

Abstract
It is generally acknowledged that the Tat protein has a pivotal role in HIV-1 replication because it stimulates transcription from the viral long terminal repeat (LTR) promoter by binding to the TAR hairpin in the nascent RNA transcript. However, a multitude of additional Tat functions have been suggested. The importance of these functions is difficult to assess in replication studies with Tat-mutated HIV-1 variants because of the dominant negative effect on viral gene expression. We therefore used an HIV-1 construct that does not depend on the Tat-TAR interaction for transcription to reevaluate whether or not Tat has a second essential function in HIV-1 replication. This HIV-rtTA variant uses the incorporated Tet-On gene expression system for activation of transcription and replicates efficiently upon complete TAR deletion. Here we demonstrated that Tat inactivation does nevertheless severely inhibit replication. Upon long-term culturing, the Tat-minus HIV-rtTA variant acquired mutations in the U3 region that improved promoter activity and reestablished replication. We showed that in the absence of a functional TAR, Tat remains important for viral transcription via Sp1 sequence elements in the U3 promoter region. Substitution of these U3 sequences with nonrelated promoter elements created a virus that replicates efficiently without Tat in SupT1 T cells. These results indicate that Tat has a versatile role in transcription via TAR and U3 elements. The results also imply that Tat has no other essential function in viral replication in cultured T cells.
INTRODUCTION
Transcription of the HIV-1 provirus starts by the binding of cellular factors, including NF-κB, Sp1, the TATA box binding protein, and RNA polymerase II, to the promoter region in the 5′ long terminal repeat (LTR). This transcription complex allows the production of a low level of viral transcripts, which are subsequently spliced and translated. One of the early viral proteins is the transcriptional activator Tat, which plays a pivotal role in HIV-1 replication because it enhances transcription through binding to the TAR hairpin at the 5′ end of newly formed RNA transcripts (reviewed in references 5 and 17). Important features in the TAR hairpin are the highly conserved 3-nucleotide (nt) pyrimidine bulge that binds the Tat protein (72) and the apical 6-nt loop to which the transcriptional elongation factor pTEFb binds in a Tat-dependent manner (62, 77). Upon TAR binding, the kinase component of pTEFb, cyclin-dependent kinase 9 (CDK9), can phosphorylate the C-terminal domain of RNA polymerase II, which enhances the processivity of the elongating polymerase (14, 58). pTEFb also directs the recruitment of TATA box binding protein to the LTR promoter and thus stimulates the assembly of new transcription complexes (61). Furthermore, Tat can recruit several chromatin-modifying proteins to remodel the promoter region (reviewed in references 30, 31, and 63).
Several other, nontranscriptional functions of Tat in the viral life cycle have been suggested (reviewed in reference 63). For example, Tat was shown to influence HIV-1 RNA splicing (38), capping (19, 20, 80), translation (15, 16, 18, 68), and reverse transcription (3, 36, 43, 44). Moreover, Tat has been proposed to modulate the expression of multiple cellular genes (reviewed in reference 63), to interact with a large number of cellular proteins (reference 32 and references therein), and to inhibit the cellular RNA interference mechanism (6, 7, 33, 67). Thus, in addition to its undisputed essential function in the Tat/TAR mechanism of transcription activation, a large array of other functions has been attributed to Tat in a variety of experimental systems, although some of these functions have been questioned (51, 66). The biologically most relevant assay system is that of the replicating virus. Most mutations in Tat severely impair viral replication, which is in agreement with the transcriptional requirement for Tat. This dominant negative effect makes it difficult to study other Tat-mediated processes in the viral replication cycle. We developed a replicating HIV-1 variant that does not depend on the Tat/TAR mechanism for transcription. This HIV-rtTA variant uses the incorporated Tet-On gene expression system for activation of transcription, and deletion of TAR is allowed in this context (22). Here we constructed Tat-deficient HIV-rtTA variants to probe the importance of other Tat-associated functions in viral replication.
MATERIALS AND METHODS
Cell and virus cultures.
SupT1 (70) and 174 × CEM cells (65) were cultured and transfected by electroporation as previously described (29). To assay virus replication, we transfected 5 × 106 cells with 5 μg of the proviral constructs and cultured them in 5 ml of medium with 1 μg/ml of doxycycline (dox; Sigma D-9891). For selection of viruses with improved replication capacity, we continued the virus-cell cultures and split the cells twice a week. When virus-induced cytopathic effects were observed, high-level virus replication was maintained by passage of the cell-free culture supernatant onto uninfected SupT1 cells. Cell and supernatant samples were stored at −80°C for subsequent analysis.
Human peripheral blood mononuclear cells (PBMCs) were isolated from fresh buffy coats by standard Ficoll-Hypaque density centrifugation. Cells were frozen in multiple vials and, when required, thawed and activated with 5 μg/ml of phytohemagglutinin (PHA; Sigma) and cultured with RPMI medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 100 U/ml of recombinant interleukin-2 (IL-2). At day 3, the PBMCs were enriched for CD4+ cells by depletion of CD8+ cells, using CD8 immunomagnetic beads (Dynal Biotech). Cells (106) in 1 ml of medium were infected with 293T-produced virus (5 ng of viral capsid protein CA-p24) on day 6 and cultured with 1 μg/ml of dox.
293T and C33A cells were cultured in 2-cm2 wells and transfected with 1 μg of a proviral construct as previously described (22). When indicated, cells were cultured in 1-cm2 wells and transfected with 3.2 μl of Nanofectin (PAA Laboratories GmbH) and 0.5 μg of proviral plasmid, 0 to 50 ng of the Tat-expressing plasmid pTat, and 50 to 0 ng of pcDNA3 (empty expression vector; the total amounts of pTat and pcDNA3 were kept constant at 50 ng). Unless stated otherwise, cells were cultured with 1 μg/ml of dox. Virus production was measured by CA-p24 enzyme-linked immunosorbent assay (ELISA) on culture medium samples (42), and values obtained in independent experiments were corrected for between-session variation (64).
Proviral DNA analysis of HIV-rtTA cultures.
Virus-infected cells were pelleted by centrifugation at 4,000 rpm for 4 min and washed with phosphate-buffered saline. DNA was solubilized by resuspending the cells in 10 mM Tris-HCl [pH 8.0], 0.1 mM EDTA, and 0.5% Tween 20, followed by incubation with 200 μg of proteinase K per ml at 56°C for 30 min and at 95°C for 10 min. Proviral DNA sequences were PCR amplified from total cellular DNA with primers tTA3 (CTGTGTCAGCAAGGCTTCTC; sense primer annealing to rtTA sequence) and U5Nar (antisense primer annealing to 3′ end of U5) (22). The PCR product was sequenced (population sequence) or ligated into a pCR2.1-TOPO TA Cloning vector (Invitrogen) followed by sequencing of several clones.
rtTA activity assay.
In the set of pHIV-LTR-2/4/6ΔtetO-X/H-luc constructs, the expression of firefly luciferase is under the control of the indicated promoter. In the plasmid pRL-CMV (Promega), the expression of Renilla luciferase is controlled by the cytomegalovirus immediate-early (CMV IE) enhancer/promoter. This plasmid was cotransfected to allow correction for differences in transfection efficiency. 293T cells were cultured in 2-cm2 wells and transfected by calcium phosphate precipitation with 20 ng of pHIV-LTR-2/4/6ΔtetO-X/H-luc, 0.4 ng of rtTA expression plasmid pCMV-rtTAF86Y A209T (29), 0.5 ng of pRL-CMV, and 1 μg of pBluescript carrier DNA as previously described (22). The cells were cultured after transfection for 48 h without or with 1 μg/ml of dox. Cells were lysed in Passive lysis buffer, and firefly and Renilla luciferase activities were determined with a Dual-Luciferase assay (Promega). The expression of firefly and Renilla luciferases was within the linear range, and no squelching effects were observed. The promoter activity was calculated as the ratio between the firefly and Renilla luciferase activities and corrected for between-session variation (64).
Tat activity assay.
Plasmid pBlue3′LTR-luc contains the complete Tat-responsive LTR promoter of the wild-type (wt) HIV-1 LAI strain (U3 region and R sequences up to position +82 relative to the transcription start site) coupled to the firefly luciferase reporter gene (41). C33A cells in 2-cm2 wells were transfected with 100 ng of the HIV-rtTA plasmids (with a variable Tat gene) or the pBluescript control plasmid, 20 ng of pBlue3′LTR-luc, 0.5 ng of pRL-CMV, and 380 ng of pBluescript (as carrier DNA) as previously described (22). After the cells were cultured for 48 h with 1 μg/ml of dox, firefly and Renilla luciferase production was measured. The Tat activity was calculated as the ratio between the firefly and Renilla luciferase activities and corrected for between-session variation (64).
The pTat vector.
The two Tat protein-coding exons (Tat1 and Tat2) of the HIV-1 LAI isolate (59) were fused by PCR and cloned into a pcDNA3 expression plasmid. Tat1 was PCR amplified from the full-length molecular clone pLAI with primers Tat-exon1 (ACGAAGCTTGGGTGTCGACATAGCA; sense primer annealing upstream of Tat AUG start codon; HindIII site underlined) and Tat-splice1 (GGGAGGTGGGTTGCTTTGATAGAGAAACTTGATG; anti-sense primer; 3′ end of Tat1 sequence underlined). Tat2 was PCR amplified from pLAI with primers Tat-splice2 (CTATCAAAGCAACCCACCTCCCAACCCCGA; sense primer; 5′ end of Tat2 sequence underlined) and Rev-exon2 (CTCCGCAGATCGTCCCAGAT; anti-sense primer annealing downstream of Tat stop codon and BamHI site). The PCR products were mixed and fused in a PCR with the outer primers Tat-exon1 and Rev-exon2. The resulting Tat1-Tat2 fragment was digested with HindIII and BamHI and ligated into the corresponding sites of pcDNA3.
The Tat-mutated HIV-rtTA variants.
Construction of the HIV-rtTA molecular clone with the TatY26A gene was described previously (28, 75). The HIV-rtTA variant used in this study (HIV-rtTAF86Y A209T-2ΔtetO) contains the optimized 2ΔtetO promoter configuration in the 5′ and 3′ LTRs (Δ represents the deletion of 15 nucleotides between tetO elements) (54), the optimized rtTAF86YA209T gene (29), and the TatY26A gene, unless stated otherwise. For the construction of HIV-rtTA-Tatwt, the NcoI-BamHI Tat1-Vpu-Env fragment from pLAI (59) was used to replace the corresponding fragment in HIV-rtTA. For the construction of other Tat-mutated HIV-rtTA variants, a shuttle plasmid was constructed in which the NcoI-BamHI fragment from HIV-rtTA was cloned into the “empty” vector resulting from NcoI and BamHI digestion of pCMV-rtTA (29). Tat was mutated by mutagenesis PCR, using the overlap extension method (56). Fragment 1 was produced by PCR amplification of pLAI with primers L5 (CATGCGGCCGCTTCTGCAACAACTGCTGTTT; annealing upstream of the SalI site in the Vpr region) and Tat-splice1. Fragment 2 was produced by PCR amplification with the mutagenic sense primer TatMut1 (CAACAGAGGAGAGCAAGAAACGGAGCCAGTAGATC; T to C mutation that inactivates the AUG translation start codon underlined), TatMut2 (GGAAGCATCCATGAAGTTAACCTAAAACTGCTTGT; mutations that create stop codons underlined), or TatMut3 (GGAAGCATCCAGGAAGTCAGCCTAAAΔCTGCTTGTACCA; Δ indicates the position of the 1-nt deletion and frameshift mutation) and the WS3 antisense primer (ATCTTAAGTTTGATCCCATAAACTGATTA; annealing downstream of the KpnI site in the Vpu region). Fragments 1 and 2 were mixed and fused by PCR with the outer primers L5 and WS3. The resulting Tat-mutated fragments were digested with SalI and KpnI and used to replace the corresponding sequence in the Tat1-Vpu-Env shuttle vector. The NcoI-BamHI fragments from these plasmids were subsequently used to replace the corresponding fragment in HIV-rtTA, which resulted in the TatmAUG, Tatstop, and Tatfs HIV-rtTA variants.
Cloning of evolved HIV-rtTA-Tatstop sequences.
For the cloning of the evolved tetO promoter configurations into the HIV-rtTA-Tatstop provirus, the SmaI-SacI U3-R fragments from the selected TA clones were used to replace the corresponding sequence in pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO-mPL (22). The BamHI-BglI fragments of these 3′-LTR-mutated plasmids were used to replace the corresponding sequences in HIV-rtTA-Tatstop variants. For the introduction of the evolved sequences in the 5′ LTR, we first constructed a pBlue-5′HIV-rtTA shuttle vector. For this, the 5′ LTR region was PCR amplified from HIV-rtTA with primers U3XbaNot (ACGTCTAGAGCGGCCGCACTGGAAGGGCTAATTCACTC; NotI site underlined) and AH-GAG-KpnI (AGTGGTACCGTTCTAGCTCCCTGCTTGCCC; KpnI/Asp718 site underlined). The product was digested with NotI and Asp718 and ligated into the corresponding sites of pBluescript SK+. The EcoRV-HindIII U3-R fragments from the selected TA clones were used to replace the corresponding region in pBlue5′HIV-rtTA. The NotI-BssHII fragments from these plasmids were subsequently used to replace the 5′-LTR sequence in the 3′-LTR-modified HIV-rtTA-Tatstop plasmids, which resulted in the HIV-rtTA-Tatstop-4tetO5′+3′ and HIV-rtTA-Tatstop-6tetO5′+3′ clones. For the construction of promoter-luciferase plasmids, the 5′-LTR region was PCR amplified with primers U3XbaNot and AD-AUG (CATGGATCCCCCGCTTAATACTGACGC; antisense primer annealing downstream of Gag translation start site). The PCR fragments were digested with EcoRV and HindIII, and the 4tetO and 6tetO fragments were ligated into the corresponding sites in pHIV-LTR-2ΔtetO-X/H-luc (22), which resulted in pHIV-LTR-4ΔtetO-X/H-luc and pHIV-LTR-6ΔtetO-X/H-luc.
Deletion of NF-κB and upstream U3 sequences.
For construction of the ΔNF variant, the LTR sequence was amplified with primers delNFkB-1 (ACGATATCCGGATGCATTCCGTCGAGTTTACCACT; sense primer annealing directly upstream of the tetO elements; EcoRV site underlined) and C(N1) (antisense primer annealing to the U5 region) (23), with plasmid pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO, which includes Env, rtTA, and 3′-LTR sequences of the HIV-rtTA genome (29), as a template. The PCR product was digested with EcoRV and SalI and used to replace the corresponding 3′-LTR fragment in pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO. The resulting shuttle plasmid pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO-dNFeco thus lacks the U3 region from downstream of the attachment (att) site (i.e., from the EcoRV site at U3 position 35) to upstream of the tetO sites. The BamHI-BglI fragment of this plasmid was used to replace the corresponding Env-rtTA-3′ LTR sequences in HIV-rtTA, which resulted in HIV-rtTA-3′dNFeco. To delete the corresponding U3 region in the 5′ LTR, the LTR region was amplified with primers delNFkB-2 (ACGTCTAGAGCGGCCGCGATATCCGTCGAGTTTACCACT; sense primer annealing directly upstream of the tetO elements; NotI site underlined) and U5Nar, with plasmid pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO as a template. After digestion with NotI and NarI, the PCR product was used to replace the corresponding 5′-LTR region in HIV-rtTA-3′dNFeco to generate the double mutant HIV-rtTA-dNF5′+3′.
Deletion of TAR.
For construction of the ΔNF ΔTAR variant, the TAR sequences were replaced with nonrelated ER3 sequences (22). The ER3-containing LTR region was PCR amplified from the TA clone that had previously been used for the construction of HIV-rtTA-ER3 (22) with primers delNFkB-2 and AD-GAG (annealing to the Gag region) (27). After digestion with NarI and NotI, the PCR product was used to replace the corresponding 5′-LTR fragment in HIV-rtTA, which resulted in HIV-rtTA-5′dNF/ER3.
Substitution of the HIV-derived promoter with nonrelated promoter sequences.
The tetO-CMV promoter configuration was developed in another HIV-rtTA study (to be published). Briefly, we constructed an HIV-rtTA variant in which the U3 region was replaced by seven tetO elements coupled to the 30-bp minimal promoter region of the CMV IE promoter and the TAR element was replaced with a 33-nt hairpin sequence that includes position +1 to +16 of the CMV transcript (with +1 indicating the transcription start site). Upon long-term culturing of this virus, we observed additional mutations in the U3-R region and the deletion of four tetO sites. This evolved U3-R promoter sequence was recloned in the HIV-rtTA plasmid. For this process, the 5′-LTR region was amplified from proviral DNA with primers delNFkB-2 and AD-GAG. The NarI- and NotI-digested PCR product was used to replace the corresponding 5′-LTR sequences in HIV-rtTA. Moreover, the 3′-LTR region was amplified from the cellular proviral DNA with primers tTA3 and U5Nar. After digestion with NdeI and HindIII, the PCR product was used to replace the corresponding fragment in pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO-mPL. The BamHI-BglI fragment of this 3′-LTR-mutated pBlue3′LTRext-ΔU3-rtTAF86Y A209T-tetO-CMV plasmid was used to replace the corresponding sequence in the 5′-LTR-mutated HIV-rtTA variant to construct HIV-rtTA-tetO-CMV5′+3′. This proviral plasmid thus contains three tetO elements and a CMV-derived minimal promoter/hairpin sequence in both the 5′ and 3′ LTRs. In this plasmid, the AatII-BglI vector fragment downstream of the 3′ LTR was replaced with the corresponding fragment from HIV-rtTA-SV40, which contains the simian virus 40 (SV40) polyadenylation signal (76). The TatY26A gene in the HIV-rtTA-tetO-CMV5′+3′ construct was replaced with the Tatwt and Tatstop genes through substitution of the NcoI-DraIII Tat-Vpu fragment with the corresponding fragment from HIV-rtTA-Tatwt and HIV-rtTA-Tatstop. The 5′ LTR in these HIV-rtTA-tetO-CMV5′+3′-Tatwt and HIV-rtTA-tetO-CMV5′+3′-Tatstop plasmids was replaced with other promoter configurations through the replacement of the NotI-NarI fragment with the corresponding fragments from HIV-rtTA, HIV-rtTA-dNF5′+3′, and HIV-rtTA-5′dNF/ER3.
Reintroduction of Sp1 binding sites.
For the construction of the tetO-CMV-Sp1 variant, the EcoRV-HindIII tetO-CMV LTR fragment was used to replace the corresponding region in pBlue-5′HIV-rtTA to construct pBlue-5′HIV-rtTA-tetO-CMV. The Sp1 sequences from HIV-rtTA-dNF5′+3′ were PCR amplified with primers pKP5′seq (CCACCTCTGACTTGAGCGTC; annealing to vector sequences upstream of the 5′ LTR) and Sp1-XhoI-rev (TCTCTCGAGTCGCCACTCCCCAGTCCCG; antisense primer annealing to the Sp1 sites; XhoI site underlined). After digestion with SalI (site located upstream of Sp1 sites) and XhoI, the PCR product was ligated into the compatible SalI site of pBlue-5′HIV-rtTA-tetO-CMV, which resulted in the pBlue-5′HIV-rtTA-tetO-CMV-Sp1 plasmid. The NotI-BssHII fragment from this plasmid was subsequently used to introduce the new promoter configuration into the 5′ LTRs of HIV-rtTA-tetO-CMV5′+3′-Tatwt and HIV-rtTA-tetO-CMV5′+3′-Tatstop. The tetO-CMV-Sp1 configuration was subsequently also introduced into the 3′ LTR of these plasmids. For this process, the EcoRV-HindIII 5′-LTR fragment derived from these plasmids was used to replace the corresponding 3′-LTR region in pBlue3′LTRext-ΔU3-rtTAF86Y A209T-2ΔtetO-dNFeco, which resulted in pBlue3′LTRext-ΔU3-rtTAF86Y A209T-tetO-CMV-Sp1. The NruI-AatlI fragment from these 3′-LTR-mutated plasmids was used to replace the corresponding region in the 5′-LTR-mutated tetO-CMV-Sp1 plasmids, which resulted in HIV-rtTA-tetO-CMV-Sp15′+3′-Tatwt and HIV-rtTA-tetO-CMV-Sp15′+3′-Tatstop. All mutations were verified by sequence analysis.
RESULTS
Tat mutations in the HIV-rtTA variant.
We previously presented a doxycycline-controlled HIV-1 variant in which the Tat/TAR transcription activation mechanism was inactivated and functionally replaced by the dox-inducible Tet-On gene expression system (Fig. 1 A) (54, 75). TAR was inactivated through several nucleotide changes in the bulge and loop elements, which prevent the binding of Tat and cyclin T1, respectively. To integrate the Tet-On system, the gene encoding the rtTA transcriptional activator protein was inserted in place of the 3′-terminal nef gene, and tet operator (tetO) binding sites were introduced in the LTR promoter. Administration of dox induces a conformational switch in the rtTA protein, which enables binding to the tetO-LTR promoter and activation of transcription. Thus, transcription and replication of HIV-rtTA are critically dependent on the addition of dox.
Fig. 1.

Mutations in Tat. (A) Schematic of the HIV-rtTA proviral DNA genome, with the LTR region subdivided in the U3, R, and U5 domains. The Tat-TAR axis of transcription regulation was inactivated by multiple nucleotide substitutions in the bulge and loop sequences of TAR (TARm; crossed boxes). Transcription and replication of the virus were made dox dependent by the introduction of tetO elements in the U3 promoter region and replacement of the Nef gene by the rtTA gene. We constructed different HIV-rtTA variants with either a wild-type or a mutated Tat gene. (B) Mutations in Tat. The sequence of the first Tat-coding exon from splice acceptor 3 (SA3) to the splice donor site (SD) is indicated with the translated amino acid sequence. The mAUG, stop, fs, and Y26A mutations in the Tat gene are boxed in gray. These mutations do not affect the Vpr and Rev amino acid sequences (the Vpr TAG stop codon and the Rev AUG start codon are underlined), splice acceptor (SA) sites, or known splice enhancer (ESE) and silencer (ESS) motifs. (C) Activity of Tat mutants. The HIV-rtTA plasmids and the control plasmid pBluescript (−) were cotransfected with a Tat-responsive HIV-1 LTR promoter/luciferase reporter construct into 293T cells. Luciferase production was measured after the cells were cultured with 1 μg/ml of dox for 48 h. Average values obtained in multiple experiments are shown, with the wild-type level set at 100% and the error bars indicating standard deviations (n = 8 for HIV-rtTA plasmids; n = 4 for pBluescript). Statistical analyses (two-sided unpaired sample t tests) demonstrated that all mutated constructs and the pBluescript control differed significantly from the wild-type construct (P < 0.01). Furthermore, the wt, Y26A, and mAUG constructs differed significantly from the pBluescript control, while the stop and fs mutants did not. (D) Replication of HIV-rtTA variants. SupT1 T cells were transfected with the different HIV-rtTA plasmids and cultured with 1 μg/ml of dox. CA-p24 levels in the culture supernatants were measured by ELISA. No virus replication was observed in the absence of dox (data not shown). The replication curves shown are representative of data obtained in four experiments.
To investigate additional roles of Tat in HIV-1 replication, we generated a set of HIV-rtTA variants with different Tat genes (Fig. 1B). The Tatwt construct encodes wild-type Tat protein. A tyrosine-to-alanine substitution was introduced at position 26 in the TatY26A variant. This mutation has previously been shown to strongly reduce Tat-mediated activation of the HIV-1 LTR promoter (74). In the Tatfs virus, a 1-nt deletion causes an early frameshift in the Tat open reading frame, which results in a polypeptide of 42 amino acids with only 19 N-terminal residues of Tat. The Tat translation start codon was mutated (AUG to ACG) in the TatmAUG variant to reduce Tat production. In the Tatstop virus, two translation stop codons were introduced at positions 15 and 17 in the Tat open reading frame, which causes premature termination of translation. All mutations were carefully chosen such that they do not affect the overlapping Vpr and Rev open reading frames or known splice signals (indicated in Fig. 1B) (1, 2, 39, 46, 69, 79). We introduced a 3-nt codon change in TatY26A (UAU to GCC) and two stop codons in Tatstop (GGA to UGA and CAG to UAA) to restrict the likelihood of simple reversion to the wild-type sequence in virus evolution experiments. For both mutants, restoration of the wild-type Tat amino acid sequence would require at least two nucleotide substitutions (8, 10).
To evaluate the effect of these mutations on Tat protein production and activity, 293T cells were transfected with the HIV-rtTA plasmids and an HIV-1 LTR-luciferase reporter construct and cultured with dox for 48 h. The LTR promoter in this construct has a wild-type TAR element and is fully Tat responsive. The Tatwt construct did indeed result in a high level of luciferase expression (Fig. 1C). The TatY26A variant showed an approximately 4-fold-reduced luciferase level, which confirms that the Y26A mutation significantly reduces Tat activity. In agreement with their inability to produce functional Tat protein, the Tatstop and Tatfs constructs resulted in very low luciferase levels that are indistinguishable from the background level obtained upon cotransfection of a control plasmid instead of HIV-rtTA. Mutation of the AUG translation start codon apparently did not fully prevent the production of Tat protein, as a considerable luciferase level (∼50% of that of the Tatwt construct) was measured for the TatmAUG construct. This observation is in agreement with previous studies that indicated that ACG codons can be used for translation initiation in viral mRNAs, although less efficiently than AUG codons (reference 49 and references therein). The relatively high level of activity of this ACG start codon may be due to the presence of G nucleotides at positions −3 and +4 relative to the start codon, which is favorable for the binding of the translation initiation complex (60).
Tat inactivation impairs HIV-rtTA replication.
We next analyzed the replication capacity of the HIV-rtTA variants in the SupT1 T cell line. None of these viruses replicated in the absence of dox (data not shown), which is in agreement with the fact that gene expression of HIV-rtTA is strictly controlled by the introduced Tet-On system. The Tatwt, TatY26A, and TatmAUG variants replicated efficiently in the presence of dox, resulting in a rapid increase in the levels of viral capsid protein (CA-p24) in the culture media (Fig. 1D) and the appearance of syncytia (data not shown). In contrast, the Tatfs mutant replicated poorly, as illustrated by delayed CA-p24 production and syncytia formation, and the Tatstop mutant did not replicate. These latter results indicate that Tat remains important for HIV-rtTA replication, despite the functional replacement of the Tat-TAR transcriptional axis with the Tet-On system. Sequence analysis revealed that the Tatfs virus rapidly acquired a 1-nt insertion that restored the wild-type Tat sequence and function, which underscores the requirement for a functional Tat protein. The 1-nt insertion most likely resulted from template slippage during the reverse transcription process on the homopolymeric A stretch (AAA to AAAA), which is a common reverse transcriptase error. The fast-replicating TatY26A and TatmAUG viruses stably maintained the introduced mutations, which suggests that 25% Tat activity suffices for HIV-rtTA replication.
Improved promoter activity can rescue replication of a Tat-negative HIV-rtTA variant.
The Tat-lacking Tatstop mutant did not cause a spreading infection (Fig. 1D), but we were able to select replication-competent variants in two independent long-term cultures. Sequence analysis demonstrated that the introduced stop codons were stably maintained in the Tat gene, but an increase in the number of tetO elements in the LTR promoter region was observed in both cultures. Whereas the original mutant has two tetO sites, sequencing of the virus population in culture I after 108 days revealed the presence of four tetO elements (Fig. 2). At 206 and 272 days, a heterogeneous virus population was present and analysis of individually cloned LTR fragments indicated that the number of tetO elements varied from 3 to 7. Similarly, the number of tetO elements increased in culture II and the dominant virus species contained three tetO sites at 55 and 130 days and four tetO sites at 283 days (Fig. 2). Multiplication and deletion of repeated sequence elements has previously been observed during HIV-1 evolution and is likely due to template slippage during the reverse transcription process (9, 13, 48, 50, 54). To test whether these LTR modifications improve viral replication, we recloned the 4- and 6-tetO configurations in the HIV-rtTA-Tatstop variant (Fig. 3A). Whereas the original Tatstop virus with the 2-tetO configuration did not replicate in SupT1 cells (Fig. 3B), the 4-tetO virus replicated weakly and the 6-tetO virus replicated fairly well, although not as well as the Tatwt and TatY26A viruses. Similar results were obtained when we tested replication in primary PBMCs (Fig. 3C) and 174 × CEM cells (fusion of a B and T cell line; Fig. 3D). The Tatwt virus replicated efficiently in these cells, whereas the Tatstop mutation prevented replication and replication was partially restored by tetO multiplication.
Fig. 2.

Evolution of HIV-rtTA-Tatstop. (A) Comparison of the U3-R regions from HIV-1 and HIV-rtTA. The HIV-rtTA variants have two tetO elements inserted between the NF-κB and Sp1 binding sites and several nucleotide substitutions in TAR (TARm). (B) Mutations observed in the tetO region of the HIV-rtTA-Tatstop mutant upon culturing with dox for up to 272 days (culture I) or 283 days (culture II). Cellular proviral DNA was isolated at 108, 206, and 272 days (culture I) and at 55, 130, and 283 days (culture II), and the LTR region was subsequently PCR amplified and either directly sequenced (population [pop] sequence; indicated on the left) or cloned into the TA-cloning vector followed by sequencing of three to six TA clones for each sample (with the frequency at which each sequence was observed indicated on the left). The nucleotide sequences of the tetO region between the NF-κB binding sites and the SpI binding sites are shown for HIV-rtTA-Tatstop (upper line) and the evolved viruses. The tetO core sequence is boxed in gray. The number of tetO elements present in each sequence is indicated on the right, with asterisks indicating the sequences that were recloned into HIV-rtTA-Tatstop (Fig. 3). Nucleotide substitutions are indicated with lowercase characters. Δ, nucleotide deletion.
Fig. 3.
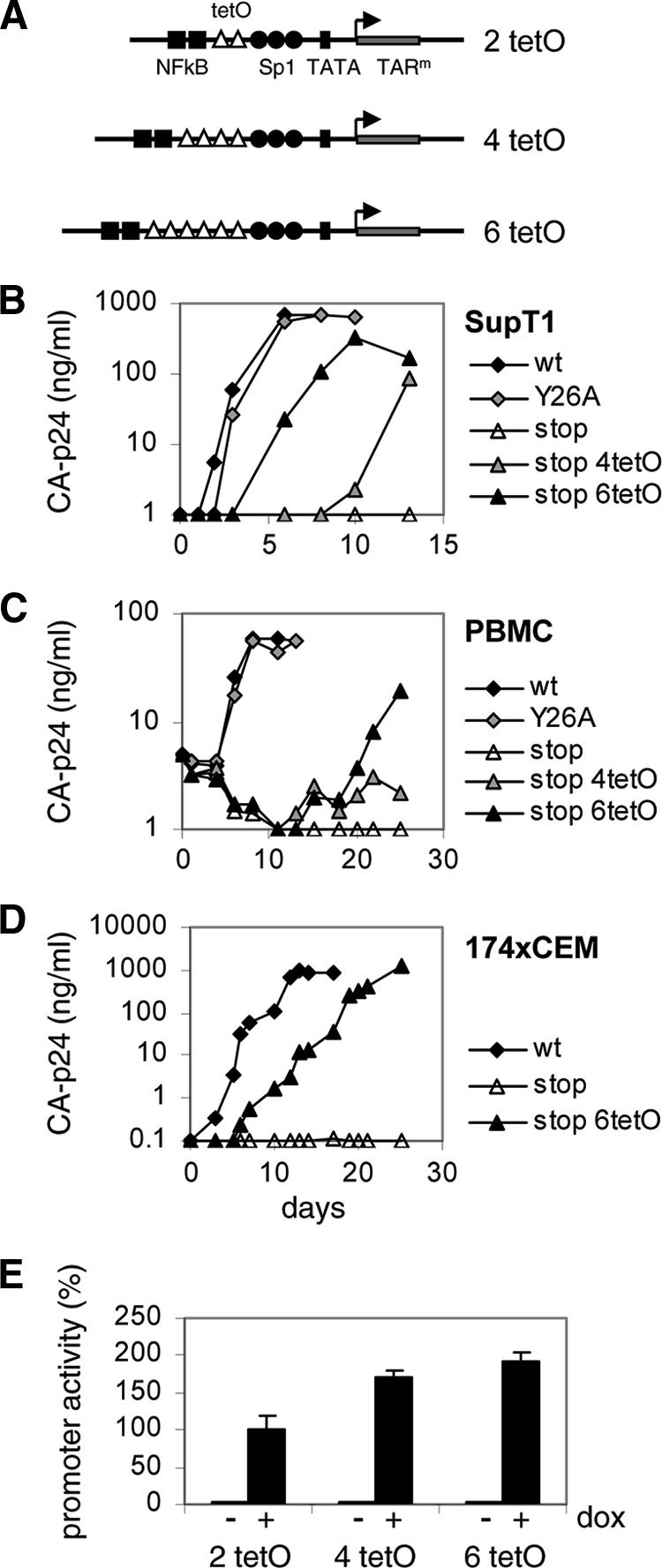
Multiplication of tetO elements rescues replication of HIV-rtTA-Tatstop. (A) The HIV-rtTA-Tatstop mutant has two tetO sites in the LTR promoter region. The 4- and 6-tetO configurations that were observed upon long-term culturing (sequences indicated with asterisks in Fig. 2) were recloned into the 5′ and 3′ LTRs of HIV-rtTA-Tatstop. (B to D) Replication of the HIV-rtTA-Tatstop variants with different tetO configurations was assayed in SupT1 cells (B), PBMCs (C), and 174 × CEM cells (D). Cells were transfected with the different HIV-rtTA plasmids (B and D) or infected with 293T-produced virus (C) and cultured with 1 μg/ml of dox. Replication was monitored by CA-p24 ELISA on culture supernatant samples. For comparison, we included HIV-rtTA-Tatwt and HIV-rtTA-TatY26A. The replication curves shown are representative of data obtained in four (B), three (C), or two (D) experiments. (E) LTR-tetO-promoter/luciferase reporter constructs with two, four, or six tetO sites were cotransfected with an rtTA-expressing plasmid into 293T cells. Cells were cultured without or with 1 μg/ml of dox for 2 days, after which luciferase production was quantified to measure promoter activity. Average values obtained in eight experiments are shown, with error bars indicating standard deviations. The average activity of the 2-tetO construct with dox was set at 100%. Statistical analyses (two-sided unpaired sample t tests) demonstrated that the promoter activity of the 2-tetO, 4-tetO, and 6-tetO constructs with dox differed significantly (2 tetO versus 4 tetO, 2 tetO versus 6 tetO, and 4 tetO versus 6 tetO; P < 0.01).
We also made reporter constructs with the LTR-tetO promoter containing two, four, or six tetO sites. 293T cells were transfected with these constructs and an rtTA-expressing plasmid, cultured without or with dox for 2 days, after which luciferase production was quantified to measure promoter activity (Fig. 3E). In the absence of dox, the luciferase levels were similarly low for all tetO configurations, which demonstrates that the tetO changes do not affect background promoter activity. The addition of dox resulted in high luciferase levels, which was arbitrarily set at 100% for the initial 2-tetO construct. The induced transcription level was significantly increased (∼160%) for the 4-tetO construct and further elevated for the 6-tetO construct (∼180%). This result demonstrates that a larger number of tetO sites does improve promoter activity, which coincides with increased virus replication capacity. Possibly, HIV-rtTA gene expression is very sensitive to this increase in promoter strength because the rtTA transcription activator is produced from this tetO promoter in an autoregulatory loop. Taken together, these results demonstrate that multiplication of the tetO elements improves LTR promoter activity and rescues replication of the Tat-deficient HIV-rtTA-Tatstop virus.
HIV-rtTA gene expression requires Tat activity.
Because the Tat requirement of HIV-rtTA can be compensated for by a more active LTR promoter, it seems plausible that Tat remains important for optimal viral gene expression. We therefore analyzed the effect of Tat on virus production. 293T cells were transfected with the different virus constructs and an increasing amount of a wild-type Tat-expressing plasmid (0 to 50 ng of pTat). 293T cells do not express the CD4 receptor that is required for HIV-1 infection, but their high level of transfection competence and capacity to express transfected virus constructs allowed us to measure transient virus production after culturing the cells with dox for 48 h (Fig. 4). Without additional wild-type Tat, virus production was high for the Tatwt and TatY26A variants, 2-fold reduced for the TatmAUG mutant, and further reduced for the Tatstop and Tatfs mutants. Complementation with wild-type Tat fully restored gene expression of the TatmAUG, Tatstop, and Tatfs viruses. These results demonstrate that HIV-rtTA gene expression is stimulated by the Tat protein in trans. It seems unlikely that Tat exerts this function through the TAR motif, because this element is inactivated by multiple mutations. Furthermore, complete deletion of TAR does not inhibit HIV-rtTA gene expression and replication (22). Thus, Tat can activate HIV-rtTA gene expression in trans in a TAR-independent manner.
Fig. 4.
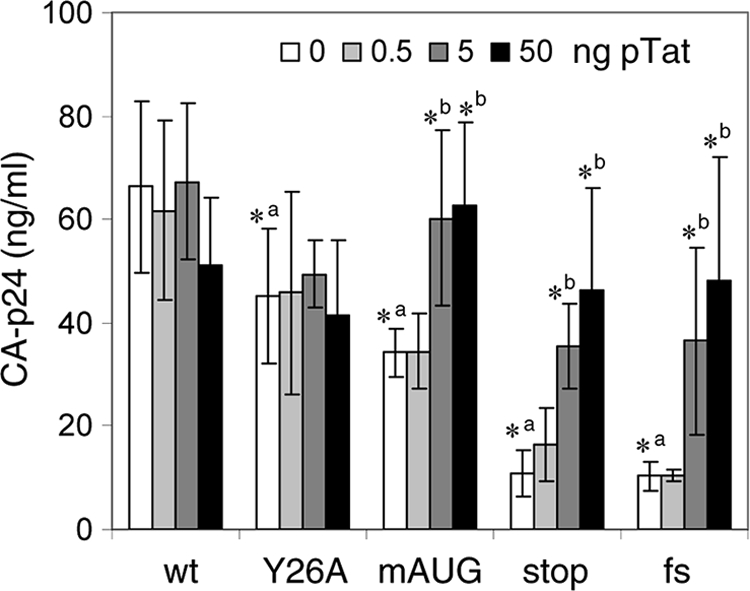
Tat activates HIV-rtTA gene expression. 293T cells were transfected with the HIV-rtTA constructs and 0 to 50 ng of pTat. After the cells were cultured with dox for 48 h, the CA-p24 levels in the culture supernatants were measured by ELISA. Average values obtained in multiple experiments are shown (n = 6 to 9), with error bars indicating standard deviations. A two-sided unpaired-sample t test was used to identify statistically significant differences (P < 0.01) between the wt and Tat mutants at 0 ng of pTat (a, asterisk indicates a statistically significant difference between the wt and mutant) and between the HIV-rtTA construct at 0 ng of pTat and the same construct at 0.5, 5, or 50 ng of pTat (b, asterisk indicates a statistically significant difference between the constructs without and with pTat). When no asterisk is shown, the difference was not statistically significant (P > 0.01).
The CA-p24 expression levels of the HIV-rtTA variants correlated well with their Tat activity levels (Fig. 1C), with the exception of those of the TatY26A variant, which demonstrated a high level of viral gene expression despite 4-fold-reduced Tat activity. Whereas the reporter construct in the Tat activity assay contains a wild-type TAR element and measures Tat/TAR-mediated activation of transcription, the HIV-rtTA construct has an inactivated TAR element and measures TAR-independent activation of gene expression. Apparently, the Y26A mutation affects the TAR-dependent but not the TAR-independent activity of Tat. We did indeed observe that gene expression of the HIV-rtTA-Tatstop variant was similarly stimulated by TatY26A and Tatwt when administered in trans (data not shown).
The Sp1 binding sites confer Tat responsiveness.
To identify the region in the HIV-rtTA promoter that mediates Tat stimulation, we made a set of novel HIV-rtTA variants in which 5′-LTR promoter sequences were removed or replaced by non-HIV promoter sequences (Fig. 5). The NF-κB sites and upstream U3 region were deleted in the ΔNF variant (Fig. 5B). We additionally removed the TAR hairpin in the ΔNF ΔTAR construct (Fig. 5C). We previously demonstrated that the HIV-1 RNA genome requires a stable stem-loop structure at the 5′ end and therefore replaced TAR with the unrelated ER3 hairpin (22). Because further truncation would result in the removal of essential tetO elements and the HIV-1-derived TATA box region, we constructed an additional HIV-rtTA variant in which the 5′-U3 and TAR region was replaced with the tetO-CMV promoter that does not contain any HIV-1-derived sequences (Fig. 5D). This promoter was developed in another HIV-rtTA study (to be published) and consists of three tetO elements coupled to the TATA box region of the CMV IE promoter and a new sequence designed to fold an RNA hairpin structure (Fig. 6).
Fig. 5.

Tat activates gene expression via U3 elements. HIV-rtTA-Tatwt (wt) and HIV-rtTA-Tatstop (stop) constructs with different 5′-LTR-promoter configurations were cotransfected with 0 to 50 ng of pTat into 293T cells. CA-p24 levels in the culture supernatants were measured after the cells were cultured with dox for 48 h. Average values obtained in multiple experiments are shown, with error bars indicating standard deviations. (A) HIV-rtTA promoter configuration with two tetO sites placed between the NF-κB and Sp1 binding sites (n = 12 to 16). (B) ΔNF constructs in which the NF-κB sites and upstream U3 region were deleted (n = 3). (C) In ΔNF ΔTAR constructs, the TAR hairpin was replaced with the unrelated ER3 hairpin (n = 6) (22). (D) tetO-CMV constructs in which the 5′ U3 and TAR regions were replaced by a promoter sequence consisting of three tetO elements coupled to a 30-bp minimal TATA box region from the CMV IE promoter and a hairpin that is based on the +1/+16 region of the CMV promoter (n = 12 to 16). (E) In the tetO-CMV-Sp1 constructs, the HIV-1 Sp1 sites were cloned between the tetO and TATA box regions of tetO-CMV (n = 3). All constructs have the tetO-CMV configuration in the 3′ LTR. A two-sided unpaired sample t test was used to identify statistically significant differences (P < 0.01) between the wt and corresponding stop constructs at 0 ng of pTat (a, asterisk indicates a statistically significant difference between the wt and stop constructs) and between the construct at 0 ng of pTat and the same construct at 0.5, 5, or 50 ng of pTat (b, asterisk indicates a statistically significant difference between the constructs without and with pTat). When no asterisk is shown, the difference was not statistically significant (P > 0.01). CA-p24 production of all HIV-rtTA constructs was very low (>10-fold reduced) in the absence of dox (data not shown).
Fig. 6.

The tetO-CMV promoter. (A) In the tetO-CMV HIV-rtTA variant, the U3-TAR promoter region was replaced with a promoter consisting of three tetO elements coupled to the 30-bp minimal TATA box region from the CMV IE promoter and a hairpin sequence that is based on the +1/+16 sequence of the CMV transcript. This promoter was developed in another HIV-rtTA study (to be published). The EcoRV and HindIII restriction enzyme sites in the U3 and R regions, respectively, are indicated. HIV-1-derived nucleotides are shown in uppercase and non-HIV nucleotides in lowercase. Reintroduction of the three Sp1 sites from the HIV-1 LAI strain between the tetO and TATA elements resulted in the tetO-CMV-Sp1 variant. (B) Secondary structure of the new 5′-hairpin element that replaces TAR, as predicted with MFold RNA-folding software (81).
To assay the Tat responsiveness of the different promoter configurations, we constructed all HIV-rtTA variants with either Tatwt or Tatstop. Cells were transfected with these plasmids and an increasing amount of the wild-type Tat vector (0 to 50 ng of pTat). CA-p24 production was measured after cells were cultured with dox for 48 h. As described above (Fig. 4), gene expression of the construct with the original HIV-rtTA promoter was significantly reduced by the Tatstop mutation (Fig. 5A, compare HIV-rtTA Tatwt and Tatstop at 0 ng of pTat) and could be restored by trans-complementation with wild-type Tat (compare HIV-rtTA Tatwt and Tatstop at 50 ng of pTat). Similarly, virus production of the ΔNF (Fig. 5B) and ΔNF ΔTAR constructs (Fig. 5C) was reduced by Tat inactivation (compare Tatwt and Tatstop variants at 0 ng of pTat) and could be restored by Tat trans-complementation (compare Tatwt and Tatstop variants at 50 ng of pTat). In contrast, the tetO-CMV construct was not responsive to Tat, as a similar CA-p24 level was observed for the Tatwt and Tatstop variants and complementation with pTat had no effect (Fig. 5D). These results demonstrate that the Sp1 binding sites or the TATA box region, both of which are present in ΔNF ΔTAR but not in tetO-CMV, mediated the Tat responsiveness of the HIV-rtTA promoter.
We made the additional tetO-CMV-Sp1 construct in which the Sp1 sites of the wild-type HIV-1 promoter were introduced between tetO and the TATA box region of tetO-CMV (Fig. 6A). These Sp1 sites greatly improved CA-p24 production (Fig. 5D and E, compare tetO-CMV and tetO-CMV-Sp1 constructs, respectively). Moreover, the Sp1 sites restored the Tat responsiveness of this promoter, as introduction of the Tat stop codon mutation reduced CA-p24 production 3-fold (Fig. 5E, compare Tatwt and Tatstop variants at 0 ng of pTat) and gene expression could be partially restored by the administration of Tat in trans (Fig. 5E, compare Tatwt and Tatstop variants at 50 ng of pTat). These experiments thus identified the three Sp1 sites as the domain in the HIV-rtTA promoter that is responsive to Tat.
Novel HIV-rtTA variant that replicates efficiently without Tat.
Tat does not influence transcription from the tetO-CMV promoter that lacks Sp1 sites (Fig. 5D). If Tat is also not required for another process in the viral life cycle, the corresponding virus should replicate efficiently in the absence of Tat. We therefore tested the replication of HIV-rtTA variants with the tetO-CMV promoter configuration and a wild-type or mutated Tat gene. Because the U3 promoter sequence in the 3′ LTR is inherited during virus replication, these viruses had the tetO-CMV configuration in both the 5′ and 3′ LTRs. The Tatwt and Tatstop tetO-CMV5′+3′ viruses do indeed replicate similarly well in SupT1 cells (Fig. 7A). Replication of both variants was somewhat delayed compared to that of wild-type HIV-rtTA, which is in agreement with their reduced promoter activity (Fig. 5A and D). Because introduction of the Sp1 sites in the tetO-CMV promoter restored the Tat responsiveness of the LTR promoter (Fig. 5E), we also constructed tetO-CMV-Sp15′+3′ viruses with a Tatwt or Tatstop gene. The Tatwt tetO-CMV-Sp15′+3′ virus replicates more efficiently than the corresponding Tatstop variant (Fig. 7A), which is in agreement with the restored Tat response. Remarkably, the Tatstop virus replicated less efficiently than the corresponding variant without Sp1 sites (Fig. 7A, compare tetO-CMV-Sp15′+3′-Tatstop with tetO-CMV5′+3′-Tatstop), while gene expression analysis showed that the tetO-CMV-Sp1 promoter is more active than the tetO-CMV promoter, even in the absence of Tat (Fig. 5D and E, compare tetO-CMV-Tatstop and tetO-CMV-Sp1- Tatstop). However, the constructs tested in Fig. 5 differ only in 5′-LTR configuration and have identical 3′ LTRs, whereas both the 5′ and 3′ LTRs were modified in the viruses tested in Fig. 7. The inefficient replication of the tetO-CMV-Sp15′+3′-Tatstop virus compared to that of the tetO-CMV5′+3′-Tatstop virus thus indicates that the introduction of Sp1 sites in the 3′ tetO-CMV LTR inhibits virus replication. We did indeed observe that introduction of these sequence elements in the 3′ LTR of a tetO-CMV5′+3′-Tatstop construct reduced viral gene expression (data not shown). Possibly, introduction of the Sp1 sites in the 3′ LTR affects the RNA secondary structure of the U3-R region of the viral transcript, which may influence polyadenylation of the RNA (24, 47). Alternative explanations are, however, possible, and further experimentation will be needed to understand this effect of 3′-Sp1 insertion on the gene expression and replication of the tetO-CMV virus. The Tatwt variant of the tetO-CMV-Sp15′+3′ virus replicates very efficiently in SupT1 cells, which demonstrates that the replication-inhibiting effect of the 3′-Sp1 sequences is more than compensated for by the stimulatory effect of the Tat-Sp1 interaction at the 5′ LTR.
Fig. 7.

The tetO-CMV virus replicates efficiently without Tat. Replication of HIV-rtTA variants with the tetO-CMV or tetO-CMV-Sp1 promoter configuration in both the 5′ and 3′ LTRs and with the Tatwt or Tatstop gene (wt and stop, respectively) was tested in SupT1 cells (A) and PBMCs (B) cultured with 1 μg/ml of dox. The wild-type HIV-1 LAI strain (cultured without dox) and HIV-rtTA-Tatwt (cultured with 1 μg/ml of dox) were included for comparison. SupT1 cells (A) were transfected with 5 μg of the proviral constructs. PBMCs (B) were infected with equal amounts of 293T-produced virus (5 ng of CA-p24). The CA-p24 levels in the culture supernatants were measured by ELISA. The replication curves shown are representative of data obtained in three (A) or two (B) experiments.
Replication of the new viruses was also tested in primary PBMCs. In these cells, HIV-rtTA replicates less efficiently than HIV-1 LAI (Fig. 7B). Neither the Tatwt nor the Tatstop tetO-CMV5′+3′ virus showed any replication. The tetO-CMV promoter was developed through HIV-rtTA evolution in SupT1 cells, and its activity may be insufficient to drive replication in PBMCs. Importantly, the Tatwt tetO-CMV-Sp15′+3′ virus does replicate in PBMCs, whereas the corresponding Tatstop variant does not, which demonstrates that the Tat-Sp1 interaction rescues replication in these cells.
DISCUSSION
The HIV-1 Tat protein enhances transcription from the proviral genome by binding to the TAR hairpin that is formed at the 5′ end of the nascent transcript. Tat recruits transcription elongation factor pTEFb (14, 58), TATA box binding protein (61), and chromatin-modifying proteins (30, 31, 63) to the LTR promoter, which results in RNA polymerase II phosphorylation, assembly of new transcription complexes, and chromatin remodeling, respectively. Here we demonstrated that Tat can also stimulate viral transcription in a TAR-independent way, which requires the presence of the Sp1 sites in the LTR promoter region. Furthermore, we constructed a replicating HIV-1 variant in which the TAR, Sp1, and other HIV-1 promoter sequences were replaced by nonrelated promoter sequences. Gene expression and replication of this virus is no longer influenced by Tat, while reintroduction of the Sp1 sites restored the Tat responsiveness of the LTR promoter. These results demonstrate that Tat has no other essential function in HIV-1 replication than a versatile trans-activating role in transcription via TAR and Sp1 sites. Our studies were limited to experiments in several commonly used cell types (293T, SupT1, and PBMCs) and we cannot exclude that Tat may have an accessory function in other cell types or in vivo.
TAR-independent activation of transcription by Tat in cells derived from the central nervous system (CNS) and phorbol ester-treated Jurkat cells has previously been suggested (4, 34, 73). However, whereas earlier studies indicated that binding of NF-κB to the LTR promoter is required for this Tat effect in CNS cells (78), our analysis indicates that the Sp1 sites are needed in T cells. Several other studies have demonstrated the importance of the Sp1 sites in the HIV-1 LTR promoter region for Tat trans-activation (11, 12, 35, 45). Studies on the interaction between Tat and Sp1 have yielded contradictory results, and both direct and indirect interactions between these transcription factors have been suggested (40, 52, 53, 57). Moreover, Tat was shown to stimulate phosphorylation of Sp1, which enhances its transcriptional activity (21), resulting in increased expression from the HIV-1 LTR.
We ascertained the TAR-independent, Sp1-dependent transcription-enhancing activity of Tat through the analysis of gene expression and replication of dox-inducible HIV-1 variants that varied in the LTR sequence and Tat gene (Fig. 5). Smith et al. previously constructed similar dox-inducible HIV-1 constructs with and without Sp1 sites in the U3 promoter region (71). Although these virus variants were not fully replication competent, gene expression from the proviral constructs could be measured and was found to be similar for both types of constructs despite the presence of a wild-type Tat gene. However, the Sp1-positive construct carried Sp1 sites in both the 5′ and 3′ LTRs, and the positive effect of the 5′ Sp1 sites on gene expression may have been concealed by a negative effect of the 3′ Sp1 sites. We did indeed observe that the introduction of Sp1 sites in the 5′ LTR of our tetO-CMV HIV-rtTA construct stimulated gene expression and restored Tat responsiveness (Fig. 5E), whereas introduction of the Sp1 sites in the 3′ LTR reduced gene expression and virus replication (Fig. 7 and data not shown). Remarkably, we could not measure a similar stimulatory Tat effect on gene expression when the Sp1-containing LTR-2tetO-luciferase reporter construct (Fig. 3A) was cotransfected with plasmids expressing Tat and rtTA in trans (data not shown). Possibly, the Tat-Sp1 interaction requires other HIV-encoded factors or virus-induced cellular factors that are not produced in the reporter assay, but these hypotheses will need further investigation.
Several other functions for Tat in HIV-1 replication have been suggested based on diverse in vitro biochemical assays and virus gene expression and replication studies in cell types similar to those we used in this study. Our results do strongly enforce the idea that Tat has no essential nontranscriptional role in HIV-1 replication. The Tat concentration and other parameters used in the biochemical assays may not reflect the physiological conditions during virus replication, which could lead to improper conclusions. Studies with Tat-mutated viruses are obviously complicated by the dominant negative effect of the Tat mutations on viral transcription. Previous studies with Tat-deficient HIV-1 variants in which transcription was activated by other transcriptional activators did suggest an important nontranscriptional function for Tat in virus replication (37). Here we used a dox-dependent HIV-1 construct in which the Tat-TAR transcription axis was functionally replaced with the Tet-On transcription mechanism to study other Tat functions. We developed a novel virus variant with a non-HIV promoter that does not depend on nor respond to Tat. This virus replicates efficiently despite complete Tat inactivation, which demonstrates that Tat has no other essential function in HIV-1 replication, although we cannot exclude an accessory function under specific conditions.
Virus evolution experiments played an important role in this study. First, the Tatfs mutant replicated poorly and rapidly reverted to the wild-type sequence. This evolutionary pressure suggested an important function for Tat in HIV-rtTA replication. Second, evolution of the poorly replicating Tatstop mutant resulted in the multiplication of tetO elements in the U3 region, which improved the LTR promoter activity and rescued viral replication. This observation provided the first indication that HIV-rtTA still needs Tat for the transcription process, despite gene expression being controlled by the incorporated Tet-On system. Multiplication and deletion of repeated sequence motifs in the HIV-1 genome has been observed previously (9, 13, 50, 54). Remarkably, evolution of the original HIV-rtTA construct with eight tetO elements and the TatY26A gene (75) resulted in the deletion of six tetO elements and a truncation of the spacer region between the two remaining tetO sites (54). These modifications fine-tuned the LTR promoter activity and optimized viral replication (55). A likely explanation for the reversion of the direction of evolution from two to three to seven tetO motifs as observed for the Tatstop variants comes from the observation that the Y26A mutation in Tat does not affect its stimulatory effect on the HIV-rtTA promoter. Apparently, two tetO elements are sufficient to drive gene expression and replication of the HIV-rtTA-TatY26A variant, because transcription is stimulated by the TatY26A protein. In the absence of Tat help, two tetO elements do not, however, suffice, which explains the replication defect of the HIV-rtTA-Tatstop variant, and tetO multiplication can sufficiently restore the level of transcription to allow viral replication.
We previously also constructed a dox-dependent simian immunodeficiency virus (SIV) SIVmac variant in which the Tat/TAR mechanism was functionally replaced by the Tet-On system (25, 26). HIV and SIV Tat proteins have a similar modular structure despite differences in sequence and size, and the tyrosine at position 26 in the highly conserved cysteine-rich domain of HIV-1 Tat corresponds with the tyrosine at position 55 in SIV Tat. Strikingly, whereas the HIV-rtTA variant was fully replication competent with the Y26A Tat mutation, the SIV-rtTA variant did not tolerate the equivalent Y55A change. Unlike the HIV-1 Tat mutation, the Y55A mutation in SIV Tat may have abolished an interaction via Sp1 sites in the promoter. Alternatively, the Y55A mutation may have affected a yet-unidentified underlying sequence element in the viral RNA and thus caused the replication defect. We can also not exclude the possibility that Tat has another essential function in SIV replication. Further experimentation with novel SIV-rtTA variants with altered Tat or promoter configurations are needed to discriminate between these possibilities.
ACKNOWLEDGMENTS
We thank Stephan Heynen for performing CA-p24 ELISA. The following reagent was obtained from Peter Cresswell through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: 174 × CEM.
This research was funded by the Dutch AIDS Foundation (Aids Fonds Netherlands grant 2005022) and NWO-Chemical Sciences (TOP grant).
Footnotes
Published ahead of print on 13 July 2011.
REFERENCES
- 1. Amendt B. A., Hesslein D., Chang L.-J., Stoltzfus C. M. 1994. Presence of negative and positive cis-acting RNA splicing elements within and flanking the first tat coding exon of human immunodeficiency virus type 1. Mol. Cell. Biol. 14:3960–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amendt B. A., Si Z.-H., Stoltzfus C. M. 1995. Presence of exon splicing silencers within human immunodeficiency virus type 1 tat exon 2 and tat-rev exon 3: evidence for inhibition mediated by cellular factors. Mol. Cell. Biol. 15:4606–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Apolloni A., Meredith L. W., Suhrbier A., Kiernan R., Harrich D. 2007. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr. HIV Res. 5:473–483 [DOI] [PubMed] [Google Scholar]
- 4. Bagasra O., Khalili K., Seshamma T., Taylor J. P., Pomerantz R. J. 1992. TAR-independent replication of human immunodeficiency virus type 1 in glial cells. J. Virol. 66:7522–7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bannwarth S., Gatignol A. 2005. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr. HIV Res. 3:61–71 [DOI] [PubMed] [Google Scholar]
- 6. Bennasser Y., Jeang K. T. 2006. HIV-1 Tat interaction with Dicer: requirement for RNA. Retrovirology 3:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennasser Y., Le S. Y., Benkirane M., Jeang K. T. 2005. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22:607–619 [DOI] [PubMed] [Google Scholar]
- 8. Berkhout B., Das A. T., Beerens N. 2001. HIV-1 RNA editing, hypermutation and error-prone reverse transcription. Science 292:7. [DOI] [PubMed] [Google Scholar]
- 9. Berkhout B. 2009. A new Houdini act: multiple routes for HIV-1 escape from RNAi-mediated inhibition. Future Microbiol. 4:151–154 [DOI] [PubMed] [Google Scholar]
- 10. Berkhout B., de Ronde A. 2004. APOBEC3G versus reverse transcriptase in the generation of HIV-1 drug-resistance mutations. AIDS 18:1861–1863 [DOI] [PubMed] [Google Scholar]
- 11. Berkhout B., Gatignol A., Rabson A. B., Jeang K. T. 1990. TAR-independent activation of the HIV-1 LTR: evidence that Tat requires specific regions of the promoter. Cell 62:757–767 [DOI] [PubMed] [Google Scholar]
- 12. Berkhout B., Jeang K. T. 1992. Functional roles for the TATA promoter and enhancers in basal and Tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 66:139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berkhout B., Verhoef K., van Wamel J. L., Back N. K. 1999. Genetic instability of live, attenuated human immunodeficiency virus type 1 vaccine strains. J. Virol. 73:1138–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bieniasz P. D., Grdina T. A., Bogerd H. P., Cullen B. R. 1999. Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc. Natl. Acad. Sci. U. S. A. 96:7791–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braddock M., Powell R., Blanchard A. D., Kingsman A. J., Kingsman S. M. 1993. HIV-1 TAR RNA-binding proteins control TAT activation of translation in Xenopus oocytes. FASEB J. 7:214–222 [DOI] [PubMed] [Google Scholar]
- 16. Braddock M., et al. 1990. A nuclear translational block imposed by the HIV-1 U3 region is relieved by the Tat-TAR interaction. Cell 62:1123–1133 [DOI] [PubMed] [Google Scholar]
- 17. Brady J., Kashanchi F. 2005. Tat gets the “green” light on transcription initiation. Retrovirology 2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Charnay N., et al. 2009. Mechanism of HIV-1 Tat RNA translation and its activation by the Tat protein. Retrovirology 6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiu Y. L., Coronel E., Ho C. K., Shuman S., Rana T. M. 2001. HIV-1 Tat protein interacts with mammalian capping enzyme and stimulates capping of TAR RNA. J. Biol. Chem. 276:12959–12966 [DOI] [PubMed] [Google Scholar]
- 20. Chiu Y. L., et al. 2002. Tat stimulates cotranscriptional capping of HIV mRNA. Mol. Cell 10:585–597 [DOI] [PubMed] [Google Scholar]
- 21. Chun R. F., Semmes O. J., Neuveut C., Jeang K.-T. 1998. Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J. Virol. 72:2615–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das A. T., Harwig A., Vrolijk M. M., Berkhout B. 2007. The TAR hairpin of human immunodeficiency virus type 1 can be deleted when not required for Tat-mediated activation of transcription. J. Virol. 81:7742–7748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das A. T., Klaver B., Berkhout B. 1998. The 5′ and 3′ TAR elements of the human immunodeficiency virus exert effects at several points in the virus life cycle. J. Virol. 72:9217–9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Das A. T., Klaver B., Berkhout B. 1999. A hairpin structure in the R region of the human immunodeficiency virus type 1 RNA genome is instrumental in polyadenylation site selection. J. Virol. 73:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Das A. T., et al. 2008. Optimization of the doxycycline-dependent simian immunodeficiency virus through in vitro evolution. Retrovirology 5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Das A. T., et al. 2007. Construction of a doxycycline-dependent simian immunodeficiency virus reveals a nontranscriptional function of Tat in viral replication. J. Virol. 81:11159–11169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Das A. T., Klaver B., Klasens B. I. F., van Wamel J. L. B., Berkhout B. 1997. A conserved hairpin motif in the R-U5 region of the human immunodeficiency virus type 1 RNA genome is essential for replication. J. Virol. 71:2346–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das A. T., Verhoef K., Berkhout B. 2004. A conditionally replicating virus as a novel approach toward an HIV vaccine. Methods Enzymol. 388:359–379 [DOI] [PubMed] [Google Scholar]
- 29. Das A. T., et al. 2004. Viral evolution as a tool to improve the tetracycline-regulated gene expression system. J. Biol. Chem. 279:18776–18782 [DOI] [PubMed] [Google Scholar]
- 30. Easley R., et al. 2010. Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim. Biophys. Acta 1799:275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gatignol A. 2007. Transcription of HIV: Tat and cellular chromatin. Adv. Pharmacol. 55:137–159 [DOI] [PubMed] [Google Scholar]
- 32. Gautier V. W., et al. 2009. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology 6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haasnoot J., et al. 2007. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 3:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrich D., Garcia J., Mitsuyasu R., Gaynor R. 1990. TAR independent activation of the human immunodeficiency virus in phorbol ester stimulated T lymphocytes. EMBO J. 9:4417–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harrich D., et al. 1989. Role of Sp1-binding domains in in vivo transcriptional regulation of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 63:2585–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harrich D., Ulich C., Garcia-Martinez L. F., Gaynor R. B. 1997. Tat is required for efficient HIV-1 reverse transcription. EMBO J. 16:1224–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang L. M., Joshi A., Willey R., Orenstein J., Jeang K. T. 1994. Human immunodeficiency viruses regulated by alternative trans-activators: genetic evidence for a novel non-transcriptional function of Tat in virion infectivity. EMBO J. 13:2886–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jablonski J. A., Amelio A. L., Giacca M., Caputi M. 2010. The transcriptional transactivator Tat selectively regulates viral splicing. Nucleic Acids Res. 38:1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jacquenet S., et al. 2001. A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of tat mRNA and binds protein hnRNP H. J. Biol. Chem. 276:40464–40475 [DOI] [PubMed] [Google Scholar]
- 40. Jeang K. T., et al. 1993. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J. Virol. 67:6224–6233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeeninga R. E., et al. 2000. Functional differences between the long terminal repeat transcriptional promoters of HIV-1 subtypes A through G. J. Virol. 74:3740–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jeeninga R. E., Jan B., van den Berg H., Berkhout B. 2006. Construction of doxycycline-dependent mini-HIV-1 variants for the development of a virotherapy against leukemias. Retrovirology 3:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kameoka M., et al. 2002. The Tat protein of human immunodeficiency virus type 1 (HIV-1) can promote placement of tRNA primer onto viral RNA and suppress later DNA polymerization in HIV-1 reverse transcription. J. Virol. 76:3637–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kameoka M., Rong L., Götte M., Liang C., Russell R. S., Wainberg M. A. 2001. Role for human immunodeficiency virus type 1 Tat protein in suppression of viral reverse transcriptase activity during late stages of viral replication. J. Virol. 75:2675–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamine J., Chinnadurai G. 1992. Synergistic activation of the human immunodeficiency virus type 1 promoter by the viral Tat protein and cellular transcription factor Sp1. J. Virol. 66:3932–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kammler S., et al. 2006. The strength of the HIV-1 3′ splice sites affects Rev function. Retrovirology 3:89–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klasens B. I. F., Thiesen M., Virtanen A., Berkhout B. 1999. The ability of the HIV-1 AAUAAA signal to bind polyadenylation factors is controlled by local RNA structure. Nucleic Acids Res. 27:446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koken S. E. C., van Wamel J. L., Goudsmit J., Berkhout B., Geelen J. L. 1992. Natural variants of the HIV-1 long terminal repeat: analysis of promoters with duplicated DNA regulatory motifs. Virology 191:968–972 [DOI] [PubMed] [Google Scholar]
- 49. Kozak M. 1989. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol. Cell. Biol. 9:5073–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leonard J. N., Shah P. S., Burnett J. C., Schaffer D. V. 2008. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe 4:484–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin J., Cullen B. R. 2007. Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J. Virol. 81:12218–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loregian A., Bortolozzo K., Boso S., Caputo A., Palu G. 2003. Interaction of Sp1 transcription factor with HIV-1 Tat protein: looking for cellular partners. FEBS Lett. 543:61–65 [DOI] [PubMed] [Google Scholar]
- 53. Loregian A., et al. 2003. The Sp1 transcription factor does not directly interact with the HIV-1 Tat protein. J. Cell. Physiol. 196:251–257 [DOI] [PubMed] [Google Scholar]
- 54. Marzio G., Verhoef K., Vink M., Berkhout B. 2001. In vitro evolution of a highly replicating, doxycycline-dependent HIV for applications in vaccine studies. Proc. Natl. Acad. Sci. U. S. A. 98:6342–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marzio G., Vink M., Verhoef K., de Ronde A., Berkhout B. 2002. Efficient human immunodeficiency virus replication requires a fine-tuned level of transcription. J. Virol. 76:3084–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mikaelian I., Sergeant A. 1992. A general and fast method to generate multiple site directed mutations. Nucleic Acids Res. 20:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pagtakhan A. S., Tong-Starksen S. E. 1997. Interactions between Tat of HIV-2 and transcription factor Sp1. Virology 238:221–230 [DOI] [PubMed] [Google Scholar]
- 58. Parada C. A., Roeder R. G. 1996. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 384:375–378 [DOI] [PubMed] [Google Scholar]
- 59. Peden K., Emerman M., Montagnier L. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185:661–672 [DOI] [PubMed] [Google Scholar]
- 60. Pisarev A. V., et al. 2006. Specific functional interactions of nucleotides at key −3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 20:624–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Raha T., Cheng S. W., Green M. R. 2005. HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biol. 3:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Richter S., Ping Y. H., Rana T. M. 2002. TAR RNA loop: a scaffold for the assembly of a regulatory switch in HIV replication. Proc. Natl. Acad. Sci. U. S. A. 99:7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Romani B., Engelbrecht S., Glashoff R. H. 2010. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J. Gen. Virol. 91:1–12 [DOI] [PubMed] [Google Scholar]
- 64. Ruijter J. M., et al. 2006. Factor correction as a tool to eliminate between-session variation in replicate experiments: application to molecular biology and retrovirology. Retrovirology 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Salter R. D., Howell D. N., Cresswell P. 1985. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics 21:235–246 [DOI] [PubMed] [Google Scholar]
- 66. Sanghvi V. R., Steel L. F. 2011. A re-examination of global suppression of RNA interference by HIV-1. PLoS One 6:e17246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schnettler E., et al. 2009. The NS3 protein of rice hoja blanca virus complements the RNAi suppressor function of HIV-1 Tat. EMBO Rep. 10:258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. SenGupta D. N., Berkhout B., Gatignol A., Zhou A. M., Silverman R. H. 1990. Direct evidence for translational regulation by leader RNA and Tat protein of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 87:7492–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Si Z., Amendt B. A., Stoltzfus C. M. 1997. Splicing efficiency of human immunodeficiency virus type 1 tat RNA is determined by both a suboptimal 3′ splice site and a 10 nucleotide exon splicing silencer element located within tat exon 2. Nucleic Acids Res. 25:861–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith S. D., et al. 1984. Monoclonal antibody and enzymatic profiles of human malignant T- lymphoid cells and derived cell lines. Cancer Res. 44:5657–5660 [PubMed] [Google Scholar]
- 71. Smith S. M., Khoroshev M., Marx P. A., Orenstein J., Jeang K. T. 2001. Constitutively dead, conditionally live HIV-1 genomes. Ex vivo implications for a live virus vaccine. J. Biol. Chem. 276:32184–32190 [DOI] [PubMed] [Google Scholar]
- 72. Tan R., Brodsky A., Williamson J. R., Frankel A. D. 1997. RNA recognition by HIV-1 Tat and Rev. Semin. Virol. 8:186–193 [Google Scholar]
- 73. Taylor J. P., et al. 1992. TAR-independent transactivation by Tat in cells derived from the CNS: a novel mechanism of HIV-1 gene regulation. EMBO J. 11:3395–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Verhoef K., Koper M., Berkhout B. 1997. Determination of the minimal amount of Tat activity required for human immunodeficiency virus type 1 replication. Virology 237:228–236 [DOI] [PubMed] [Google Scholar]
- 75. Verhoef K., Marzio G., Hillen W., Bujard H., Berkhout B. 2001. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J. Virol. 75:979–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vrolijk M. M., Harwig A., Berkhout B., Das A. T. 2009. Destabilization of the TAR hairpin leads to extension of the polyA hairpin and inhibition of HIV-1 polyadenylation. Retrovirology 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wei P., Garber M. E., Fang S.-M., Fisher W. H., Jones K. A. 1998. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92:451–462 [DOI] [PubMed] [Google Scholar]
- 78. Yang L., et al. 1997. Distinct transcriptional pathways of TAR-dependent and TAR-independent human immunodeficiency virus type-1 transactivation by Tat. Virology 235:48–64 [DOI] [PubMed] [Google Scholar]
- 79. Zahler A. M., Damgaard C. K., Kjems J., Caputi M. 2004. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J. Biol. Chem. 279:10077–10084 [DOI] [PubMed] [Google Scholar]
- 80. Zhou M., et al. 2003. The Tat/TAR-dependent phosphorylation of RNA polymerase II C-terminal domain stimulates cotranscriptional capping of HIV-1 mRNA. Proc. Natl. Acad. Sci. U. S. A. 100:12666–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]