

Abstract
The cellular ALIX protein functions within the ESCRT pathway to facilitate intralumenal endosomal vesicle formation, the abscission stage of cytokinesis, and enveloped virus budding. Here, we report that the C-terminal proline-rich region (PRR) of ALIX folds back against the upstream domains and auto-inhibits V domain binding to viral late domains. Mutations designed to destabilize the closed conformation of the V domain opened the V domain, increased ALIX membrane association, and enhanced virus budding. These observations support a model in which ALIX activation requires dissociation of the autoinhibitory PRR and opening of the V domain arms.
TEXT
Retroviral Gag polyproteins contain short sequence motifs, termed “late domains,” that facilitate virus budding by recruiting components of the cellular ESCRT pathway (4, 38). For example, the HIV-1 p6Gag protein contains “PTAP” and “YPXL” late domains (designated by their consensus sequences), that bind directly to the TSG101 and ALIX proteins, respectively (6, 9, 19, 33, 39). ALIX, in turn, binds the CHMP4 subunits of the ESCRT-III complex, resulting in recruitment of the VPS4 ATPase, membrane fission, and virus release (10, 27).
ALIX contains three distinct structural elements: an N-terminal Bro1 domain, a central V domain, and a C-terminal proline-rich region (PRR). The boomerang-shaped Bro1 domain binds CHMP4 proteins (7, 13, 20), the V domain comprises two extended three-helix bundles and binds YPXL late domains (7, 16, 44, 45), and the PRR binds a series of other proteins but is predicted to lack a persistent secondary or tertiary structure (7, 8, 24). Like other ESCRT factors, ALIX must cycle between soluble (inactive) and membrane-associated (active) states. Several lines of evidence suggest that conformational changes accompany (or induce) these transitions. First, recombinant ALIX proteins can form stable monomers and dimers (7, 23), and biochemical evidence suggests that the dimer is the active conformation (5, 7, 23, 29). Second, small-angle X-ray scattering (SAXS) profiles indicate that the two arms of the V domain may open and associate in an antiparallel fashion when the protein dimerizes (29). Third, recent reports show that the PRR can inhibit ALIX binding to conformationally sensitive monoclonal antibodies, CHMP4 proteins, and viral Gag proteins (46–48). However, previous studies have not characterized the structure or conformational transitions of pure, full-length ALIX, because this protein has not been available.
Although we were unable to express full-length human ALIX protein in Escherichia coli, we could produce multimilligram quantities of pure recombinant ALIX in insect cells using a baculoviral expression system. Briefly, 2 liters of SF21 cells were infected with a BaculoDirect (Invitrogen) expression vector, which encoded His6-ALIX (ALIX residues 1 to 868, WISP10-643). The cells were lysed by sonication 48 h postinfection (300 mM NaCl, 10 mM imidazole, 5% [vol/vol] glycerol, 1% [vol/vol] Triton X-100, 50 mM Tris [pH 8.0]). ALIX was purified from the clarified lysate by Ni2+ chromatography (Qiagen; elution with 250 mM imidazole), anion-exchange chromatography (0.025 to 1.0 M NaCl gradient, 25 mM Tris [pH 8.8] [Q Sepharose; GE Healthcare]), and size exclusion chromatography (monomeric species collected [Superdex 200; GE Healthcare]) (Fig. 1A). This procedure typically yielded 5 mg of pure monomeric ALIX, and the protein identity was confirmed by electrospray ionization mass spectrometry. ALIX constructs that lacked the PRR (ALIX residues 1 to 698, denoted ALIXBro1-V [WISP10-648] and ALIXBro1-V,R649E [WISP11-296]) were expressed and purified in E. coli as described previously (7).
Fig. 1.

Small-angle X-ray scattering analyses of recombinant ALIX. (A) SDS-PAGE analysis (Coomassie blue staining) showing the stepwise expression and purification of full-length human His6-ALIX protein following baculoviral expression in SF21 insect cells (lane 1), nickel affinity chromatography (lane 2), anion-exchange chromatography (lane 3), and gel filtration chromatography (lane 4). (B) Log I(q) versus q solution small-angle X-ray scattering profile for ALIX (circles) and ALIXBro1-VR649E (gray diamonds). Fits to the ALIX scattering data are shown in red (models depicted in panels D and E) or blue (model depicted in F). For clarity, the data have been offset on the I(q) axis. (C) Probable atom-pair distance distributions [P(r) versus r] for ALIX (open circles) and the crystal structure of ALIXBro1-V (black squares) and ALIXBro1-V,R649E (gray diamonds). The crystal structure P(r) was calculated as for the experimental data except that the intensity profile was generated using CRYSOL26 (34) and the coordinates of ALIXBro1-V (7). The areas under the P(r) curves are proportional to I(0) and correctly scaled according to the ratios of the square of the molecular masses of the proteins. (D and E) ALIX models that fit the scattering data (red lines in panel B). The Bro1 domain is shown in light blue, the two arms of the V domain are shown in green and blue, respectively, and PRR dummy atoms are shown in magenta. (F) A model in which the PRR projects into solution does not fit the SAXS data (blue line in panel B).
SAXS experiments were performed to examine the conformation of the C-terminal PRR. These measurements were performed using an Anton Paar SAXSess line-collimation instrument as described previously (11). Analysis of the forward scattering intensity, I(0) (26), gave a solution molecular mass for ALIX (102.5 kDa) (see Table S1 in the supplemental material) that agreed well with the calculated monomeric mass (100.8 kDa), indicating that the protein was monodisperse and monomeric. The log I(q) versus q SAXS profile for ALIX (open circles) is shown in Fig. 1B. Indirect Fourier transformation of the SAXS profile using the program GIFT (3) yielded the probable atom-pair distance distribution within ALIX [P(r) versus r plot] (Fig. 1C), the radius of gyration (Rg), 44.8 ± 1.5Å, and the maximum linear dimension (Dmax), 155 ± 5 Å. These structural parameters are very similar to the Rg (45.3 Å) and Dmax (158 Å) values calculated from the P(r) profile derived from the ALIXBro1-V crystal structure (Fig. 1C, black squares). The overall shapes of the experimentally derived and crystal structure-based P(r) curves are also similar, indicating that the mass of the PRR must pack near the center of mass of the Bro1-V core structure. In contrast, a recent SAXS analysis of an ALIX construct that lacked the Bro1 domain and the final 108 PRR residues indicated that in this context the truncated PRR projected away from the V domain (32). These observations support a role for the Bro1 domain and/or the C-terminal two-thirds of the PRR in mediating the fold-back structure.
Atomic models for ALIX were generated using the ALIXBro1-V crystal structure (7) and dummy atom representations of PRR residues. The ALIXBro1-V structure was fixed as a single rigid body and the PRR positioned by refinement against the SAXS data using the program BUNCH (28). In the absence of any distance constraints, the PRR consistently refined toward the second arm of the V domain (Fig. 1D; red line fit in Fig. 1B) (χ2 of 0.54, calculated using CRYSOL26; 34) (see Fig. S1 in the supplemental material). Models in which the PRR center was constrained to be within 5 Å of the center of the Bro1 domain fit the scattering data equally well (Fig. 1E; red line fit in Fig. 1B) (χ2 of 0.54). In contrast, models in which the PRR was rotated away from the V domain and into solution did not fit the experimental data (Fig. 1F; blue line fit in Fig. 1B) (χ2 of 4.70). Thus, the SAXS data indicate that the ALIX PRR lies close to the Bro1-V core in solution, probably interacting with arm2 of the V domain and possibly also interacting with the Bro1 domain.
Isothermal titration calorimetry was performed to test whether the PRR influenced ALIX binding to a high-affinity late-domain peptide from equine infectious anemia virus (EIAV) p9Gag (19TQNLYPDLSEIKK31; bold residues contact arm2 of the ALIX V domain) (44). As expected, the control ALIXBro1-V protein bound the EIAV p9Gag peptide with an equilibrium dissociation constant (KD) of 3.6 μM (Fig. 2, black squares), which matches our previous biosensor-based analyses (KD of 6 μM; see reference 44). In contrast, full-length ALIX did not bind detectably to the EIAV p9Gag peptide under these conditions (open circles). Similarly, the EIAV p9Gag peptide bound approximately 100-fold less tightly to ALIX than to ALIXBro1-V in biosensor binding experiments (data not shown). In contrast, the two ALIX constructs bound with similar affinities to a peptide that corresponded to the binding epitope on CHMP4B (205KKKEEEDDDMKELENWAGSM224; bold residues contact the ALIX Bro1 domain [see reference 20]) (ALIX KD was 112 ± 57 μM, and ALIXBro1-V KD was 75 ± 24 μM [n = 6]). In this respect, our results differ from those of Zhou et al., who observed that the PRR also inhibited CHMP4 binding (48). However, our experiments were performed using pure monomeric ALIX and CHMP4B peptides, whereas theirs were performed with full-length proteins in crude extracts and could have been influenced by additional factors, such as avidity effects resulting from protein oligomerization. Our data indicate that the PRR does not significantly alter the intrinsic affinity of the CHMP4 binding site on the Bro1 domain but strongly inhibits YPXL late-domain binding to arm2 of the V domain.
Fig. 2.

ALIX protein binding to an EIAV p9Gag late-domain peptide. Isothermal calorimetry titrations of an EIAV p9Gag peptide (19TQNLYPDLSEIKK31, 750 μM) into 70 μM ALIXBro1-V (filled squares) or ALIX (open circles) in a solution of 20 mM sodium phosphate (pH 7.2), 150 mM NaCl, and 1 mM dithiothreitol (DTT) (25°C). The solid line shows the theoretical curve for a 1:1 peptide: ALIXBro1-V complex with a dissociation constant of 3.6 μM (N was 1.01 ± 0.004, ΔG25°C was −7.42 ± 0.02 kcal/mol, ΔH25°C was −5.55 ± 0.03 kcal/mol, and ΔS25°C was 6.30 ± 0.17 entropy units [eu], where N, G, H, and S are ligand-binding stoichiometry, free energy, enthalpy, and entropy, respectively [MicroCal Origin software]). The peptide was added in 39 0.5-μl injections (180-s intervals) using a MicroCal iTC200.
The two arms of the ALIX V domain are juxtaposed at an acute angle of ∼30° in crystal structures of the monomeric protein (termed the “closed” conformation) (7, 16, 44, 45). The suggestion that the linker connecting the two arms functions as a hinge that opens further under some conditions (7) is consistent with SAXS data, which indicate that that the two arms are indeed more open in the ALIX dimer (29). We therefore hypothesized that mutations that destabilize the closed conformation might promote ALIX dimerization, membrane association, binding to the oligomeric Gag protein, and virus budding. This idea was tested by examining whether mutation of ALIX Arg649 localized the protein to cellular membranes or stimulated virus release. Arg649 forms an array of hydrogen bonding interactions that connect the three linker strands in the closed conformation, and the Arg649Glu mutation is therefore expected to destabilize the closed conformation of the V domain. Structural parameters derived from SAXS data collected from ALIXBro1-V,R649E (see Table S1 and Figure S2 in the supplemental material) show that there is a mass redistribution in the mutant compared to the ALIXBro1-V crystal structure as indicated by a change in the probable atom-pair distribution (Fig. 1 C, gray diamonds), which extends out to ∼190 Å. The Rg of ALIXBro1-V,R649E is ∼8 Å greater than the ALIXBro1-V crystal structure (53.5 Å versus 45.3 Å), while the average radius of gyration of cross section (Rgc) of the mutant (calculated in PRIMUS) (14) is smaller (Rgc of ∼12 Å versus ALIXBro1-V of ∼14.5 Å). These data show that the Arg649Glu mutation causes ALIXBro1-V to extend and become “thinner,” indicating that the mutation causes the V-domain to occupy a more “open” configuration.
Membrane flotation experiments were performed as previously described (25) to test whether the Arg649Glu mutation enhanced ALIX membrane association. Briefly, transfected 293T cells were collected 6 h posttransfection, washed three times with cold NTE buffer (150 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA), and suspended in 800 μl NTE buffer containing 6% (wt/vol) sucrose and a protease inhibitor cocktail (Sigma). Cells were disrupted by sonication (Fig. 3A, lane 1, Lysate), and nuclei and protein aggregates were removed by low-speed centrifugation (800 × g for 15 min; resolubilized in 10% triton/NTE buffer) (Fig. 3A, lane 3, Pellet). ALIX and ALIXR649E remained in the nonpelleted fractions (Fig. 3A, lane 2, Supernatant). This fraction was adjusted to 80% (wt/vol) sucrose in NTE and placed on the bottom of a 14- by 89-mm centrifuge tube (331372; Beckman), and the membrane fractions were “floated” by sedimentation (35,000 rpm for 24 h at 4°C; Beckman SW41 rotor) through layers of 65% sucrose (6 ml) and 10% sucrose (2.5 ml). Fractions containing membrane-bound proteins (4 ml, lane 4) and soluble proteins (5 ml, lane 6) and an intermediate fraction (3 ml, lane 5) were collected, and their protein contents were analyzed by Western blotting. Control soluble (aldolase) and integral membrane (cadherin) proteins concentrated in the soluble and membrane fractions, respectively, as expected (Fig. 3A, rows 3 and 4, respectively). The wild-type FLAG-ALIX protein remained predominantly (79%) in the soluble fraction (Fig. 3A, row 1, compare lanes 4 and 6), whereas most (65%) of the ALIXR649E mutant associated with membranes (Fig. 3A, row 2, compare lanes 4 and 6). Thus, these data indicate that V domain opening activates ALIX for membrane binding.
Fig. 3.
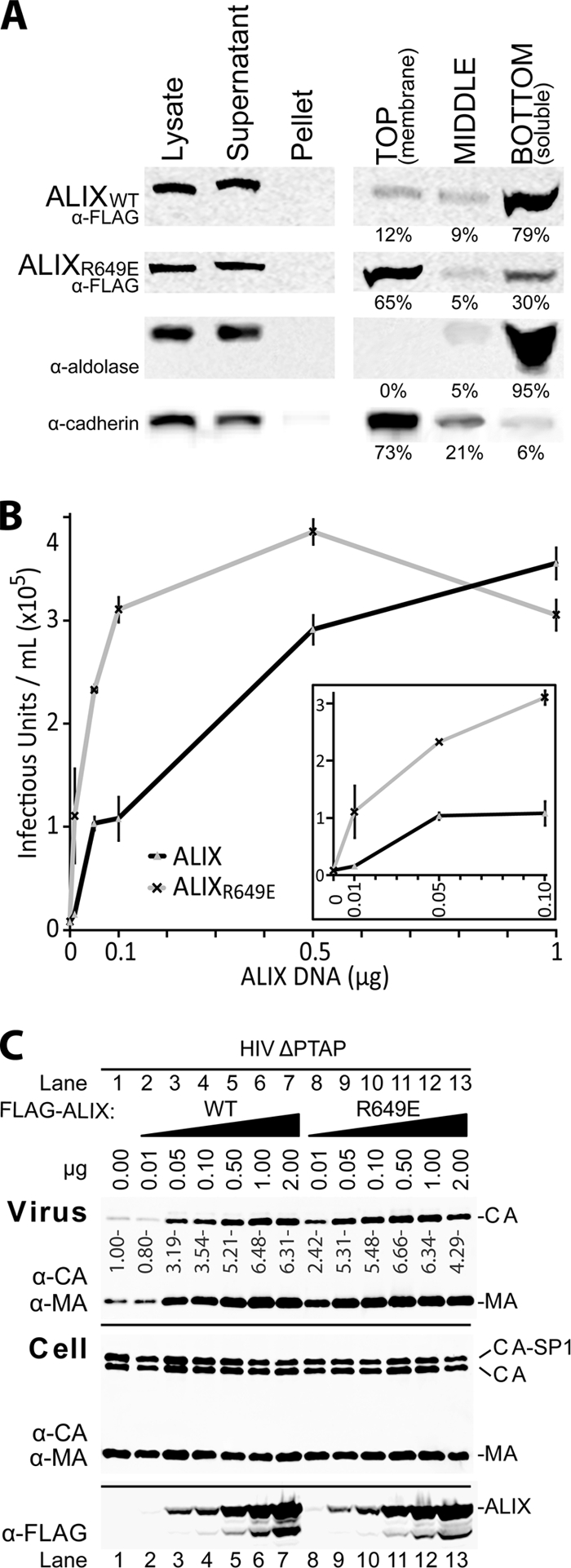
The Arg649Glu mutation activates ALIX for membrane association and virus budding. (A) Flotation analysis showing the degree of membrane association of ALIX and ALIXR649E. Lanes 1 to 3 show the crude fractionation of 293T cell lysates (lane 1) expressing either ALIX (row 1, WISP03-308), ALIXR649E (row 2, WISP06-180), or no protein (control, rows 3 and 4). The data demonstrate that neither ALIX nor ALIXR649E forms insoluble aggregates (compare lanes 2 and 3 and see text for details). Lanes 4 to 6 show the percentages of ALIX (row 1) ALIXR649E (row 2), aldolase (soluble protein control, row 3), and cadherin (integral membrane protein control, row 4) that partitioned into the membrane-containing (lane 4), soluble (lane 6), or intermediate (row 5) fractions. (B) HIV-1 ΔPTAP viral titers released by 293T cells (six-well plates, 1 μg plasmid DNA) cotransfected with an empty vector control or with the indicated quantities of pCl-neo-FLAG vectors expressing wild-type ALIX (gray triangles, black line) or ALIXR649E (black crosses, gray line). Titers were measured in triplicate using single-cycle MAGIC infectivity assays. (C) Western blots of supernatants and cells corresponding to the experiment described in panel B, showing levels of virion-associated CA and MA released into the media (panel 1), and cellular levels of viral Gag, (anti-CA [α-CA] and α-MA, panel 2), and ALIX protein levels (α-FLAG, panel 3).
To test whether the ALIXR649E mutant was also hyperactive in stimulating virus release, we measured the release and infectivity of an HIV-1NL4-3 virus that could not bind TSG101 (HIV-1 ΔPTAP) and was therefore highly dependent upon expression of exogenous ALIX for budding (7, 37). As shown in Fig. 3B and C, ALIXR649E was more potent than the wild-type protein in stimulating virus release and infectivity, particularly when ALIX levels were limiting. For example, viral infectivity was 7-fold higher when cells were transfected with 0.01 μg of the ALIXR649E expression construct (Fig. 3B, inset) even though the two proteins were expressed at equivalent levels (Fig. 3C, panel 3, compare lanes 2 and 8). Similarly, 0.1 μg of the ALIXR649E expression construct stimulated virus release and infectivity to levels that were comparable to those induced by 1.0 μg of the wild-type ALIX expression construct (Fig. 3B, inset; Fig. 3C, panel 3, compare lanes 7 and 11). Thus, the Arg649Glu mutation activates ALIX to facilitate HIV-1 release, further supporting the idea that destabilizing the closed conformation of the ALIX V domain produces a constitutively active protein.
Together with previous reports (5, 29, 48), our studies support a model in which ALIX is activated to facilitate virus budding through a series of conformational changes that (i) release the PRR from the Bro1-V domains and expose the YPXL late-domain binding site, (ii) open the V domain, (iii) stimulate membrane recruitment of ALIX, and (iv) induce protein dimerization (Fig. 4). These conformational changes are likely to be concerted and mutually reinforcing, because (i) ALIX dimerization appears to require V domain opening (29) and to be regulated by PRR residues (5), (ii) membrane recruitment will tend to increase local ALIX concentrations and thereby drive dimerization, and (iii) ALIX dimerization will tend to promote binding to oligomeric Gag complexes owing to avidity. Thus, all of these effects likely combine to stimulate ALIX recruitment during viral Gag protein assembly at the plasma membrane. Other factors that may also contribute to ALIX activation include ubiquitin association (12), phosphorylation (31), the ubiquitin E3-ligase POSH (1, 36, 42), and factors that bind the PRR, such as CEP55, endophilins, TSG101, ALG-2, PYK2, Src kinases, and the Cbl-SET/CIN85-endophilin complex (5, 7, 18, 21, 22, 30, 32, 33, 36, 40, 41).
Fig. 4.
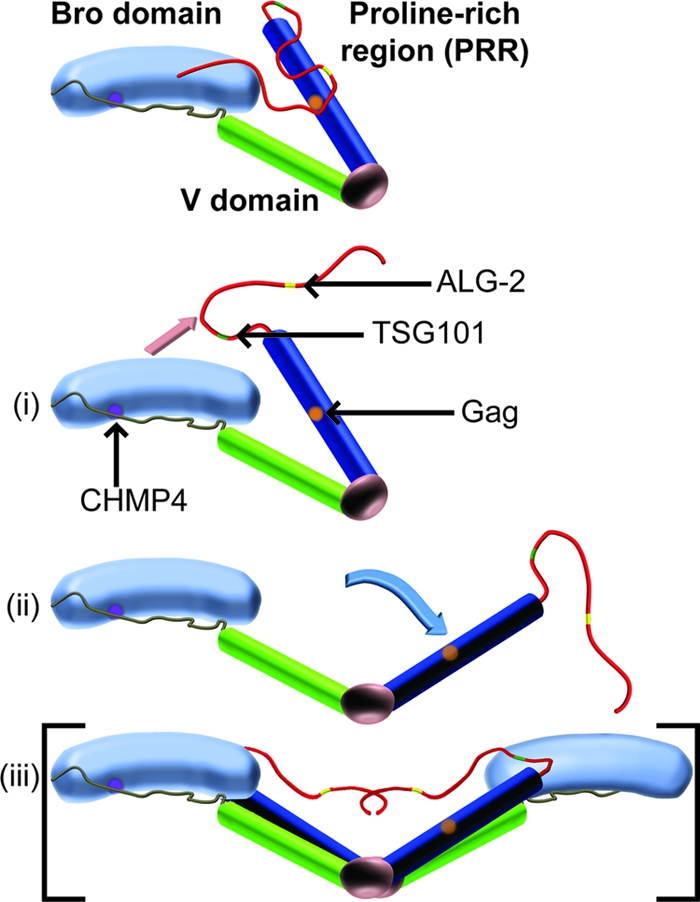
Model depicting different stages of ALIX activation. (i) Monomeric ALIX adopts an autoinhibited state in the cytosol in which the two V domain arms (blue and green) adopt a “closed” conformation, and the PRR (red) folds back onto the V domain to occlude the YPXL late-domain binding site (orange) and onto the Bro1 domain (light blue with the CHMP4 binding site shown in purple). ALIX activation requires dissociation of the PRR from the Bro1-V core (ii) (pink arrow), opening of the V domain (iii) (blue arrow), and protein dimerization (iv), denoted in brackets because dimeric species were characterized in references 5 and 29, not in the present study.
In addition to providing a regulatable step in ESCRT complex assembly, ALIX dimerization may nucleate the assembly of two strands of CHMP4, which is thought to form filaments within the necks of budding vesicles (10, 27). During yeast intralumenal endosomal vesicle formation, the ESCRT-II complex performs an analogous function in nucleating the polymerization of two CHMP4 strands (35). In that case, two CHMP4 filaments are formed because the ESCRT-II complex contains two copies of the ESCRT-III binding protein Vps25p (10, 27). Similarly, the mechanism of PRR autoinhibition described here for ALIX is analogous to autoinhibition of the ESCRT-III proteins, whose C-terminal tails also fold back on the body of the protein to prevent protein oligomerization and membrane binding until they are released by binding to upstream factors (2, 15, 17, 43). Once the proteins are opened, the oligomerization domains can polymerize, and the C-terminal tails are free to recruit additional downstream factors. Thus, different ESCRT factors employ common principles to cycle on and off membranes and maintain the sequential protein assembly pathways required for regulated membrane fission.
Supplementary Material
Acknowledgments
We thank David Myszka and Rebecca Rich, University of Utah HSC Protein Interactions Core Research Facility, for assistance with the biosensor binding experiments.
This work was supported by NIH grants AI051174 (to W.I.S.) and P50 082545 (to C.P.H.).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 29 June 2011.
REFERENCES
- 1. Alroy I., et al. 2005. The trans-Golgi network-associated human ubiquitin-protein ligase POSH is essential for HIV type 1 production. Proc. Natl. Acad. Sci. U. S. A. 102:1478–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bajorek M., et al. 2009. Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16:754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bergmann A., Fritz G., Glatter O. 2000. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA). J. Appl. Crystallogr. 33:1212–1216 [Google Scholar]
- 4. Bieniasz P. D. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe 5:550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlton J. G., Agromayor M., Martin-Serrano J. 2008. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc. Natl. Acad. Sci. U. S. A. 105:10541–10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Demirov D. G., Ono A., Orenstein J. M., Freed E. O. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. U. S. A. 99:955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fisher R. D., et al. 2007. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128:841–852 [DOI] [PubMed] [Google Scholar]
- 8. Fujii K., Hurley J. H., Freed E. O. 2007. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 5:912–916 [DOI] [PubMed] [Google Scholar]
- 9. Garrus J. E., et al. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65 [DOI] [PubMed] [Google Scholar]
- 10. Hurley J. H., Hanson P. I. 2010. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11:556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jeffries C. M., Whitten A. E., Harris S. P., Trewhella J. 2008. Small-angle X-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J. Mol. Biol. 377:1186–1199 [DOI] [PubMed] [Google Scholar]
- 12. Joshi A., Munshi U., Ablan S. D., Nagashima K., Freed E. O. 2008. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 9:1972–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim J., et al. 2005. Structural basis for endosomal targeting by the Bro1 domain. Dev. Cell 8:937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Konarev P. K., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36:1277–1282 [Google Scholar]
- 15. Lata S., et al. 2008. Structural basis for autoinhibition of ESCRT-III CHMP3. J. Mol. Biol. 378:818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee S., Joshi A., Nagashima K., Freed E. O., Hurley J. H. 2007. Structural basis for viral late-domain binding to Alix. Nat. Struct. Mol. Biol. 14:194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin Y., Kimpler L. A., Naismith T. V., Lauer J. M., Hanson P. I. 2005. Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7-1 with itself, membranes, and the AAA+ ATPase SKD1. J. Biol. Chem. 280:12799–12809 [DOI] [PubMed] [Google Scholar]
- 18. Martin-Serrano J., Yaravoy A., Perez-Caballero D., Bieniasz P. D. 2003. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. U. S. A. 100:12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin-Serrano J., Zang T., Bieniasz P. D. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313–1319 [DOI] [PubMed] [Google Scholar]
- 20. McCullough J., Fisher R. D., Whitby F. G., Sundquist W. I., Hill C. P. 2008. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc. Natl. Acad. Sci. U. S. A. 105:7687–7691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Missotten M., Nichols A., Rieger K., Sadoul R. 1999. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 6:124–129 [DOI] [PubMed] [Google Scholar]
- 22. Morita E., et al. 2007. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26:4215–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munshi U. M., Kim J., Nagashima K., Hurley J. H., Freed E. O. 2007. An Alix fragment potently inhibits HIV-1 budding: characterization of binding to retroviral YPXL late domains. J. Biol. Chem. 282:3847–3855 [DOI] [PubMed] [Google Scholar]
- 24. Odorizzi G. 2006. The multiple personalities of Alix. J. Cell Sci. 119:3025–3032 [DOI] [PubMed] [Google Scholar]
- 25. Ono A., Freed E. O. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orthaber D., Bergmann A., Glatter O. 2000. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Applied Crystallogr. 33:218–225 [Google Scholar]
- 27. Peel S., Macheboeuf P., Martinelli N., Weissenhorn W. 2011. Divergent pathways lead to ESCRT-III-catalyzed membrane fission. Trends Biochem. Sci. 36:199–210 [DOI] [PubMed] [Google Scholar]
- 28. Petoukhov M. V., Svergun D. I. 2005. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89:1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pires R., et al. 2009. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure 17:843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt M. H., Chen B., Randazzo L. M., Bogler O. 2003. SETA/CIN85/Ruk and its binding partner AIP1 associate with diverse cytoskeletal elements, including FAKs, and modulate cell adhesion. J. Cell Sci. 116:2845–2855 [DOI] [PubMed] [Google Scholar]
- 31. Schmidt M. H., Dikic I., Bogler O. 2005. Src phosphorylation of Alix/AIP1 modulates its interaction with binding partners and antagonizes its activities. J. Biol. Chem. 280:3414–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi X., et al. 2010. Identification and biophysical assessment of the molecular recognition mechanisms between the human haemopoietic cell kinase Src homology domain 3 and ALG-2-interacting protein X. Biochem. J. 431:93–102 [DOI] [PubMed] [Google Scholar]
- 33. Strack B., Calistri A., Craig S., Popova E., Gottlinger H. G. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699 [DOI] [PubMed] [Google Scholar]
- 34. Svergun D., Barberato C., Koch M. H. J. 1995. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773 [Google Scholar]
- 35. Teis D., Saksena S., Judson B. L., Emr S. D. 2010. ESCRT-II coordinates the assembly of ESCRT-III filaments for cargo sorting and multivesicular body vesicle formation. EMBO J. 29:871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsuda M., Seong K. H., Aigaki T. 2006. POSH, a scaffold protein for JNK signaling, binds to ALG-2 and ALIX in Drosophila. FEBS Lett. 580:3296–3300 [DOI] [PubMed] [Google Scholar]
- 37. Usami Y., Popov S., Gottlinger H. G. 2007. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 81:6614–6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Usami Y., et al. 2009. The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 37:181–184 [DOI] [PubMed] [Google Scholar]
- 39. VerPlank L., et al. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55Gag. Proc. Natl. Acad. Sci. U. S. A. 98:7724–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vito P., Pellegrini L., Guiet C., D'Adamio L. 1999. Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J. Biol. Chem. 274:1533–1540 [DOI] [PubMed] [Google Scholar]
- 41. von Schwedler U. K., et al. 2003. The protein network of HIV budding. Cell 114:701–713 [DOI] [PubMed] [Google Scholar]
- 42. Votteler J., et al. 2009. Exploring the functional interaction between POSH and ALIX and the relevance to HIV-1 release. BMC Biochem. 10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xiao J., et al. 2009. Structural basis of Ist1 function and Ist1-Did2 interaction in the multivesicular body pathway and cytokinesis. Mol. Biol. Cell 20:3514–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhai Q., et al. 2008. Structural and functional studies of ALIX interactions with YPXnL late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol. 15:43–49 [DOI] [PubMed] [Google Scholar]
- 45. Zhai Q., Landesman M. B., Robinson H., Sundquist W. I., Hill C. P. 2011. Identification and structural characterization of the ALIX-binding late domains of simian immunodeficiency virus SIVmac239 and SIVagmTan-1. J. Virol. 85:632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou X., et al. 2009. The CHMP4b- and Src-docking sites in the Bro1 domain are autoinhibited in the native state of Alix. Biochem. J. 418:277–284 [DOI] [PubMed] [Google Scholar]
- 47. Zhou X., et al. 2008. The HIV-1 p6/EIAV p9 docking site in Alix is autoinhibited as revealed by a conformation-sensitive anti-Alix monoclonal antibody. Biochem. J. 414:215–220 [DOI] [PubMed] [Google Scholar]
- 48. Zhou X., Si J., Corvera J., Gallick G. E., Kuang J. 2010. Decoding the intrinsic mechanism that prohibits ALIX interaction with ESCRT and viral proteins. Biochem. J. 432:525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.