

Abstract
TRIM5αrh is a cytosolic protein that potently restricts HIV-1 before reverse transcription. TRIM5αrh is composed of four different domains: RING, B-box 2, coiled coil, and B30.2(SPRY). The contribution of each of these domains to restriction has been extensively studied, with the exception of the RING domain. The RING domain of TRIM5α exhibits E3-ubiquitin ligase activity, but the contribution of this activity to the restriction of HIV-1 is not known. To test the hypothesis that the E3-ubiquitin ligase activity of the RING domain modulates TRIM5αrh restriction of HIV-1, we correlated the E3-ubiquitin ligase activity of a panel of TRIM5αrh RING domain variants with the ability of these mutant proteins to restrict HIV-1. For this purpose, we first solved the nuclear magnetic resonance structure of the RING domain of TRIM5α and defined potential functional regions of the RING domain by homology to other RING domains. With this structural information, we performed a systematic mutagenesis of the RING domain regions and tested the TRIM5α RING domain variants for the ability to undergo self-ubiquitylation. Several residues, particularly the ones on the E2-binding region of the RING domain, were defective in their self-ubiquitylation ability. To correlate HIV-1 restriction to self-ubiquitylation, we used RING domain mutant proteins that were defective in self-ubiquitylation but preserve important properties required for potent restriction by TRIM5αrh, such as capsid binding and higher-order self-association. From these investigations, we found a set of residues that when mutated results in TRIM5α molecules that lost both the ability to potently restrict HIV-1 and their self-ubiquitylation activity. Remarkably, all of these changes were in residues located in the E2-binding region of the RING domain. Overall, these results demonstrate a role for TRIM5α self-ubiquitylation in the ability of TRIM5α to restrict HIV-1.
INTRODUCTION
Several newly discovered proteins that are endogenously expressed in primates show the ability to dominantly block retroviral infection and cross-species transmission by interfering with the early phase of viral replication (3, 33, 57, 63). Of particular interest are members of the tripartite motif (TRIM) family of proteins. Splicing variant alpha of TRIM5 from rhesus macaques (TRIM5αrh) is an ∼53-kDa cytosolic protein that potently restricts HIV-1 (28, 61). TRIM5αrh blocks HIV-1 and certain other retroviruses soon after viral entry but prior to reverse transcription (28, 63). The retroviral capsid protein is the viral determinant for susceptibility to restriction by TRIM5α (48). Studies on the fate of the HIV-1 capsid in the cytosol of infected cells have correlated restriction with a decrease amount of cytosolic particulate capsid (11, 51, 64).
TRIM5αrh is composed of four different domains: RING, B-box 2, coiled coil, and B30.2(SPRY) (53). The RING domain of TRIM5αrh is an E3-ubiquitin ligase (13, 27, 37, 43, 68); however, a role for the really interesting new gene (RING) domain's E3-ubiquitin ligase activity in HIV-1 restriction by TRIM5αrh has not been established. The B-box 2 domain of TRIM5α and other TRIM proteins, such as TRIM63, self-associates, forming dimeric complexes that are important for TRIM5α higher-order self-association and capsid binding avidity; these B-box 2 domain functions are essential for full and potent restriction of HIV-1 (12, 15, 19, 25, 44, 49). The coiled-coil domain enables TRIM5αrh dimerization (27, 37), which is critical for the interaction of the B30.2(SPRY) domain with the HIV-1 capsid (58, 64). The B30.2(SPRY) domain, which provides the capsid recognition motif, dictates the specificity of restriction (45, 56, 62, 65, 70).
The specific interaction of substrates with other TRIM proteins, such as TRIM8 and TRIM11, results in RING domain-dependent ubiquitylation and proteasomal degradation of the target protein (24, 46, 66). The RING domain, originally termed the A-box domain, is involved in protein-protein interactions (17). This domain binds two Zn2+ atoms tetrahedrally in a cross-brace conformation (5, 21). RING domains in other proteins play the role of molecular scaffolds, allowing the formation of supramolecular complexes by self-association of the RING domain, which in some cases improves E3-ubiquitin ligase activity (4, 29, 30, 36, 52). Interestingly, RING-RING interactions have been reported to be functionally important for genomic stability, as in the heterodimer formed by the RING domain of BRCA1 with the RING domain of BARD-1 (6).
TRIM5αrh exhibits an intrinsic rapid turnover of 50 to 60 min that is dependent on an intact RING domain, but this property is apparently not important for restriction (11, 13, 68). However, TRIM5αrh degrades at a higher rate than its normal turnover when in the presence of the HIV-1 capsid (54). Interestingly, this capsid-dependent degradation is inhibited by the use of proteasome inhibitors. Disruption of proteasome function alters TRIM5αrh localization and allows the completion of HIV-1 late reverse transcription during infection (1, 13, 67); however, inhibitors of the proteasome do not alleviate restriction (1, 50, 54, 64, 67). Altogether these results suggest a role for the RING domain and proteasome in restriction.
We present a nuclear magnetic resonance (NMR) structure of the TRIM5 RING domain. Alteration of the different functional regions of the RING domain revealed structures important for TRIM5 self-ubiquitylation, HIV-1 restriction, higher-order self-association, and capsid binding. To understand which RING functions contribute to retroviral restriction, the relationship among these TRIM5 properties was investigated. We found that alteration of the RING domain self-ubiquitylation activity correlated with a loss of restriction potency. These results suggested a contribution of the RING domain self-ubiquitylation activity to the restriction of HIV-1 by TRIM5.
MATERIALS AND METHODS
Sample preparation of the TRIM5 RING domain.
The DNA fragment encoding the RING domain of TRIM5αhu (amino acid residues 1 to 78, Swiss-Prot accession no. Q9C035) was amplified via PCR from Invitrogen Japan K. K. clone IOH 14670 and cloned into the plasmid vector pCR2.1 (Invitrogen, Carlsbad, CA) as a fusion with an N-terminal His tag and a tobacco etch virus protease cleavage site. The 13C- and 15N-labeled protein was synthesized by a cell-free protein expression system (32). Purification was performed by a standard procedure (39). For structure determination, a single 1.25 mM uniformly 13C- and 15N-labeled sample was prepared in a mixture of 20 mM Tris-HCl buffer at pH 7.0, 100 mM NaCl, 1 mM dithiothreitol (DTT), 0.02% NaN3, 0.05 mM ZnCl2, and 1 mM iminodiacetic acid, with the addition of D2O to 10% (vol/vol). The engineered protein sample used for the NMR measurements includes seven additional residues (GSSGSSG) as a tag linker.
NMR spectroscopy, structure determination, and analysis.
All of the NMR spectra for structure determination were recorded at 23°C on Bruker AVANCE 600 and 800 spectrometers equipped with a pulse-field gradient triple-resonance probe. Sequence-specific resonance assignments were made using the standard triple-resonance techniques. The backbone assignment was achieved by the combined analysis of HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and CBCA(CO)NH spectra. The side chain resonances were identified by the combined use of HBHA(CO)NH, (H)CC(CO)NH, HCCH correlation spectroscopy, HCCH total correlation spectroscopy (TOCSY), (H)CCH TOCSY, and two-dimensional 1H and15N heteronuclear single quantum coherence (HSQC) and 1H-13C-HSQC spectra. Nuclear Overhauser effect (NOE) data for structure determination were extracted from three-dimensional 15N- and 13C-edited NOE spectra recorded with a mixing time of 150 ms. The stereospecific assignments for prochiral b-methylene protons were carried out with HN(CO)HB and HNHB. The 1H-15N-HSQC spectra for concentration-dependent experiments were recorded at 25°C on a Varian INOVA 800 spectrometer equipped with a pulse-field gradient triple-resonance probe. The NMRpipe software package (8) and the program KUJIRA (34), created on the basis of NMRView (26), was employed for optimal visualization and spectral analysis. Automated NOE cross-peak assignments (22) and structure calculations with torsion angle dynamics were performed using the software package CYANA 2.0.17. Dihedral angle restrains were derived using the program TALOS (7). A total of 100 conformers were calculated independently. The 20 conformers with the lowest final CYANA target function values were finally selected. The structures were validated using PROCHECK-NMR (38). The program MOLMOL (35) was used to analyze the resulting 20 conformers and to prepare drawings of the structures, unless noted otherwise in the figure legends. The 20 selected conformers have been deposited in the Protein Data Bank (PDB; entry 2ECV).
Creation of cells stably expressing TRIM5α variants.
Retroviral vectors encoding wild-type or mutant rhesus monkey TRIM5αrh proteins were created using the pLPCX vector. The TRIM5αrh proteins contained an influenza hemagglutinin (HA) epitope tag at the C terminus or a FLAG epitope tag at the N terminus. Recombinant viruses were produced in 293T cells by cotransfecting the pLPCX plasmids with the pVPack-GP and pVPack-VSV-G packaging plasmids (Stratagene). The pVPack-VSV-G plasmid encodes the vesicular stomatitis virus (VSV) G envelope glycoprotein, which allows efficient entry into a wide range of vertebrate cells. Cf2Th canine thymocytes were transduced and selected in 5 μg/ml puromycin (Sigma).
Infection with viruses expressing green fluorescent protein (GFP).
Recombinant HIV-1 and equine infectious anemia virus (EIAV) expressing GFP were prepared as described previously (14). All recombinant viruses were pseudotyped with the VSV G glycoprotein. For infections, 3 × 104 Cf2Th cells seeded in 24-well plates were incubated at 37°C with virus for 24 h. Cells were washed and returned to culture for 48 h and then subjected to fluorescence-activated cell sorter (FACS) analysis with a FACScan (BD). HIV-1 and EIAV stocks were titrated by serial dilution on Cf2Th cells to determine the concentration of infectious viruses.
Protein analysis.
Cellular proteins were extracted with radioimmunoprecipitation assay buffer (10 mM Tris [pH 7.4], 100 mM NaCl, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1% NP-40, 2 mg/ml aprotinin, 2 mg/ml leupeptin, 1 mg/ml pepstatin A, 100 mg/ml phenylmethylsulfonyl fluoride). The cell lysates were analyzed by SDS-PAGE (10% acrylamide), followed by blotting onto nitrocellulose membranes (Amersham Pharmacia Biotech). Detection of protein by Western blotting utilized monoclonal antibodies directed against the HA epitope tags (Roche) and FLAG epitope tags (Sigma) and monoclonal antibodies to β-actin (Sigma) directly conjugated to horseradish peroxidase (HRP). Proteins were detected by enhanced chemiluminescence (NEN Life Science Products) using the FluorChem FC2 detection system (Alpha Innotech). Signals were acquired as an image (TIFF) file and quantified by the Quantity One software (Bio-Rad Laboratories).
TRIM5α self-ubiquitylation.
Human 293T cells were transfected with plasmids encoding FLAG-tagged mutant and wild-type TRIM5αrh proteins. Forty-eight hours later, the cells expressing each TRIM5αrh variant were lysed in 1 ml of whole-cell extract buffer (50 mM Tris [pH 8.0], 280 mM NaCl, 0.5% octylphenoxypolyethoxyethanol [IGEPAL]–10% glycerol, 1 mM DTT, protease inhibitor cocktail [Roche]). Lysates were centrifuged at 14,000 rpm for 1 h at 4°C. Postspin lysates were then precleared using protein A-agarose (Sigma) for 1 h at 4°C. Precleared lysates were incubated with anti-FLAG-agarose beads (Sigma) for 2 h at 4°C to precipitate the FLAG-tagged proteins. Beads containing the immunoprecipitate were washed four times in whole-cell extract buffer. Subsequently, immune complexes were eluted using 200 μg/ml FLAG tripeptide in whole-cell extract buffer. The eluted samples were separated by SDS-PAGE and analyzed by Western blotting using anti-FLAG antibodies conjugated to HRP. Subsequently, similar amounts of mutant and wild-type TRIM5α were supplemented with 5 μM ubiquitin aldehyde, a potent inhibitor of all ubiquitin C-terminal hydrolases, ubiquitin-specific proteases, and deubiquitylating enzymes (BostonBiochem). The inhibitor-treated fractions containing mutant and wild-type TRIM5αrh were incubated in a final reaction mixture containing 200 nM E1 (human recombinant UBE1; BostonBiochem), 100 nM E2 (human recombinant UbcH5b; BostonBiochem), 200 μM ubiquitin tagged with a myc epitope (human recombinant ubiquitin), and ATP (energy regeneration solution containing MgCl2, ATP, and ATP-regenerating enzymes to recycle hydrolyzed ATP; BostonBiochem). The reaction mixture was incubated at 37°C for 1 h, and collected fractions were analyzed by Western blotting using HRP-conjugated antibodies against FLAG and myc. Similar reactions were performed in the absence of recombinant E1 and E2 enzymes to determine the contribution of endogenous E1 and E2 enzymes to TRIM5αrh ubiquitylation.
Higher-order self-association of TRIM5α.
Human 293T cells were independently transfected with plasmids encoding FLAG-tagged and HA-tagged mutant or wild-type TRIM5αrh proteins. Forty-eight hours later, the cells expressing each TRIM5αrh variant were lysed in 1 ml of whole-cell extract buffer (50 mM Tris [pH 8.0], 280 mM NaCl, 0.5% octylphenoxypolyethoxyethanol–10% glycerol, 1 mM DTT, protease inhibitor cocktail [Roche]). Lysates were centrifuged at 14,000 rpm for 1 h at 4°C. Postspin lysates were then precleared using protein A-agarose (Sigma) for 1 h at 4°C; a small aliquot of each of these lysates was stored as an input sample. Precleared lysates containing the differently tagged proteins were mixed at a 1:1 ratio and incubated with anti-FLAG–agarose beads (Sigma) for 2 h at 4°C to precipitate the FLAG-tagged proteins. Beads containing the immunoprecipitate were washed four times in whole-cell extract buffer. Subsequently, immune complexes were eluted using 200 μg/ml FLAG tripeptide in whole-cell extract buffer. The eluted samples were separated by SDS-PAGE and analyzed by Western blotting using anti-HA or anti-FLAG antibodies conjugated to HRP.
HIV-1 capsid-nucleocapsid (CA-NC) expression and purification.
The HIV-1 CA-NC protein was expressed, purified, and assembled as previously described (18, 20). The pET11a expression vector (Novagen) expressing the CA-NC protein of HIV-1 was used to transform Escherichia coli BL-21(DE3). CA-NC expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when the culture reached an optical density at 600 nm of 0.6. After 4 h of induction, the cells were harvested and resuspended in a mixture of 20 mM Tris-HCl (pH 7.5), 1 μM ZnCl2, 10 mM 2-mercaptoethanol, and protease inhibitors (Roche). Lysis was performed by sonication, and debris was pelleted for 30 min at 35,000 × g. Nucleic acids were stripped from the solution by using 0.11 equivalent of 2 M (NH4)2SO4 and the same volume of 10% polyethylenimine. Nucleic acids were removed by stirring and centrifugation at 29,500 × g for 15 min. The protein was recovered by addition of 0.35 equivalent of saturated (NH4)2SO4. The protein was centrifuged at 9,820 × g for 15 min and resuspended in a mixture of 100 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1 μM ZnCl2, and 10 mM 2-mercaptoethanol. The CA-NC protein was dialyzed against a mixture of 50 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1 μM ZnCl2, and 10 mM 2-mercaptoethanol and stored at −80°C.
In vitro assembly of CA-NC complexes.
HIV-1 CA-NC particles were assembled in vitro by diluting the CA-NC protein to a concentration of 0.3 mM in a mixture of 50 mM Tris-HCl (pH 8.0), 0.5 M NaCl, and 2 mg/ml DNA oligo-(TG)50. The mixture was incubated at 4°C overnight and centrifuged at 8,600 × g for 5 min. The pellet was resuspended in assembly buffer (50 mM Tris-HCl [pH 8.0], 0.5 M NaCl) at a final protein concentration of 0.15 mM (18, 20) and stored at 4°C until needed.
Binding of TRIM5αrh variants to HIV-1 capsid complexes.
293T cells were transfected with plasmids expressing wild-type or mutant TRIM5αrh proteins. Forty-eight hours after transfection, cell lysates were prepared as follows. Previously washed cells were resuspended in hypotonic lysis buffer (10 mM Tris [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT). The cell suspension was frozen, thawed, and incubated on ice for 10 min. Afterwards, the lysate was centrifuged at full speed in a refrigerated Eppendorf microcentrifuge (∼14,000 × g) for 5 min. The supernatant was supplemented with 1/10 volume of 10× phosphate-buffered saline (PBS) and then used in the binding assay. In some cases, samples containing the TRIM5αrh variants were diluted with extracts prepared in parallel from untransfected cells. To test binding, 5 μl of CA-NC particles assembled in vitro was incubated with 200 μl of cell lysate at room temperature for 1 h. A fraction of this mixture was stored (input). The mixture was spun through a 70% sucrose cushion (70% sucrose, 1× PBS, 0.5 mM DTT) at 100,000 × g in an SW55 rotor (Beckman) for 1 h at 4°C. After centrifugation, the supernatant was carefully removed and the pellet was resuspended in 1× SDS-PAGE loading buffer (pellet). The level of TRIM5αrh proteins was determined by Western blotting with an anti-HA antibody as described above. The level of HIV-1 CA-NC protein in the pellet was assessed by Western blotting with an anti-p24 CA antibody.
Quantitative real-time PCR.
Cf2Th cells expressing wild-type and mutant TRIM5αrh proteins were challenged with HIV-1–GFP at a multiplicity of infection (MOI) of 0.2. Viruses were pretreated with DNase to prevent contamination from carryover plasmid DNA. An infection using a virus that was heat inactivated (60°C for 30 min) was performed as a control for carryover plasmid DNA in the PCR. After 6 h, the cells were lysed and DNA was extracted using a Qiagen Blood Tissue DNA extraction kit. PCRs were prepared using the QuantiTect probe PCR kit. Each sample contained 100 ng of total cellular DNA. PCR was carried out using two primers that amplify a 263-bp fragment of GFP (GFP-fwd, 5′-GAC GTA AAC GGC CAC AAG-3′; GFP-rev, 5′-GGT CTT GTA GTT GCC GTC GT-3′; GFP-Probe, 5′-56-FAM-CCT ACG GCA AGC TGA CCC TGA-36-TAMRA-3′). The calibration curve was prepared using an HIV-1–GFP plasmid.
Protein structure accession number.
The coordinates and structure factors for the RING domain of human TRIM5α have been deposit in the PDB under accession number 2ECV.
RESULTS
Solution structure of the TRIM5α RING domain.
A fragment of human TRIM5α that encompasses the RING domain (residues 1 to 78) was expressed and labeled in a cell-free system. The solution structure of the RING domain was solved using multidimensional NMR spectroscopy (PDB entry 2ECV). The solution structures were well defined from residues 10 to 70 (Fig. 1). Residues 10 to 61 form a core structure that interacts with the C-terminal region composed of residues 62 to 70; however, the interaction between the core and C-terminal regions did not form a single conformation. The root mean square deviations (RMSD) from the mean structure were 0.70 ± 0.18 Å for backbone atoms and 1.05 ± 0.15 Å for heavy atoms in the defined region of the domain. The structural statistics are summarized in Table 1. The RING domain of TRIM5α adopts a ββα RING fold; however, it contains shorter β-strands and a longer α-helix than typical RING domain folds (9). Most hydrophobic residues are packed in the core region of the RING domain between the β-hairpin and α-helix. The hydrophobic core is partly exposed to solvent and forms two hydrophobic patches (Fig. 1).
Fig. 1.

Solution structure of the human TRIM5α RING domain. (A) Superposition of 20 NMR structures showing the α-helix in red, β-strands in blue, and Zn2+ in green. (B) Ribbon diagram of the NMR structure shown in panel A from the same perspective. The Zn2+ coordinating residues are also shown in green. (C) Electrostatic mapping of the RING domain surface highlighting the positions of positive (blue), negative (red), and neutral (white) charges. Dotted circles indicate the location of the putative E2-binding site for the RING domain of TRIM5α. (D) A Corey-Pauling-Koltun model of the RING domain of human TRIM5α is shown with labels of the visualized residues. Acidic and basic residues are shown in red and blue, respectively. Hydrophobic residues are shown in green.
Table 1.
Summary of conformational constraints and statistics of the final 20 structures of the RING domain of TRIM5α
| Parameter | Value |
|---|---|
| NOE upper distance restraints | |
| Intraresiduea | 345 |
| Medium rangeb | 613 |
| Long rangec | 349 |
| Total | 1,307 |
| Dihedral angle restraints (φ and ψ) | 26 |
| CYANA target function value (Å2) | 0.20 ± 0.01 |
| No. of violations | |
| Distance violations (>0.30 Å) | 0 |
| Dihedral angle violations (>5.0°) | 0 |
| RMSD from averaged coordinates (Å)d | |
| Backbone atoms | 0.70 ± 0.18 |
| Heavy atom | 1.05 ± 0.15 |
| Ramachandran plot (%)d | |
| Residues in most-favored regions | 71.7 |
| Residues in additional allowed regions | 28.2 |
| Residues in generously allowed regions | 0.1 |
| Residues in disallowed regions | 0.0 |
|i − j| = 0.
1 ≤ |i − j| ≤ 4.
|i − j| > 4.
Values calculated for the region encompassing residues 8 to 63.
Mutational analysis of the TRIM5αrh RING surface.
Several studies suggest that RING domains exhibit two functional surfaces (9). One surface is destined to interact with an E2 enzyme, as has been shown for a large number of RING domains (9). The ability of the RING domain (E3) to interact with E2 facilitates the transfer of ubiquitin from E2 to the target substrate, which might be a protein interacting with the E3-containing protein or the E3-containing protein itself (9). In the case of the RING domain of TRIM5αrh, we named this surface the E2-binding region (Fig. 2A). Opposite to the E2-binding region, a second functional surface is destined to either self-associate, as shown for the RING domain of RAG1 (2), or associate with a related RING domain, as in the case of BRCA1-BARD1 interaction (6). We named this surface the RING-RING interaction region of the RING domain of TRIM5αrh because of the structural similarity to the RING-RING interaction region between BRCA-1 and BARD1 (Fig. 2B). We created an extensive panel of mutant proteins that included both the E2-binding and RING-RING interaction regions based on the NMR structure of the RING domain (Table 2). Additionally, we mutagenized residues that were located neither in the E2-binding region nor in the RING-RING interaction region.
Fig. 2.

The putative E2-binding and RING-RING interaction regions of the TRIM5αrh RING domain. The structure of the TRIM5αrh RING domain is based on the human TRIM5α RING domain (PDB 2ECV) and was assembled by using the SWISS-MODEL protein homology modeling program. (A) The putative E2-binding region (magenta) was identified by fitting the model structure to the Cbl-UBCH7 complex structure and by modeling the interaction between the RING domain and UBCH7. (B) The putative RING-RING interaction region (green) was identified in the same way by fitting it to the BRCA1-BARD1 RING structure for the RING domain region. Since conformational prediction of the N-terminal and C-terminal regions was difficult due to poor sequence homology, these regions were not included. Residues that interact with Zn2+ atoms are indicated by black asterisks above the sequence alignment.
Table 2.
Phenotypes of TRIM5α RING variantsa
| TRIM5αrh variant | RING domain surfaceb | Mean restriction potency ± SDc against: |
Mean % TRIM5 self-ubiquitylation ± SDd | Mean binding to HIV-1 CA-NC complexes ± SDe | Mean % reverse transcription ± SDf | Mean % higher-order self-association ± SDg | |
|---|---|---|---|---|---|---|---|
| HIV-1 | EIAV | ||||||
| Wild type | NA | 100.00 | 100.00 | 100 | 1 | 0 | 100 |
| I61E | RING-RING | 101.00 ± 0.24 | 101.44 ± 2.10 | 99.56 ± 3.24 | 1.1 ± 0.03 | 2.63 ± 0.35 | 105.32 ± 3.56 |
| P65E | Other | 100.98 ± 0.02 | 102.66 ± 0.87 | 96.79 ± 4.77 | 0.98 ± 0.08 | 4.95 ± 3.79 | 103.09 ± 5.61 |
| E66R | Other | 101.23 ± 0.45 | 103.55 ± 0.45 | 98.22 ± 9.82 | 0.81 ± 0.20 | 0.33 ± 0.05 | 40.23 ± 3.29 |
| Q64E | RING-RING | 102.20 ± 0.33 | 99.75 ± 1.70 | 97.37 ± 9.34 | 1.02 ± 0.30 | 5.89 ± 1.33 | 99.81 ± 4.77 |
| Q69E | RING-RING | 93.00 ± 3.70 | 99.34 ± 2.30 | 100.32 ± 6.55 | 0.69 ± 0.20 | 0 ± 0.15 | 45.21 ± 7.51 |
| K45E | E2 binding | 91.23 ± 1.73 | 101.34 ± 5.67 | 105.26 ± 4.24 | 0.91 ± 0.32 | 1.96 ± 0.19 | 102.26 ± 2.91 |
| M47D | Other | 89.79 ± 7.38 | 104.45 ± 0.27 | 97.05 ± 5.99 | 1.1 ± 0.05 | 5.67 ± 0.64 | 100.33 ± 3.59 |
| Y49A/K50A | E2 binding | 73.23 ± 6.75 | 22.45 ± 2.34 | 102.41 ± 11.12 | 1.15 ± 0.09 | 2.92 ± 0.45 | 75.32 ± 2.56 |
| K45A | E2 binding | 73.18 ± 2.57 | 50.39 ± 3.70 | 105.26 ± 5.78 | 0.5 ± 0.15 | 8.74 ± 3.05 | 78.36 ± 7.8 |
| P70E | RING-RING | 68.99 ± 5.64 | 95.00 ± 0.56 | 40.33 ± 7.65 | 0.65 ± 0.10 | 0.74 ± 0.15 | 100.19 ± 5.6 |
| A41E | E2 binding | 67.71 ± 3.83 | 49.92 ± 0.98 | 101.23 ± 3.15 | 0.72 ± 0.06 | 1.96 ± 2.49 | 101.25 ± 6.72 |
| Y49A | E2 binding | 63.08 ± 9.79 | 21.12 ± 1.78 | 111.58 ± 9.73 | 0.91 ± 0.35 | 0.41 ± 0.23 | 55.04 ± 9.11 |
| S62E | E2 binding | 62.99 ± 2.84 | 37.86 ± 2.51 | 114.63 ± 5.99 | 0.67 ± 0.07 | 4.5 ± 1.59 | 100.04 ± 3.11 |
| H43A | RING-RING | 58.10 ± 7.21 | 66.52 ± 3.67 | 102.26 ± 4.32 | 0.57 ± 0.14 | 2.98 ± 0.55 | 30.37 ± 4.62 |
| V74E | RING-RING | 54.18 ± 4.50 | 40.00 ± 0.97 | 32.42 ± 2.39 | 0.62 ± 0.15 | 2.84 ± 1.43 | 48.98 ± 7.21 |
| E51A | Other | 53.17 ± 2.60 | 92.70 ± 3.48 | 101.33 ± 8.4 | 0.81 ± 0.11 | 4.62 ± 2.72 | 55 ± 5.71 |
| H29A | RING-RING | 53.04 ± 5.71 | 43.43 ± 1.52 | 74.32 ± 6.39 | 0.94 ± 0.10 | 2.35 ± 0.16 | 97.51 ± 5.17 |
| R54A | Other | 49.11 ± 8.33 | 74.24 ± 1.33 | 99.39 ± 4.51 | 0.89 ± 0.08 | 6.24 ± 0.08 | 70.16 ± 2.57 |
| P57A | E2 binding | 47.48 ± 4.51 | 35.20 ± 4.71 | 27.05 ± 8.22 | 0.54 ± 0.07 | 5.89 ± 0.07 | 101.11 ± 3.18 |
| R72E | RING-RING | 45.50 ± 6.36 | 66.75 ± 2.89 | 45.79 ± 5.99 | 0.4 ± 0.03 | 0.43 ± 0.07 | 51.38 ± 7.15 |
| M47A | Other | 45.08 ± 5.55 | 93.00 ± 3.21 | 105.26 ± 5.11 | 0.71 ± 0.19 | 5.86 ± 0.061 | 56.74 ± 0.85 |
| S55E | Other | 43.35 ± 5.17 | 29.69 ± 1.76 | 49.63 ± 3.65 | 0.67 ± 0.05 | 0 ± 0.09 | 59.73 ± 3.19 |
| N42E | E2 binding | 40.81 ± 9.63 | 30.62 ± 1.44 | 44.26 ± 4.21 | 0.87 ± 0.27 | 1.98 ± 1.74 | 9.32 ± 4.73 |
| I68E | RING-RING | 40.51 ± 3.54 | 59.50 ± 2.59 | 21.02 ± 4.88 | 0.89 ± 0.04 | 1.96 ± 0.66 | 11.3 ± 4.55 |
| S46A | Other | 39.67 ± 3.30 | 83.69 ± 2.33 | 71.25 ± 2.84 | 0.98 ± 0.10 | 3.23 ± 1.19 | 89.56 ± 2.5 |
| K50E | Other | 39.52 ± 3.51 | 31.95 ± 3.72 | 103.47 ± 6.22 | 1.15 ± 0.20 | 15.72 ± 4.93 | 62.11 ± 0.34 |
| R60K | E2 binding | 38.94 ± 9.81 | 22.60 ± 0.79 | 44.74 ± 8.7 | 0.97 ± 0.05 | 14.82 ± 2.87 | 91.46 ± 3.77 |
| L48A | E2 binding | 34.99 ± 4.08 | 48.49 ± 3.21 | 49.13 ± 5.33 | 1.03 ± 0.10 | 6.46 ± 0.11 | 100.09 ± 3.87 |
| N67E | RING-RING | 32.18 ± 5.30 | 58.75 ± 2.19 | 48.69 ± 4.59 | 1.1 ± 0.02 | 19.65 ± 1.12 | 98.51 ± 6.33 |
| I17A/L19A | E2 binding | 29.94 ± 4.16 | 36.50 ± 3.66 | 0 | 0.56 ± 0.20 | 18.73 ± 12.68 | 7.83 ± 1.86 |
| K50A | Other | 29.29 ± 3.23 | 63.09 ± 1.76 | 107.88 ± 9.41 | 0.84 ± 0.18 | 2.98 ± 0.23 | 60.82 ± 4.75 |
| V58A | E2 binding | 26.23 ± 7.39 | 22.74 ± 1.79 | 0 | 1.05 ± 0.25 | 1.96 ± 0.53 | 55.29 ± 7.19 |
| A41E/N42E | E2 binding | 25.54 ± 5.00 | 20.11 ± 1.21 | 49.63 ± 6.16 | 0.94 ± 0.30 | 3.25 ± 1.3 | 90.41 ± 3.11 |
| L21K | E2 binding | 23.54 ± 3.59 | 71.67 ± 3.66 | 14.77 ± 3.98 | 0.95 ± 0.09 | 19.73 ± 0.58 | 10.95 ± 5.29 |
| V58E | E2 binding | 23.13 ± 7.25 | 20.68 ± 1.22 | 0 | 0.65 ± 0.30 | 3.93 ± 2.02 | 5.18 ± 2.77 |
| E20K | E2 binding | 21.31 ± 4.67 | 50.39 ± 1.99 | 2.11 ± 1.45 | 1.02 ± 0.14 | 2.81 ± 0.1 | 30.42 ± 3.69 |
| L48D | E2 binding | 19.21 ± 5.36 | 61.80 ± 2.51 | 31.99 ± 3.45 | 0.89 ± 0.04 | 19.38 ± 0.91 | 0h |
| R60E | E2 binding | 15.65 ± 5.16 | 14.92 ± 1.24 | 0 | 0.96 ± 0.11 | 4.95 ± 2.59 | 90.48 ± 7.92 |
| I17E | E2 binding | 14.71 ± 3.83 | 15.98 ± 0.83 | 0 | 1.1 ± 0.06 | 8.09 ± 2.53 | 88.23 ± 4.93 |
| N71E | RING-RING | 14.70 ± 3.81 | 45.33 ± 3.88 | 23.94 ± 3.11 | 0.32 ± 0.12 | 3.93 ± 0.05 | 5.7 ± 2.11 |
| K44A | E2 binding | 14.03 ± 7.03 | 46.25 ± 2.13 | 102.34 ± 6.71 | 0.62 ± 0.02 | 15.56 ± 0.57 | 97 ± 6.49 |
| E24A | Other | 12.24 ± 2.49 | 31.20 ± 2.45 | 101.45 ± 8.16 | 0.51 ± 0.05 | 5.68 ± 0.55 | 101.25 ± 4.93 |
| L19K | E2 binding | 10.47 ± 4.43 | 35.30 ± 1.66 | 0 | 1.03 ± 0.03 | 14.97 ± 1.35 | 74.23 ± 0.45 |
| R60A | E2 binding | 10.46 ± 2.18 | 33.25 ± 3.11 | 0 | 1.12 ± 0.04 | 4.11 ± 0.53 | 98.26 ± 3.72 |
| H32A | Other | 8.27 ± 1.80 | 15.40 ± 2.61 | 0 | 0.36 ± 0.09 | 1.96 ± 0.05 | 0 |
Shaded variants retain wild-type binding to HIV-1 capsid and higher-order self-association. These variants were used to establish a correlation between TRIM5αrh self-ubiquitylation and anti-HIV-1 activity (see Fig. 7).
Location of each TRIM5αrh variant on the NMR structure of the RING domain. E2 binding means that the residue is located in the E2-binding region of the RING domain. Similarly, RING-RING represents a variant located in the RING-RING interaction region. NA means not applicable. Other means location on a surface different from the E2-binding and RING-RING interaction regions.
Restriction was measured by infecting cells expressing the indicated TRIM5αrh variants with HIV-1 and EIAV expressing GFP. After 48 h, the percentage of GFP-positive cells (infected cells) was determined by flow cytometry. Restriction potency was defined here as the fraction of TRIM5αrh's RING domain variant restriction fold relative to the wild-type's restriction fold when 50% of the control cells are infected. Experiments were performed at least three times; typical results are shown.
To assay the E3-ubiquitin ligase activity of TRIM5αrh RING domain variants, the ability of these TRIM5α variants to undergo self-ubiquitylation was assayed. FLAG-tagged TRIM5α variants from transfected 293T cells semipurified by immunoprecipitation were incubated with E1, E2, myc-tagged ubiquitin, and ATP for 1 h at 37°C. Samples resulting from the incubation were analyzed by Western blotting using antibodies against FLAG and myc for the detection of TRIM5α and ubiquitin, respectively. The amount of ubiquitylated TRIM5α protein was determined by subtracting the amount of nonubiquitylated TRIM5α protein remaining in the reaction mixture that was incubated with E1 and E2 from the TRIM5α protein in the control reaction mixture, which was not incubated with E1 and E2 enzymes. The amount of nonubiquitylated TRIM5α in the reaction mixtures was quantified by using the Quantity-One software from Bio-Rad. TRIM5α self-ubiquitylation was expressed as the percentage of the total TRIM5α variant input protein. Experiments were performed at least three times.
Binding to the HIV-1 capsid complexes was determined for each TRIM5αrh variant as described in Materials and Methods. Binding is expressed as the amount of the variant TRIM5αrh bound to HIV-1 capsid complexes divided by the amount of bound wild-type TRIM5αrh at a similar input level. Experiments were repeated at least three times. Note that because the binding ratios are calculated at input levels at which some binding of the mutant TRIM5αrh protein to the HIV-1 capsid complexes can be detected, these ratios overestimate the relative capsid-binding affinities of the mutant proteins.
HIV-1 reverse transcription was measured by real-time PCR 7 h after infection as described in Materials and Methods. The value shown represents the percentage of late reverse transcripts observed in cells expressing the indicated ΤΡΙΜ5αrh variant relative to the level of late reverse transcripts in control HIV-1-infected cells transduced with the empty LPCX vector. Experiments were performed at least two times.
Each TRIM5αrh variant was assayed for higher-order self-association as described in Materials and Methods. The percentage represents the fraction of the TRIM5αrh variant coprecipitated with itself relative to the coprecipitation of wild-type TRIM5αrh with itself.
A high background level was observed for the TRIM5αrh variant in the control sample without a TRIM5α target protein; the reported values may be less reliable as a result of this background level.
Assay of the E3-ubiquitin ligase activity of TRIM5αrh RING domain variants.
The RING domain of TRIM5αrh presents self-ubiquitylation activity and could use UbcH5b as an E2-conjugating enzyme (13, 27, 37, 43, 68). Self-ubiquitylation assays are widely used as a sensitive indicator of potential E3 activity of RING domain proteins, particularly in vitro, and when the nature of the substrate is not established (69), as in the case of TRIM5αrh. To measure the ability of the TRIM5αrh RING domain variants to undergo RING domain-dependent self-ubiquitylation, we established an in vitro assay using purified, FLAG-tagged TRIM5αrh by immunoprecipitation. Purified RING domain variants and wild-type TRIM5αrh proteins were incubated with recombinant UBE-1 (E1), recombinant UbcH5b (E2), an energy regeneration system, and myc-tagged ubiquitin. TRIM5αrh self-ubiquitylation is observed only when E1 and E2 enzymes are added to the reaction mixture (Fig. 3). The amount of ubiquitylated TRIM5α protein was determined by subtracting the amount of nonubiquitylated TRIM5α protein remaining in the reaction mixture that was incubated with E1 and E2 enzymes from the amount of TRIM5α protein in the control reaction mixture, which was not incubated with E1 and E2 enzymes. The value of nonubiquitylated TRIM5 in the reaction mixtures was quantified by using the Quantity-One software from Bio-Rad. TRIM5α self-ubiquitylation was expressed as the percentage of the total TRIM5α variant input protein (Table 2).
Fig. 3.

E3-ubiquitin ligase activities of TRIM5αrh RING domain variants. Human 293T cells were transfected with plasmids encoding FLAG-tagged mutant and wild-type TRIM5αrh proteins. Forty-eight hours later, the cells expressing each TRIM5αrh variant were lysed in whole-cell extract and immunoprecipitated using anti-FLAG–agarose beads as described in Materials and Methods. Beads containing the immunoprecipitated TRIM5αrh variants were washed and eluted using 200 μg/ml FLAG tripeptide in whole-cell extract buffer as described in Materials and Methods. Samples were supplemented with 5 μM ubiquitin aldehyde, a potent inhibitor of all ubiquitin C-terminal hydrolases, ubiquitin-specific proteases, and deubiquitinating enzymes. Similar amounts of inhibitor-treated samples containing mutant and wild-type TRIM5αrh were incubated with or without 200 nM enzyme E1 (human recombinant UBE1) and 100 nM enzyme E2 (human recombinant UbcH5a) as indicated. Reaction mixtures were supplemented with 200 μM ubiquitin tagged with a myc epitope (human recombinant ubiquitin) and an energy regeneration solution containing MgCl2, ATP, and ATP-regenerating enzymes to recycle hydrolyzed ATP. The reaction mixture was incubated at 37°C for 1 h, and collected fractions were analyzed by Western blotting using HRP-conjugated antibodies against FLAG to detect the levels of TRIM5αrh variants. To detect ubiquitylated forms of TRIM5αrh variants, membranes were blotted using HRP-conjugated antibodies against myc. Purple circles and green circles indicate TRIM5αrh variants with defective self-ubiquitylation activity located on the E2-binding and RING-RING interaction region, respectively. The results of three independent experiments were similar; the result of a single experiment is shown.
On the E2-binding region of the RING domain, we identified residues that when mutated led to a loss of self-ubiquitylation activity: I17, L19, E20, L21, A41/N42, S46, L48, Y49/K50, S55, P57, V58, and R60 (Fig. 3; see Fig. S1 in the supplemental material). Among all of the mutations that affected self-ubiquitylation (Table 2), we found several residues in the E2-binding region that when mutated dramatically affected self-ubiquitylation, such as I17, L19, E20, L21, A41/N42, L48, V58, and R60, as shown by the purple filled circles in Fig. 3. These results were in agreement with those of other studies where mutation of the E2-binding region of the RING domain affected the self-ubiquitylation or ubiquitylation of a specific substrate (23, 42, 59, 71).
Several residues located in the RING-RING interaction region of the RING domain also affected the self-ubiquitylation activity of TRIM5αrh, i.e., N67, I68, Q69, P70, N71, R72, and V74 (Fig. 3; see Fig. S1 in the supplemental material). Interestingly, residues I68 and N71, represented by green filled circles in Fig. 3, were affected the most in their self-ubiquitylation ability. These results suggested that the RING-RING interaction region might also be important for self-ubiquitylation activity, as has been shown for other RING domains (4, 30, 36, 52). Altogether, these results demonstrated that the self-ubiquitylation activity of TRIM5αrh could be eliminated by mutating single residues in the E2-binding or the RING-RING interaction region.
Effect of TRIM5αrh RING changes on HIV-1 capsid binding.
The capacity of TRIM5αrh to bind the HIV-1 capsid is a property essential to its ability to block HIV-1 infection (16, 64). To identify RING domain variants that lost self-ubiquitylation but bind the HIV-1 capsid, we measured the abilities of these mutant proteins to bind in vitro-assembled HIV-1 CA-NC complexes. To measure the binding of the different TRIM5αrh RING domain variants to in vitro-assembled HIV-1 CA-NC complexes, we used our previously described quantitative binding assay (15), which adjusts the input levels of TRIM5α variants to compare capsid-binding abilities more accurately. Of all of the TRIM5αrh RING domain variants that were defective in self-ubiquitylation, those that retained wild-type binding to HIV-1 CA-NC (Fig. 4) were the I17E, L19K, E20K, L21K, A41E/N42E, S46A, L48A, L48D, Y49A/K50A, V58A, R60A, R60K, and R60E mutant proteins. The normal binding of these RING domain variants to capsid implied that the defect in self-ubiquitylation is likely to be a defect in the E2-binding region. The majority of the mutations in the E2-binding regions of different RING domains result in a defect in the recruitment of the corresponding E2 to the RING domain (9). Remarkably, most of the TRIM5αrh RING domain variants that lost self-ubiquitylation but bound HIV-1 CA-NC complexes at wild-type levels were located in the E2-binding region of the RING domain (Fig. 2A). However, a few mutations in the RING-RING interaction region, including H29A, N67E, and I68E, retained wild-type binding to capsid but were deficient in self-ubiquitylation (Fig. 4; see Fig. S2 in the supplemental material).
Fig. 4.

Binding of TRIM5αrh RING mutant proteins to assembled HIV-1 capsids. 293T cells were transfected with plasmids expressing the indicated wild-type and mutant TRIM5αrh proteins tagged with HA epitopes. Thirty-six hours after transfection, cells were lysed. The lysates were incubated at room temperature for 1 h with HIV-1 CA-NC complexes that had been assembled in vitro. The mixtures were applied to a 70% sucrose cushion and centrifuged. INPUT represents the lysates analyzed by Western blotting (WB) before being applied to the 70% cushion. The input mixtures were Western blotted for the HA tag. The pellet from the 70% cushion (PELLET) was analyzed by Western blotting using antibodies against the HA tag and HIV-1 CA-NC protein. The Western blots were quantitated as described in Materials and Methods, and binding values are shown in Table 2. The results of three independent experiments were similar; the result of a single experiment is shown.
Higher-order association of TRIM5αrh RING mutant proteins.
Higher-order self-association is important for the ability of TRIM5αrh to restrict HIV-1 (12, 15, 19, 40). The B-box 2 domain is critical to the formation of higher-order complexes, which have been shown to increase the avidity of TRIM5αrh for the HIV-1 capsid (15, 40); residue R121 in the B-box 2 domain of TRIM5αrh is essential for the ability of TRIM5αrh to form hexagonal structures on the surface of the HIV-1 capsid (12, 15, 19). Even though the B-box domain is in close contact with the RING domain, the role of the RING domain in higher-order self-association has not been tested. To more stringently analyze our mutant proteins that are deficient in self-ubiquitylation but bind the HIV-1 capsid, we measured their capacity to undergo higher-order self-association, an important property of TRIM5αrh required for potent restriction of HIV-1 (12, 15, 40). We tested higher-order self-association in the mutant proteins that lost their self-ubiquitylation activity but retained binding to the HIV-1 capsid (Fig. 5). To test the abilities of the RING mutant proteins to form higher-order complexes, we mixed cell lysates containing FLAG-tagged RING domain variants with lysates containing the same RING domain variant tagged with HA. After precipitation with an anti-FLAG antibody, the precipitates were Western blotted with antibodies directed against the FLAG and HA epitope tags. The wild-type TRIM5αrh-HA protein was efficiently coprecipitated with TRIM5αrh-FLAG using anti-FLAG antibodies (Fig. 5), consistent with the ability of wild-type TRIM5αrh to self-associate in higher-order complexes (15, 40). After studying the higher-order self-association capabilities of the RING domain mutant proteins that were defective in self-ubiquitylation but bind the HIV-1 capsid at wild-type levels, we narrowed our study down to the following 9 RING domain variants (Fig. 5 and Table 2): I17E, H29A, A41E/E42E, S46A, L48A, R60A, R60K, R60E, and N67E. These 9 RING domain variants showed wild-type capsid binding and higher-order self-association, but they were defective in self-ubiquitylation.
Fig. 5.

Higher-order self-association of TRIM5αrh RING mutant proteins. 293T cells were transfected with plasmids expressing the indicated wild-type or mutant TRIM5α proteins with a FLAG or an HA epitope tag. Cells expressing wild-type and mutant TRIM5αrh proteins were lysed 48 h after transfection. The cell lysates containing similar inputs were mixed, and the indicated mixtures were used for immunoprecipitation (IP) with an antibody directed against the FLAG epitope, as described in Materials and Methods. Elution of the immunocomplexes was performed with a FLAG tripeptide and analyzed by Western blotting (WB) using anti-HA and anti-FLAG antibodies. The results of three independent experiments were similar; the result of a single experiment is shown.
Several TRIM5αrh RING domain variants lost the ability to form higher-order complexes (Fig. 5 and Table 2; see Fig. S3 in the supplemental material). These results demonstrated that, besides the B-box 2 domain (15), an intact RING domain is also necessary for higher-order self-association. Apparently, the ability of TRIM5αrh to form higher-order complexes is sensitive to changes in different surface residues of the B-box 2 and RING domains.
Next we analyzed the abilities of this selected group of mutant proteins, which exhibit deficient self-ubiquitylation but normal capsid binding and higher-order self-association, to block HIV-1 infection.
Retroviral restriction by TRIM5αrh RING domain variants.
To examine the abilities of TRIM5αrh RING mutant proteins to inhibit retroviral infection, dog Cf2Th cells stably expressing these mutant proteins (see Fig. S4 in the supplemental material) were challenged with recombinant HIV and equine infectious anemia virus expressing GFP as a reporter abbreviated here as HIV-1–GFP and EIAV-GFP, respectively (Fig. 6; see Fig. S5 in the supplemental material) (47, 55). The TRIM5αrh RING variants exhibited a range of HIV-1-restricting abilities and are rank ordered in Table 2 according to restriction potency—defined here as the fraction of restriction by the mutant relative to the wild-type TRIM5αrh restriction when 50% of the control cells are infected.
Fig. 6.

Restriction of HIV-1 and EIAV infection by TRIM5αrh mutant proteins. Cf2Th cells were transduced with the LPCX vector expressing HA-tagged wild-type and mutant TRIM5αrh proteins. Stable cell lines were selected with 5 μg/ml puromycin, and the expression levels of mutant and wild-type TRIM5αrh proteins were assayed by Western blotting using HRP-conjugated antibodies against HA (see Fig. S4 in the supplemental material). The cells were challenged with different amounts of HIV-1–GFP (A, B) or EIAV-GFP (C, D). The percentage of GFP-positive cells was measured 48 h later by FACS. The results of three independent experiments were similar; the results of a single experiment are shown.
Infection of cells stably expressing the different RING domain variants by HIV-1–GFP and EIAV-GFP identified residues essential for restriction. Changing arginine 60 in the RING domain of TRIM5αrh to lysine, glutamic acid, or alanine drastically reduced the potency of TRIM5αrh against HIV-1 by 5- to 10-fold (Fig. 6A). Similarly, the I17E, A41E/N42E, N67E, and L48A variants showed reduced potency against HIV-1 (Fig. 6B). Remarkably, all of these residues were located in the E2-binding region of the RING domain, with the exception of N67E, which is located in the RING-RING interaction region. When cells expressing these RING domain variants were challenged with EIAV, we observed a consistent decrease in restriction (Fig. 6C and D). These results suggested a role for ubiquitylation in HIV-1 restriction by TRIM5αrh.
TRIM5αrh self-ubiquitylation correlates with HIV-1 restriction.
Using this selected group of variants that exhibit deficient self-ubiquitylation but normal capsid binding and higher-order self-association (shaded residues in Table 2), we tested the hypothesis that TRIM5α self-ubiquitylation correlates with restriction. To do so, we graphically represented HIV-1 restriction versus self-ubiquitylation for this selected group of variants. Remarkably, a strong correlation (rs = 0.9090, P < 0.001) between TRIM5α self-ubiquitylation and HIV-1 restriction by this panel of mutant proteins was observed (Fig. 7). These results support the hypothesis that the E3-ligase activity of the RING domain represents a major contribution of the RING domain to HIV-1 restriction by TRIM5α.
Fig. 7.
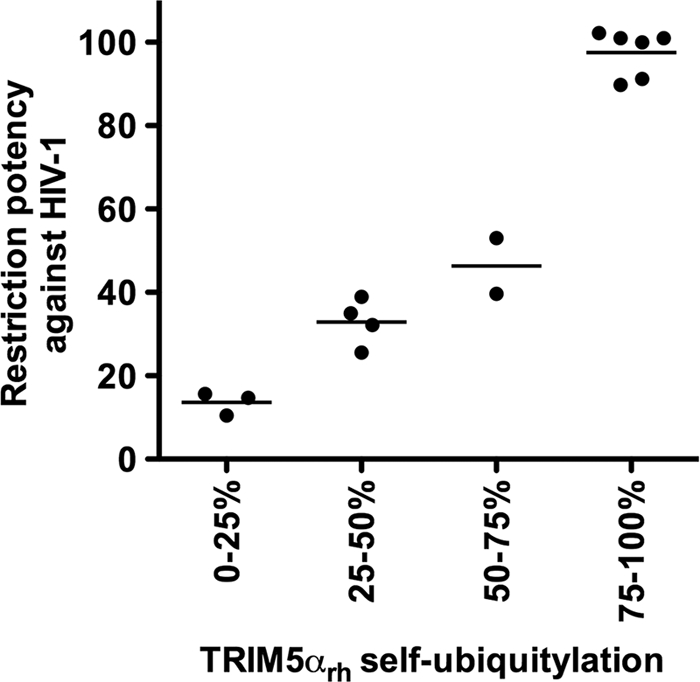
TRIM5α self-ubiquitylation activity correlates with anti-HIV-1 activity. The abilities of RING domain variants to self-ubiquitylate (TRIM5α self-ubiquitylation) and restrict HIV-1 were assessed as described in the footnotes to Table 2 and in Materials and Methods. TRIM5α RING domain variants that were not defective in binding to the HIV-1 capsid and higher-order self-association were analyzed. The Spearman rank correlation coefficient, rs, is 0.9090, with a 95% confidence interval of 0.8623 to 0.9847 (two-sided P value of < 0.0001).
Inhibition of HIV-1 reverse transcription by TRIM5αrh mutant proteins.
HIV-1 restriction by TRIM5αrh occurs prior to the initiation of reverse transcription (28, 63). However, the use of proteasome inhibitors during restriction allows the occurrence of reverse transcription without affecting the blockage of infection (1, 64, 67). To examine the ability of the TRIM5αrh RING domain variants to block HIV-1 reverse transcription, we assayed the level of late reverse transcripts in mutant-expressing cells challenged with HIV-1 (Fig. 8; see Fig. S6 in the supplemental material). HIV-1 reverse transcript levels were low in cells expressing potently restricting TRIM5αrh RING domain variants (Fig. 8 and Table 2). In contrast, in cells expressing RING domain variants that did not restrict HIV-1 infection potently, the levels of HIV-1 reverse transcripts were higher (Fig. 8 and Table 2).
Fig. 8.

Blockade of HIV-1 reverse transcription by TRIM5αrh RING mutant proteins. Cf2Th cells expressing the indicated wild-type and mutant TRIM5αrh proteins or containing the empty LPCX vector were challenged at an MOI of 0.4 with DNase-pretreated HIV-1–GFP. After 7 h, cells were lysed and total DNA was extracted. The levels of viral DNA were measured by quantitative real-time PCR using a probe against GFP as described in Materials and Methods. Similar results were obtained in three independent experiments.
Half-lives of TRIM5αrh RING domain variants.
We measured the half-lives of the TRIM5αrh RING domain variants that lost self-ubiquitylation but preserved capsid binding and higher-order self-association (HOSA) (see Fig. S7 in the supplemental material), as previously described (12, 15). Some of the RING domain variants exhibited a longer half-life than wild-type TRIM5αrh, which is ∼50 min (13). For example, the A41E/N42E, N67E, and S46A variants exhibited a half-life slightly longer than the wild-type half-life of ∼70 min; the I17E variant exhibited a half-life of ∼105 min. Other variants, such as the R60E variant, exhibit a half-life similar to that of the wild-type protein of ∼55 min. Interestingly, the R60A and R60K variants exhibited half-lives shorter than that of the wild-type protein at ∼15 min and ∼45 min, respectively. These results did not correlate self-ubiquitylation with degradation since we found mutant proteins that did not self-ubiquitylate but have shorter half-lives than wild-type TRIM5αrh. These results are in agreement with previous observations suggesting that the degradation of TRIM5αrh is modestly affected by the use of proteasome inhibitors in the absence of restricted viruses (12).
DISCUSSION
The RING domain of TRIM5α exhibits E3-ubiquitin ligase activity, but the contribution of this activity to the restriction of HIV-1 is not understood. Here we present the structure of the RING domain of human TRIM5α and use this information to direct a mutational analysis of the functional surfaces of the RING domain of TRIM5αrh. To explore the role of the E3-ubiquitin ligase in restriction, we correlated the E3-ubiquitin ligase activity of TRIM5α with the different properties of the restriction factor TRIM5α, including HIV-1 restriction, binding to the HIV-1 capsid, inhibition of reverse transcription, and the ability to form higher-order complexes. We found a distinct set of TRIM5α variants located on the E2-binding surface of the RING domain, where the loss of E3-ubiquitin ligase activity correlated with a defect in HIV-1 restriction ability. Our results demonstrate that E3-ubiquitin ligase activity has a role in HIV-1 restriction by TRIM5α, as has been previously suggested by others (1, 50, 54, 64, 67).
The RING domain of TRIM5α adopts a ββα RING fold containing shorter β-strands and a longer α-helix than the typical fold observed in RING domains. Comparison of the RING domain of TRIM5α with other RING domains revealed that this structure has two regions that are potentially important for RING domain function (Fig. 2). Similar to the RING domains of Cbl, CHIP, and cIAP2 (9), the RING domain of TRIM5α presents a distinct E2-binding region composed of similar amino acids (Fig. 2A). Opposite to the E2-binding region is the RING-RING interaction region, which is similar to the interaction region that allows BRCA1 and BARD1 RING domains to form a heterodimer (6) (Fig. 2B). The construct used to solve the NMR structure of the RING domain of TRIM5α did not include the last 10 amino acids; these residues are part of the association helix used by BRCA1 and BARD1 RING domains to heterodimerize. Longer constructs of the TRIM5α RING domain resulted in a poor HSQC spectrum.
The E2-binding region of RING domains is essential for docking of the E2 protein and allows the transfer of ubiquitin from E2 to the target protein. In order to disrupt this docking site in the RING domain of TRIM5α, we generated a series of mutations on the different surfaces of the RING domain (Table 2); these variants were tested for the different properties of the restriction factor TRIM5α. These experiments revealed that TRIM5α self-ubiquitylation requires an intact E2-binding region, which suggested that an intact E2 docking site in the RING domain is required for the self-ubiquitylation property of TRIM5α. Mutations in all of the residues of the E2-binding site of the RING domain affected self-ubiquitylation to a certain extent; in some cases, complete loss of self-ubiquitylation was observed. Mutations in the E2-binding region that resulted in a partial effect on self-ubiquitylation could be explained by the existence of complementation by a different RING domain. Hetero-oligomerization with a related RING domain could rescue ubiquitylation activity in a defective RING domain, as has been shown for ubiquitylation-deficient mutant proteins of the Mdm2 RING domain that can be rescued by hetero-oligomerization with the RING domain of MdmX (60).
To exclude RING mutations that had effects on other properties that are important for HIV-1 restriction by TRIM5α, we quantitatively measure the binding of these variants to the HIV-1 capsid, as previously shown (15). Similarly, we also measured the ability of the RING variants to undergo higher-order self-association, which is also required for potent restriction of HIV-1. Remarkably, TRIM5α self-ubiquitylation activity correlates with restriction activity on mutant proteins where binding to the HIV-1 capsid and higher-order self-association were not affected. This correlation supports the hypothesis that the E3-ubiquitin ligase activity of the RING domain is required for potent restriction of HIV-1 by TRIM5α.
Several observations have linked the restriction of HIV-1 by TRIM5α with the proteasome. The observation that proteasomal inhibitors allow the occurrence of reverse transcription without affecting restriction suggests that TRIM5α blocks restriction before and after reverse transcription (1, 64, 67). The use of proteasome inhibitors in the fate of the capsid assay showed an increase in particulate capsid during infection in the presence of TRIM5α, which also suggests a role for the proteasome in restriction and uncoating (11, 14). The Aiken laboratory has demonstrated that TRIM5α is degraded in a proteasome-dependent manner in the presence of a restricted capsid, which links the proteasome with the HIV-1 restriction by TRIM5α (54). The present work attempted to connect ubiquitylation, a process preceding proteasomal degradation, with the ability of TRIM5α to block HIV-1. Similar to what we observed for a panel of B-box 2 mutant proteins (15), the levels of HIV-1 late reverse transcripts for this panel of RING mutant proteins inversely correlated with the degree of restriction.
Mutations in the RING-RING interaction region that removed the ability to undergo self-ubiquitylation might cause a defect in RING oligomerization, which is different from affecting the docking site of the E2 enzyme. Several mutations in the RING-RING interaction region also affected the self-ubiquitylation activity of TRIM5α without affecting folding measured by HIV-1 capsid binding and higher-order self-association. This is in agreement with findings suggesting that RING domain oligomerization enhances ubiquitylation (30, 31, 52). Loss of RING domain oligomerization could account for the partial defect in self-ubiquitylation observed in some of the RING-RING interaction region variants. In some cases, loss of RING dimerization could result in complete loss of E3-ubiquitin ligase activity, as has been shown for the RING domain of RNF4 (41). Even though concentration dependence experiments to test RING domain dimerization failed to prove homodimerization (data not shown), these experiments did not exclude the possibility that the RING domain hetero-oligomerizes with a different RING domain, as shown for BRCA-1 and BARD (6).
Our results demonstrated that potent restriction of HIV-1 by TRIM5αrh requires intact self-ubiquitylation activity. One could conceive a model in which the self-ubiquitylation of TRIM5α is required to remove TRIM5α when it is forming hexagonal structures on the surface of the capsid (19, 54); removal of TRIM5α from the surface of the capsid will allow a decrease on the amount of particulate capsid during infection, assisting a rapid uncoating process (10). Further analysis destined to understand the nature of the endogenous E2 enzyme and the ubiquitylation substrate of TRIM5α will give new mechanistic insights into restriction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Steve Porcelli for critical reading of the manuscript. We thank Joe Sodroski for the initial support of this project. We thank Takashi Umehara for analytical ultracentrifugation measurements and Xu-rong Qin for concentration dependency measurements in 1H-15N-HSQC experiments. We also thank Satoru Watanabe, Takushi Harada, Takeshi Nagira, Yasuko Tomo, Masaomi Ikari, Kazuharu Hanada, Yukiko Fujikura, and Akiko Tanaka for sample preparation and help with the screening data of the human TRIM5 RING domain.
The work of the structure determination was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) of the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan. This work has also been supported by a K99/R00 Pathway to Independence Award to F.D.-G. (4R00MH086162-02) and grant R01AI7930231 from the National Institutes of Health, an American Foundation for AIDS Research Mathilde Krim fellowship phase II in basic biomedical research (amfAR research grant 107787-47-RKHF), and a Claudia Adams Barr award from the Dana-Farber Cancer Institute to F.D.-G.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 6 July 2011.
REFERENCES
- 1. Anderson J. L., et al. 2006. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 80:9754–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellon S. F., Rodgers K. K., Schatz D. G., Coleman J. E., Steitz T. A. 1997. Crystal structure of the RAG1 dimerization domain reveals multiple zinc-binding motifs including a novel zinc binuclear cluster. Nat. Struct. Biol. 4:586–591 [DOI] [PubMed] [Google Scholar]
- 3. Best S., Le Tissier P., Towers G., Stoye J. P. 1996. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382:826–829 [DOI] [PubMed] [Google Scholar]
- 4. Borden K. L. 2000. RING domains: master builders of molecular scaffolds? J. Mol. Biol. 295:1103–1112 [DOI] [PubMed] [Google Scholar]
- 5. Borden K. L. 1998. RING fingers and B-boxes: zinc-binding protein-protein interaction domains. Biochem. Cell Biol. 76:351–358 [DOI] [PubMed] [Google Scholar]
- 6. Brzovic P. S., Rajagopal P., Hoyt D. W., King M. C., Klevit R. E. 2001. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 8:833–837 [DOI] [PubMed] [Google Scholar]
- 7. Cornilescu G., Delaglio F., Bax A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13:289–302 [DOI] [PubMed] [Google Scholar]
- 8. Delaglio F., et al. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6:277–293 [DOI] [PubMed] [Google Scholar]
- 9. Deshaies R. J., Joazeiro C. A. 2009. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78:399–434 [DOI] [PubMed] [Google Scholar]
- 10. Diaz-Griffero F. 2011. Caging the beast: TRIM5α binding to the HIV-1 core. Viruses 3:423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diaz-Griffero F., et al. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology 369:400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz-Griffero F., et al. 2007. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5alpha B-box 2 domain. J. Virol. 81:10362–10378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diaz-Griffero F., et al. 2006. Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 349:300–315 [DOI] [PubMed] [Google Scholar]
- 14. Diaz-Griffero F., et al. 2008. A human TRIM5alpha B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology 378:233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diaz-Griffero F., et al. 2009. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 83:10737–10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz-Griffero F., et al. 2006. Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351:404–419 [DOI] [PubMed] [Google Scholar]
- 17. Freemont P. S., Hanson I. M., Trowsdale J. 1991. A novel cysteine-rich sequence motif. Cell 64:483–484 [DOI] [PubMed] [Google Scholar]
- 18. Ganser B. K., Li S., Klishko V. Y., Finch J. T., Sundquist W. I. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83 [DOI] [PubMed] [Google Scholar]
- 19. Ganser-Pornillos B. K., et al. 2011. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. U. S. A. 108:534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ganser-Pornillos B. K., von Schwedler U. K., Stray K. M., Aiken C., Sundquist W. I. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hennig J., et al. 2008. The fellowship of the RING: the RING-B-box linker region interacts with the RING in TRIM21/Ro52, contains a native autoantigenic epitope in Sjogren syndrome, and is an integral and conserved region in TRIM proteins. J. Mol. Biol. 377:431–449 [DOI] [PubMed] [Google Scholar]
- 22. Herrmann T., Guntert P., Wuthrich K. 2002. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24:171–189 [DOI] [PubMed] [Google Scholar]
- 23. Huang A., et al. 2009. E2-c-Cbl recognition is necessary but not sufficient for ubiquitination activity. J. Mol. Biol. 385:507–519 [DOI] [PubMed] [Google Scholar]
- 24. Ishikawa H., Tachikawa H., Miura Y., Takahashi N. 2006. TRIM11 binds to and destabilizes a key component of the activator-mediated cofactor complex (ARC105) through the ubiquitin-proteasome system. FEBS Lett. 580:4784–4792 [DOI] [PubMed] [Google Scholar]
- 25. Javanbakht H., Diaz-Griffero F., Stremlau M., Si Z., Sodroski J. 2005. The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5alpha. J. Biol. Chem. 280:26933–26940 [DOI] [PubMed] [Google Scholar]
- 26. Johnson B. A. 2004. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278:313–352 [DOI] [PubMed] [Google Scholar]
- 27. Kar A. K., Diaz-Griffero F., Li Y., Li X., Sodroski J. 2008. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J. Virol. 82:11669–11681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keckesova Z., Ylinen L. M., Towers G. J. 2004. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. U. S. A. 101:10780–10785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kentsis A., Borden K. L. 2004. Physical mechanisms and biological significance of supramolecular protein self-assembly. Curr. Protein Pept. Sci. 5:125–134 [DOI] [PubMed] [Google Scholar]
- 30. Kentsis A., Gordon R. E., Borden K. L. 2002. Control of biochemical reactions through supramolecular RING domain self-assembly. Proc. Natl. Acad. Sci. U. S. A. 99:15404–15409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kentsis A., Gordon R. E., Borden K. L. 2002. Self-assembly properties of a model RING domain. Proc. Natl. Acad. Sci. U. S. A. 99:667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kigawa T., et al. 1999. Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett. 442:15–19 [DOI] [PubMed] [Google Scholar]
- 33. Kirmaier A., et al. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kobayashi N., et al. 2007. KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J. Biomol. NMR 39:31–52 [DOI] [PubMed] [Google Scholar]
- 35. Koradi R., Billeter M., Wuthrich K. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55 [DOI] [PubMed] [Google Scholar]
- 36. Kostic M., Matt T., Martinez-Yamout M. A., Dyson H. J., Wright P. E. 2006. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J. Mol. Biol. 363:433–450 [DOI] [PubMed] [Google Scholar]
- 37. Langelier C. R., et al. 2008. Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 82:11682–11694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8:477–486 [DOI] [PubMed] [Google Scholar]
- 39. Li H., et al. 2008. Structure of the C-terminal phosphotyrosine interaction domain of Fe65L1 complexed with the cytoplasmic tail of amyloid precursor protein reveals a novel peptide binding mode. J. Biol. Chem. 283:27165–27178 [DOI] [PubMed] [Google Scholar]
- 40. Li X., Sodroski J. 2008. The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 82:11495–11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liew C. W., Sun H., Hunter T., Day C. L. 2010. Ring domain dimerization is essential for RNF4 function. Biochem. J. 431:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mace P. D., et al. 2008. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 283:31633–31640 [DOI] [PubMed] [Google Scholar]
- 43. Maegawa H., Miyamoto T., Sakuragi J., Shioda T., Nakayama E. E. 2010. Contribution of RING domain to retrovirus restriction by TRIM5alpha depends on combination of host and virus. Virology 399:212–220 [DOI] [PubMed] [Google Scholar]
- 44. Mrosek M., et al. 2008. Structural analysis of B-Box 2 from MuRF1: identification of a novel self-association pattern in a RING-like fold. Biochemistry 47:10722–10730 [DOI] [PubMed] [Google Scholar]
- 45. Nakayama E. E., Miyoshi H., Nagai Y., Shioda T. 2005. A specific region of 37 amino acid residues in the SPRY (B30.2) domain of African green monkey TRIM5alpha determines species-specific restriction of simian immunodeficiency virus SIVmac infection. J. Virol. 79:8870–8877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niikura T., et al. 2003. A tripartite motif protein TRIM11 binds and destabilizes Humanin, a neuroprotective peptide against Alzheimer's disease-relevant insults. Eur. J. Neurosci. 17:1150–1158 [DOI] [PubMed] [Google Scholar]
- 47. Olsen J. C. 1998. Gene transfer vectors derived from equine infectious anemia virus. Gene Ther. 5:1481–1487 [DOI] [PubMed] [Google Scholar]
- 48. Owens C. M., Yang P. C., Gottlinger H., Sodroski J. 2003. Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J. Virol. 77:726–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Perez-Caballero D., Hatziioannou T., Yang A., Cowan S., Bieniasz P. D. 2005. Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J. Virol. 79:8969–8978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perez-Caballero D., Hatziioannou T., Zhang F., Cowan S., Bieniasz P. D. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 79:15567–15572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perron M. J., et al. 2007. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 81:2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poyurovsky M. V., et al. 2007. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 26:90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reymond A., et al. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20:2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rold C. J., Aiken C. 2008. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 4:e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saenz D. T., Teo W., Olsen J. C., Poeschla E. M. 2005. Restriction of feline immunodeficiency virus by Ref1, Lv1, and primate TRIM5alpha proteins. J. Virol. 79:15175–15188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sawyer S. L., Wu L. I., Emerman M., Malik H. S. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. U. S. A. 102:2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sayah D. M., Sokolskaja E., Berthoux L., Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573 [DOI] [PubMed] [Google Scholar]
- 58. Sebastian S., Luban J. 2005. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shloush J., et al. 2011. Structural and functional comparison of the RING domains of two p53 E3 ligases, Mdm2 and Pirh2. J. Biol. Chem. 286:4796–4808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Singh R. K., Iyappan S., Scheffner M. 2007. Hetero-oligomerization with MdmX rescues the ubiquitin/Nedd8 ligase activity of RING finger mutants of Mdm2. J. Biol. Chem. 282:10901–10907 [DOI] [PubMed] [Google Scholar]
- 61. Song B., et al. 2005. TRIM5alpha association with cytoplasmic bodies is not required for antiretroviral activity. Virology 343:201–211 [DOI] [PubMed] [Google Scholar]
- 62. Song B., et al. 2005. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J. Virol. 79:6111–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stremlau M., et al. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 64. Stremlau M., et al. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stremlau M., Perron M., Welikala S., Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J. Virol. 79:3139–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Toniato E., et al. 2002. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 277:37315–37322 [DOI] [PubMed] [Google Scholar]
- 67. Wu X., Anderson J. L., Campbell E. M., Joseph A. M., Hope T. J. 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 103:7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamauchi K., Wada K., Tanji K., Tanaka M., Kamitani T. 2008. Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 275:1540–1555 [DOI] [PubMed] [Google Scholar]
- 69. Yang Y., Lorick K. L., Jensen J. P., Weissman A. M. 2005. Expression and evaluation of RING finger proteins. Methods Enzymol. 398:103–112 [DOI] [PubMed] [Google Scholar]
- 70. Yap M. W., Nisole S., Stoye J. P. 2005. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 15:73–78 [DOI] [PubMed] [Google Scholar]
- 71. Yin Q., Lamothe B., Darnay B. G., Wu H. 2009. Structural basis for the lack of E2 interaction in the RING domain of TRAF2. Biochemistry 48:10558–10567 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.