

Abstract
The trimeric RNA polymerase complex (3P, for PA-PB1-PB2) of influenza A virus (IAV) is an important viral determinant of pathogenicity and host range restriction. Specific interactions of the polymerase complex with host proteins may be determining factors in both of these characteristics and play important roles in the viral life cycle. To investigate this question, we performed a comprehensive proteomic analysis of human host proteins associated with the polymerase of the well-characterized H5N1 Vietnam/1203/04 isolate. We identified over 400 proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS), of which over 300 were found to bind to the PA subunit alone. The most intriguing and novel finding was the large number of mitochondrial proteins (∼20%) that associated with the PA subunit. These proteins mediate molecular transport across the mitochondrial membrane or regulate membrane potential and may in concert with the identified mitochondrion-associated apoptosis inducing factor (AIFM1) have roles in the induction of apoptosis upon association with PA. Additionally, we identified host factors that associated with the PA-PB1 (68 proteins) and/or the 3P complex (34 proteins) including proteins that have roles in innate antiviral signaling (e.g., ZAPS or HaxI) or are cellular RNA polymerase accessory factors (e.g., polymerase I transcript release factor [PTRF] or Supt5H). IAV strain-specific host factor binding to the polymerase was not observed in our analysis. Overall, this study has shed light into the complex contributions of the IAV polymerase to host cell pathogenicity and allows for direct investigations into the biological significance of these newly described interactions.
INTRODUCTION
The trimeric RNA (3P) influenza virus polymerase complex (PA-PB1-PB2) assembles in the nucleus, where influenza virus replication occurs. Several events inside the host cell must take place in order to support viral replication, such as the regulation of early antiviral host responses, host protein shutoff, and nuclear transport of viral RNA and proteins (21, 29, 63). These events require the viral RNA polymerase to interact with cellular factors, some of which may also be important for host range specificity but have yet to be identified (17, 44). A range of RNA interference (RNAi)-based, genome-wide screens have been published recently that identified host factors involved in influenza virus replication (4, 22, 31, 35, 52, 56). Although the degree of overlap between these studies was minimal, similar biological pathways and functional classifications were identified, suggesting their importance in the influenza virus infectious cycle. Watanabe et al. (67) compiled a list of 128 proteins that affected influenza virus replication in at least two out of the six screens. These included proteins involved in the nuclear import of viral ribonucleoproteins (vRNPs), such as importin family members and nuclear pore complex proteins.
Other studies have attempted to directly identify proteins that interact with either the influenza virus polymerase or the viral ribonucleoprotein complex, using a yeast two-hybrid system or proteomics approaches such as fractionation by SDS-PAGE followed by mass spectrometry (MS) (10, 12, 14, 20, 25, 27, 33, 41, 43, 45, 46, 49, 61, 65, 66). However, these approaches have been performed only at relatively small scale, and few interacting proteins have been identified to date. The largest study in which host factors were identified by mass spectrometry was described by Mayer et al. (41). A total of 41 proteins were found to bind to influenza virus vRNP, but only three proteins (HSP90, RanBP5 [IPO5], and PARP-1) were found to bind to the polymerase complex alone.
One of our goals was to identify novel cellular interaction partners to PA and the trimeric complex. PA was recently described as a virulence factor for H5N1 avian influenza viruses in ducks (54) and pandemic H1N1/2009 influenza viruses in mice (57). However, the mechanism by which this occurs remains unknown. Other major virulence factors of the influenza A viruses (IAVs) are the well-studied PB1-F2 protein and the PB2 protein. Both have been described to have roles in the induction of apoptosis during influenza virus infection (9, 14, 21, 46, 69). PB2 has also been found to interact with IPS-1 at the mitochondria, where it may be involved in controlling virus-induced apoptosis as well as regulating innate antiviral immune responses (21, 29). Together, these findings suggest an important role for the mitochondria in the influenza virus life cycle. We were interested in determining whether PA may have similar functions as PB1-F2 or PB2 that may help understand the underlying mechanism that contributes to virulence.
Another interest was to identify factors that bind to only the heterotrimeric form of the polymerase complex since only a few 3P-binding partners have been described. Thus far, it is known that the 3P complex interacts with the large subunit of the cellular RNA polymerase II (Pol II) (12, 63). It is thought that the scavenging of RNA Pol II by the 3P complex serves multiple purposes in the influenza virus life cycle, such as the induction of premature termination of RNA polymerase II transcription, which ultimately leads to host protein synthesis shutoff and may help the 3P complex to gain access to polymerase accessory factors such as capped mRNA fragments and splicing factors (12, 63).
We utilized a comprehensive approach to identify the cellular factors that bind to the influenza virus polymerase complex or its subunits. In this approach, we purified the H5N1 (Vietnam/1203/04 [VN/1203]) polymerase (3P) complex and its subunit components (PA and PA-PB1) from human lung epithelial cells (A549 cell line) infected with adenovirus (Ad) vectors encoding these proteins. This novel expression system allows for the expression and purification of high levels of polymerase proteins (2). We analyzed proteins in the purified fractions by mass spectrometry, either before or after nuclease treatment to remove copurified RNA (and to dissociate protein complexes held together through an RNA intermediate). Using a subtractive analysis, we were able to identify host factors that bind to PA, PA-PB1, or the 3P complex. It has to be stressed that factors identified to bind, e.g., to PA or PA-PB1 may ultimately also bind to PA-PB1 and/or the 3P complex, respectively, since host factor binding may occur prior to complex formation.
In addition to published factors (10, 14, 17, 31, 33, 35, 43, 45, 48, 52, 59, 61, 66), such as chaperonins and members of the Ran signaling pathway, we found a novel association of PA with mitochondrial proteins. Particularly, we found a strong association of PA with the apoptosis-inducing factor (AIFM1), suggesting a potential role of PA during apoptosis. Further, we identified RNA polymerase accessory factors that were preferentially associated with the 3P complex, including polymerase I transcript release factor (PTRF), Tat-SF1, and Supt5H (30, 68, 70), which may be important for viral polymerase function. Finally, a comparison of the proteomic content among 3P complexes from different virus isolates (VN/1203/04/H5N1, WSN/33/H1N1 [WSN], and CA/04/09 [CA/04], a recent pandemic H1N1 isolate) indicated that these cellular factors may be important in the life cycle of all three viral strains since they did not interact with the viral polymerase in a strain-specific manner.
MATERIALS AND METHODS
Cell lines and viruses.
Human lung epithelial cells (A549) and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 IU of penicillin ml−1, 50 μg of streptomycin ml−1, and 2 mM glutamine. The polymerase genes of the H1N1 (A/WSN/33) and H5N1 (A/Vietnam/1203/04) viruses were assembled as described previously (2). The polymerase genes from the H1N1 virus of a recent pandemic (CA/04/09) were human codon optimized and assembled from synthetic oligonucleotides by EpochBiolabs (Missouri City, TX); protein identifiers (IDs) for polymerase genes of CA/04 were ACP44156.1, ACP41103.1, and ACP41102.1. The adenoviral vectors were constructed as described previously (2). Briefly, PA from each viral strain was tagged at the C terminus with a tandem affinity purification (TAP) tag, and the viral polymerase genes were cloned into adenoviral expression vectors as described for A/Vietnam/1203/04. All adenovirus infections were performed at a multiplicity of infection (MOI) of 1,000, except for recombinant Ad type 5 expressing the VN/1203 PA (rAd5-PAVN/1203), where an MOI of 100 was used (2).
Influenza virus polymerase purification.
Briefly, A549 cells were infected with adenovirus vectors encoding influenza virus polymerase proteins either alone (PA-TAP) or in combination with PB1 (PA-PB1), PB2 (PA-PB2), or PB1 and PB2 (3P complex); uninfected A549 cells were used as a control. Cell lysates were prepared 48 h postinfection (hpi) and left untreated or treated with RNase A (100 μg/ml) and DNase I (50 μg/ml) on ice for 30 min prior to purification by affinity chromatography using IgG-Sepharose beads, as described previously (2). Total cell lysates were either left untreated or treated, prior to IgG affinity purification. Successful nuclease treatment of the lysates was verified by agarose gel electrophoresis and ethidium bromide staining (data not shown). Elution fractions were pooled and concentrated in 100 mM NH4HCO3 using Amicon Ultra (3,000-molecular-weight cutoff [MWCO]) centrifugation devices. Concentrated fractions contained approximately 1 to 2 μg of total protein per μl.
Vector construction for host factor coimmunoprecipitations (co-IPs).
The pShCMV-based expression vectors encoding PA, PB1, and PB2 were constructed as described previously (2), except PAVN/1203, which was cloned without a TAP tag, and PAWSN, which was cloned with a His6 tag only. To obtain host factor expression plasmids coding for Flag-tagged proteins, the coding regions of Supt5H (NM_001111020), PTRF (NM_012232), and AIFM1 (NM_004208) were PCR amplified from cDNA. The cDNA of Supt5H or PTRF was cloned into the XhoI and BglII sites of the expression vector pCAGGS-NH2 Flag, and AIFM1 was cloned into EcoRI and SmaI sites of expression vector pCAGGS-COOH Flag. The pCAGGs Flag vectors were kindly provided by Luis Martinez-Sobrido (University of Rochester, Rochester, NY).
Antibodies and coimmunoprecipitations.
To analyze binding of transiently expressed proteins, expression plasmids (indicated in the relevant figure legends) were transfected into HEK293T cells (∼90% confluence, 12-well plate) using Lipofectamine 2000 (Invitrogen). At approximately 24 h posttransfection, cells were washed with phosphate-buffered saline (PBS) and lysed in 200 μl of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM sodium orthovanadate, 1 mM sodium fluoride) supplemented with complete protease inhibitor (Complete Mini; Roche). Cellular debris was pelleted by centrifugation, and whole-cell lysate (supernatant) was incubated with 1 μg of anti-Flag antibody (Sigma) for 1 h at 4°C, with rotation. Lysate was then incubated with protein G agarose beads (Roche) overnight, and beads were washed three times with wash buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% NP-40, and protease inhibitor mix). Bound proteins were eluted by boiling for 5 min with 1× SDS-polyacrylamide gel loading buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 100 mM dithiothreitol [DTT], 0.01% bromophenol blue).
For co-IP of proteins from cells infected with influenza virus, a confluent well of A549 cells (2 × 106 cell equivalent) in a six-well plate was infected with influenza A/WSN/33 virus (MOI of 10). The cells were collected at various times postinfection (24, 30, and 42 h), washed with PBS, and lysed in 500 μl of RIPA lysis buffer (supplemented as described above). Lysates were cleared of cellular debris by centrifugation for 5 min at 4°C (13,000 rpm). A total of 200 μl of the cleared lysate was transferred into a new tube and subjected to immunoprecipitation using 1 μg of anti-H1N1 PA rabbit polyclonal antibody. After a 1-h rotation at 4°C, 20 μl of protein A agarose beads (Santa Cruz) was mixed with the antibody-antigen complexes and rotated overnight. Beads were recovered by centrifugation at 2,500 rpm for 5 min and washed three times with wash buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% NP-40, 1 mM PMSF) and then resuspended in 1× SDS sample buffer. Bound proteins were eluted by boiling for 5 min and separated by electrophoresis through a 7.5% polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane and analyzed by Western blotting using PA- and AIFM1-specific antibodies. Proteins were visualized with an enhanced chemiluminescence (ECL) detection kit (Amersham Life Science). The WSN virus (A/WSN/33), a generous gift from T. Takimoto (University of Rochester, Rochester, NY), was propagated in MDCK cells as described previously (5).
Antibodies were obtained from the following sources: anti-H1N1 PA rabbit polyclonal antibody (ABIN398948) was purchased from Antibodies-Online GmbH (Atlanta, GA); PB1-specific (vK-20), PB2-specific (vN-19), and anti-goat IgG horseradish peroxidase (HRP)-conjugated (Sc-2033) specific antibodies were obtained from Santa Cruz; ECL anti-rabbit IgG-HRP (NA934V) and ECL anti-mouse IgG-HRP (NA931V) were purchased from Amersham, and anti-Penta-His (mouse) was from Qiagen. The following antibodies to detect specific cellular host factors were obtained from Santa Cruz: goat polyclonal antibodies directed against AIFM1 (sc-9416), IPO5 (sc-11369), and IPO7 (sc-55230) and a mouse monoclonal directed against PRKDC (protein kinase, DNA-activated catalytic subunit; sc-56090).
Mass spectrometry.
Samples were prepared with PPS Silent Surfactant (Protein Discovery, Inc., Knoxville, TN) based on the manufacturer's recommended protocol and digested with trypsin (treated with sequencing-grade tosylsulfonyl phenylalanyl chloromethyl ketone [TPCK]; Promega). Briefly, trypsin digestion was carried out in 50 mM ammonium bicarbonate buffer, pH 7.8, overnight at 37°C at a trypsin to protein ratio of 1:50. The peptides were analyzed on an LTQ-Orbitrap mass spectrometer (Thermo Scientific). The reverse-phase column consisted of a trap column (100 μm by 1.5 cm) of Magic C18AQ resin in an IntegraFrit (New Objective) connected in-line to an analytical column (75 μm by 27 cm) of Magic C18AQ resin in a PicoFrit. The liquid chromatography-tandem MS (LC-MS/MS) acquisition consisted of a full mass spectrum followed by up to five data-dependent MS/MS spectra of the five most abundant ions. The full mass spectrum scan range was 400 to 1,800 m/z, and the instrument resolution was set to 60,000. The data-dependent scans were collected using the following settings: repeat count of 1, repeat duration of 30, exclusion list size of 100, exclusion duration of 45, and exclusion mass width of 0.55 m/z low and 1.55 m/z high. Charge state screening was used allowing analysis of +2, +3, and +4 and higher charge states while rejecting analysis of +1 and unassigned charge states. Each sample was analyzed in three technical replicates (unless otherwise indicated) of the total 1 to 2 μg of protein. The LC-MS/MS data were searched against the human International Protein Index (IPI) database (version 3.70) appended with IAV-specific protein sequences. The data processing and database search were performed in CPAS (https://www.labkey.org/wiki/home/Documentation/page.view?name = ms2) with the X!Tandem search engine. Peptide Prophet (34) was used to evaluate peptide assignment at a false-discovery rate (FDR) of <5%, and Protein Prophet (47) was used to group peptides into proteins. Only peptides that passed the 5% FDR threshold for Peptide Prophet were used for the grouping. Contaminant proteins (evident from intermittent blank runs) were removed from further analysis.
We also allowed for up to 10% of carryover in the negative-control lysate; this ensured that we did not erroneously filter out more abundant peptides that were observed at much smaller amounts in the control lysate. In addition, only proteins with multiple peptide occurrences (across all conditions) were analyzed further. The protein occurrence among polymerase fractions was analyzed on the Entrez Gene level, using the Venn diagram tool provided in the Resolver software (Rosetta). Functional enrichment analysis was performed in DAVID (Database for Annotation, Visualization, and Integrated Discovery) Bioinformatics Resources, version 6.7, and Ingenuity Pathway analysis (IPA). Protein interaction networks were extracted from the human proteome database present in the Ingenuity Pathway Knowledge Base (IPKB). The P values for each canonical pathway were calculated with a right-tailed Fisher's exact test, which compares the number of proteins that participate in a given pathway to the total number of protein annotations stored in the IPKB for that particular pathway.
RESULTS
Purification of IAV polymerase complex and associated host factors from A549 cells.
Since the influenza A virus (IAV) heterotrimeric (3P) polymerase complex is an RNA-dependent RNA polymerase (RdRp) complex with RNA binding activity, we set out to identify host cellular factors that associated with the polymerase complex both in the presence or absence of nucleic acid. Lysates were prepared from A549 cells that were infected with adenoviruses expressing either PA (PA-TAP), PA and PB1 (PA-PB1), or PA, PB1, and PB2 (3P complex); uninfected A549 cells were used as a control. Affinity purification was directed against the TAP tag; therefore, only polymerase proteins that associated with PA were precipitated by this method (Fig. 1A). Affinity-purified complexes precipitated from two sets of independently generated cell lysates, described as set 1 and set 2, were analyzed by liquid chromatography-mass spectrometry (LC-MS). Set 1 consisted of polymerase components from the VN/1203 strain (PA, PA-PB1, and 3P) that were either treated with nuclease or left untreated. Samples from set 2 consisted of an independent repeat experiment of untreated polymerase components of the VN/1203 strain as well as 3P complexes from the WSN and CA/04 strains (Fig. 1A).
Fig. 1.

Purification of the influenza virus polymerase complex and subunit components from A549 cells. (A) Overview of the influenza virus polymerase purification and expression technique. A549 human lung epithelial cells were infected with adenoviruses (rAd5) expressing the indicated proteins (PA-TAP, PB1, and PB2). Control A549 cell lysates or A549 cell lysates expressing PA, PA-PB1, or the 3P complex from VN/1203 (H5N1), WSN (H1N1), or CA/04 (H1N1) were left untreated (−) or treated (+) with nucleases (DNase and RNase A). The polymerase complex (PA-TAP/PB1/PB2) or the subunit components (PA-TAP, PA-TAP/PB1, and PA-TAP/PB2) were purified by IgG affinity chromatography and elution with tobacco etch virus protease. (B) A549 cells were transduced with adenovirus vectors, encoding the indicated VN/1203 polymerase subunits (top). After 48 h, cell lysates were prepared and subjected to IgG affinity chromatography and elution with tobacco etch virus protease. Nuclease-treated, affinity-purified polymerase subunits were then electrophoretically separated on a 7.5% polyacrylamide gel and detected by silver staining. The polymerase subunits (PB1, PA, and PB2) are indicated, as are the positions of molecular mass markers (left). There were no obvious differences in banding patterns compared to samples not treated with nuclease (data not shown).
Affinity-purified proteins were prepared as described previously (2) and separated by polyacrylamide gel electrophoresis (PAGE), and purified proteins were detected by silver staining (Fig. 1B). All viral polymerase proteins were detected, as indicated in Fig. 1B, along with other unidentified cellular proteins. The viral polymerase proteins were also verified by Western blot analysis (data not shown) and resulted in one single discrete band for each viral protein. In contrast to the purified polymerase complexes, few cellular proteins were visible in the negative-control sample from untreated A549 cells. No obvious differences in protein staining patterns were observed between polymerase complexes purified from untreated versus nuclease-treated cell lysates (data not shown).
In our approach, affinity-purified protein complexes from untreated or nuclease-treated cell lysates were then subjected to a proteomic analysis, with the objective of distinguishing protein complexes held together mainly through an RNA intermediate versus those held together by direct protein-protein binding. Table 1 describes the underlying quality control (QC) matrix for the VN/1203 polymerase components (set 1), showing the relative abundance of viral proteins across the experimental conditions. A reasonable sequence coverage for each viral protein was achieved (60 to 74%), with 40 to 55 unique peptides detected for each viral protein (Table 1). The abundance of each viral protein was measured by the total number of spectral counts that match peptides to this particular viral protein (Table 1). All viral proteins were readily detected in samples from both experimental sets (see Table S1 in the supplemental material for the complete QC matrix on set 2). As expected, PB2 was detected only in the purified 3P complex while PB1 was detected only in the purified PA-PB1 and 3P complexes; PA was found in all purified complexes (also, as expected) (Table 1).
Table 1.
MS data of the H5N1 influenza A virus polymerase complex (set 1)
| gi no.a | Protein nameb | Sequence coverage (%) | No. of unique peptides | No. of spectral counts of the indicated proteinc |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Without nuclease |
With nuclease |
||||||||
| PA | PA-PB1 | 3P | PA | PA-PB1 | 3P | ||||
| 50313055 | PA | 74.47 | 55 | 1,354 | 1,300 | 856 | 1,102 | 978 | 732 |
| 50296440 | PB1 | 60.11 | 40 | 0 | 409 | 300 | 1d | 385 | 332 |
| 50296548 | PB2 | 66.14 | 43 | 0 | 1d | 536 | 5d | 7d | 506 |
NCBI gi number (sequence record identifier).
From influenza virus A/Vietnam/1203/2004 (H5N1).
Counts for each protein were averaged across technical replicates. PA-PB1 contains PA and PB1; 3P contains PA, PB1, and PB2.
The nonzero values are most likely due to a slight carryover in subsequent LC-MS runs.
To identify nonspecific protein interactions with IgG-Sepharose beads as well as potential carryover during the LC-MS analysis (28), we included control cell lysates in our analysis. From all proteins identified by LC-MS (see master tables for each set in Table S2 in the supplemental material), we filtered out the nonspecific binders first, which were defined as proteins that were present in the purified viral polymerase fractions as well as the control lysate. A background list with the nonspecific binders was established, which included proteins such as tubulin, actin, keratin, EEF1, and serum albumin (see Table S2). To further increase confidence in protein assignments, only proteins for which multiple peptides were detected (across all conditions) were included in the list of polymerase binding partners. With this method, we detected a total of 509 proteins in set 1 and 743 proteins in set 2 as PA-, PA-PB1-, or 3P-binding partners. The union of these two data sets yielded 859 unique cellular host factors that were associated with the VN/1203 polymerase or subcomponents thereof.
Identification of VN/1203 PA-binding partners.
Of the 859 cellular factors, 651 were found to bind to PA (Fig. 2). We identified 390 proteins in set 1 that bound to PA in untreated and nuclease-treated samples combined. Of these, 373 proteins bound to PA in the absence of nuclease treatment, and 207 proteins bound to PA after nuclease treatment (Fig. 2, PA −N and PA +N, respectively). Another 576 PA-interacting proteins were identified in set 2 (Fig. 2). All three protein groups overlapped by 166 proteins which we considered to represent proteins that bound to PA both in the presence and absence of any associated RNA (termed here “shared” factors).
Fig. 2.
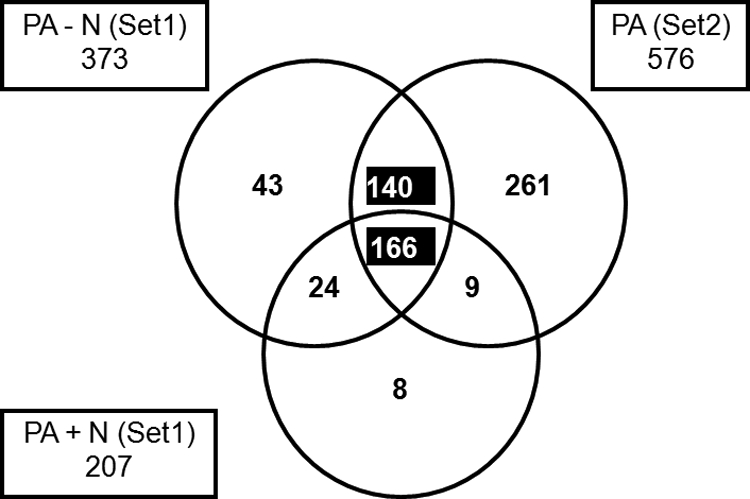
Cellular PA-binding partners. Venn diagram, representing 651 cellular proteins identified in affinity-purified complexes with PA in the absence of nuclease treatment (−N, Set 1; Set 2) and after nuclease treatment (+N, Set 1). We attribute the identification of more proteins in set 2 than in set 1 to slight differences in sample preparation between experiments. Numbers in the various boxes/sectors denote numbers of unique cellular proteins identified in each category. Highlighted in the center of this figure are the 166 cellular proteins that we defined as shared RNA-independent PA-binding partners (i.e., proteins which bound to PA both in the presence and absence of PA-associated nucleic acid). Highlighted in the top center of this figure are the 140 cellular proteins that we defined as RNA-dependent binding partners (i.e., proteins which bound to PA only when PA was also complexed with nucleic acid). All proteins derived from set 2 were not treated with nucleases.
Overall, the shared PA-binding proteins constituted more than 80% of the total complement of proteins identified in the nuclease-treated PA complex (207 total proteins) (Fig. 2, PA +N). In addition, we identified 140 proteins that appeared to bind to PA in an RNA-dependent fashion (Fig. 2) and were present in the PA fractions from both experimental sets. The top five canonical pathways associated with the 166 shared RNA-independent and the 140 RNA-dependent PA-binding partners, as well as both data sets combined (306 proteins), were determined by Ingenuity Pathway Analysis and are listed in Table 2. The number of molecules detected in each particular pathway is indicated together with their P values, calculated using a Benjamini-Hochberg multiple testing corrections. Pathways such as Ran signaling and caveolar-mediated endocytosis signaling have been described before, albeit not with regard to PA alone. Interesting and novel was the enrichment for proteins involved in mitochondrial properties such as mitochondrial dysfunction or oxidative phosphorylation. The association of PA with these proteins occurred mainly in the presence of nucleic acid (the RNA-dependent 140 proteins) and remained the most significant pathway when both RNA-dependent and -independent factors were combined.
Table 2.
PA-binding partners and top canonical pathway association
| Protein group (n)a | Canonical pathwayb | No. of molecules detected | P valuec |
|---|---|---|---|
| RNA-independent proteins (166) | Ran signaling | 5 | 8.76E−05 |
| Fatty acid biosynthesis | 4 | 5.27E−04 | |
| Caveolar-mediated endocytosis signaling | 7 | 6.90E−04 | |
| Aminoacyl-tRNA biosynthesis | 4 | 4.31E−02 | |
| Purine metabolism | 10 | 4.56E−02 | |
| RNA-dependent proteins (140) | Oxidative phosphorylation | 10 | 1.04E−04 |
| Mitochondrial dysfunction | 8 | 1.11E−03 | |
| Citrate cycle | 4 | 5.86E−03 | |
| PXR/RXR activationd | 5 | 1.14E−02 | |
| Aminoacyl-tRNA biosynthesis | 4 | 1.14E−02 | |
| Combined groups (306) | Mitochondrial dysfunction | 14 | 1.81E−05 |
| Oxidative phosphorylation | 15 | 1.81E−05 | |
| Aminoacyl-tRNA biosynthesis | 8 | 6.64E−05 | |
| Ran signaling | 5 | 4.94E−04 | |
| Caveolar-mediated endocytosis signaling | 9 | 4.94E−04 |
n, number of proteins.
Ingenuity Pathway analysis category.
As determined by Benjamini-Hochberg multiple testing correction.
PXR, pregnane X receptor; RXR, retinoid X receptor.
We next focused on the 166 shared RNA-independent PA-binding partners since these proteins were present in all three purified sample preparations (Fig. 2, PA set 1 with and without nuclease treatment and set 2) and are therefore likely to be true PA-binding partners. Figure 3 shows the main PA interaction network for the 166 cellular proteins with the major canonical pathways that were identified under Table 2. Interestingly, besides the Ran signaling, the aminoacyl-tRNA synthesis and the caveolar-mediated endocytosis pathways, we identified epidermal growth factor receptor (EGFR) as one of the major connecting points between these pathways (Fig. 3). Indeed, EGFR is also part of the caveolar-mediated endocytosis pathway and has recently been speculated to be important for influenza virus endocytosis (11). Also highlighted are those molecules (Fig. 3, orange symbols) that are involved in molecular transport such as protein and RNA transport. Overall, the findings summarized in Fig. 3 (see Table S3 in the supplemental material for a complete list of the 166 proteins) and Table 2 are broadly consistent with previous reports on biological pathways and host factors required for the influenza virus infectious cycle (31, 35). Additional information on each reported molecule such as amino acid coverage, peptide composition, and the relative abundance as measured by spectral counts can be found in the supplemental data (see Table S2).
Fig. 3.

The cellular PA-interaction networks. The cellular interaction networks of PA-specific binding partners are shown. Represented in this analysis is a subset of the 166 cellular proteins which bound to PA in an RNA-independent manner and which were identified in all three protein preparations studied (Fig. 2). The analysis shown here reveals a novel interaction of PA with members of the Ran signaling pathway, the caveolar endocytosis-mediated pathway, and the aminoacyl-tRNA biosynthesis pathway. Orange symbols represent molecules that are involved in molecular transport. Highlighted in green are mitochondrial proteins, and highlighted in yellow are molecules chosen for further analysis (IPO7 and PRKDC) or discussed further in the text (EGFR). Nodes depict proteins in the network, and solid lines represent direct whereas dotted lines represent indirect relationships between the proteins. An arrow pointing from one node to the next signifies an activation event, such as phosphorylation or methylation. For simplicity, self-binding or autoregulation events were not included in this figure.
Next, we used DAVID Bioinformatics resources to perform a functional enrichment analysis that condenses two identified gene lists (166 RNA-dependent genes and the combined gene list of 306 genes) into functional groups across the Gene Ontology (GO) categories (26). Table 3 shows the biological processes and the molecular functions in which these 166 shared factors (RNA independent) are involved. The main cellular components with which these proteins associated were the nuclear pore/envelope, the COPI vesicle coat, the mitochondrion, and endoplasmic reticulum (ER). The counts of the molecules associated with these cellular structures are shown in Table 3 along with their calculated P values (defined by DAVID Bioinformatics). These cellular structures have been previously reported to be important in the influenza virus life cycle; however, their role in the context of PA-binding proteins has not been defined. Of particular interest is the novel association of PA with mitochondrial proteins since only PB2 and PB1-F2 have been linked to mitochondria in the past (6, 9). In fact, the mitochondrion category even gained significance when the combined list of molecules from RNA-dependent and -independent factors (306 factors) were examined, suggesting that some functions of PA at the site of the mitochondria are RNA dependent.
Table 3.
Properties of PA-binding partners as determined by DAVID
| GO term | DAVID functional annotationd | Group of 166 proteinsa |
Combined group of 306 proteinsb |
||
|---|---|---|---|---|---|
| No. of molecules | P valuec | No. of molecules | P valuec | ||
| Biological process | Intracellular transport | 32 | 1.40E−09 | 46 | 3.10E−10 |
| Intracellular protein transport | 20 | 2.20E−06 | 24 | 6.80E−05 | |
| COPI coating of Golgi vesicle | 6 | 4.00E−06 | 7 | 2.10E−06 | |
| Nucleocytoplasmic transport | 12 | 5.60E−05 | 15 | 1.10E−04 | |
| Molecular function | Nucleotide binding | 54 | 2.10E−07 | 108 | 1.20E−19 |
| Protein transporter activity | 9 | 1.20E−04 | 11 | 1.40E−04 | |
| ATPase activity | 13 | 5.60E−03 | 32 | 1.20E−11 | |
| Cellular component | Organelle envelope | 35 | 3.60E−13 | 60 | 5.40E−22 |
| Pore complex | 13 | 2.50E−08 | 15 | 1.70E−07 | |
| COPI vesicle coat | 6 | 1.70E−06 | 7 | 7.00E−07 | |
| Mitochondrion | 33 | 2.30E−06 | 66 | 1.30E−14 | |
| Endoplasmic reticulum | 27 | 1.30E−04 | 42 | 5.20E−05 | |
RNA-independent factors.
Union of RNA-dependent and -independent factors (166 + 140 proteins).
P values were determined by a Benjamini-Hochberg multiple testing correction.
The mitochondrion category is highlighted as being of particular importance (see text).
From the 166 shared factors, we identified 33 PA-binding factors that located to mitochondria (P = 2.3E−06) (Table 3). Next, we compiled a heat map of these 33 factors across both experimental data sets which represents their relative abundance as measured by the average of individual spectral counts detected by LC-MS (Fig. 4). Most of the mitochondrial proteins identified were from the inner mitochondrial membrane and are involved in transmembrane transport. AIFM1 (apoptosis inducing factor, mitochondrial), one of the PA-associated mitochondrial proteins located at the inner mitochondrial membrane, was also one of the most readily detected PA-interacting partners, as measured by LC-MS.
Fig. 4.

Heat map of 33 mitochondrial proteins that bind to PA. The relative abundance of the 33 mitochondrial proteins that are present in the shared RNA-independent fraction of VN/1203 PA-binding partners is shown here across all samples from set 1 and set 2 in the form of a heat map. AIFM1 was among the most abundant mitochondrial proteins. The molecules are arranged according to their mitochondrial location. Molecules involved in transmembrane transport are indicated. T.T., transmembrane transport; n.d., not defined to specific compartment in mitochondrion.
Next to AIFM1, other readily detected, nonmitochondrial proteins were PRKDC (protein kinase, DNA activated) as well as the beta-importin IPO7. The relative abundance of these factors together with IPO5 (RanBP5), a previously identified binding partner of PA-PB1 (10), is shown in Fig. 5A in the form of a heat map. IPO5 is omitted from the complete list of PA- and PA-PB1-binding partners (see Table S3 in the supplemental material) because of the stringent exclusion criteria that were applied in analyzing our data. However, the heat map (Fig. 5A) clearly shows that IPO5 associates chiefly with the PA-PB1 dimer, which is in accordance with published data (10). We verified the binding of these four cellular host factors to H5N1 PA by Western blot analysis of affinity-purified complexes of polymerase or polymerase subcomponents (Fig. 5B). AIFM1, IPO5, and, to a somewhat lesser extent, IPO7 were detected in complex with PA alone as well as in the PA-PB1- and 3P-containing complexes (which also contain PA). Both IPO5 and IPO7 predominantly bound to PA-PB1 (Fig. 5B), which is consistent with our LC-MS results (Fig. 5A).
Fig. 5.

Association of human β-importins (IPO5 and IPO7), AIFM1, and PRKDC with PA and 3P complexes from various strains. (A) The relative abundance (spectral counts) of a subset of PA-binding partners (shared fraction) is shown in form of a heat map, with PRKDC being the most abundant protein. The spectral counts are shown from samples across both experimental sets (set 1 and set 2). (B) A549 cells were transduced with adenovirus vectors encoding the indicated VN/1203 (H5N1) polymerase proteins (3P denotes the PA-PB1-PB2 complex). Non-nuclease-treated PA-containing protein complexes were then purified by affinity chromatography, along with a negative-control eluate (A549), as described in Fig. 1A. Protein content in these complexes was evaluated by Western blotting, using antisera specific for IPO5, IPO7, and AIFM1 as well as anti-PA, anti-PB1, and anti-PB2. (C) A549 cells were transduced with adenovirus vectors encoding the 3P complex from the indicated influenza viruses. Non-nuclease-treated 3P complexes were then purified by affinity chromatography, along with a negative-control eluate (A549), as described in Fig. 1A. The purified polymerase complexes from influenza virus strains WSN, VN/1203, and CA/04 were separated by PAGE and detected by silver staining (top panel). The purified protein samples were also separated by SDS-PAGE and analyzed by immunoblotting with antibodies specific for IPO5, IPO7, AIFM1, and PRKDC.
The LC-MS results (Fig. 5A) also suggested that these host cell factors interacted with the viral polymerase components in a strain-independent fashion since they were also detected in affinity-purified 3P complexes from the H1N1 strains WSN and CA/04. These results were further confirmed by Western blotting (Fig. 5C) where IPO5, IPO7, AIFM1, and PRKDC were detected in 3P complexes from all three of the viral strains tested (Fig. 5C).
We next focused our attention on AIFM1 since the interaction of PA with mitochondrial proteins has not been described before. We tested whether AIFM1, one of the most readily detected mitochondrial proteins, could also be detected in a complex with PA by coimmunoprecipitation (co-IP) experiments. Since AIFM1 binding to the viral polymerase did not occur in a strain-specific fashion, we transfected 293T cells with vectors encoding polymerase proteins (PA, PB1, and PB2) from the WSN strain together with Flag-tagged AIFM1. Cells were harvested at 24 h posttransfection, and AIFM1 was immunoprecipitated with an anti-Flag antibody. The polymerase proteins and AIMF1 were readily detected in the total cell extract (Fig. 6, lower panel). AIFM1 efficiently coprecipitated PA when PA was either expressed alone (Fig. 6, lane 2, top panel) or together with PB1 and PB2 (Fig. 6, lane 4, top panel). As expected, no polymerase components were detected in the absence of AIFM1-Flag (Fig. 6, lanes 1 and 3). These data are consistent with the notion that AIFM1 binds chiefly to PA (Fig. 5A and B) and suggest that AIFM1 binding to PA also occurs when in a complex with PB1 and PB2 (3P).
Fig. 6.

AIFM1 interacts with the viral polymerase in transfected and influenza virus-infected cells. The interaction of AIFM1 to PA or the 3P complex was confirmed by coimmunoprecipitation experiments. (A) HEK293T cells were transfected with an expression construct (0.2 μg) encoding AIFM1-Flag (lanes 2 and 4) or empty vector (lanes 1 and 3) and PA (lanes 1 and 2) or the trimeric polymerase complex (0.6 μg each) (lanes 3 and 4). The viral proteins were derived from the A/WSN/33 strain. Whole-cell extract was prepared at 24 h posttransfection and used for immunoprecipitation (IP) with Flag-specific rabbit polyclonal antibody. The presence of AIFM1-Flag and PA, PB1, and PB2 in the precipitates (upper panel) as well as cell extract (lower panel) was analyzed by Western blotting using Flag (AIFM1-Flag), His (PA-His), and polymerase-specific antibodies (PB1 and PB2). (B) AIFM1 binds to PA in influenza virus (A/WSN/33)-infected A549 cells. Mock-infected (M)and WSN-infected A549 cells were lysed at various time points postinfection (24, 30, and 42 hpi), and total protein lysates (left panel) or PA-coimmunoprecipitated proteins (right panel) were subjected to immunoblot analysis using antibodies specific to AIFM1 and PA. Note that the IgG used to immunoprecipitate PA is also apparent in right panel.
We next tested whether AIFM1 could also be detected in a complex with PA in A549 cells that were infected with wild-type influenza virus. We infected A549 cells with A/WSN/33 (H1N1) virus and prepared cell lysates at various times postinfection (24, 30, and 42 hpi). We then performed a coimmunoprecipitation assay using anti-PA antibody. PA of the WSN virus and AIFM1 were readily detected in the total cell extract (Fig. 6B, top and middle of left panel). AIFM1 was also detected in the protein complex that was immunoprecipitated with the PA-specific antibody (Fig. 6B, middle of right panel) at all times postinfection (24, 30, and 42 hpi); it was not, however, detected in immunoprecipitated complexes from mock-infected cells. These findings confirm that the interaction between AIFM1 and PA also occurs in the context of a virally infected cell.
Identification of PA-PB1 binding partners.
Having identified cellular proteins that interact with PA, we were interested in proteins that bind to larger subcomponents of the influenza virus polymerase. We turned our attention first to the PA-PB1 dimer and looked for proteins that bound to the PA-PB1 dimer but were not found in the PA fraction. Since the PA-PB1 fraction contains unbound monomeric PA (2), the PA-specific binding partners were subtracted, presumably leaving only the PA-PB1-interacting partners. To be conservative in this analysis, we also subtracted out all proteins that were detected in association with PA from a second, independent set of experiments (set 2). We identified a total of 68 proteins that were present in the PA-PB1 fraction (with or without nuclease-treated fractions combined) but not in the PA fraction (Fig. 7A). These proteins could be specific interactors with the PA-PB1 dimer or with PB1 (remaining bound to PB1 when it is in a complex with PA).
Fig. 7.

Identification of 68 cellular binding partners to PA-PB1 (A) and 34 to the 3P (B) complex. (A) A Venn diagram illustrates cellular proteins identified in affinity-purified complexes with PA (set 1 and set 2) or PA-PB1 (set 1). Most of the proteins that copurified with PA (Set 1 and set 2) also copurified with the PA-PB1 complex from the same virus. Numbers in the various boxes/sectors denote numbers of unique cellular proteins identified in each category. Highlighted in the lower panel of this figure are the 68 cellular proteins that interacted with the H5N1 PA-PB1 heterodimer. The boxes at the bottom of the figure denote proteins that were detected only in complexes not treated with nuclease (−N), only in complexes treated with nuclease (+N), or regardless of nuclease treatment (−/+N). (B) Venn diagram, representing cellular proteins identified in affinity-purified complexes with PA alone or PA-PB1 or the complete heterotrimeric polymerase complex (3P) from the A/Vietnam/1203/2004 (H5N1) virus. Most of the proteins identified in association with the 3P complex from A/Vietnam/1203/2004 were also detected in complex with PA plus PA-PB1. However, 34 proteins associated uniquely with the 3P complex. Numbers in the various boxes/sectors denote numbers of unique cellular proteins identified in each category. Highlighted in the lower panel of this figure are the 34 cellular proteins that interacted uniquely with the H5N1 3P. The boxes denote proteins that were detected only in complexes not treated with nuclease (−N), only in complexes treated with nuclease (+N), or regardless of nuclease treatment (−/+N). An asterisk next to the protein symbol depicts proteins previously reported to associate with the influenza virus polymerase. The proteins were grouped according to their functions. Proteins that are underlined were also detected in the corresponding PA-PB1 or 3P (H5N1) fraction from set 2.
We then separated out proteins that were present in only the untreated fraction (Fig. 7, −N), in only the nuclease-treated fraction (Fig. 7, +N), or present in both. We identified 23 proteins that were present in both the untreated and nuclease-treated samples (Fig. 7, −/+N), 34 proteins that were present in only the untreated sample, and 11 proteins that were present in only the nuclease-treated samples. In the untreated (−N) fraction, we mainly detected proteins that are involved in translation (such as ribosomal proteins), protein folding, and transcriptional regulation (Fig. 7A). We further detected cell surface proteoglycans (GPC1 and SDC1) which may have a potential role in influenza virus entry. However, the exact role of viral polymerase binding to these proteins remains uncertain. Another protein we detected was the CCCH-type zinc finger protein ZC3HAV1 (also called ZAP), an antiviral protein that was reported to protect cells from infection by retroviruses as well as certain alphaviruses and filoviruses (1, 19). We detected four isoforms of the ZAP protein, including ZAPS (isoform 2) (see Table S2 in the supplemental material). ZAPS facilitates the interaction of RIG-I with IPS-1, leading to the induction of antiviral gene expression, such as interferon beta (IFN-β) (24). It has been previously shown that PB2 of the viral polymerase can bind to IPS-1 and inhibit IFN-β production (29). Our findings also indicate that PA-PB1 (and possibly also the 3P complex) may have important functions in regulating innate antiviral immune responses by binding to ZAPS.
In the combined (Fig. 7, −/+N) fraction, we detected some ribosomal proteins, chaperones, proteins involved in proteolysis (proteasomal proteins), and the RNA polymerase II (Pol II) and I (Pol I) transcription termination factor (TTF2). In the nuclease-treated (+N) fraction, we detected proteasomal proteins, protein kinases, and transcriptional regulators. Among the last were the Pol II subunit POLR2C and the Pol I transcript release factor PTRF. These transcriptional regulators may directly be involved in the modulation of influenza virus RNA polymerase function.
Identification of 3P-binding partners.
After completing our analysis of cellular proteins that interact with the PA-PB1 heterodimer, we next proceeded to identify proteins that interact specifically with the complete heterotrimeric polymerase complex (3P), comprising PA-PB1-PB2 (Fig. 7B). We subtracted out proteins that bound to PA alone or to PA-PB1 alone under both conditions (with and without nuclease treatment) (Fig. 7B). This analysis revealed a total of 34 proteins that bound uniquely to the 3P complex and not to PA or PA-PB1. These proteins could be specific interactors with the trimeric polymerase complex or with PB2 (remaining bound to PB2 when it is in a complex with PA).
Ten proteins bound specifically to the 3P complex, regardless of nuclease treatment (Fig. 7B, −/+N). These included proteins that may have important roles in the shuttling of the 3P complex from the nucleus to the cytoplasm (alpha-importins KPNA3 and KPNA4 [importin α-4 and α-3, respectively]) or in COPI-mediated transport (SDPR [serum deprivation response]). The importance of alpha-importins in the influenza A virus life cycle has been recognized before and has been attributed mainly to enhancement of transcription and replication, mediated by binding to PB2 and the nucleoprotein (NP) (17, 18, 65). However, the association of these particular importins with specifically the 3P complex is novel and may point to a specific nucleo-cytoplasmic transport mechanism of the 3P complex compared to its subunit components. Further, we detected the recently identified cap-binding protein GEMIN5 (3) and the main subunit of the RNA polymerase II (POLR2A), a previously identified binding partner of the 3P complex (31), which again may be directly involved in modulating influenza virus RNA polymerase function.
Twelve cellular proteins bound specifically to the 3P complex but only if the complex was not treated with nuclease (Fig. 7B, −N). These included some ribosomal proteins, transcriptional activators, and the DNA topoisomerase. Finally, 12 factors bound specifically to the 3P complex but only when the complex was exposed to nuclease treatment (Fig. 7B, +N). In addition to the well-described influenza virus antiviral protein MxA (Mx1), we identified four transcriptional regulators that belong to the Pol II regulatory network (HIV Tat-specific factor [HTat-SF1], PAF1, PRMT5, and Supt5H). These factors may directly affect viral polymerase function at the level of transcription and/or replication.
Overall, these studies showed that one major interaction network among the cellular host factors identified in association with the PA-PB1 heterodimer and the full 3P polymerase complex is the RNA-polymerase II interaction network (Fig. 8A). We next selected two Pol II-interacting proteins (Supt5H and HTat-SF1) and one Pol I-interacting protein (PTRF) to be analyzed further. Supt5H and PTRF are cellular factors that have not been previously described to associate with the influenza virus polymerase; HTat-SF1 has been reported to bind only to the viral nucleoprotein and not to the polymerase itself (45). Figure 8b shows a heat map of the relative abundance of these proteins as identified by LC-MS. It can be readily appreciated from Fig. 8b that all three proteins mainly associate with the trimeric polymerase complex; however, in the case of PTRF some binding to PA-PB1 is observed as well.
Fig. 8.

Pol II interaction network of PA-PB1- and 3P-associated host factors. (A) Proteins shown were identified in association with the following affinity-purified polymerase subunits/preparations: PA-PB1 fraction, blue; 3P fraction, green; PA fraction, white. Nodes depict proteins in the network, and solid lines represent direct whereas dotted lines represent indirect relationships between the proteins. An arrow pointing from one node to the next signifies an activation event, such as phosphorylation or methylation. (B) The relative abundances (spectral counts) of Pol II and Pol I factors are shown in the form of a heat map. The spectral counts are shown from samples across both experimental sets (set 1 and set 2).
We next confirmed the interaction of Supt5H and PTRF with the polymerase complex by reciprocal coimmunoprecipitation studies. We transfected Flag-tagged Supt5H and PTRF into HEK293T cells along with vectors encoding PA, PB1, and PB2. Cells were harvested at 24 h posttransfection, and total cell extracts were analyzed by immunoblotting for the presence of the Flag-tagged proteins by using an anti-Flag antibody as well as for the presence of individual polymerase proteins using PA-, PB1-, and PB2-specific antibodies (Fig. 9A, bottom panel). Viral proteins were detected in the control samples transfected with empty vector (lanes 1 and 3) as well as in samples transfected with the individual Flag-tagged constructs (lanes 2 and 4). A coimmunoprecipitation assay was performed with an anti-Flag antibody to precipitate protein complexes containing Supt5H and PTRF (Fig. 9A, top panel). PTRF and Supt5H (lanes 2 and 4, respectively) were detected in those samples that were transfected with the corresponding constructs, but not in the empty vector control samples (lanes 1 and 3). Proteins that coimmunoprecipitated with PTRF and Supt5H included PA, PB1, and PB2, consistent with their association with the 3P complex (Fig. 7B). These results are in accordance with the LC-MS data presented in the heat map under Fig. 8B, in which Supt5H and PTRF were found to associate preferentially with the trimeric complex rather than PA-PB1 alone. In contrast to the LC-MS data, we did not observe any differences in the amount of viral proteins that coprecipitated with PTRF or Supt5H in the absence or presence of nuclease treatment; this could reflect the overall low peptide abundance for these particular proteins as determined by LC-MS analysis (see Table S2 in the supplemental material).
Fig. 9.

Interaction of Supt5H and PTRF with the viral polymerase complex and polymerase subunits in transfected HEK293T cells. (A) To confirm the interaction between Supt5H and PTRF to the 3P complex by coimmunoprecipitation experiments, HEK293T cells were transfected with expression constructs (0.2 μg) encoding PTRF-Flag (lane 2), Supt5H (lane 4), or empty vector (lanes 1 and 2) and the components of the VN/1203 (H5N1) polymerase complex (PA, PB1, and PB2) (0.6 μg each). Whole-cell extract was prepared at 24 h posttransfection and used for immunoprecipitation (IP) with Flag-specific rabbit polyclonal antibody. The presence of Supt5H-Flag and PTRF-Flag, PA, PB1, and PB2 in the precipitates (upper panel) as well as cell extract (lower panel) was analyzed by Western blotting using Flag and polymerase-specific antibodies. Approximately 3% of total cell extract used for IP was loaded. (B) Flag-tagged Supt5H expression vector (lanes 4 to 6) or empty vector (lanes 1 to 3) was transfected into HEK293T cells with plasmid vector encoding PA alone, PA and PB1, or 3P from the VN/1203 strain (H5N1). Whole-cell extract was prepared 24 h posttransfection and used for immunoprecipitation with Flag-specific rabbit polyclonal antibody. The presence of Supt5H-Flag and PA, PB1, and PB2 in the precipitates (upper panel) as well as cell extract (lower panel) was analyzed by Western blotting using Flag and polymerase-specific antibodies. Approximately 3% of total cell extract used for IP was loaded. Numbers at the top of lanes 4 to 6 denote the relative ratios of PA to Supt5H, as determined by densitometric analysis of the immunoprecipitates.
Since our LC-MS analysis revealed that Supt5H associated only with the full trimeric polymerase complex (3P) (Fig. 8B), we performed an additional experiment to examine the specificity of this interaction. We transfected Flag-tagged Supt5H into HEK293T cells that were transfected with expression plasmids encoding PA, PA-PB1, or PA-PB1-PB2 (3P). Cells were harvested at 24 h posttransfection, and total cell extracts were analyzed by immunoblotting for the presence of the Flag-tagged proteins by using an anti-Flag antibody as well as for the presence of individual polymerase proteins using PA-, PB1-, and PB2-specific antibodies (Fig. 9B, bottom panel). Viral proteins were detected in the control samples transfected with empty vector (lanes 1 to 3) and in samples transfected with the Flag-tagged Supt5H constructs (lanes 4 to 6). A coimmunoprecipitation assay was performed with an anti-Flag antibody to precipitate protein complexes containing Supt5H (Fig. 9B, top panel). Supt5H was detected in those samples that were transfected with the corresponding expression construct (lanes 4 to 6) but not in the empty vector control samples (lanes 1 to 3). Supt5H most efficiently coprecipitated the complete 3P complex (lane 6), but it also precipitated the PA-PB1 complex and, weakly, PA alone. Densitometry was performed to quantitate the ratio of PA to Supt5H in the immunoprecipitated fractions. The ratio of PA/Supt5H was set to 1 for the 3P fraction (Fig. 9B, lane 6) and compared with that of PA alone (lane 4) and PA with PB1 (lane 5). A smaller fraction of PA (0.39) was pulled down by Supt5H when it was expressed alone or together with PB1 (0.64), suggesting preferential binding of Supt5H to the 3P complex. Collectively, the results in Fig. 9 corroborate our LC-MS data and confirm that PTRF and Supt5H are, indeed, polymerase binding partners with the ability to interact with the 3P complex.
DISCUSSION
In this study, we conducted an unbiased proteomic approach to identify host cell factors that bind to the influenza virus polymerase; our goal was to provide a framework that will guide future studies in dissecting out the mechanism of influenza virus polymerase function. In our study, we put a particular focus on cellular factors that bind to PA, PA-PB1, and the full trimeric complex.
An important advantage of our analytical method was the very high sensitivity of the comprehensive LC-MS approach. As a result, we identified over 400 potential polymerase binding partners, some of which have been described previously to interact with influenza virus viral proteins and/or be important in the influenza virus life cycle (see Table S4 in the supplemental material). This large number of putative binding partners includes several proteins (62) that have been previously identified in genome-wide screens (see Table S4A) and are known to regulate viral replication. Some of these interactions affect viral replication by targeting influenza virus entry and hemagglutinin (HA) trafficking (COPI coat complex), by inducing the degradation of host cell proteins (proteasome complex), by targeting nuclear import and export of viral proteins (XPO1, XPO5, CSE1L, and TNPO3), or by interfering with the innate immune signaling pathway (Toll-like receptor signaling) (see Table S4A). Our findings complement and extend these earlier genome-wide RNAi knockdown studies, by identifying specific viral proteins (notably PA) which may interact with these essential host factors. Our findings together with published data also reveal a subset of host proteins that are capable of interacting with both the viral polymerase complex and additional virus proteins, such as NS1 (STAU1, TOP2A, and KPNA3) (13, 42, 52), NS2 (AIMP2 [JTV1]), M1 (STAU1), HA, and neuraminidase ([NA] RPS6KA3) (52). These interactions suggest that some host factors may have multiple roles in the influenza virus life cycle.
The large number of putative binding partners identified in our analysis excludes a number of previously identified polymerase binding factors. This is because of our rigorous approach to excluding proteins that might nonspecifically copurify with the polymerase. Thus, we excluded all factors that were detected in the control lysate fraction in one or more experiments. The exclusion of these proteins from our analysis should not be interpreted as definitive evidence that they do not interact with the polymerase. Rather, it is simply a reflection of our conservative analytical approach. For example, IPO3 (TNPO2) and PARP-1, two of the 3P binders identified by Mayer et al. (41), were detected only in our set 2 sample and not in our set 1 sample and were therefore also excluded from further analysis. It should also be noted that proteins listed as associating only with PA or PA-PB1 may also bind to the 3P complex since host factor binding to the subunits may occur prior to complex formation.
Given the large number of polymerase binding factors that were identified in our analysis, it is not possible to discuss more than a few of them here. We have chosen to focus on those which reveal previously unappreciated cellular pathways that may be targeted by the influenza virus polymerase and which may provide insight into the virus life cycle. For a small subset of these host proteins, we went further and experimentally validated their ability to interact with the influenza virus polymerase (e.g., PTRF and Supt5H). Future studies will be required to fully explore and validate all of the pathways and interactions reported here. We have therefore provided a complete list of all identified factors together with their peptide composition and spectral counts in the supplemental data section (see Table S2 in the supplemental material).
Our results suggest that PA has a range of functions in the influenza virus life cycle in addition to its previously described roles in the proteolytic degradation of viral and host proteins, the endonucleolytic cleavage of capped RNA primers, and transcript elongation (15, 16, 23, 51). Indeed, the large number of cellular proteins that were associated with PA alone was somewhat unexpected. Particularly interesting was the association of PA with over 30 mitochondrial proteins. An association of influenza virus proteins with mitochondrial factors has been reported before for PB1-F2, a regulator of influenza A virus-mediated apoptosis (8), and PB2, which interacts with IPS-1 in mitochondria (21), leading to the inhibition of interferon beta and antiviral signaling. To our knowledge, PA has not been found to associate with mitochondrial proteins before. The fact that PA associated with mitochondrial proteins that mediate outer (voltage-dependent anion channel protein 3 [VDAC3]) or inner membrane permeabilization (transmembrane transport proteins) (Fig. 4) suggests that PA may have a role in the regulation of apoptosis during influenza virus infection. The general role of VDACs in the induction of apoptosis is evident and has been extensively reviewed in the literature (50, 60). It is also conceivable that binding of these cellular proteins to PA could inhibit their function in a manner similar to the postulated antiapoptotic effects mediated by binding of NS1 to apoptosis-inducing proteins (39).
In addition, our study revealed the mitochondrial protein AIFM1 as one of the most readily detected host factor proteins in the PA fraction. This observation could be due to an overall higher cellular expression of AIFM1 and/or due to more specific binding of PA to AIFM1 than to other cellular factors that were identified. In healthy cells, AIFM1 is located on the inner membrane of mitochondria. However, upon apoptosis induction, nuclear translocation of AIFM1 occurs via a caspase-independent pathway. This pathway has recently been shown to be activated by influenza virus (H5N1), leading to mitochondrial dysfunction and cell death through the induction of an extracellular Ca2+ influx (62). However, the viral component involved in the activation of this caspase-independent pathway is unknown. Our results indicate a possible role for PA in this process.
We also examined the association of cellular factors with PA-PB1 and the 3P complex and discovered that the Pol II complex represents a major interaction network. Pol II itself has been previously reported to associate with the viral 3P complex. However, to our knowledge none of the Pol II-associated proteins have been shown to associate with the polymerase complex. HTat-SF1 has been described before only as an NP binding partner (45). It remains possible that Pol II is the main interactor with the viral polymerase and that associated factors such as Supt5H and HTat-SF1 interact indirectly with the viral polymerase via Pol II. However, it is intriguing to speculate that some of these proteins may interact directly with the viral polymerase. For example, DNA damage-induced protein kinase DNA-PK (PRKDC), an abundant factor present in the PA fraction, is important for DNA repair and the regulation of innate immune responses after viral infection through the phosphorylation of interferon regulatory factor 3 (IRF-3) (32). DNA-PK has also been implicated in regulating transcription by RNA polymerase II (53). In addition, HTat-SF1 may also be an important interacting protein since it has previously been described as an NP binding partner (45).
An important aspect of the virus life cycle, which is also related at least in part to interactions with Pol II, is virus-mediated shutoff of host protein synthesis. Our analysis revealed several host factor interactions which may contribute to this shutoff. For example, in addition to its interaction with members of the Pol II network (Fig. 8A), the viral polymerase was also associated with proteasomal proteins and E3 ubiquitin ligases (such as PJA2) (Fig. 7A and B). This supports the hypothesis that the viral polymerase may induce ubiquitination and degradation of Pol II (64). In addition, we found a novel association of PA with the aminoacyl-tRNA biosynthesis pathway, suggesting a previously unrecognized mechanism by which the virus may induce host-protein synthesis shutoff.
Our studies also revealed that several representatives of the caveolar-mediated endocytosis pathway, including members of the COPI coat complex (Fig. 3), are PA-binding partners. COPI components are important for HA trafficking (4), and depletion of some of these members (COPG and ARCN1) has also been shown to block influenza virus infection at the entry step (4, 35). Moreover, epidermal growth factor receptor (EGFR), another PA-associated factor and member of this endocytic pathway, has been shown to promote uptake of influenza viruses (11). Interestingly, EGFR signaling and trafficking are also regulated by a number of other viruses, including hepatitis C virus (40), hepatitis E virus (7), human papillomavirus (55, 58), and herpes simplex virus type-1 (38), and this pathway may regulate postentry steps in the viral life cycle, including viral RNA synthesis, nuclear export of vRNP, or release of virions (36). This finding underscores the notion that influenza virus may be susceptible to inhibition by small-molecule inhibitors of EGFR tyrosine kinase activity (36), possibly including antitumor drugs such as gefitinib (37).
In conclusion, the present analysis provides new insight into the cellular factors that associate with the influenza A virus RNA polymerase and suggests that the viral polymerase may significantly perturb cellular pathways involved in endocytosis, protein and RNA transport, and the regulation of apoptosis. Our data also reveal an unexpected interaction of polymerase proteins with mitochondrial proteins (including AIFM1) and with the RNA polymerase II machinery. These results suggest previously unappreciated roles for the influenza virus polymerase in regulating virus replication and pathogenesis and point the way to new antiviral strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shikha Chakraborty-Sett (University of Rochester) and Jason Hogan (FHCRC Proteomics) for excellent technical assistance and Marcus Korth and Lynn Law for critically reading the manuscript. We also thank Toru Takimoto (University of Rochester) for providing a titered stock of influenza A/WSN/33 virus.
This project has been funded in part by federal funds from the Department of Health and Human Services, under contracts HHSN272200800060C (M.G.K.) and HHSN266200700008C (S.D.), the National Institute of Allergy and Infectious Diseases under U54 AI081680 (M.G.K.), and the National Center for Research Resources under P51RR000166 (M.G.K.).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 29 June 2011.
REFERENCES
- 1. Bick M. J., et al. 2003. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 77:11555–11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bradel-Tretheway B. G., et al. 2008. The human H5N1 influenza A virus polymerase complex is active in vitro over a broad range of temperatures, in contrast to the WSN complex, and this property can be attributed to the PB2 subunit. J. Gen. Virol. 89:2923–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradrick S. S., Gromeier M. 2009. Identification of gemin5 as a novel 7-methylguanosine cap-binding protein. PLoS One 4:e7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brass A. L., et al. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bussey K. A., Bousse T. L., Desmet E. A., Kim B., Takimoto T. 2010. PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. J. Virol. 84:4395–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carr S. M., Carnero E., Garcia-Sastre A., Brownlee G. G., Fodor E. 2006. Characterization of a mitochondrial-targeting signal in the PB2 protein of influenza viruses. Virology 344:492–508 [DOI] [PubMed] [Google Scholar]
- 7. Chandra V., Kar-Roy A., Kumari S., Mayor S., Jameel S. 2008. The hepatitis E virus ORF3 protein modulates epidermal growth factor receptor trafficking, STAT3 translocation, and the acute-phase response. J. Virol. 82:7100–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen C. J., et al. 2010. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 84:10051–10062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W., et al. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7:1306–1312 [DOI] [PubMed] [Google Scholar]
- 10. Deng T., et al. 2006. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 80:11911–11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eierhoff T., Hrincius E. R., Rescher U., Ludwig S., Ehrhardt C. 2010. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 6:e1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engelhardt O. G., Smith M., Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 79:5812–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falcon A. M., Fortes P., Marion R. M., Beloso A., Ortin J. 1999. Interaction of influenza virus NS1 protein and the human homologue of Staufen in vivo and in vitro. Nucleic Acids Res. 27:2241–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fislova T., Thomas B., Graef K. M., Fodor E. 2010. Association of the influenza virus RNA polymerase subunit PB2 with the host chaperonin CCT. J. Virol. 84:8691–8699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fodor E., et al. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76:8989–9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fodor E., Mingay L. J., Crow M., Deng T., Brownlee G. G. 2003. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase promotes the generation of defective interfering RNAs. J. Virol. 77:5017–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gabriel G., Herwig A., Klenk H. D. 2008. Interaction of polymerase subunit PB2 and NP with importin α1 is a determinant of host range of influenza A virus. PLoS Pathog. 4:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gabriel G., et al. 2011. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao G., Guo X., Goff S. P. 2002. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297:1703–1706 [DOI] [PubMed] [Google Scholar]
- 20. Gonzalez S., Ortin J. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J. 18:3767–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Graef K. M., et al. 2010. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 84:8433–8445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao L., et al. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hara K., et al. 2001. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells 6:87–97 [DOI] [PubMed] [Google Scholar]
- 24. Hayakawa S., et al. 2011. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 12:37–44 [DOI] [PubMed] [Google Scholar]
- 25. Honda A., Okamoto T., Ishihama A. 2007. Host factor Ebp1: selective inhibitor of influenza virus transcriptase. Genes Cells 12:133–142 [DOI] [PubMed] [Google Scholar]
- 26. Huang D. W., Sherman B. T., Lempicki R. A. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 27. Huarte M., Sanz-Ezquerro J. J., Roncal F., Ortin J., Nieto A. 2001. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J. Virol. 75:8597–8604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes N. C., Wong E. Y., Fan J., Bajaj N. 2007. Determination of carryover and contamination for mass spectrometry-based chromatographic assays. AAPS J. 9:E353–E360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwai A., et al. 2010. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 285:32064–32074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jansa P., Burek C., Sander E. E., Grummt I. 2001. The transcript release factor PTRF augments ribosomal gene transcription by facilitating reinitiation of RNA polymerase I. Nucleic Acids Res. 29:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karlas A., et al. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822 [DOI] [PubMed] [Google Scholar]
- 32. Karpova A. Y., Trost M., Murray J. M., Cantley L. C., Howley P. M. 2002. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. U. S. A. 99:2818–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawaguchi A., Nagata K. 2007. De novo replication of the influenza virus RNA genome is regulated by DNA replicative helicase, MCM. EMBO J. 26:4566–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392 [DOI] [PubMed] [Google Scholar]
- 35. Konig R., et al. 2010. Human host factors required for influenza virus replication. Nature 463:813–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar N., Liang Y., Parslow T. G. 2011. Receptor tyrosine kinase inhibitors block multiple steps of influenza A virus replication. J. Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langhammer S., Koban R., Yue C., Ellerbrok H. 2011. Inhibition of poxvirus spreading by the anti-tumor drug gefitinib (Iressa). Antiviral Res. 89:64–70 [DOI] [PubMed] [Google Scholar]
- 38. Liang Y., Kurakin A., Roizman B. 2005. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Proc. Natl. Acad. Sci. U. S. A. 102:5838–5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu H., et al. 2010. The ESEV PDZ-binding motif of the avian influenza A virus NS1 protein protects infected cells from apoptosis by directly targeting Scribble. J. Virol. 84:11164–11174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mankouri J., Griffin S., Harris M. 2008. The hepatitis C virus non-structural protein NS5A alters the trafficking profile of the epidermal growth factor receptor. Traffic 9:1497–1509 [DOI] [PubMed] [Google Scholar]
- 41. Mayer D., et al. 2007. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J. Proteome Res. 6:672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melen K., et al. 2007. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol. 81:5995–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Momose F., et al. 2001. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J. Virol. 75:1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moncorge O., Mura M., Barclay W. S. 2010. Evidence for avian and human host cell factors that affect the activity of influenza virus polymerase. J. Virol. 84:9978–9986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Naito T., et al. 2007. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 104:18235–18240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Naito T., Momose F., Kawaguchi A., Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 81:1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75:4646–4658 [DOI] [PubMed] [Google Scholar]
- 48. O'Neill R. E., Palese P. 1995. NPI-1, the human homolog of SRP-1, interacts with influenza virus nucleoprotein. Virology 206:116–125 [DOI] [PubMed] [Google Scholar]
- 49. Perez-Gonzalez A., Rodriguez A., Huarte M., Salanueva I. J., Nieto A. 2006. hCLE/CGI-99, a human protein that interacts with the influenza virus polymerase, is a mRNA transcription modulator. J. Mol. Biol. 362:887–900 [DOI] [PubMed] [Google Scholar]
- 50. Rostovtseva T. K., Tan W., Colombini M. 2005. On the role of VDAC in apoptosis: fact and fiction. J. Bioenerg. Biomembr. 37:129–142 [DOI] [PubMed] [Google Scholar]
- 51. Sanz-Ezquerro J. J., Zurcher T., de la Luna S., Ortin J., Nieto A. 1996. The amino-terminal one-third of the influenza virus PA protein is responsible for the induction of proteolysis. J. Virol. 70:1905–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shapira S. D., et al. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sheppard H. M., Liu X. 2000. Transcription by RNA polymerase II in DNA-PK deficient SCID mouse cells. Biochim. Biophys. Acta 1493:41–47 [DOI] [PubMed] [Google Scholar]
- 54. Song J., et al. 2011. The PA protein directly contributes to the virulence of H5N1 avian influenza viruses in domestic ducks. J. Virol. 85:2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Straight S. W., Hinkle P. M., Jewers R. J., McCance D. J. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 67:4521–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sui B., et al. 2009. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology 387:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun Y., et al. 2011. High genetic compatibility and increased pathogenicity of reassortants derived from avian H9N2 and pandemic H1N1/2009 influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 108:4164–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suprynowicz F. A., et al. 2010. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 84:10619–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tarendeau F., et al. 2007. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 14:229–233 [DOI] [PubMed] [Google Scholar]
- 60. Tsujimoto Y., Shimizu S. 2002. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie 84:187–193 [DOI] [PubMed] [Google Scholar]
- 61. Turan K., et al. 2004. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 32:643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ueda M., et al. 2010. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J. Virol. 84:3068–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vreede F. T., Chan A. Y., Sharps J., Fodor E. 2010. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 396:125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vreede F. T., Fodor E. 2010. The role of the influenza virus RNA polymerase in host shut-off. Virulence 1:436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang P., Palese P., O'Neill R. E. 1997. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 71:1850–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang P., et al. 2009. Nuclear factor 90 negatively regulates influenza virus replication by interacting with viral nucleoprotein. J. Virol. 83:7850–7861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Watanabe T., Watanabe S., Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu-Baer F., Lane W. S., Gaynor R. B. 1998. Role of the human homolog of the yeast transcription factor SPT5 in HIV-1 Tat-activation. J. Mol. Biol. 277:179–197 [DOI] [PubMed] [Google Scholar]
- 69. Zamarin D., Garcia-Sastre A., Xiao X., Wang R., Palese P. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou Q., Sharp P. A. 1996. Tat-SF1: cofactor for stimulation of transcriptional elongation by HIV-1 Tat. Science 274:605–610 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.