

Abstract
Inhibition of translation is an integral component of the innate antiviral response and is largely accomplished via interferon-activated phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). To successfully infect a host, a virus must overcome this blockage by either controlling eIF2α phosphorylation or by utilizing a noncanonical mode of translation initiation. Here we show that enterovirus RNA is sensitive to translation inhibition resulting from eIF2α phosphorylation, but it becomes resistant as infection progresses. Further, we show that the cleavage of initiation factor eIF5B during enteroviral infection, along with the viral internal ribosome entry site, plays a role in mediating viral translation under conditions that are nonpermissive for host cell translation. Together, these results provide a mechanism by which enteroviruses evade the antiviral response and provide insight into a noncanonical mechanism of translation initiation.
INTRODUCTION
Initiation of cap-dependent translation occurs in three initial steps. The canonical first step is recruitment of an eIF2–GTP–Met-tRNAiMet ternary complex to the 40S subunit in a binding step stabilized by initiation factors eIF3 (eukaryotic initiation factor 3), eIF1, and eIF1A to form a 43S complex. The 43S complex is competent to bind mRNA complexed with eIF4F and eIF4B bound to the 5′ cap structure to form 48S initiation complexes that are capable of scanning for initiator codons, aided by eIF4A/eIF4B RNA helicase functions (20). Stable 48S complexes arise with formation of codon-anticodon base pairing in the P site of the 40S ribosome subunit. Proper placement of the tRNA anticodon is mediated by eIF2, which is aided by eIF1 and perhaps eIF5B in rejection of poor-context triplets (20, 28). Upon proper codon-anticodon base pairing, eIF2 hydrolyzes its bound GTP, followed by eIF5B-mediated ejection of factors from the 40S subunit and 60S subunit joining to form the complete 80S ribosome (20, 42).
Before 60S subunit joining, hydrolyzed eIF2-GDP is released from the 48S complex and must be recycled by interaction with the guanine nucleotide exchange factor eIF2B to exchange the GDP for GTP for use in the next round of translation initiation. Exposure to several types of stress stimuli restricts translation through the activation of eukaryotic initiation factor 2 (eIF2α) kinases PKR (protein kinase R), PERK (double-stranded RNA-activated ER kinase), HRI (heme-regulated inhibitor), and GCN2 (general control nonderepressing 2). Phosphorylation of eIF2α turns eIF2 into a competitive inhibitor of eIF2B function through an increased affinity of phospho-eIF2 for eIF2B, causing the accumulation of eIF2-GDP and a corresponding global inhibition of translation initiation (25, 51).
Infection of mammalian cells with viruses often induces an antiviral response that can produce interferon and activate the eIF2α kinase PKR, causing a suppression of translation by eIF2α phosphorylation (34). However, poliovirus (PV), a member of the Picornaviridae family and Enterovirus genus, is resistant to type I interferon treatment in vitro (4, 5, 36). Inhibition of translation by eIF2α phosphorylation also results in the formation of cytoplasmic mRNA stress granules in uninfected cells (reviewed in references 24 and 59). Several viruses both induce the formation of stress granules (31, 43, 55) and modulate the stress granule pathway (33, 35, 49). During infection with PV, stress granule formation is induced by an eIF2α-independent mechanism (31) and eIF2α phosphorylation is suppressed until late times postinfection (8).
The single-stranded, positive-sense RNA genome of poliovirus is preferentially translated due to inhibition of cap-dependent cellular translation by cleavage of initiation factors eIF4GI, eIF4GII, and poly(A)-binding protein (PABP) (reviewed in reference 29) and the cap-independent recruitment of ribosomes by the viral internal ribosome entry site (IRES). IRES-driven translation is also important for the preferential expression of proteins during cellular conditions of reduced translation such as apoptosis and mitosis (17, 50). Several viral IRESs require fewer canonical initiation factors than canonical cap-dependent initiation, and most do not require eIF4E. The poliovirus IRES is a type 1 IRES that requires eIF4G, eIF4A, eIF4B, and eIF2 (14, 37, 38); however, initiation occurs at a downstream canonical AUG start codon that is reached by ribosome scanning after binding internally on the IRES.
Recently, some IRES elements were shown to translate independently of eIF2, including the cricket paralysis virus intergenic region IRES (CrPV-IGR) (21, 22), the hepatitis C virus (HCV) IRES (26, 52), the classical swine fever virus (CSFV) IRES (41), and the cellular Src IRES (1). The latter three require initiation at AUG codons; thus, Met-tRNAiMet recruitment occurs via novel alternate mechanisms. In the case of CSFV, it was determined that eIF5B/eIF3 substitutes for eIF2/eIF3 function in the formation of 48S ribosome/mRNA complexes (41).
Previous work by our laboratory revealed that poliovirus inhibits the formation of stress granules by the cleavage of Ras-GTPase-activating protein (Ras-GAP) Src homology 3 (SH3)-domain-binding protein 1 (G3BP) with virus 3C proteinase (3Cpro) and that the rescue of stress granule formation by expression of a cleavage-resistant G3BP mutant reduced PV progeny virion production (55). When we investigated the mechanism by which cleavage-resistant G3BP inhibits PV replication, we determined that under conditions of oxidative stress, PV translation becomes increasingly resistant to eIF2α phosphorylation as infection progresses. Translation resistance to stress was not due to cleavage of G3BP. We found that eIF2-independent translation is not an inherent property of enteroviral IRES elements in the absence of viral infection. Further, a cleavage fragment of eIF5B can rescue IRES translation under extreme stress conditions; thus, eIF5B cleavage may play an important role in translation resistance to eIF2α phosphorylation during infection.
MATERIALS AND METHODS
Cloning and DNA constructs.
pcDNA3.1-HA-eIF5B was a kind gift of C. U. Hellen. pcDNA3.1-HA-eIF5BQ478E was produced by site-directed mutagenesis of pcDNA3.1-HA-eIF5B (sense strand 5′-GAAGTTATGGAAGAAGGAGTACCAGAAAAGG-3′ and antisense strand 5′-CCTTTTCTGGTACTCCTTCTTCCATAACTTC-3′), and pcDNA3.1-HA-eIF5B479-1220 was produced by inverse PCR of pcDNA3.1-HA-eIF5B (sense strand 5′-GGAGTACCAGAAAAGGAAGAGACACC-3′ and antisense strand 5′-GGCGTAGTCGGGCACGTC-3′). pTRE2-PV-Fluc (Fluc stands for firefly luciferase) was produced by PCR amplification of the poliovirus (PV) internal ribosome entry site (IRES) from pT7-PV-IRES-Fluc (9) with primers containing SacII sites and was inserted into the SacII site of pTRE2-luc (Clontech). pTRE2-CVB3-Fluc (CVB3 stands for coxsackievirus B3) was produced by insertion of the SalI and XbaI fragment purified from pRL-CVB3-FL (FL stands for firefly luciferase) (obtained from B. Semler) and inserted into pTRE2 (Clontech) digested with the same enzymes. The plasmid pREP-G3BP1-GFP (GFP stands for green fluorescent protein) encoding hamster Ras-GTPase-activating protein (Ras-GAP) Src homology 3 (SH3) domain-binding protein 1 (G3BP1) (which does not contain the consensus 3C proteinase [3Cpro] cleavage sequence) was a kind gift from Ilya Frolov.
Firefly luciferase reporter expression.
HeLa TetON cells (Clontech Laboratories) were maintained in Dulbecco modified Eagle medium (DMEM) (Cellgro) supplemented with 10% fetal bovine serum, penicillin-streptomycin, and l-glutamine at 37°C in 5% CO2. A total of 1 × 105 HeLa TetON cells were transfected with FugeneHD (Roche) for 36 h to allow for expression of eIF5B479-1220 and overnight with pTRE2-Fluc reporters in 24-well plates. Reporter expression was induced by the addition of 1 μg/ml tetracycline in the presence and absence of sodium enite for 3 h at 37°C. Cells were then lysed in passive lysis buffer or TRIzol reagent (Invitrogen) for analysis of firefly luciferase or reporter mRNA, respectively, by reverse transcription (RT)-PCR to standardize the results. Reporters for electroporation were transcribed in vitro using T7 RNA polymerase (Invitrogen) according to the manufacturer's protocols from ApaI-linearized pT7-FLuc, HindIII-linearized pT7-PV 5′-FLuc-3′ A71 (9) and NaeI-linearized pCMV-RLuc-IGR-FLuc (CMV stands for cytomegalovirus). HeLa S3 and 293T cells were electroporated using the Neon electroporation system (Invitrogen) according to the manufacturer's protocols prior to treatment with sodium arsenite (Sigma) or Sal003 (kind gift of Mauro Costa-Mattioli) at the indicated concentrations. Nuclease-treated rabbit reticulocyte lysate (RRL) (Promega) was supplemented with HeLa ribosomal salt wash and then pretreated with increasing doses of poly(I · C) (Sigma) for 30 min at 34°C before programming with in vitro RNA reporters and incubation at 34°C for 45 min.
Poliovirus infection and stress experiments.
Poliovirus was grown and purified as previously described (23), and coxsackievirus B3 (CVB3) was precipitated from infected HeLa cell cultures with 2.2% NaCl–7% polyethylene glycol 6000. Infections were conducted at a multiplicity of infection (MOI) of 20 for 293T cells or at an MOI of 30 for HeLa cells. To stress cells, 50 to 500 μM sodium arsenite (Sigma-Aldrich) in medium was incubated with cells for the final 30 min of infection before the cells were pulse-labeled with 30 μCi/ml of [35S]methionine-cysteine (Tran35S-label; MP Biomedicals). Some cell cultures were heat shocked at 44°C for 30 min before pulse-labeling. Labeled cells were pelleted and lysed in NP-40 lysis buffer (10 mM Tris-Cl [pH 7.4], 100 mM NaCl, 1 mM EDTA, and 1% NP-40), and S10 fractions were frozen until analysis. For various experiments, 293T cells were transfected for 36 h prior to infection with control sheared salmon sperm DNA or pEGFP-N1 (EGFP stands for enhanced green fluorescent protein) (Clontech) DNA. Experimental groups were transfected with pcDNA3.1-G3BP, pcDNA3.1-G3BPQ326E, pcDNA3.1-HA-eIF5B, pcDNA3.1-HA-eIF5BQ478E, or pcDNA3.1-eIF5B479-1220 using Lipofectamine 2000 (Invitrogen) in DMEM according to the manufacturer's protocols.
Autoradiography and immunoblot analysis.
Radiolabeled S10 lysates were resolved on 10% polyacrylamide gels and dried or transferred to nitrocellulose membranes. Dried gels were exposed to film (Kodak), and viral translation was quantified by densitometry of the indicated viral bands (ImageJ). Western blot analysis of hemagglutinin (HA)-tagged eIF5B (HA-eIF5B) expression by anti-HA (Y-11; Santa Cruz Biotechnology) (diluted 1:1,000), anti-eIF5B (kind gift from T. Dever) (1:1,500), phospho-eIF2α (119A11; Cell Signaling Technologies) (1:1,500), anti-eIF2α (FL-315; Santa Cruz Biotechnology), (1:1,000), eIF4GI (10a) (1:1,000), G3BP1 (55) (1:2,000), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore; 1:1,000) was conducted in phosphate-buffered saline (PBS) containing 4% powdered milk and 1% Triton X-100 overnight at 4°C. Detection of primary antibody by anti-rabbit IgG (Bio-Rad; 1:4,000) was visualized with Pico or Femto substrate (Pierce) exposure to X-ray film.
RESULTS
Poliovirus translation is resistant to eIF2α phosphorylation.
Viral infection results in the activation of the antiviral response as well as cellular stress responses that induce stress granules containing translationally silenced mRNA (2, 6). PV infection results in an eIF2α-independent induction of stress granules (31, 55), although the level of induction is cell type dependent and is inhibited at late time points postinfection (55). To determine the effects of induction of the cellular stress response and near complete phosphorylation of eIF2α on viral infection, we subjected PV-infected cells to oxidative stress with sodium arsenite (Ars) treatment at various times through the infection and measured viral translation by pulse-label/autoradiography. Arsenite treatment induced high-level phosphorylation of eIF2α as expected (Fig. 1B), which also resulted in a drastic inhibition of cellular translation in mock-infected cells (Fig. 1A, lane 2). However, as PV infection progresses, apparent viral translation became evident at 3 to 6 h postinfection (hpi) despite induction of high-level phosphorylation of eIF2α, suggesting that PV translation is resistant to Ars stress (Fig. 1A). Viral translation was relatively efficient under severe stress conditions and reached levels that were >50% of those in unstressed cells between 4 and 6 hpi.
Fig. 1.
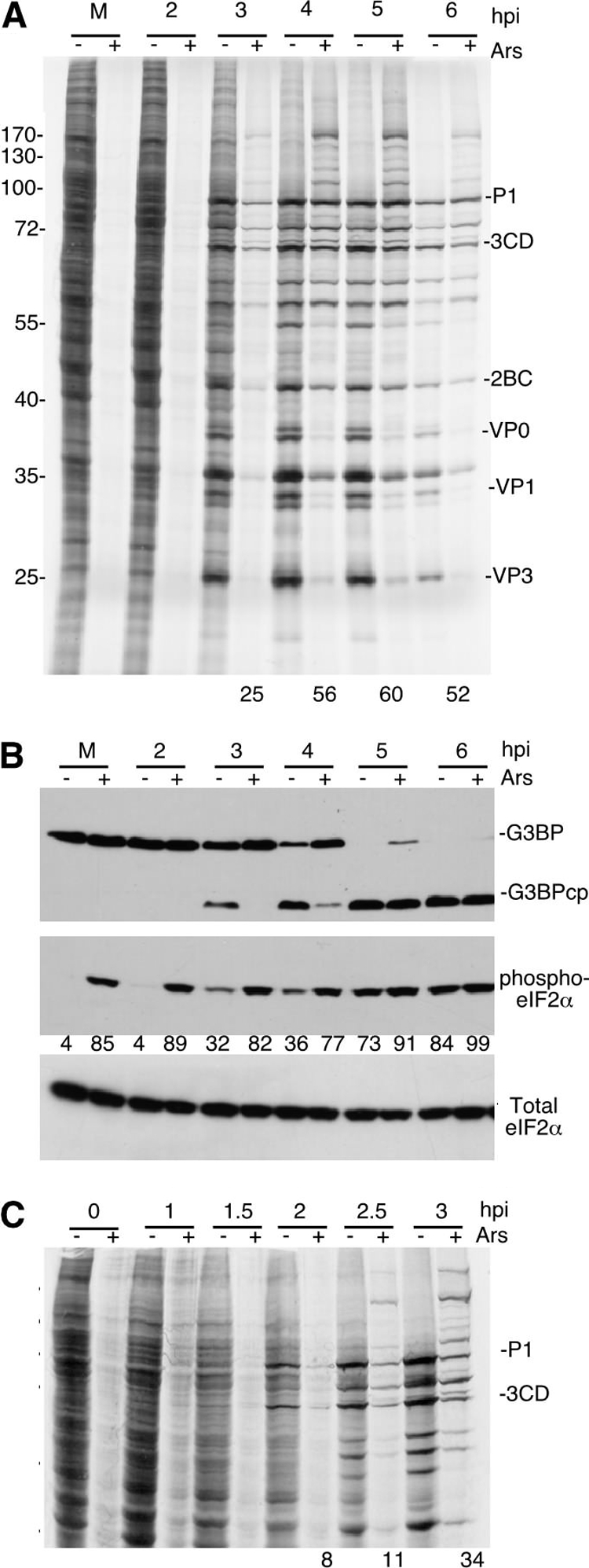
Poliovirus (PV) translation becomes resistant to sodium arsenite (Ars) stress. (A and C) HeLa cells infected with PV were stressed with 0.5 mM Ars (+) or not stressed with Ars (−) for 30 min prior to the time points indicated (hpi, hours postinfection). Cells were then pulse-labeled for 30 min with Tran35S-label before analysis by SDS-PAGE and autoradiography. The numbers below the gel in lanes 3 to 6 indicate the percentage of translation in Ars-stressed lanes compared to unstressed conditions as determined by densitometry as a function of time. The migration positions of molecular mass standards (in kilodaltons) are shown to the left of the gel, and the migration positions of PV proteins are shown to the right of the gel. (B) Western blot analysis of PV-infected cells to determine the cleavage of G3BP and phosphorylation of eIF2α using specific antibodies as indicated for each panel. Values indicate percentage of maximal eIF2α phosphorylation. G3BPcp, G3BP cleavage product.
Previous work has shown that in unstressed cells, PV restricts eIF2α phosphorylation until late points in the infectious cycle partly through degradation of PKR (8, 39). However, in Ars-stressed cells, PV infection was unable to prevent high-level eIF2α phosphorylation (Fig. 1B), which is mediated by another eIF2α kinase, HRI (32). Poliovirus infection alone did not induce the maximal eIF2α phosphorylation that was achieved with Ars, even at the end of the replicative cycle (6 hpi).
To further refine the kinetics of resistance to Ars stress, we infected cells for 0 to 3 h at 30-min intervals and found that resistance is not apparent at 2 hpi (Fig. 1C; follow prominent viral polypeptides P1 and 3CD). Arsenite-resistant translation first appears at 2.5 hpi and became readily apparent at 3 hpi (Fig. 1C). To determine whether 3Cpro activity or cleavage of G3BP correlates with the resistance to Ars stress, we monitored the cleavage of G3BP by 3Cpro via immunoblotting and found that as with the emergence of resistance, the first measurable cleavage of G3BP in unstressed cells, and therefore 3Cpro activity, is observed at 3 hpi (Fig. 1B). Application of Ars also restricts 3Cpro activity (Fig. 1B and 2D). Taken together, these data suggest that PV translation is resistant to eIF2α phosphorylation and that this resistance correlates with 3Cpro activity and possibly G3BP cleavage.
Fig. 2.

Expression of cleavage-resistant G3BP does not cause resistance to Ars stress. (A) HeLa cells expressing hamster pREP-G3BP-GFP were infected, stressed, and pulse-labeled with Tran35S-label and analyzed as described in the legend to Fig. 1A. The migration positions of molecular mass standards (in kilodaltons) are indicated to the left of the gel. (B) Immunoblot analysis of lysates for total and phosphorylated eIF2α. (C) Immunoblot analysis for G3BP showing the cleavage of endogenous G3BP but not G3BP-GFP. (D) HeLa S10 lysates were treated with a constant concentration of Ars (1 mM) plus various concentrations (1 μg to 10 ng) of recombinant viral 2Apro or 3Cpro to determine the effects of Ars treatment on proteinase activity. The height of the triangle above the lanes indicates the relative amount of 2Apro or 3Cpro. 2A and 3C proteinase activities were monitored by the cleavage of eIF4GI and G3BP, respectively. (E) 293T cells transfected with control DNA or plasmids expressing His-G3BP or His-G3BPQ326E were infected with PV, translating proteins were pulse-labeled at 4 or 6 hpi and analyzed by SDS-PAGE/autoradiography and densitometry of the indicated viral polypeptide bands. Data are expressed as percent incorporation relative to untransfected control cells (% Cont).
Expression of cleavage-resistant G3BP does not block poliovirus resistance to Ars stress.
Previously we showed that overexpression of both wild-type G3BP and cleavage-resistant G3BPQ326E increased stress granules in Ars-stressed infected cells and inhibited PV replication (55). Since G3BP may act as an inhibitor of translation (3, 40), we sought to determine whether cleavage of G3BP plays a role in resistance of virus translation to Ars stress. Hamster G3BP1 does not contain the consensus 3Cpro cleavage site; thus, we expressed cleavage-resistant hamster G3BP-GFP in HeLa cells and repeated infections with and without Ars stress. Although expression of cleavage-resistant G3BP (Fig. 2C) did not change the kinetics of resistance to oxidative stress relative to a control infection, it did have a general suppressive effect on viral replication and/or translation, reducing the output of pulse-labeled viral proteins by about 2-fold (Fig. 2A). Once again, translation resistance to eIF2α phosphorylation emerged by 3 hpi, which was when G3BP cleavage was first detected. This indicates that stress resistance correlates with 3Cpro activity but not with G3BP cleavage. Further, expression of cleavage-resistant G3BP did not diminish the high phosphorylation of eIF2α under Ars stress. Similarly, when 293T cells were transfected with wild-type and cleavage-resistant G3BPQ326E and infected with PV, incorporation of [35S]Met/Cys radiolabel into major 3CD, 2C, and VP3 bands was reduced compared to control transfected cells (Fig. 2E). Thus, apparent viral RNA translation was modestly inhibited by G3BP expression.
We repeatedly observed a dysfunction in polyprotein processing in cells treated with Ars as seen in the appearance of high-molecular-weight radiolabeled protein species with concomitant reduction of processed PV proteins (Fig. 1A, 1C, 2A, and later figures). Attempts to verify the identity of the high-molecular-weight bands with pulse-chase were unsuccessful (data not shown), although the polypeptide pattern resembles that observed in cell-free translation reaction mixtures treated with hydantoin (54). To determine whether Ars treatment can lead to the direct inhibition of the viral 2A and 3C proteinases and thus restrict polyprotein processing, we treated HeLa S10 lysates with decreasing doses of recombinant 2Apro and 3Cpro in the presence of a constant concentration of arsenite and measured activity by cleavage of eIF4GI and G3BP substrates, respectively (Fig. 2D). Arsenite treatment differentially inhibited viral proteinase activity in vitro, with 1 mM arsenite inhibiting a significantly greater amount of 3Cpro (1 μg) than 2Apro (0.01 μg) cleavage activity. This suggests that both proteinases are sensitive to arsenite-mediated inhibition, but 3Cpro is restricted more significantly, thus providing an explanation for the defective polyprotein processing observed after Ars administration to cells.
To determine whether PV translation was resistant to additional stress mechanisms, we subjected infected HeLa cells to heat shock for 30 min, which also results in eIF2α phosphorylation and inhibition of host translation (Fig. 3A and D). Under these conditions, PV translation was also evident at late time points, and expression levels were similar to those achieved during Ars stress. Interestingly, defects in viral polyprotein processing were not apparent under heat shock conditions. Since arsenite and heat shock may inhibit PV RNA replication or translation via other pathways not related to eIF2a phosphorylation, we treated infected cells with Sal003, a derivative of salubrinal that inhibits eIF2α phosphatases (10). Sal003 treatment increased levels of phospho-eIF2α and effectively shutoff host translation (Fig. 3B and D). However, PV translation at 4 to 6 hpi was evident, similar to Ars- and heat shock-treated cells, further demonstrating that the observed PV translation resistance is eIF2α specific. Sal003 treatment did not result in significant viral polyprotein processing defects observed with Ars.
Fig. 3.

Enterovirus translation is resistant to stressors. (A to C and E) SDS-PAGE/autoradiographs of cell proteins labeled with Tran35S-label. (A) PV-infected HeLa cells were infected with PV and stressed with heat shock 30 min prior to labeling at the indicated time points. (B) PV-infected 293T cells were treated with 20 μM Sal003 or dimethyl sulfoxide (DMSO) vehicle for 90 min prior to 30 min pulse-labeling. (C) HeLa cells infected with CVB3 were stressed as described in the legend to Fig. 1. (D) Immunoblots showing phospho-eIF2α in the panels. (E) PV-infected cells pretreated with alpha interferon (IFNα) (1,000 U) for 16 h. Control infection without IFN at 4 h is shown.
To determine whether translation by other enteroviruses is resistant to eIF2α phosphorylation, we examined cells infected with coxsackievirus B3 (CVB3). Application of Ars showed that CVB3 translation was similarly resistant to oxidative stress at the mid and late phases of infection (Fig. 3C). Taken together, these results suggest that a wider range of enteroviruses may be resistant to several types of stress that result in eIF2α phosphorylation. As expected, PV translated efficiently in interferon-treated cells despite increased phospho-eIF2α observed at the beginning of infection (Fig. 3D and E).
Enterovirus IRES translation is blocked by eIF2α phosphorylation.
Recently some viral IRESs have been shown to be refractory to the phosphorylation of eIF2α and can support translation when ternary complexes are depleted (26, 44, 52, 56). To determine whether enterovirus IRES elements are similarly resistant to eIF2α phosphorylation and ternary complex depletion, we tested translation of firefly luciferase (FL) reporters containing viral IRES elements under stress conditions. First, reporters were expressed in cells from tetracycline (Tet)-inducible constructs. Figure 4A shows that translation of FL RNAs containing PV and CVB3 IRESs were strongly inhibited by increasing doses of Ars, and the levels of inhibition were comparable to the levels of control FL RNA translated by canonical cap-dependent initiation. To substantiate this result, HeLa cells were electroporated with in vitro-transcribed FL reporter RNAs. When Ars stress was applied to these cells, translation of both capped FL and PV-IRES-FL RNA were similarly inhibited in a dose-dependent manner that correlated with increased eIF2α phosphorylation (Fig. 4B and E). In contrast, a reporter containing the cricket paralysis virus intergenic region IRES (IGR) was slightly stimulated by application of Ars stress (Fig. 4B). This was expected, since the IGR-IRES is known to translate independently of all initiation factors, including eIF2 (56).
Fig. 4.

Enteroviral IRES reporter RNAs are inhibited by eIF2α phosphorylation. (A) HeLa TetON cells were transfected with luciferase (pTRE2-FL) reporters containing PV or coxsackievirus B3 (CVB3) IRES elements upstream of the firefly luciferase (FL) open reading frame (ORF). Expression of IRES-FL reporters and stress was coinduced for 3 h with constant exposure to tetracycline and Ars, respectively. Firefly luciferase (FL) activity is shown in relative light units (RLU). (B) In vitro-transcribed capped FL or uncapped IRES-FL reporter RNAs were electroporated into HeLa S3 cells prior to treatment with Ars for 90 min and analysis of FL expression. (C) Rabbit reticulocyte lysates supplemented with HeLa ribosomal salt wash initiation factors were pretreated with water or poly(I · C) for 30 min before being programmed with FL reporter RNAs. FL expression was measured after 45 min of translation at 34°C. (D) In vitro-transcribed capped FL or PV-FL reporter RNAs were electroporated into cells, which were then treated with 10 μm Sal003 for 90 min prior to analysis of FL expression. Results were compiled from three independent experiments, and error bars represent standard errors. (E) Immunoblots showing eIF2α or phospho-eIF2α (P-eIF2α) in the experiments above.
Next, rabbit reticulocyte lysates were treated with increasing doses of poly(I · C) to activate an alternate eIF2α kinase, PKR. An equivalent dose-dependent inhibition of capped FL and PV-IRES-FL occurred with increased eIF2α phosphorylation; however, the IGR-FL RNA was not inhibited (Fig. 4C). Last, 293T cells were treated with Sal003 to block eIF2α dephosphorylation. Sal003 decreased translation of both capped FL and PV-IRES-FL but did not alter IGR-FL translation (Fig. 4D). All treatments increased eIF2α phosphorylation as intended (Fig. 4E). Taken together, these data demonstrate that enteroviral IRES elements are fully susceptible to translation inhibition resulting from activation of eIF2α kinases, indicating that the viral IRES is not by itself sufficient to mediate eIF2-independent translation initiation observed during virus infection. Since eIF2-independent translation did correlate with increased 3Cpro activity (Fig. 1), we hypothesized that proteolytic modification of a translation factor may play a role in this gain of IRES translation during stress.
Expression of eIF5B modulates poliovirus translation under stress.
Initiation factor eIF5B is conserved throughout eukaryotes and is the orthologue of prokaryotic initiation factor 2 (IF2) (27, 57). eIF5B mediates the joining of the 40S and 60S ribosomal subunits (42), interacts with eIF1A (12, 30), and stabilizes the initiator tRNAiMet after eIF2 release (12) to aid in the stringency of AUG start codon selection. The GTPase activity of eIF5B acts as a checkpoint for completion of initiation on the correct start codon (47). eIF5B is also cleaved during infection by enterovirus 3Cpro upstream of the four conserved functional domains contained in the C terminus (13). The kinetics of eIF5B cleavage also correlate with resistance of PV translation to Ars stress (Fig. 5A), although kinetics of cleavage in arsenite-treated groups are expected to lag 30 min because of 3Cpro arsenite sensitivity (Fig. 2D). Thus, we investigated whether cleavage of eIF5B may provide a potential mechanism to relieve the block to PV-IRES translation imposed by eIF2α phosphorylation (Fig. 4). To test this hypothesis, we created a 3Cpro cleavage-resistant mutant by replacing the required glutamine at the scissile bond with glutamate (eIF5BQ478E). We expressed HA-tagged wild-type eIF5B and eIF5BQ478E in cells and monitored PV translation during subsequent infection in the presence and absence of Ars stress. Analysis of autoradiographs from PV-infected cells shows that expression of eIF5B in cells subjected to Ars stress causes a modest increase in apparent viral translation during the primary phase of replication (3 to 5 hpi; Fig. 5B and C). Whereas expression of wild-type eIF5B caused a sustained increase in translation pulse-label at 6 hpi, expression of eIF5BQ478E inhibited pulse-labeling of virus proteins at 6 hpi. This suggested that viral translation in stressed cells at late time points postinfection may be more dependent on cleavage of eIF5B. Ectopically expressed wild-type eIF5B was cleaved with kinetics similar to that of endogenous eIF5B during infection (13), and mutant eIF5BQ478E was resistant to cleavage during PV infection as expected (Fig. 5A and E). Expression of eIF5B did not inhibit eIF2α phosphorylation in response to Ars stress, and thus, the observed increases in viral polypeptides may result from increases in eIF2-independent translation (Fig. 5D).
Fig. 5.

Expression of eIF5B modulates PV translation under stress. (A) (Top) Schematic depicting the 3Cpro cleavage site on eIF5B. (Bottom) Gel showing kinetics of cleavage of endogenous eIF5B during PV infection in the presence or absence of Ars. frag., fragment. (B) 293T cells expressing wild-type eIF5B or cleavage-resistant eIF5BQ478E were infected and stressed with 0.5 mM Ars before pulse-labeling translating proteins and analysis by SDS-PAGE and autoradiography. (C) Stimulation of PV translation by eIF5B expression was quantified by densitometry of autoradiographs. Results are an aggregate of three independent experiments, and error bars indicate standard errors. (D) Immunoblot analysis of eIF2α and phosphorylated eIF2α. (E) HA-specific immunoblot indicating expression of HA-eIF5B transgenes.
Expression of the C-terminal eIF5B fragment stimulates PV translation.
Cleavage of eIF5B could impact IRES translation either by reduction in intact eIF5B, and thus its normal functions, or by creation of N- or C-terminal cleavage products that may have novel functions. To determine whether the cleavage product produced by the cleavage of eIF5B by 3Cpro is functional, we expressed the C-terminal 741-amino-acid peptide released by 3Cpro cleavage (HA-eIF5B479-1220). Analysis of autoradiographs of PV-infected, 35S-labeled lysates (Fig. 6A and B) reveals that expression of HA-eIF5B479-1220 stimulated apparent virus translation relative to control infections in cells expressing green fluorescent protein (GFP). Expression of HA-eIF5B479-1220 produced a larger stimulation of pulse-labeling of viral proteins under stress conditions, ranging from a 1.7- to 2.0-fold increase during the peak replicative phase of the infection (Fig. 6A and B). Immunoblot analyses confirmed that equal expression of eIF5B479-1220 occurred throughout the infection and that the eIF5B fragment did not inhibit eIF2α phosphorylation in response to Ars treatment (Fig. 6C). Rather, expression of eIF5B479-1220 resulted in reproducible increases of eIF2α phosphorylation, whose translation restriction activity would be expected to counteract translation stimulation provided by eIF5B expression.
Fig. 6.

Expression of eIF5B479-1220 stimulates PV translation. (A) 293T cells expressing control GFP or eIF5B479-1220 were infected with PV and stressed with 0.5 mM Ars for 30 min, and then translating proteins were pulse-labeled and analyzed by SDS-PAGE/autoradiography. (B) Densitometric analysis of three independent experiments showing stimulation of translation of PV RNA in the presence of eIF5B479-1220 with and without Ars treatment relative to controls (Cont) expressing GFP. Error bars indicate the standard errors. (C) Immunoblot analysis of cell lysates for HA-tagged eIF5B479-1220 and phospho-eIF2α (p-eIF2α) as indicated.
While it is crucial to test effects of eIF5B expression on authentic virus translation during infection, such apparent translation is the cumulative result of viral RNA replication and translation. To avoid potential stimulatory effects of eIF5B expression on viral RNA replication, we transfected reporter RNA into cells expressing GFP or HA-eIF5B479-1220 and tested whether virus IRES translation was rescued during Ars stress. Figure 7A shows that expression of eIF5B479-1220 caused a 2-fold decrease in capped FL RNA translation in unstressed cells and did not rescue translation blocks imposed by Ars-induced eIF2α phosphorylation. In contrast, expression of HA-eIF5B479-1220 stimulated PV-FL translation in unstressed cells. Incubation with 50 or 100 μM Ars caused a decrease in translation of both capped FL and PV-FL RNA. However, translation of PV-FL was inhibited to a lesser degree in cells expressing eIF5B479-1220. When plotted as fold stimulation of translation over GFP-expressing control cells (Fig. 7B), expression of eIF5B479-1220 increased the relative translation activity of PV-IRES-FL RNA up to 3-fold in Ars-stressed cells. Interestingly, the stimulation or partial rescue of translation was maximal at 50 mM Ars, and eIF5B479-1220 expression did not overcome translation inhibition at the highest Ars levels (200 μM), though eIF2α phosphorylation did not further increase significantly. This suggests that at the highest levels, Ars may induce additional mechanisms that restrict translation other than through eIF2α phosphorylation. As expected (15), translation of control IGR-FL was not stimulated by HA-eIF5B479-1220 expression. Immunoblot analysis of cell lysates with eIF5B-specific antibody showed that the expression level of HA-eIF5B479-1220 was higher than endogenous eIF5B in these experiments and was unaltered by Ars treatment (Fig. 7C).
Fig. 7.

Expression of eIF5B479-1220 rescues PV IRES translation activity. (A) 293T cells expressing GFP or eIF5B479-1220 were electroporated with FL reporter RNAs and treated with 50, 100, and 200 μM Ars for 90 min, and then lysates were analyzed for FL activity. Data are an aggregate of three experiments, and error bars represent the standard errors. (B) Data are expressed as fold stimulation from eIF5B expression versus GFP expression. (C) Immunoblot analysis of eIF5B479-1220 expression and phospho-eIF2α in 293T cells electroporated with FL reporters. Blots used eIF5B-specific antibody and phospho-eIF2α antibody as indicated.
DISCUSSION
In this report, we show that enteroviruses can switch from an eIF2-dependent to an eIF2-independent mode of translation initiation during the course of infection. We show that although translation of enteroviral RNA is sensitive to sodium arsenite (Ars) treatment, translation initiation becomes resistant to inhibition by eIF2α phosphorylation and that this switch correlates with 3Cpro activity. Overall, the model of arsenite stress and high-level eIF2α phosphorylation employed in this study is not completely artificial, as PV infection induces stress and resulting eIF2α phosphorylation that reaches high levels late in infection (Fig. 1B). Investigation into the role of eIF5B cleavage by 3Cpro revealed that the C-terminal eIF5B cleavage fragment produced during enteroviral infection may play a role in rescuing viral IRES-mediated translation under restrictive conditions of eIF2α phosphorylation and near complete inhibition of host cell translation. The ability to cleave eIF5B is conserved among enteroviruses (13), thus providing an important mechanism to ensure continued virus translation after interferon activation of PKR or stress activation of HRI (Fig. 3E).
In previous work, we showed that PV infection results in the inhibition of the formation of cytoplasmic stress granules and that expression of cleavage-resistant G3BP rescued stress granule formation and inhibited viral replication approximately 7- to 8-fold (55). Since the kinetics of stress granule (SG) inhibition and G3BP cleavage correlate well with the observation of eIF2-independent viral translation (Fig. 1B), we expressed cleavage-resistant hamster G3BP, which has a proline residue in place of the glutamine residue at the P1′ position of the 3Cpro cleavage site, and monitored resistance to arsenite stress (Fig. 2). We observed an overall decrease in viral translation in the presence of G3BP; however, we did not observe a change in the kinetics of the development of eIF2 independence or the phosphorylation of eIF2α, indicating that the cleavage of G3BP does not play a role in the development of eIF2-independent translation and that eIF2 independence correlates better with 3Cpro activity. We also note that high-molecular-weight protein species appear in enterovirus-infected cells subjected to Ars stress (Fig. 1, 2, 3, 5, and 6) but not heat shock or Sal003 treatment (Fig. 3A and B). Given the direct inhibition of 3Cpro cleavage activity exerted by 1 mM Ars (Fig. 2D), the observed lack of processed viral proteins and the drastic inhibition of host translation that occurs during enteroviral infection (29), we propose that these high- molecular-weight proteins are unprocessed polyprotein forms.
Induction of the host antiviral response induces many changes in cell functions, with a crucial effect being the suppression of translation via the activation of the eIF2α kinase PKR and the subsequent phosphorylation of serine 51 of eIF2α (53, 58). Additionally, another eIF2α kinase, GCN2, provides an antiviral response against RNA viruses (7). Phosphorylation of eIF2α results in an increase in the affinity of eIF2 for its guanine exchange factor, eIF2B, turning eIF2 into a competitive inhibitor of eIF2B function (45), thereby restricting the availability of ternary complex (eIF2-GTP-tRNAiMet) and causing an inhibition of translation initiation (25). Several virus IRESs have recently been shown to be refractory to eIF2α phosphorylation, including cricket paralysis virus (CrPV), hepatitis C virus (HCV), and classical swine fever virus (CSFV) (41, 52, 56). However, despite the similarity in the use of noncanonical, nonscanning IRES elements for initiation, the requirements for initiation factors differ between these viral IRESs. The CrPV intergenic region IRES does not require cellular initiation factors (56), whereas the related HCV and CSFV IRES elements require eIF3 (41, 52). IRES elements of enteroviruses utilize several initiation factors, including eIF4G, eIF3, eIF4A, and eIF4B (14, 37). Our data suggest that enteroviral IRES elements require eIF2 only initially for optimal translation and can switch to an eIF2-independent mode of initiation as infection matures (Fig. 1 and 3). In agreement with previously published work (52), we show that the activation of different eIF2α kinases, HRI and PKR, results in the suppression of enteroviral IRES-containing RNA both in vitro and in vivo in the absence of viral infection (Fig. 4) but that the introduction of the C-terminal cleavage fragment of eIF5B produced by enterovirus 3Cpro can partly rescue translation initiation by the viral IRES (Fig. 7). This mechanism also departs from previously described mechanisms of eIF2-independent translation on the HCV IRES in that the PV and CVB3 IRES elements require scanning of the recruited 40S ribosome to reach the AUG start codon as opposed to direct placement of the AUG codon in the ribosomal P site.
eIF5B is the eukaryotic homologue of prokaryotic initiation factor 2 (IF2), and like IF2, it is required for ribosomal subunit joining during translation initiation (42). eIF5B also interacts with eIF1A, the eukaryotic homologue of IF1, to mediate efficient and stringent selection of the AUG start codon (11, 12). However, in contrast to IF2, eIF5B does not under normal conditions directly recruit the tRNAiMet to the ribosome, although the recruitment of eIF5B stabilizes the tRNAiMet in the ribosomal P site upon release of eIF2 (11, 46, 47). Archaeal initiation factor 5B (aIF5B) and N-terminally truncated eIF5B from Saccharomyces cerevisiae have been shown to interact with Met-tRNAiMet in solution, albeit less efficiently and with lower affinity than eIF2 (18). Intriguingly, Guillon et al. (18) showed that N-terminally truncated eIF5B can also interact with noninitiator tRNA. In the case of enterovirus infection, increasing levels of eIF5B in cells resulted in a modest stimulation of IRES-mediated translation. This was also reflected in a two- to threefold increase in virus titer from low-multiplicity infections in cells expressing eIF5B (data not shown). Enterovirus-mediated cleavage of eIF5B occurs upstream of the conserved functional domains located in the C terminus (13), producing a similar N-terminally truncated form of eIF5B. Here we show that this fragment has stimulatory effects on viral translation under conditions of restricted eIF2, although the exact mechanism by which translation is stimulated is unknown. N-terminally truncated eIF5B is known to be functional in the joining of ribosomal subunits during initiation in vitro (42) as well as in initiation of translation in cell-free translation extracts from eIF5B knockout yeast (27). Our data suggest that the de novo formation of a comparable C-terminal fragment in enterovirus-infected cells maintains its function and can act to suppress translation inhibition by phosphorylated eIF2α, potentially through the recruitment of Met-tRNAiMet or the use of alternative tRNA initiating at non-AUG start codons. This indicates that the removal of the N-terminal region of eIF5B produces a fragment that behaves more like prokaryotic IF2 and is significant in that it allows for enteroviral escape from a major obstacle of the antiviral response and successful replication.
Some of our data also suggest that the cleavage of eIF5B has an inhibitory effect on cap-dependent translation (Fig. 7A), giving the cleavage of eIF5B a 2-fold role in the enteroviral life cycle, as cleavage may further inhibit host cell translation in conjunction with eIF4G and poly(A)-binding protein (PABP) cleavage mediated by virus proteinases (29). It is also significant in that it provides another mechanism of eIF2-independent initiation separate from the recently described activity of ligatin and MCT-1/DENR (16, 48), which was only functional on RNA molecules that directly place the AUG start codon in the P site of the 40S ribosome. Thus, the cleavage of eIF5B provides a mechanism by which IRES-recruited 40S ribosomes that require scanning to reach the AUG start codon (such as PV) can initiate independently of eIF2. This mechanism, along with ligatin and MCT-1/DENR-mediated tRNA recruitment, may provide alternative mechanisms of IRES initiation that could also be utilized in other conditions where translation of IRES-containing cellular transcripts dominates and cap-dependent translation is inhibited, such as during apoptosis (data not shown). Further work is required to elucidate the mechanism by which eIF5B479-1220 mediates translation initiation, including determination of the tRNA utilized for initiation and the precision by which the modified translational machinery can correctly initiate at the primary AUG start codon. Since the rescue observed in our results was incomplete, modification of other factors may also be required. PV 2A proteinase may cause additional required modifications, since it not only cleaves eIF4GI to produce the C-terminal fragment that is required for optimal enteroviral IRES activity (14, 19), but it is also required for translation of viral RNA in the presence of phospho-eIF2α induced by PV infection alone (39). Additionally, eIF1 has been shown to destabilize initiation complexes formed in the absence of eIF2 by the CSFV IRES (41). Future work should determine whether the eIF1-eIF5B interaction occurs after eIF5B cleavage and can destabilize initiation complexes on enteroviral IRES elements. Together, these findings provide insight into an alternative mode of translation that allows for enteroviruses to evade the antiviral and stress responses and sustain high-level translation and replication.
ACKNOWLEDGMENTS
We are grateful to Mauro Costa-Mattioli for thoughtful discussions and providing Sal003. For gifts of plasmids, we thank Ilya Frolov for pREP-GFP-G3BP1, Bert Semler for pRL-CVB3-FL, Sunnie Thompson for pCMV-Rluc-IGR-FLuc, and Christopher Hellen for pcDNA3.1-HA-eIF5B, and we also thank Tom Dever for antibody against eIF5B.
This work was supported by NIH Public Health Service grant AI 50237. J.P.W. and L.C.R. were partly supported by NIH training grant T32 AI07471.
Footnotes
Published ahead of print on 22 June 2011.
REFERENCES
- 1. Allam H., Ali N. 2010. Initiation factor eIF2-independent mode of c-Src mRNA translation occurs via an internal ribosome entry site. J. Biol. Chem. 285:5713–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson P., Kedersha N. 2009. Stress granules. Curr. Biol. 19:R397–R398 [DOI] [PubMed] [Google Scholar]
- 3. Atlas R., Behar L., Elliott E., Ginzburg I. 2004. The insulin-like growth factor mRNA binding-protein IMP-1 and the Ras-regulatory protein G3BP associate with tau mRNA and HuD protein in differentiated P19 neuronal cells. J. Neurochem. 89:613–626 [DOI] [PubMed] [Google Scholar]
- 4. Barral P. M., et al. 2007. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81:3677–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barral P. M., et al. 2009. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity. Pharmacol. Ther. 124:219–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beckham C. J., Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berlanga J. J., et al. 2006. Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 25:1730–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Black T. L., Safer B., Hovanessian A., Katze M. G. 1989. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J. Virol. 63:2244–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonderoff J. M., Larey J. L., Lloyd R. E. 2008. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J. Virol. 82:9389–9399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boyce M., et al. 2005. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939 [DOI] [PubMed] [Google Scholar]
- 10a. Byrd M. P., Zamora M., Lloyd R. E. 2005. Translation of eukaryotic translation initiation factor 4GI (eLF4GI) proceeds from multiple mRNAs containing a novel cap-dependent internal ribosome entry site (IRES) that is active during poliovirus infection. J. Biol. Chem. 280:18610–18622 [DOI] [PubMed] [Google Scholar]
- 11. Choi S. K., Lee J. H., Zoll W. L., Merrick W. C., Dever T. E. 1998. Promotion of met-tRNAiMetbinding to ribosomes by yIF2, a bacterial IF2 homolog in yeast. Science 280:1757–1760 [DOI] [PubMed] [Google Scholar]
- 12. Choi S. K., et al. 2000. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol. Cell. Biol. 20:7183–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Breyne S., Bonderoff J. M., Chumakov K. M., Lloyd R. E., Hellen C. U. 2008. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 378:118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Breyne S., Yu Y., Unbehaun A., Pestova T. V., Hellen C. U. 2009. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc. Natl. Acad. Sci. U. S. A. 106:9197–9202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deniz N., Lenarcic E. M., Landry D. M., Thompson S. R. 2009. Translation initiation factors are not required for Dicistroviridae IRES function in vivo. RNA 15:932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dmitriev S. E., et al. 2010. GTP-independent tRNA delivery to the ribosomal P-site by a novel eukaryotic translation factor. J. Biol. Chem. 285:26779–26787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graber T. E., Holcik M. 2007. Cap-independent regulation of gene expression in apoptosis. Mol. Biosyst. 3:825–834 [DOI] [PubMed] [Google Scholar]
- 18. Guillon L., Schmitt E., Blanquet S., Mechulam Y. 2005. Initiator tRNA binding by e/aIF5B, the eukaryotic/archaeal homologue of bacterial initiation factor IF2. Biochemistry 44:15594–15601 [DOI] [PubMed] [Google Scholar]
- 19. Hambidge S. J., Sarnow P. 1992. Translational enhancement of the poliovirus 5′ noncoding region mediated by virus-encoded polypeptide-2A. Proc. Natl. Acad. Sci. U. S. A. 89:10272–10276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson R. J., Hellen C. U., Pestova T. V. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jan E., Kinzy T. G., Sarnow P. 2003. Divergent tRNA-like element supports initiation, elongation, and termination of protein biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 100:15410–15415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jan E., et al. 2001. Initiator Met-tRNA-independent translation mediated by an internal ribosome entry site element in cricket paralysis virus-like insect viruses. Cold Spring Harb. Symp. Quant. Biol. 66:285–292 [DOI] [PubMed] [Google Scholar]
- 23. Jones C., Ehrenfeld E. 1983. The effect of poliovirus infection on the translation in vitro of VSV messenger ribonucleoprotein particles. Virology 129:415–430 [DOI] [PubMed] [Google Scholar]
- 24. Kedersha N., et al. 2002. Evidence that ternary complex (eIF2-GTP-tRNAiMet)-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell 13:195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krishnamoorthy T., Pavitt G. D., Zhang F., Dever T. E., Hinnebusch A. G. 2001. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21:5018–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lancaster A. M., Jan E., Sarnow P. 2006. Initiation factor-independent translation mediated by the hepatitis C virus internal ribosome entry site. RNA 12:894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee J. H., Choi S. K., Roll-Mecak A., Burley S. K., Dever T. E. 1999. Universal conservation in translation initiation revealed by human and archaeal homologs of bacterial translation initiation factor IF2. Proc. Natl. Acad. Sci. U. S. A. 96:4342–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee J. H., et al. 2002. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc. Natl. Acad. Sci. U. S. A. 99:16689–16694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lloyd R. 2006. Translational control by viral proteinases. Virus Res. 119:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marintchev A., Kolupaeva V. G., Pestova T. V., Wagner G. 2003. Mapping the binding interface between human eukaryotic initiation factors 1A and 5B: a new interaction between old partners. Proc. Natl. Acad. Sci. U. S. A. 100:1535–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mazroui R., et al. 2006. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 17:4212–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McEwen E., et al. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280:16925–16933 [DOI] [PubMed] [Google Scholar]
- 33. McInerney G. M., Kedersha N. L., Kaufman R. J., Anderson P., Liljeström P. 2005. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 16:3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meurs E. F., et al. 1992. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor-2 and partial resistance to encephalomyocarditis virus growth. J. Virol. 66:5805–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montero H., Rojas M., Arias C. F., Lopez S. 2008. Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J. Virol. 82:1496–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrison J. M., Racaniello V. R. 2009. Proteinase 2Apro is essential for enterovirus replication in type I interferon-treated cells. J. Virol. 83:4412–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ochs K., et al. 2002. Interaction of translation initiation factor eIF4B with the poliovirus internal ribosome entry site. J. Virol. 76:2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ochs K., et al. 2003. Impaired binding of standard initiation factors mediates poliovirus translation attenuation. J. Virol. 77:115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Neill R. E., Racaniello V. R. 1989. Inhibition of translation in cells infected with a poliovirus 2Apro mutant correlates with phosphorylation of the alpha subunit of eucaryotic initiation factor 2. J. Virol. 63:5069–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ortega A. D., Willers I. M., Sala S., Cuezva J. M. 2010. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J. Cell Sci. 123:2685–2696 [DOI] [PubMed] [Google Scholar]
- 41. Pestova T. V., de Breyne S., Pisarev A. V., Abaeva I. S., Hellen C. U. 2008. eIF2-dependent and eIF2-independent modes of initiation on the CSFV IRES: a common role of domain II. EMBO J. 27:1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pestova T. V., et al. 2000. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403:332–335 [DOI] [PubMed] [Google Scholar]
- 43. Piotrowska J., et al. 2010. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 84:3654–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robert F., et al. 2006. Initiation of protein synthesis by hepatitis C virus is refractory to reduced eIF2.GTP.Met-tRNAiMet ternary complex availability. Mol. Biol. Cell 17:4632–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rowlands A. G., Panniers R., Henshaw E. C. 1988. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 263:5526–5533 [PubMed] [Google Scholar]
- 46. Shin B. S., et al. 2011. Structural integrity of alpha-helix H12 in translation initiation factor eIF5B is critical for 80S complex stability. RNA 17:687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shin B. S., et al. 2002. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111:1015–1025 [DOI] [PubMed] [Google Scholar]
- 48. Skabkin M. A., et al. 2010. Activities of ligatin and MCT-1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev. 24:1787–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith J. A., et al. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spriggs K. A., Stoneley M., Bushell M., Willis A. E. 2008. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell 100:27–38 [DOI] [PubMed] [Google Scholar]
- 51. Sudhakar A., et al. 2000. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2 alpha) promotes complex formation between eIF2 alpha(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 39:12929–12938 [DOI] [PubMed] [Google Scholar]
- 52. Terenin I. M., Dmitriev S. E., Andreev D. E., Shatsky I. N. 2008. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 15:836–841 [DOI] [PubMed] [Google Scholar]
- 53. Thomis D. C., Samuel C. E. 1992. Mechanism of interferon action-autoregulation of RNA-dependent P1/eIF-2alpha protein kinase (PKR) expression in transfected mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 89:10837–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Verlinden Y., Cuconati A., Wimmer E., Rombaut B. 2000. The antiviral compound 5-(3,4-dichlorophenyl) methylhydantoin inhibits the post-synthetic cleavages and the assembly of poliovirus in a cell-free system. Antiviral Res. 48:61–69 [DOI] [PubMed] [Google Scholar]
- 55. White J. P., Cardenas A. M., Marissen W. E., Lloyd R. E. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305 [DOI] [PubMed] [Google Scholar]
- 56. Wilson J. E., Pestova T. V., Hellen C. U. T., Sarnow P. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511–520 [DOI] [PubMed] [Google Scholar]
- 57. Wilson S. A., et al. 1999. Cloning and characterization of hIF2, a human homologue of bacterial translation initiation factor 2, and its interaction with HIV-1 matrix. Biochem. J. 342(Part 1):97–103 [PMC free article] [PubMed] [Google Scholar]
- 58. Wu S. Y., Kaufman R. J. 1997. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem. 272:1291–1296 [DOI] [PubMed] [Google Scholar]
- 59. Yamasaki S., Anderson P. 2008. Reprogramming mRNA translation during stress. Curr. Opin. Cell Biol. 20:222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]