result in death for approximately one-third of patients within 5 years of diagnosis (19, 20, 50). A number of oncolytic viruses have been developed for targeting carcinomas derived from endodermal and ectodermal epithelia and parenchyma (e.g., breast, colon, lung) (16, 26). In contrast, sarcomas have received relatively little focused analysis with oncolytic viruses, which may have the capacity to target these tumors.
Each malignant tumor has an untransformed cell of origin, and the lineage of this cell influences both the epigenetic and genetic features of the resulting tumor and thus has important consequences for tumor biology, including response to treatment (14). One of the earliest points of divergence in cell lineage is between the three primary germ layers during development, and the resulting epigenetic differences can be expected to have a substantial influence on oncogenesis and tumor behavior. For example, compared to tumors from other germ layers, mesodermal tumors are characterized by a higher frequency of fusion gene-mediated transformations, a different pattern of angiogenesis, and a tendency for hematogenous, rather than lymphatic, metastasis (6, 13, 29, 31, 42). Because of their distinct pedigree, sarcomas may differ from other tumor types in innate immune status, availability of host proteins that support viral replication, or other factors affecting the efficacy of viral oncolysis. Additionally, the diversity encountered within sarcomas, which themselves derive from a wide variety of cells of origin, raises the possibility that not all sarcomas will respond equally to any particular oncolytic virus.
Vesicular stomatitis virus (VSV) has many properties useful in an oncolytic virus, including the absence of genome integration, innately oncoselective replication and killing, and an ability to replicate even in hypoxic areas of tumors, and accordingly has shown significant efficacy in a variety of carcinomas in vitro and in vivo (4, 10, 25, 40). The oncoselectivity of VSV derives at least in part from its sensitivity to the effects of interferon (IFN), combined with the fact that various features of the IFN response are disrupted in many cancers (39, 46).
VSV-G/green fluorescent protein (GFP) is a recombinant VSV featuring the addition, between the G and L genes, of a gene encoding a VSV-G/GFP fusion protein. This strain expresses both the native viral G protein, allowing it to maintain a fitness comparable to that of the wild type, and the fusion protein, which localizes to the plasma membrane to allow easy visualization of infected cells (43). An oncolytically enhanced mutant of VSV-G/GFP is VSV-rp30a, isolated by positive selection after multiple passages on human glioblastoma (48). VSV-rp30a, which has 4 mutations relative to VSV-G/GFP (see Discussion), showed enhanced tumor targeting and destroyed multifocal glioblastoma xenografts in the periphery and in the brains of mice following vascular virus delivery (33). One important question we address here is whether the enhanced oncolytic potential of VSV-rp30a is limited to glioblastoma (a neural ectoderm-derived tumor), or whether it generalizes to nonrelated cancers of mesodermal origin. Although sarcomas rarely originate in the brain, they can metastasize into the brain.
Here we address the potential of VSV to effectively target and kill sarcomas. We tested against a diverse panel of 13 human sarcoma lines. The selectivity of VSV for sarcoma was tested in vitro and in vivo. In a subcutaneous xenograft model, intravenous VSV-rp30a specifically infected and arrested rapidly growing sarcoma tumors. A notable exception to sarcoma susceptibility to VSV was the synovial sarcoma SW982, which was extremely resistant to infection. We determined the mechanism of synovial sarcoma resistance to be based on an upregulated IFN system; pharmacological attenuation of IFN signaling led to enhanced VSV infection and cell death.
MATERIALS AND METHODS
Cell culture.
From the American Type Culture Collection (ATCC, Manassas, VA), we obtained human fibrosarcoma HT-1080 (CCL-121), human osteosarcoma SJSA-1 (CRL-2098), human liposarcoma SW-872 (HTB-92), and baby hamster kidney cells (BHK). Normal human fibroblasts were purchased from Cambrex (Walkersville, MD). Osteosarcoma Saos-2 cells were provided by Dan DiMaio (Yale University). Human synovial sarcoma SW982 (ATCC: HTB-93) and human bladder carcinoma T24 cells were provided by the Yale Cancer Center. Timothy Cripe (Children's Hospital Medical Center, Cincinnati, OH) generously provided human malignant fibrous histiosarcoma MFH-1, human osteosarcoma Osteomet/143.98.2 (CRL-11226), human rhabdomyosarcomas Rh30 and RD, malignant peripheral nerve sheath (MPNS) tumors S462-TY and STS-26T, and human Ewing's sarcoma family of tumors (ESFT) A637 and 5838 (7). Normal human decidual cells were a gift of F. Schatz and C. Lockwood (Yale University) (27). Primary mouse brain vascular endothelial (mBVE) cells were a gift of J. Madri (Yale University) (24). Primary human glial cell cultures derived from patients undergoing epilepsy surgery were described previously (47).
Isolation of subclones of synovial sarcoma.
Single-cell-derived subclone populations of SW982, i.e., SW-S and SW-R, were isolated in individual wells of a 96-well plate. SW982 cells were diluted to 0.5 cells per 100 μl in medium that was 50% conditioned, and 100 μl was seeded per well. Wells with individual colonies were propagated and cultures of the desired virus-resistant and virus-susceptible phenotypes were identified.
Propagation.
All cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), nonessential amino acids, 25 mM HEPES, and penicillin-streptomycin (pen-strep), except MFH1, Rh30, and 5838 cells, which were grown in RPMI with 10% FBS and pen-strep. Cells were grown at 37°C with 5% CO2.
Viral preparations.
VSV-G/GFP, a gift of J. Rose, expresses both the native G protein as well as a G/GFP fusion protein (between G and L genes) (43). VSV-rp30a, a derivative of VSV-G/GFP, was generated by positive selection and plaque purification on glioblastoma as previously described and contains two silent mutations and two missense mutations, one in P and one in L (47, 48). Human cytomegalovirus (CMV) with a GFP reporter, from J. Vieira (University of Washington, Seattle, WA), was used as previously described (44). Sindbis virus expressing GFP was a gift of J. M. Hardwick (Johns Hopkins University, Baltimore, MD) and used as previously reported (48).
Infectivity assays.
Cells (5 × 104 per well) were seeded in 24-well dishes and incubated overnight. GFP reporter-expressing virus was added at the indicated multiplicity of infection (MOI). At the indicated times postinfection, the percentages of cells positive for GFP fluorescence were assayed by phase-contrast and fluorescence microscopy with an Olympus Optical (Tokyo, Japan) IX71 fluorescence microscope with a SPOT-RT camera (Diagnostic Instruments, Sterling Heights, MI). Each condition was assayed in triplicate wells. Within each well, total and GFP+ cells were counted in three adjacent fields using a 20× microscope objective. For longer-duration experiments in which cells were preincubated for 4 days with anti-IFN agents, 1 × 104 cells were seeded instead.
Virus-mediated killing.
Cells (5 × 104 per well) were seeded in 24-well dishes and incubated overnight. Medium was changed to minimal essential medium (MEM) without phenol red, with 10% FBS and pen-strep, and VSV-G/GFP or VSV-rp30a was added at 5 PFU/cell. At 36 h postinfection (hpi), ethidium homodimer (EtHD) (Invitrogen, Carlsbad, CA) was added to a final concentration of 4 μM. After 30 min at 37°C, total cells and EtHD-positive (red fluorescent) cells were counted as for the infectivity assay.
Replication assay.
Cells (1 × 105 per well) were seeded in 12-well dishes and incubated overnight. VSV-G/GFP or VSV-rp30a was added at 1 PFU/cell in 500 μl and incubated for 30 min at 37°C. Each well was washed 1 time with phosphate-buffered saline (PBS), and then 1 ml of growth medium was added. One-hundred-microliter samples of medium were taken at 24 hpi and stored at −80°C until the time of analysis. Each experimental condition was replicated in triplicate wells. PFU/ml for each sample was determined by serial dilution and plaque assay on BHK cells, as previously described (43), performed in duplicate for each sample.
Mouse procedures.
Animal experiments were approved by and performed in accordance with institutional guidelines of the Yale University Animal Care and Use Committee. Immunodeficient homozygous CB17-SCID mice 7 to 8 weeks of age were obtained from Taconic Farms (Germantown, NY). Bilateral subcutaneous flank tumors were established by injection of 5 ×105 sarcoma cells in 100 μl of PBS. One dose of 5 ×107 PFU of VSV-rp30a in 100 μl was injected via tail vein when tumor volume averaged approximately 120 mm3. Tumor dimensions were measured by caliper, and tumor volume (V) was estimated by the formula for the volume of an ellipsoid of length a and uniform width b, i.e., V = 4/3 π(a/2)(b/2)(b/2). For histology, animals were killed with a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde in PBS.
Cell association assay.
Cell association of VSV-rp30a was measured under four different conditions as described in the text and in a previous report (33). In all cases, 12-well dishes were seeded with 200,000 cells per well, exposed in quadruplicate to 10 PFU/cell of VSV-rp30a under the conditions specified, and washed five times with PBS plus 5 mM Ca2+ before RNA harvest and genome titration by quantitative RT-PCR (qRT-PCR) (see below). To assess membrane binding under conditions that inhibit endocytosis, virus and cells were incubated for 20 min at 4°C (“binding”). To assess binding and endocytosis together, infected cells were incubated 30 min at 37°C (“30 min”). To measure kinetics of early cell cycle events beyond 30 min, cells were washed after a 30-min incubation at 37°C and incubated an additional hour at 37°C (“90 min”). Finally, to assess cell association under conditions that block endosomal acidification and therefore viral escape from the endosome, the 90-min conditions were replicated but in the presence of 5 mM ammonium chloride (“endosomal”). Cells were washed 5 times, total RNA was harvested, and VSV-genome concentration was measured by quantitative RT-PCR.
Quantitative RT-PCR measurement of VSV genomes, IFN-β expression, and interferon-stimulated gene (ISG) expression.
RNA was extracted from cell lysates by using TRIzol (Invitrogen, Carlsbad, CA) and reverse transcribed by random hexamer priming by using the Super Script III reverse transcriptase kit (Invitrogen). For host cell gene expression analysis, TaqMan gene expression assays (Applied Biosystems, Foster City, CA) for hIFN-β, ISG-15, MxA, OAS-1, ISG-56, and β-actin were acquired and used as recommended by the manufacturer. For measurement of VSV genomes, a TaqMan assay was used in which primers span the junction between N and P genes; gene spanning reduces detection of mRNA transcripts, which are monocistronic. Each sample was measured in triplicate PCRs, and the result was internally normalized to β-actin expression levels in that sample, also measured in triplicate.
Cytogenetic analysis.
Cytogenetic analysis of SW982, SW-S, and SW-R, six metaphases each, was performed by the Yale University Cytogenetics Laboratory under the directorship of Peining Li.
Exogenous agents.
Valproate (VPA) and universal IFN-αA/D were purchased from Sigma (St. Louis, MO) and Jak inhibitor 1 was from Calbiochem/EMD Chemicals (Gibbstown, NJ). Vaccinia virus B18R protein preparation was generously provided by Michael Robek (Yale University) (3). B18R was used at a concentration sufficient to neutralize 10 ng of type I IFN per milliliter.
Statistical analysis.
Comparisons were made by unpaired t tests, using KaleidaGraph software v3.6 (Synergy Software). Ratios were analyzed after logarithmic conversion of data [x′ = log(x + 1)] as recommended for statistical analysis of ratios (36).
RESULTS
Infectivity of VSV-G/GFP and VSV-rp30a in human sarcomas.
Because sarcomas are diverse in their genetics and histology, we tested a substantial number of sarcoma types to address the questions below. A panel of 13 human sarcoma lines was tested, representing seven sarcoma types: two Ewing's sarcoma family tumors (ESFTs), three osteosarcomas, two malignant peripheral nerve sheath (MPNS) tumors, two rhabdomyosarcomas, two fibrosarcomas, one liposarcoma, and one synovial sarcoma.
All sarcomas were assessed after infection at an MOI of 5 with VSV-G/GFP or VSV-rp30a. Based on expression of the viral reporter gene, GFP, VSV-rp30a infected 100% of sarcoma cells within 36 hpi in 11 of the 13 sarcomas (Fig. 1B); in comparison, the infectivity of VSV-G/GFP was lower (Fig. 1A). At 12 hpi, the infectivity advantage of VSV-rp30a over VSV-G/GFP was universal (13 of 13 sarcomas) and averaged 3.6-fold (± 0.53 standard error of the mean [SEM]) better across all sarcomas.
Fig. 1.

Infectivity, killing and replication of VSV-G/GFP and VSV-rp30a in a diverse panel of human sarcomas. (A and B) Infectivity. Thirteen human sarcoma lines were infected at 5 PFU/cell with either VSV-G/GFP (A) or VSV-rp30a (B). The percentage of cells expressing GFP was assessed at 12 hpi (white bars) and at 36 hpi (black bars). (C) Killing. Sarcomas were mock infected (white bars) or infected at 5 PFU/cell with VSV-G/GFP (gray bars) or VSV-rp30a (black bars). At 36 hpi, cells were incubated with ethidium homodimer (EtHD-1) and the percentage of cells fluorescing red was assessed. (D) Replication. Sarcomas were infected with VSV-G/GFP (gray bars) or VSV-rp30a (black bars) at 1 PFU/cell, washed, and incubated. Supernatants taken at 24 hpi were plaque titered in duplicate on BHK cells. All results were assessed in triplicate. Error bars, SEM. ESFT, Ewing's sarcoma family of tumors; MPNS, malignant peripheral nerve sheath tumor.
There was no clear correlation between sarcoma tissue type and susceptibility to infection. The percentage of cells infected 12 hpi by VSV-rp30a averaged between 80% and 95% for all but three sarcoma lines (Fig. 1B, white bars), demonstrating that the high infectivity of VSV-rp30a generalizes to multiple sarcomas. Liposarcoma cells and fibrosarcoma MFH-1 cells were only moderately infected by VSV-rp30a at this time point, but by 36 hpi, all liposarcoma cells and the majority of MFH-1 expressed GFP, demonstrating successful infection.
In contrast, synovial sarcoma SW982 was only 0.5% infected by VSV-rp30a at 12 hpi, and 0.8% infected at 36 hpi, demonstrating a unique and strong resistance to infection.
Cytolysis of human sarcomas by VSV-G/GFP and VSV-rp30a.
Ethidium homodimer (EtHD) was used as a label for dead cells. VSV-G/GFP-infected, VSV-rp30a-infected, and mock-infected cells were assessed 36 hpi (Fig. 1C). In 12 of 13 sarcoma cultures, infection with VSV-rp30a resulted in cell death for the majority of cells within 36 h. VSV-G/GFP also killed sarcoma cells in 12 out of 13 lines but with a rate that was statistically significantly lower than that for VSV-rp30a (P < 0.05). For both viruses, the number of dead sarcoma cells at 36 hpi correlated well with the percentage of cells expressing GFP 36 hpi after infection with the same MOI (Fig. 1A and B, black bars); EtHD-positive cells typically represented between 70 and 90% of infected GFP-expressing cells at this time point. We did not find any sarcoma cells that expressed the GFP reporter that did not eventually die.
In contrast to these findings, only a small minority of infection-resistant synovial sarcoma SW982 cells were dead at 36 hpi. Only 1 to 2% of cells were EtHD positive, and this percentage was not different (P > 0.3) from the percentage of cells dead (1%) in mock-infected cultures.
Replication of VSV-G/GFP and VSV-rp30a in human sarcomas.
VSV progeny production was studied at 24 hpi (Fig. 1D). The three sarcomas with the highest amounts of viral replication were ESFT A673, osteosarcoma 132.98.2, and MPNS S462-TY, which were among those with high infection susceptibility (Fig. 1A and B). The three sarcomas with the least replication efficiency were also those with the poorest infectivity: liposarcoma SW872, fibrosarcoma MFH-1, and synovial sarcoma SW982.
VSV-rp30a showed a replication advantage (P < 0.05) over VSV-G/GFP in 9 of 13 sarcomas and a trend toward advantage in 3 additional lines. This advantage averaged 2.0-fold (± 0.26 SEM).
In infection-resistant synovial sarcoma, progeny titers were so low (<250 PFU/ml) that they might represent only trace inoculum that remained on the cells despite washing, rather than true progeny. These titers are 800-fold (VSV-G/GFP) and 11,000-fold (VSV-rp30a) lower than those seen in MFH-1, otherwise the cell least supportive of VSV replication.
VSV-rp30a is selective for sarcoma over normal mesoderm.
Cells or tissues with intact innate immunity can limit the number of rounds of replication that VSV can successfully achieve. To assess the oncoselectivity of VSV in normal versus sarcoma mesoderm, we infected a panel of three normal mesoderm-derived cells and three sarcomas. Normal primary human fibroblasts, primary human decidua, and primary mouse brain vascular endothelium (mBVE) cultures, as well as three human sarcoma lines, were infected with VSV-rp30a (0.2 PFU/cell), and the percentage of cells infected was assessed 20 hpi (Fig. 2A). Infection rates in the three sarcomas averaged 9.9-fold more than in the three primary cell cultures, demonstrating oncoselectivity. mBVE cells were particularly resistant (<0.1% infected), an important finding in light of the fact that vascular endothelium is exposed to high virus titers during intravenous administration, as in our in vivo model below.
Fig. 2.
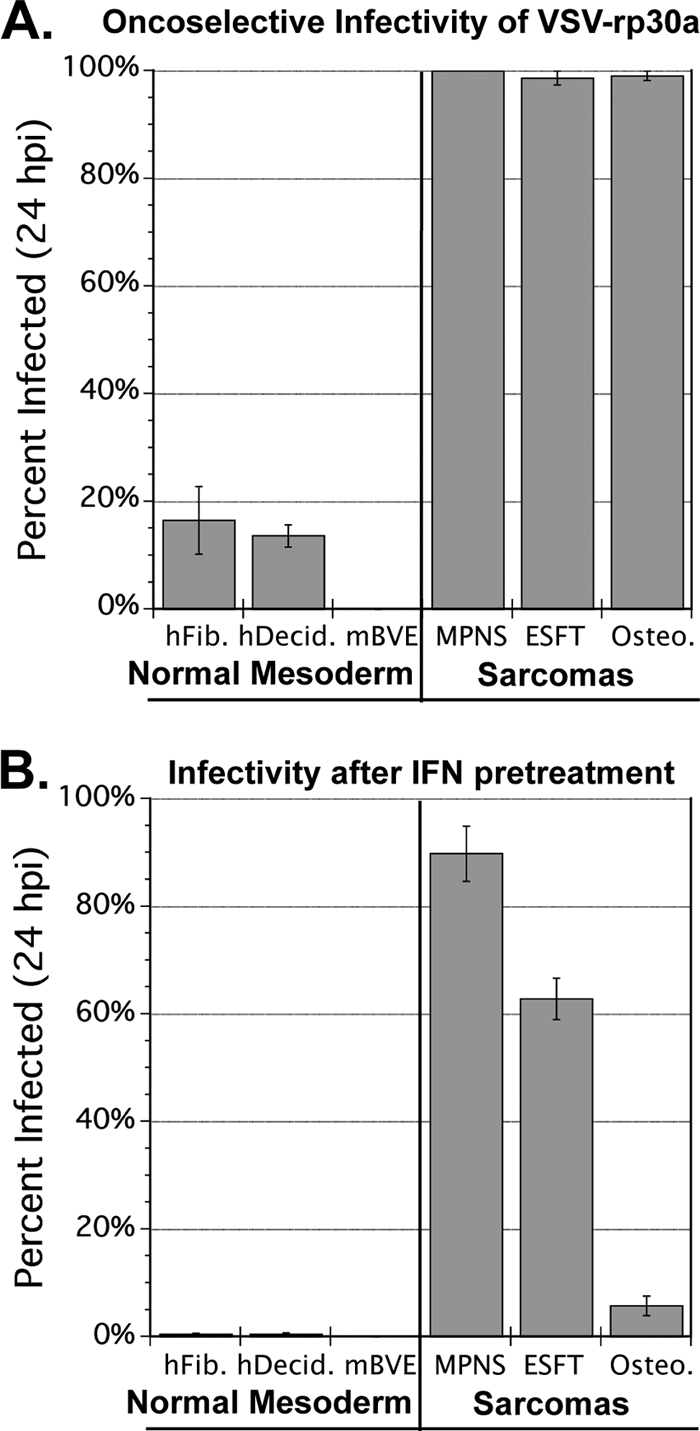
Oncoselectivity and IFN pretreatment effects. Three primary mesoderm-derived cell cultures and three sarcomas were infected with VSV-rp30a at MOI 0.2 PFU/cell. Cells were either infected without (A) or with (B) IFN pretreatment (100 U/ml for 6 h). The percentage of cells expressing GFP was assessed in triplicate wells for each condition, and bars represent standard errors. hFib, normal human fibroblasts; hDecid, normal human decidua; mBVE, mouse brain vascular endothelium; MPNS, malignant peripheral nerve sheath tumor; ESFT, Ewing's sarcoma family tumor; Osteo., osteosarcoma 132.98.2.
Within normal tissues and within tumor tissues, interferon secretion may be initiated by virus infection and may protect nearby cells. We therefore tested the infectivity of VSV-rp30a (MOI 0.2 PFU/cell) against our panel of normal and transformed cells after preincubation for 6 h with 100 units/ml of universal IFN-α (Fig. 2B). Normal primary cells were all VSV resistant after IFN pretreatment, with fewer than 1% of the cells expressing GFP at 20 hpi. Sarcomas were all infected but to different degrees. MPNS tumor S462-TY (100% infected in the absence of IFN) was 90% infected, representing only a small degree of IFN-mediated protection. ESFT A673, 98.8% infected in the absence of IFN, was 62.9% infected. Osteosarcoma 132.98.2, 99.1% infected in the absence of IFN, was only 5.8% infected, indicating a high degree of IFN protection in early stages of infection. Thus, sarcomas are highly variable in the degree to which they can be protected from infection by exogenous IFN.
All cultures were observed for additional time. Unprotected sarcoma cells were 100% infected and lysed by 2 days postinfection (dpi). IFN-protected sarcoma cells were 100% infected and lysed by 3 dpi, and cell death was confirmed by EtHD staining. In contrast, a significant portion of each of the three unprotected normal mesodermal cultures remained uninfected 3 dpi, and IFN-protected cultures were all less than 1% infected. The percentages of unprotected human fibroblasts, human decidua, and mBVE cells infected 3 dpi were approximately 30%, 50%, and 1%, respectively. Overall, these data are evidence that over time, sarcomas are unable to evade complete destruction after low MOI infection by VSV, even when protected in advance with IFN, whereas normal mesodermal cells in culture are VSV resistant to varying degrees without IFN pretreatment and are nearly 100% protected by IFN pretreatment.
VSV-rp30a selectively infects subcutaneous sarcoma xenografts and arrests tumor growth in vivo.
To assess the ability of systemic VSV-rp30a to specifically infect sarcoma in vivo, we performed bilateral subcutaneous implantations of fibrosarcoma HT1080 and ESFT A673 in SCID mice. When tumors averaged approximately 120 mm3, 5 × 107 PFU of VSV-rp30a was injected via tail vein. Mice bearing HT1080 and A673 tumors were euthanized 6 or 7 dpi. Of nine established fibrosarcoma tumors, VSV-rp30 infection was found in all nine. Infection was generally widespread within the tumor, infecting approximately 90% of the cells. Adjacent normal tissue was consistently uninfected (Fig. 3A to C). Similarly, liver and spleen tissue showed no sign of infection at the same time point. Figure 3X is a high-magnification image of a region of the tumor with a high density of GFP expression, indicated in Fig. 3C. GFP labeling of cell membranes, as shown, is characteristic of G/GFP fusion protein expression. Regions of the tumor with only weak fluorescence at low magnification often can be seen to display this infection-specific pattern of GFP expression as well, as shown in Fig. 3Y, a magnified image of the relatively dim region indicated in Fig. 3C, suggesting these areas are at an early stage of infection. Of 11 established A673 tumors, all 11 were infected, although to a lesser degree than HT1080 tumors at the same time postinfection. The observed selectivity of VSV for sarcomas in this model is significant because VSV is not a species-restricted virus and can infect both mouse and human cells.
Fig. 3.

Infection and suppression of subcutaneous sarcoma xenografts by intravenous VSV-rp30a. (A to C) SCID mice with subcutaneous human fibrosarcoma HT1080 xenografts were infected via tail vein with a single dose of 5.0E7 PFU of VSV-rp30a. Histological images of a representative tumor 7 dpi are shown: phase contrast showing two lobules of the tumor outlined in red (A), green fluorescence (C), and image overlay (B). (X and Y) High-magnification green fluorescence micrographs of two regions of the tumor indicated by white squares in C. (D) SCID mice with subcutaneous (s.c.) A673 xenografts were left uninfected (dotted line) or injected intravenously (i.v.) with a single dose of 5.0E7 PFU of VSV-rp30a (solid line) on day zero. Data points represent the mean volume, relative to day zero, for tumors under each condition. Error bars, SEM.
To assess the ability of intravenous VSV-rp30a to affect the growth potential of infected sarcoma xenografts, bilateral subcutaneous A673 tumors were established in 10 SCID mice. Successful tumor development in the majority of implantations, combined with injection of VSV-rp30a in five mice on day 13 after sarcoma implantation, was followed by daily measurement of tumor volume for 8 virus-treated tumors versus 6 untreated tumors. Mean volume of these tumors on the day of injection was 128 mm3 for untreated mice and, similarly, 116 mm3 for treated mice (nonsignificant, P > 0.05). Mean changes in tumor volume (each tumor compared to its own volume on the day of injection) for 9 days following treatment are shown in Fig. 3D for treated versus untreated tumors. Untreated A673 tumor size increased by 10.8-fold, in significant contrast (P = 0.0003) to treated tumors, which did not grow, and further had a trend toward decreasing size (90% of original volume). This experiment demonstrates the ability of intravenous VSV-rp30a to find, selectively infect, and arrest the growth of an otherwise rapidly growing sarcoma in vivo.
The studies above show that the vast majority of a representative panel of human sarcoma cell lines are highly susceptible to VSV-induced oncolysis, especially to VSV-rp30. In the following, we address the mechanisms underlying the resistance of synovial sarcoma to VSV.
VSV-resistant synovial sarcoma SW982 are permissive for viral binding and endocytosis.
The paucity of GFP expression among VSV-infected synovial sarcoma cells suggested a significant block to an early step in the viral life cycle, perhaps binding or cell entry. We tested this possibility by measuring the efficiency of cell association of VSV-rp30a under four different conditions, as described in Materials and Methods and in a previous report (33). We compared synovial sarcoma to highly VSV-susceptible sarcoma S462-TY, after addition of VSV-rp30a at 10 PFU/cell.
Binding of VSV-rp30a to synovial sarcoma SW982 was slightly more efficient than binding to MPNS tumor S462-TY (1.5-fold), indicating that lack of viral receptor does not explain the resistance of SW982. Under the 30-min and 90-min conditions, there were slightly more genomes associated with MPNS than with synovial sarcoma (1.4-fold and 1.8-fold, respectively). However, under the set of experimental conditions in which viral escape from the endosome is blocked (“endosomal”), genomes in infected synovial sarcoma and MPNS were equivalent (Fig. 4A). Taken together, these data indicate that VSV-resistant synovial sarcoma SW982 cells are not substantively deficient for VSV binding or endocytosis relative to a highly susceptible sarcoma.
Fig. 4.

Synovial sarcoma SW982 resistance to infection. (A) Cell association of VSV-rp30a. Virus-resistant SW982 (white bars) and virus-susceptible S462-TY (black bars) were incubated with 10 PFU/cell of virus under four different conditions (as described in Materials and Methods) designed to measure the efficiency of binding and internalization. After washing, cell-associated genomes from quadruplicate wells were assessed by quantitative RT-PCR normalized to β-actin. Results are normalized to SW982. Error bars, SEM. (B) Resistance to high MOI. Synovial sarcoma SW982 (top) or control sarcoma SJSA-1 (bottom) cells were infected in triplicate by VSV-G/GFP or VSV-rp30a at MOI 0.5, 5, or 50 PFU/cell. The percentage of cells positive for GFP was assessed 12 hpi (gray bars) and 36 hpi (black bars). Error bars, SEM. (C) Resistance to other viruses. SW982, osteosarcoma SJSA-1, or ESFT A673 cells were infected with three recombinant GFP-expressing viruses: VSV, Sindbis virus, or human CMV. Representative micrographs 24 hpi are shown.
Synovial sarcoma SW982 is profoundly resistant to high-titer VSV, Sindbis, and cytomegalovirus.
Among all the sarcomas we tested, the resistance to VSV observed in synovial sarcoma SW982 stood apart as unique. Even when we raised the MOI to a very high level of 50 PFU/cell, VSV-G/GFP and VSV-rp30a still infected only a small fraction of cells, less than 1% for each virus, at both 12 and 36 hpi (Fig. 4B, top panel). At each of these MOIs, by 36 hpi, VSV-G/GFP and VSV-rp30a both infected 100% of other sarcoma cells—osteosarcoma SJSA-1 infected in parallel (Fig. 4B, bottom panel). We explored further this rare degree of VSV resistance.
To test the hypothesis that synovial sarcoma has a high resistance to viral infection in general, we tried infecting the VSV-resistant synovial sarcoma with other unrelated viruses. To ensure that these viruses were capable of infection, we also tested three other sarcomas that were susceptible to VSV (SJSA-1, A673, and S462-TY). We used GFP-expressing human cytomegalovirus, a double-strand DNA virus in the family Herpesviridae, and Sindbis virus, a positive-RNA-strand alpha virus in the family Togaviridae. The synovial sarcoma was highly resistant to both Sindbis and CMV (Fig. 4C); only a small minority (<1%) of cells showed signs of infection at 24 and 48 hpi. In contrast, CMV and Sindbis efficiently infected other sarcoma cells tested, the majority of cells being GFP positive by 48 hpi. ESFT A673 showed a level of resistance to CMV comparable to that seen in synovial SW982, despite the susceptibility of these cells to VSV and Sindbis. A demonstrative subset of our results at 24 hpi are shown in Fig. 4C. Together, these results indicate a general resistance of this synovial sarcoma to multiple types of viruses unrelated to VSV.
VSV-resistant synovial sarcoma SW982 has high basal expression of interferon-stimulated genes.
Given the uniform resistance of SW982 to highly divergent viruses, and verification that binding and endocytosis were intact, we postulated that innate immunity may be responsible for this unique degree of resistance. One hypothesis is that VSV infection of these cells induces a stronger upregulation of IFN-β and downstream interferon-stimulated gene (ISG) expression in response to VSV, compared to susceptible cells. However, in measuring the mRNA levels of IFN-β, ISG-15, and MxA 6 hpi in VSV-infected versus mock-infected synovial sarcoma, and in fibroblasts, we saw a greater relative postinfection upregulation of all three genes in normal fibroblasts compared to the relatively resistant SW982 (data not shown). Surprisingly, we observed that the basal, i.e., preinfection expression levels of ISGs MxA and ISG-15 were both remarkably higher in SW982 than in normal cells (90-fold and 36-fold, respectively) (Fig. 5A and B).
Fig. 5.
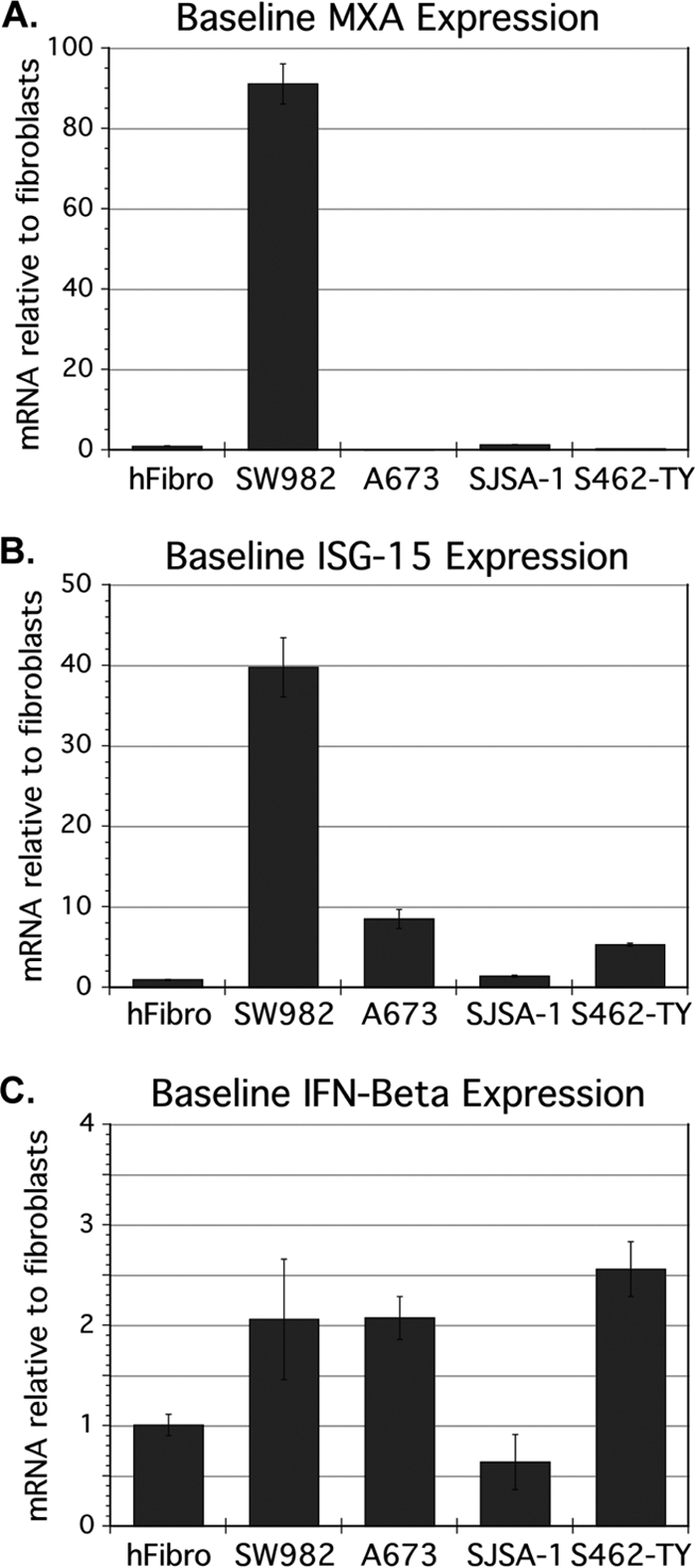
Baseline expression of interferon-stimulated genes (ISGs) and interferon β in human fibroblasts and sarcomas. mRNA was harvested from uninfected cultures of human fibroblasts (hFibro), VSV-resistant synovial sarcoma SW982, and three VSV-susceptible sarcomas (ESFT A673, Osteosarcoma SJSA1, and MPNS tumor S562-TY). mRNA concentrations for two ISGs, MxA (A) and ISG-15 (B), and for IFN-β (C) were measured from triplicate samples by quantitative RT-PCR and normalized to β-actin. Error bars, SEM.
This finding led to the hypothesis that relative to various susceptible sarcomas, SW982 might be unique in having aberrantly high expression of ISGs prior to infection, and that this may account for the resistance phenotype. We measured basal expression of MxA and ISG-15 in three VSV-susceptible sarcomas (A673, SJSA-1, and S462-TY) and found that uninfected SW982 expressed both of these ISGs at much greater levels than did uninfected VSV-susceptible sarcomas: on average, synovial sarcoma cells showed a 270-fold higher expression for MxA (P = 0.002) and 12-fold higher expression for ISG-15 (P = 0.03) (Fig. 5A and B).
If ISGs were globally upregulated in SW982 in advance of infection, the direct antiviral activities of several of these proteins might abrogate the viral life cycle very early in infection, thus preventing GFP reporter expression. The MxA protein, for example, blocks rhabdoviruses early in the life cycle by binding nucleocapsids (37). The expression of ISGs is regulated by JAK/STAT signaling downstream of receptors for IFN (34), raising the possibility that SW982 cells secrete increased IFN constitutively. Using specific primers for IFN-β, we did not detect elevated basal expression levels in SW982 cells relative to the average expression in other cells tested (P = 0.33) (Fig. 5C). The possibility remained that SW982 could be secreting another IFN, such as one of the multiple alpha interferons, or that ISGs were upregulated by an IFN-independent mechanism.
The ability of medium from cultured cells to inhibit VSV infection is a commonly employed bioassay for interferon (28). However, when we conditioned medium on synovial sarcoma overnight and exposed human fibroblasts to this medium for 4 h, we observed no effect on the percentage of cells infected 24 h postinfection with VSV-G/GFP (1 PFU/cell), as compared to fibroblasts infected after exposure to fresh medium or medium conditioned on fibroblasts, despite the ability of IFN-α-spiked medium (200 U/ml) to completely protect these cells from VSV after the same 4-h incubation period (data not shown). This result suggested that SW982 cells do not show enhanced interferon secretion at a level sufficient to protect other cells from VSV.
Rare VSV-susceptible cells within SW982 can be grown into single-cell subclones with significant and stable VSV susceptibility.
A small minority (<1%) of the SW982 population were VSV susceptible, expressing GFP at high levels despite the absence of GFP in the great majority (>99%) of the culture. This heterogeneity of phenotype raised the question of whether the susceptible subpopulation might harbor a genetic anomaly. Intratumoral diversity at the genetic level often results in variability in cell phenotype within the population, including variability in susceptibility to tumor killing (15). We adopted the strategy of growing single-cell-derived subclones of tumor cell lines to study this diversity, for example, as performed by Held et al. (17). Two phenotype-homogenous cell lines were expanded. The SW-S subclone was highly susceptible to VSV. In contrast, the SW-R was very resistant to infection; even with very high MOIs (50 and 100 MOI) of VSV, we found no infected cells (Fig. 6A).
Fig. 6.

Resistant SW982 subclone SW-R can be made susceptible by blocking IFN signaling. (A) Susceptibility of SW-S, SW-R, and SW-R after treatment with IFN-abrogating agents. Representative micrographs 0 hpi and 24 hpi (VSV-rp30a, MOI 5) are shown. Single-cell subclone cell lines of SW982, SW-S, and SW-R are VSV sensitive and VSV resistant, respectively. SW-R cells were treated with valproate (VPA), vaccinia B18R, or JAK-inhibitor 1 (JAK-I) starting at the indicated time points relative to infection. (B) qRT-PCR was used to measure mRNA of the four indicated interferon-stimulated genes in SW-S, SW-R, SW-R pretreated for 14 h with VPA, and SW-R pretreated with vaccinia B18R for 4 days. Triplicate samples were analyzed. Error bars, SEM. (C) Triplicate human glial cultures were exposed to fresh medium, fresh medium with 200 U/ml of IFN, or conditioned medium. Medium was conditioned for 1, 2, or 3 days on SW-R. After 6 h of exposure, glial cell RNA was harvested and analyzed for MxA mRNA. Error bars, SEM.
Cytogenetic analysis of clonal synovial sarcoma lines (and the parental line, SW-P) was performed to verify their identity as synovial sarcoma rather than a contaminating cell line, often an unrecognized problem in cultured cells (21). Also, we sought to determine if there were any unique cytogenetic features. The Yale Cytogenetics Laboratory analyzed six metaphases each of SW-P, SW-R, and SW-S and found that all three lines shared multiple karyotypic alterations, specifically 47, XX, t(1;4;9) (q12;q11,p24), del(5) (q31q33), der(9,13) (q10q10), +der(20) t(5;20) (q11.2:p13), +mar[6]. Rouleau et al. (35) reported nearly identical karyotype features for SW982: 47-49, XX, t(1;4;9) (q12;q11;p24), del(5) (q22q33), der(9;13) (q10;q10), +der(20) t(5;20) (q11.1;p13). The karyotype analysis supports the view that the susceptible subpopulation, SW-S, is not a contaminating cell line but instead is closely related to the parent SW982 cells. Additionally, SW-S had three unique features, namely, the addition of unidentified chromosomal material to a long arm of chromosomes 7, 11, and 18, any of which might be related to its enhanced susceptibility, although a cytogenetically undetectable mutation may also be the causative mutation specific to these cells.
Preadministration, but not coadministration, of agents that antagonize interferon signaling renders VSV-resistant sarcoma cells susceptible.
In the resistant synovial sarcoma cells, we found elevated basal ISG expression rather than a robust ISG response to infection. If an upregulated ISG system is the mechanism responsible for the remarkable resistance of synovial sarcoma cells to VSV, then reducing the baseline ISG expression should increase the level of infection in resistant cells. We therefore tested three converging approaches to interfering with ISG expression. These included disrupting gene regulation with histone deacetylase inhibitors, disrupting second messenger signaling after IFN binds to its receptor with a JAK1 inhibitor, and reducing IFN binding to type I IFN receptors with poxvirus B18R protein.
Histone deacetylase inhibitors such as valproic acid (VPA) disrupt gene expression and can impact cellular resistance to viral infection (11, 30, 32). We added VPA (30 mM) to the medium of SW-R cells at different times relative to infection. Neither cotreatment nor 14-h pretreatment increased susceptibility (Fig. 6A), despite the finding that a 14-h preincubation with VPA reduced basal mRNA levels for three out of four ISGs in SW-R to levels comparable to those in SW-S (Fig. 6B). As a longer preincubation may be necessary to fully reduce ISG protein levels, we used a 4-day preincubation. This 4-day VPA treatment resulted in a dramatic reversal of the VSV-resistant phenotype of SW-R (Fig. 6A) and greatly enhanced infection.
Signaling downstream of the type I IFN receptor and resultant ISG expression are dependent on tyrosine kinase JAK1 (34). As an additional test of our hypothesis, we exposed SW-R to JAK inhibitor 1 (JAK-I) (500 nM) either coincident with infection or 4 days preinfection. Consistent with our previous results, interdiction of IFN signaling had no profound effect when initiated at the time of infection, whereas JAK-I added 4 days prior to infection resulted in profound VSV susceptibility in this otherwise resistant cell line (Fig. 6A).
Vaccinia virus B18R protein attenuates signaling by the type I IFN receptor (2). Similar to our results with VPA and JAK-I, addition of B18R at the time of infection of SW-R did not increase susceptibility (Fig. 6A). In contrast, a 4-day preincubation with B18R resulted in profound susceptibility (Fig. 6A). Consistent with our hypothesis that basal ISG expression correlates with phenotype, 4-day exposure of SW-R to B18R reduced basal ISG mRNA expression levels, including MxA, ISG-15, OAS-1, and ISG-56, even below those of SW-S (Fig. 6B). B18R acts as a decoy IFN receptor, thereby reducing IFN responses. This suggests that the SW-R cells may indeed release small amounts of IFN; a low basal level of IFN release has been proposed as a general property of other cell types (41). Although we had not found evidence of IFN secretion in our bioassay described above, when we harvested culture medium after a 3-day incubation with the SW-R cells, this conditioned medium (compared with unconditioned control medium) did increase MxA expression 180-fold in human astrocytes (Fig. 6C). A similar induction was not seen after a 1-day incubation. These data are consistent with the view that SW-R cells may secrete a low level of IFN in the absence of virus infection.
VSV-resistant cells show reduced level of IFN-β induction.
As described above, when we initially studied the induction of IFN-β by virus in VSV-resistant SW982 cells, we found basal mRNA levels comparable to those observed in susceptible cells, and only a moderate induction of IFN-β mRNA postinfection. Now having tools in hand to reverse the phenotype of SW-R, we revisited the issue of IFN response to infection. We looked in SW-S, SW-R, and SW-R made VSV susceptible by a 4-day pretreatment with B18R and measured IFN-β mRNA 10 h after infection (or mock infection) with VSV (MOI, 1). Interestingly, we found a robust postinfection upregulation of IFN-β expression in VSV-sensitive cells: specifically, we found a 3,300-fold upregulation in SW-S and a 6,300-fold upregulation in SW-R pretreated with B18R (Fig. 7). However, in contrast, we observed only a modest 23-fold increase of IFN-β in VSV-resistant SW-R not treated with B18R, suggesting that early abrogation of infection in highly resistant cells, mediated by constitutively expressed ISG effectors of innate immunity, precludes stimulation of the signaling cascades that would otherwise lead to upregulation of IFN-β mRNA. These results support the view that it is upregulated basal ISG levels, and not an exaggerated de novo response to infection, that enhances VSV resistance in SW982.
Fig. 7.
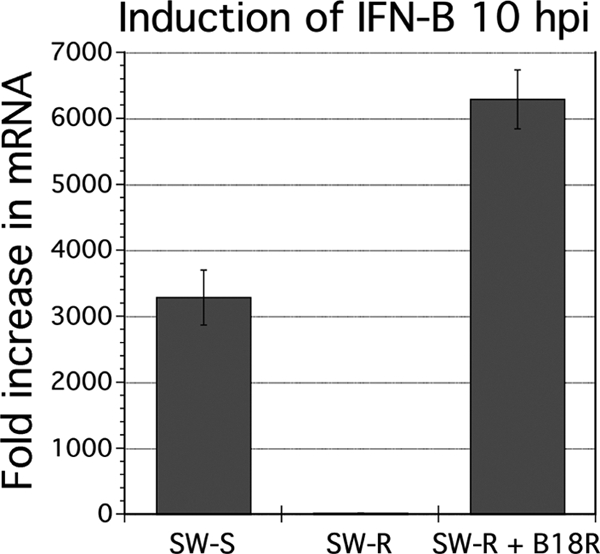
Induction of IFN-β by viral infection of SW-S, SW-R, or SW-R + B18R (4 days). IFN-β expression was assessed by qRT-PCR at 0 hpi and 10 hpi from parallel triplicate cultures infected with VSV-rp30a so that the fold induction after infection could be determined. Measurements were normalized to β-actin. Error bars, SEM.
JAK inhibitor enhances VSV infection of VSV-resistant liposarcoma and bladder carcinoma.
To investigate whether the mechanism of attenuation of SW-R resistance generalized to any other mesodermal tumors, the effect of JAK-I on relatively VSV-resistant liposarcoma was studied. To ask if the mechanism is restricted to sarcomas or would generalize to nonmesodermal carcinomas, we also tested a relatively VSV-resistant bladder carcinoma, T24. We measured infectivity 20 hpi with VSV-rp30a, employing SW-R as a control. Similar to SW-R, both the liposarcoma and the bladder carcinoma were rendered more susceptible to VSV-rp30a by JAK-I pretreatment (4 days) but not by cotreatment (Fig. 8). Pretreatment increased the percentage of cells infected 4.0-fold for the liposarcoma (P = 0.001) and 5.1-fold for the bladder carcinoma (P = 0.0001), whereas cotreatment had no significant effect.
Fig. 8.
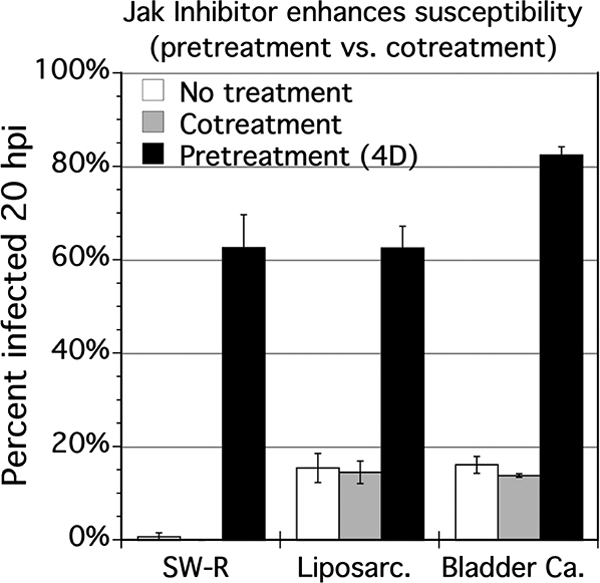
Timing of JAK inhibitor and increasing VSV susceptibility of sarcoma and carcinoma. SW-R, liposarcoma SW872, and bladder transitional cell carcinoma T24 were exposed to 500 nM Jak-1 inhibitor at the time of infection with VSV-rp30a (“cotreatment”), 4 days in advance of infection (“pretreatment”), or not at all (“untreated”). Cells were infected with VSV-rp30a at an MOI of 1 PFU/cell, and the percentage of cells infected was assessed 20 hpi. Triplicate wells were analyzed. Error bars, SEM.
DISCUSSION
VSV selectively targets and kills a diverse range of human sarcomas in vitro and in vivo.
VSV was effective at rapidly infecting and killing most types of sarcoma and was 10-fold more infective in sarcomas than in normal mesoderm. IFN-pretreatment completely protected normal mesoderm, but after a partial initial attenuation of infection, all sarcoma cells were dead 3 days postinfection. In an in vivo mouse model, a single intravenous inoculation of VSV-rp30 a led to infection of all tumors from two types of sarcoma and blocked sarcoma tumor growth. Normal mouse tissue showed no obvious infection.
Enhanced oncoselectivity of VSV-rp30 a generalizes from glioblastoma to sarcoma.
VSV-G/GFP and VSV-rp30a were highly infectious and cytolytic to 12 of 13 sarcomas tested. VSV-rp30a showed a 3.6-fold greater infectivity than VSV-G/GFP and 2.0-fold greater replication. VSV-rp30 was originally selected as a particularly effective mutant VSV after growing the parent VSV-G/GFP for many generations on neuroectoderm-derived human glioblastoma (48). The finding that enhanced infection and cytolysis shown by VSV-rp30 generalize from glioblastoma to mesoderm-derived tumors suggests a non-tissue-specific mechanism. With multiple mutations, including those in the phosphoprotein (P) and polymerase (L) genes, the mechanism of VSV-rp30-enhanced oncolytic action is under investigation.
VSV may act differently on different lines of the same tumor type.
VSV showed no consistent preference for one sarcoma tissue type over another. Different lines of sarcoma of the same tissue origin sometimes showed different sensitivities to VSV infection, similar to earlier observations on different glioblastoma brain tumors (48) and consistent with another report in which VSV showed strong infection of one derivation of prostate cancer, whereas another prostate cancer line was resistant to infection (9). In parallel with this heterogeneity of response, the infectivity of some sarcomas was not substantively affected by interferon, whereas others showed an attenuated initial infection, a finding consistent with previous reports in other cell types (39). The specific types and degrees of innate immune defect in cancer cells appear to be a major determinant of sensitivity to VSV, independent of the tissue origin of the tumor. However, the cell's ability to respond to virus by secreting IFN-β may not correlate with the cell's ability to respond to IFN, as these two functions are distinct phases of the innate immune response which may be affected differently by mutations in tumors (40). Thus, for example, cancer cells with a defect in VSV detection machinery may be sensitive to VSV despite their ability to be protected from VSV by exogenous IFN.
Sarcoma diversity and other oncolytic viruses.
Oncolytic herpes simplex virus has been tested as an oncolytic agent and worked well on three rhabdomyosarcomas. HSV showed intermediate susceptibility for two osteosarcomas and significant resistance in four of four Ewing's sarcomas tested (7). Interestingly, MFH-1 cells were highly susceptible to HSV oncolysis, whereas VSV, as mentioned above, was relatively less efficient in this sarcoma. This is consistent with the view that although VSV-rp30a works very well for many types of cancer, other unrelated oncolytic viruses may work better in some cases.
Rapid oncolysis and other oncolytic viruses.
VSV in general infects and replicates fairly rapidly, with progeny virus beginning to emerge within only a few hours (4). VSV-rp30a infection progressed more rapidly than VSV-G/GFP. Viruses that act quickly, as does VSV, have a potential advantage in completion of tumor targeting and destruction prior to the upregulation of antiviral T and B cells that eventually eliminate the virus. Oncolytic reovirus inhibited proliferation in vitro of one rhabdomyosarcoma, two ESFTs, one osteosarcoma, and one synovial sarcoma HSSY-II (18). Reovirus inhibited sarcoma cell proliferation by 50% after 5 days. In contrast, at comparable MOIs, in the present study we show that VSV-rp30a completely infected 12 of 13 sarcomas within only 36 h, and sarcoma cultures were 100% killed by 48 h even at the low MOI of 0.2 PFU/cell. Intravenous reovirus inhibited growth of these sarcomas in vivo; however, this required multiple doses over many days (18), as opposed to VSV, which effectively arrested sarcoma growth after a single dose.
Elevated basal IFN signaling characterizes virus-resistant cells.
Synovial sarcoma SW982 cells were unique and interesting for their remarkable degree of VSV resistance. The SW982 virus resistance was not due to a deficiency in VSV binding or endocytosis and resistance generalized to human CMV and Sindbis virus, indicating a mechanism not selective for VSV but universal for multiple unrelated viruses. Uninfected SWR cells had an elevated baseline expression of ISGs relative to other sarcomas. The relatively high differential observed for MxA (270-fold) is consistent with evidence that this gene is among those with a highly dynamic range of response to IFN, one reason the MxA promoter is often used in reporter constructs designed to assay for IFN (28). The magnitude of the MxA expression is comparable to the 300-fold induction of MxA observed after incubation of glial cells with IFN (Fig. 6C). PC3, a prostate cancer cell, was VSV resistant relative to a VSV-susceptible LNCaP prostate cancer and also showed elevated levels of ISG expression; however, PC3 was still synchronously infected by an MOI of 50 (9), whereas the synovial sarcoma subclone SW-R here showed no infection even at a higher VSV concentration, indicating a remarkable degree of virus resistance. This difference could be due to a greater upregulation of ISG expression in SW982 cells than in PC3 cells. Parallel to our findings in SW982, the kinetics of binding and endocytosis for resistant and susceptible prostate cells were similar.
Although SW982 showed a low basal IFN secretion, an increased ISG state could also be due to IFN-independent mechanisms; in mutated tumor cells these genes might be constitutively activated even in the absence of an IFN stimulus (22, 38). PC3 prostate cancer showed elevated ISGs in the absence of evidence of IFN signaling (9). Basal IFN secretion, though abnormal in degree in SW982, may be a characteristic of some normal cells (5, 41, 45). We found that preadministration but not coadministration of JAK inhibitor enhanced susceptibility not only for SW982 but also for moderately resistant liposarcoma SW872 as well as a human bladder carcinoma, T24. Together, these results are consistent with the possibility that elevated basal ISG expression may be one mechanism underlying cellular resistance to viral oncolysis in other cancers.
Histone deacetylase inhibitor VPA added in advance of infection lowered basal ISG levels and rendered the resistant cells highly susceptible to infection. An inhibitor of JAK1, which signals downstream of the IFN receptor, also made these cells susceptible. Preadministration of B18R, which binds secreted type I IFNs, also increased VSV infection of SW982, supporting the view that the cells secrete a low level of IFN that enhances the resistance of these cells. Preadministration of B18R also increased IFN-β upregulation in response to VSV infection by over three orders of magnitude, suggesting that removing the block to infection in these cells allows infection to proceed to a point where it more efficiently stimulates the IFN-β response. Together, our data indicate a mechanism of elevated basal expression of antiviral ISGs in these cells which effectively blocks VSV infection at a postendocytotic, previral gene expression step in the viral life cycle.
Using viruses, including VSV, to enhance a systemic immune attack on cancer cells is showing considerable promise (1, 8, 12, 49). However, in terms of direct oncolytic action on cancer cells, the opposite approach also has merit, that is, attenuating the immune response to the virus in order to enhance the direct oncolytic action. The ability of some tumors to mount an effective ISG response after infection raises the potential for use of anti-IFN compounds to enhance viral oncolysis in vivo. Histone deacetylase inhibitor MS275 administered to nude mice successfully enhanced oncolysis of subcutaneous tumor xenografts by VSV (30). Similarly, valproate enhanced survival of glioblastoma-bearing mice treated intratumorally with oncolytic HSV (32). If one aim of administering an IFN-abrogating agent, however, is to reduce basal ISG expression in advance of infection, rather than simply mitigate postinfection responses, this consideration is relevant to decisions about the timing of drug administration. IFN-blocking agents have the potential to enhance oncolytic efficacy not only by preventing the generation of IFN-stimulated, VSV-resistant tumor cells consequent to the IFN response to oncolytic virus infection but also by optimizing the initial round of tumor infection by eliminating preexisting VSV resistance.
The efficacy of several oncolytic viruses against tumors may benefit from exposure to IFN-reducing agents. This raises the question of whether an attenuated IFN response might lead to unwanted side effects by enhancing infection of normal tissue. This was not a problem with VSe1, an IFN blocker; in an immunocompetent model of murine colon cancer, the systemic coadministration of oncolytic VSV-Δ51 and VSe1 resulted in an enhanced retardation of tumor growth, with little sign of infection in normal tissues (11). Neither virus nor VSe1 alone was effective against the tumor. Similarly, coinfection with vaccinia virus which expresses the IFN-blocking protein B18R also enhanced VSV-Δ51 actions with no reduction in animal survival (23). Thus, anti-IFN compounds can be used to enhance oncolytic efficiency in vivo with minimal increase in infection of normal tissue.
ACKNOWLEDGMENTS
In addition to those mentioned in Materials and Methods for their kind contributions, we particularly thank Timothy Cripe for provision of multiple sarcoma lines; Yang Yang and Vitaliy Rogulin for excellent technical facilitation; Guido Wollmann, Mike Robek, Dan DiMaio, and William Edward for many helpful suggestions; and Ellen Vollmers, John N. Davis and Jill Paglino for critical manuscript review. Some facilities were provided by the Yale Comprehensive Cancer Center.
A.N.V.D.P. has an equity interest in Azgardabio, which focuses on the clinical use of viruses.
Grant support was provided by NIH RO1 CA 124737 and NS48854.
Footnotes
Published ahead of print on 6 July 2011.
REFERENCES
- 1. Agarwalla P. K., Barnard Z. R., Curry W. T., Jr 2010. Virally mediated immunotherapy for brain tumors. Neurosurg. Clin. N. Am. 21:167–179 doi:10.1016/j.nec.2009.08.013 [DOI] [PubMed] [Google Scholar]
- 2. Alcami A., Symons J. A., Smith G. L. 2000. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J. Virol. 74:11230–11239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bandi P., Pagliaccetti N. E., Robek M. D. 2010. Inhibition of type III interferon activity by orthopoxvirus immunomodulatory proteins. J. Interferon Cytokine Res. 30:123–134 doi:10.1089/jir.2009.0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barber G. N. 2004. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 17:516–527 doi:10.1089/vim.2004.17.516 [DOI] [PubMed] [Google Scholar]
- 5. Basagoudanavar S. H., et al. 2011. Distinct roles for the NF-κB RelA subunit during antiviral innate immune responses. J. Virol. 85:2599–2610 doi:10.1128/JVI.02213-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berman J. J. 2004. Tumor classification: molecular analysis meets Aristotle. BMC Cancer 4:10 doi:10.1186/1471-2407-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bharatan N. S., Currier M. A., Cripe T. P. 2002. Differential susceptibility of pediatric sarcoma cells to oncolysis by conditionally replication-competent herpes simplex viruses. J. Pediatr. Hematol. Oncol. 24:447–453 [DOI] [PubMed] [Google Scholar]
- 8. Bridle B. W., et al. 2010. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 18:1430–1439 doi:10.1038/mt.2010.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carey B. L., Ahmed M., Puckett S., Lyles D. S. 2008. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J. Virol. 82:12104–12115 doi:10.1128/JVI.01508-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Connor J. H., Naczki C., Koumenis C., Lyles D. S. 2004. Replication and cytopathic effect of oncolytic vesicular stomatitis virus in hypoxic tumor cells in vitro and in vivo. J. Virol. 78:8960–8970 doi:10.1128/JVI.78.17.8960-8970.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diallo J. S., et al. 2010. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol. Ther. 18:1123–1129 doi:10.1038/mt.2010.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz R. M., et al. 2007. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 67:2840–2848 doi:10.1158/0008-5472.CAN-06-3974 [DOI] [PubMed] [Google Scholar]
- 13. Friedl P., Wolf K. 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3:362–374 doi:10.1038/nrc1075 [DOI] [PubMed] [Google Scholar]
- 14. Garraway L. A., Sellers W. R. 2006. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer 6:593–602 doi:10.1038/nrc1947 [DOI] [PubMed] [Google Scholar]
- 15. Gerlinger M., Swanton C. 2010. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 103:1139–1143 doi:10.1038/sj.bjc.6605912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hammill A. M., Conner J., Cripe T. P. 2010. Oncolytic virotherapy reaches adolescence. Pediatr. Blood Cancer 55:1253–1263 doi:10.1002/pbc.22724 [DOI] [PubMed] [Google Scholar]
- 17. Held M. A., et al. 2010. Characterization of melanoma cells capable of propagating tumors from a single cell. Cancer Res. 70:388–397 doi:10.1158/0008-5472.CAN-09-2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hingorani P., et al. 2011. Systemic administration of reovirus (Reolysin) inhibits growth of human sarcoma xenografts. Cancer 117:1764–1774 doi:10.1002/cncr.25741 [DOI] [PubMed] [Google Scholar]
- 19. Jawad M. U., Cheung M. C., Clarke J., Koniaris L. G., Scully S. P. 2011. Osteosarcoma: improvement in survival limited to high-grade patients only. J. Cancer Res. Clin. Oncol. 137:597–607 doi:10.1007/s00432-010-0923-7 [DOI] [PubMed] [Google Scholar]
- 20. Jemal A., Thomas A., Murray T., Thun M. 2002. Cancer statistics, 2002. CA Cancer J. Clin. 52:23–47 [DOI] [PubMed] [Google Scholar]
- 21. Lacroix M. 2008. Persistent use of “false” cell lines. Int. J. Cancer 122:1–4 doi:10.1002/ijc.23233 [DOI] [PubMed] [Google Scholar]
- 22. Leaman D. W., Salvekar A., Patel R., Sen G. C., Stark G. R. 1998. A mutant cell line defective in response to double-stranded RNA and in regulating basal expression of interferon-stimulated genes. Proc. Natl. Acad. Sci. U. S. A. 95:9442–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le Boeuf F., et al. 2010. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 18:888–895 doi:10.1038/mt.2010.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Q., Michaud M., Canosa S., Kuo A., Madri J. A. 2011. GSK-3beta: a signaling pathway node modulating neural stem cell and endothelial cell interactions. Angiogenesis 14:173–185 doi:10.1007/s10456-011-9201-9 [DOI] [PubMed] [Google Scholar]
- 25. Lichty B. D., Power A. T., Stojdl D. F., Bell J. C. 2004. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol. Med. 10:210–216 doi:10.1016/j.molmed.2004.03.003 [DOI] [PubMed] [Google Scholar]
- 26. Liu T. C., Kirn D. 2008. Gene therapy progress and prospects cancer: oncolytic viruses. Gene Ther. 15:877–884 doi:10.1038/gt.2008.72 [DOI] [PubMed] [Google Scholar]
- 27. Lockwood C. J., et al. 2011. Decidual hemostasis, inflammation, and angiogenesis in pre-eclampsia. Semin. Thromb. Hemost. 37:158–164 doi:10.1055/s-0030-1270344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meager A. 2002. Biological assays for interferons. J. Immunol. Methods 261:21–36 [DOI] [PubMed] [Google Scholar]
- 29. Mitelman F., Johansson B., Mertens F. 2007. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7:233–245 doi:10.1038/nrc2091 [DOI] [PubMed] [Google Scholar]
- 30. Nguyen T. L., et al. 2008. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. U. S. A. 105:14981–14986 doi:10.1073/pnas.0803988105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oda Y., Tsuneyoshi M. 2008. Recent advances in the molecular pathology of soft tissue sarcoma: implications for diagnosis, patient prognosis, and molecular target therapy in the future. Cancer Sci. 100:200–208 doi:10.1111/j.1349-7006.2008.01024.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Otsuki A., et al. 2008. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 16:1546–1555 doi:10.1038/mt.2008.155 [DOI] [PubMed] [Google Scholar]
- 33. Ozduman K., Wollmann G., Piepmeier J. M., van den Pol A. N. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882–1893 doi:10.1523/JNEUROSCI.4905-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Randall R. E., Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 doi:10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 35. Rouleau C., et al. 2008. Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clin. Cancer Res. 14:7223–7236 doi:10.1158/1078-0432.CCR-08-0499 [DOI] [PubMed] [Google Scholar]
- 36. Ryder E. F., Robakiewicz P. 2001. Statistics for the molecular biologist: group comparisons. Curr. Protoc. Mol. Biol. Appendix 3I. doi:10.1002/0471142727.mba03is43 [DOI] [PubMed] [Google Scholar]
- 37. Sadler A. J., Williams B. R. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568 doi:10.1038/nri2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sen G. C., Sarkar S. N. 2007. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr. Top. Microbiol. Immunol. 316:233–250 [DOI] [PubMed] [Google Scholar]
- 39. Stojdl D. F., et al. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825 doi:10.1038/77558 [DOI] [PubMed] [Google Scholar]
- 40. Stojdl D. F., et al. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 41. Taniguchi T., Takaoka A. 2001. A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2:378–386 doi:10.1038/35073080 [DOI] [PubMed] [Google Scholar]
- 42. Tomlinson J., et al. 1999. Different patterns of angiogenesis in sarcomas and carcinomas. Clin. Cancer Res. 5:3516–3522 [PubMed] [Google Scholar]
- 43. van den Pol A. N., Dalton K. P., Rose J. K. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 76:1309–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van den Pol A. N., et al. 2007. Cytomegalovirus induces interferon-stimulated gene expression and is attenuated by interferon in the developing brain. J. Virol. 81:332–348 doi:10.1128/JVI.01592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J., et al. 2010. NF-kappa B RelA subunit is crucial for early IFN-beta expression and resistance to RNA virus replication. J. Immunol. 185:1720–1729 doi:10.4049/jimmunol.1000114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wollmann G., Robek M. D., van den Pol A. N. 2007. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J. Virol. 81:1479–1491 doi:10.1128/JVI.01861-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wollmann G., Rogulin V., Simon I., Rose J. K., van den Pol A. N. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 doi:10.1128/JVI.02040-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wollmann G., Tattersall P., van den Pol A. N. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005–6022 doi:10.1128/JVI.79.10.6005-6022.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wongthida P., et al. 2011. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum. Gene Ther. [Epub ahead of print.] doi:10.1089/hum.2010.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zagars G. K., et al. 2003. Prognostic factors for patients with localized soft tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 97:2530–2543 doi:10.1002/cncr.11365 [DOI] [PubMed] [Google Scholar]