

Abstract
Several previously known transcription cofactors have been demonstrated in vitro recently to be histone acetyltransferases and deacetyltransferases, suggesting that remodeling of chromatin through histone acetylation plays a fundamental role in gene regulation. Clear evidence has not yet been obtained, however, to demonstrate that histone acetylation is required for gene activation in vivo. In this study we performed an alanine-scan mutagenesis through the HAT (histone acetyltransferase) domain identified previously by deletion mapping in recombinant yeast Gcn5. We identified multiple substitution mutations that eliminated completely Gcn5’s ability to potentiate transcriptional activation in vivo. Strikingly, each of these mutations was also critical for free and nucleosomal histone acetylation by Gcn5 functioning within the native yeast HAT complexes, Ada, and SAGA. Moreover, the growth phenotypes of these mutations as measured by colony size and liquid growth assay closely tracked transcription and HAT activities. In contrast, mutations that did not affect in vivo function of Gcn5 were able to acetylate histones. These data argue strongly that acetylation is required for gene regulation by Gcn5 in vivo, and support previous arguments that nucleosomal histones are among the physiological substrates of acetylation by Gcn5.
Gene expression is a highly regulated process at the level of transcriptional activation. Recruitment of the transcriptional basal machinery by activators and cofactors has been shown to be an important component of gene regulation in vitro (Zawel and Reinberg 1995) and in vivo (Struhl 1995). Transcriptional activator proteins facilitate association of components of the basal machinery with the TATA box and start site of transcription (Triezenberg 1995). Under physiological conditions, DNA is complexed with histones forming nucleosomes that assemble into higher order chromatin (Wolffe 1992). Chromatin assembly strongly inhibits transcription (Owen-Hughes and Workman 1994; Paranjape et al. 1994), probably by blocking binding of transcription factors to their cognate DNA-binding sites. Therefore, in vivo, transcription factors must overcome the DNA access problem posed by nucleosomes. Moreover, genetic manipulations of histones confirm their important role in regulation of transcription (Grunstein 1990).
Both transcriptional activators and cofactors have critical roles in altering the repressive chromatin structure to promote binding of basal factors. Activation domains have been shown to be involved in remodeling chromatin (Svaren et al. 1994), and nucleosome structural changes occur in the absence of transcription suggesting that activators may induce transcription by causing chromatin changes before initiation (Fascher et al. 1993). Transcriptional cofactors also activate or repress transcription by destablizing or stablizing the repressive nucleosome structure (Kingston et al. 1996). For example, the SWI/SNF family of protein complexes activates transcription by disrupting the nucleosome structure in an ATP-dependent manner to increase the binding of transcription factors (Cote et al. 1994; Peterson et al. 1994).
Nucleosomes are composed of ∼146 bp of DNA wrapped around the histone octamer, consisting of two molecules each of the histones H2A, H2B, H3, and H4 (Wolffe 1992). Each core histone contains a globular domain and extended basic amino-terminal tail that stabilizes histone octamer association with DNA through ionic interactions. Covalent modifications of nucleosomal histones have been correlated with modulation of chromatin structure and function (Bradbury 1992). These modifications include phosphorylation, ubiquitination and, in particular, acetylation of ε amino groups of lysines in the histone tails. Acetylation is associated with both gene activation (hyperacetylation) and gene silencing (hypoacetylation) (for review, see Loidl 1994; Turner and O’Neill 1995). Acetylation of histones increases the affinity of transcription factors for nucleosomal DNA (Lee et al. 1993; Vettese-Dadey et al. 1996).
The recent identification of histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes as previously recognized transcription cofactors (Pazin and Kadonaga 1997; Wade and Wolffe 1997) suggests a direct mechanistic connection between the activity of transcription factors and changes in chromatin structure that precede initiation. The first nuclear HAT enzyme, p55, was purified and cloned from Tetrahymena, and was found to be closely related to yeast Gcn5 (Brownell et al. 1996), a known transcription coactivator/adaptor (Guarante 1995). The ability of yeast Gcn5, and its binding partner yeast Ada2, to associate with activation domains (Barlev et al. 1995; Chiang et al. 1996; Silverman et al. 1994) and with the TATA-binding protein (TBP; Barlev et al. 1995), suggests that acetylation of nucleosomal histones through activation domain recruitment of Gcn5 may be a fundamental and novel feature of gene regulation. Supporting this notion were subsequent revelations that several other transcription coactivators/adaptors possess intrinsic HAT activity, including additional Gcn5 family members [hGcn5 (Wang et al. 1997; Yang et al. 1996) and P/CAF (Yang et al. 1996)] as well as unrelated CBP/p300 (Bannister and Kouzarides 1996; Ogryzko et al. 1996) and TAFII250 (Mizzen et al. 1996).
Conversely, the first HDAC was identified (Taunton et al. 1996) and strikingly, it also was a previously known transcription factor, Rpd3 (Vidal and Gaber 1991), possessing transcriptional repression activity. Several other transcriptional repressors have been shown to be HDACs, including Sin3 (Hassig et al. 1997; Kadosh and Struhl 1997; Laherty et al. 1997; Zhang et al. 1997) and the corepressor N-CoR/SMRT (Alland et al. 1997; Heinzel et al. 1997). Despite these provocative observations, however, direct evidence has not yet been obtained that acetylation or deacetylation of histones has a direct role in gene regulation.
Although recombinant proteins have been tested for histone acetylation and deacetylation activity, recent studies demonstrate that, in vivo, the HATs (Grant et al. 1997; Horiuchi et al. 1997; Saleh et al. 1997) and HDACs (Rundlett and Grunstein 1996; Zhang et al. 1997) function as components of high-molecular-weight complexes. For example, yeast Gcn5 is the catalytic HAT subunit present in two complexes, termed Ada and SAGA (Spt, Ada, Gcn5 acetyltransferase; Grant et al. 1997). The Ada complex also contains Ada2 and Ada3, which were similarly isolated as suppressors of growth toxicity caused by overexpression of the potent chimeric activator composed of the yeast Gal4 DNA-binding domain fused to the herpes virus VP16 activation domain (Berger et al. 1992; Piña et al. 1993; Marcus et al. 1994). In addition to the Ada proteins, the SAGA complex contains Spt7 (Gansheroff et al. 1995) and Spt20 (Roberts and Winston 1996) proteins, which were isolated as suppressors of Ty element insertions. Interestingly, Spt20/Ada5 was identified in both Ada and Spt genetic selections (Marcus et al. 1996; Roberts and Winston 1996) and is present in the SAGA complex (Grant et al. 1997). The importance of determining acetylation activity within these native complexes is illustrated by the observation that recombinant yeast Gcn5 acetylates only free histones, whereas, in association with Ada and SAGA complexes, Gcn5 acetylates nucleosomal histones (Grant et al. 1997). In addition, genetic studies demonstrate that SAGA contains functions involved in transcriptional activation distinct from those mediated by the Gcn5/Ada2/Ada3 group (Roberts and Winston 1997), underscoring further the importance of testing HAT activity in the context of native complexes.
The boundaries of the HAT domain in recombinant yeast Gcn5 were mapped and the region was shown to be required for Gcn5’s role in transcriptional activation in vivo (Candau et al. 1997). This region of the Gcn5 family members Tetrahymena p55 (Brownell et al. 1996), yeast Gcn5 (Georgakopoulos and Thireos 1992), human Gcn5 (Candau et al. 1996), and human P/CAF (Yang et al. 1996) is remarkably conserved in primary sequence (Brownell et al. 1996). Certain sequence motifs have been identified in these HAT domains and are found in other acetyltransferases (Reifsnyder et al. 1996; Neuwald and Lansman 1997). Paradoxically, the other transcriptional coactivators possessing intrinsic HAT activity, CBP/p300 and TAFII250, display no obvious sequence homology within the putative HAT domain compared with the Gcn5 family (Bannister and Kouzarides 1996; Mizzen et al. 1996; Ogryzko et al. 1996). In fact, recent data demonstrates that, in addition to histones, p300 acetylates the transcriptional activator p53, and, surprisingly, with nearly equal strength (Gu and Roeder 1997).
The data described above raise important questions—what are the critical residues for acetylation of histone substrates in Gcn5 and are these residues required for Gcn5’s role in transcriptional activation? If so, this would argue that first, acetylation is a critical and direct determinant of gene regulation in vivo, and second, that histones are likely to be a critical physiological substrate. In the current study, we have undertaken alanine-scan mutagenesis to address these issues. The role of specific mutations was tested on Gcn5 functioning within the context of native yeast Ada and SAGA complexes. Our results demonstrate clearly that critical residues for histone acetylation in Gcn5 are also required for its in vivo function in growth and as a transcription cofactor. These data provide compelling evidence that acetylation by Gcn5 is crucial for transcriptional activation in vivo.
Results
Construction of alanine-scan mutants within the putative HAT domain of Gcn5
The four Gcn5 family members that have been identified are yeast Gcn5, human Gcn5, human P/CAF, and Tetrahymena p55. Although the overall sequence identity between the four proteins is <40%, the amino acid sequence in the putative HAT domain is 67% identical and 82% similar (Fig. 1A). The primary structural similarity is reflected in conservation of function between the human and yeast HAT domains, whereas no other domain of human Gcn5 is capable of function in yeast (Wang et al. 1997). The HAT domain of Gcn5 can be divided into four subdomains, I, II, III, and IV (Brownell et al. 1996), based on exceptionally strong sequence identity between all family members (Fig. 1A).
Figure 1.

Amino acid sequence within the putative HAT domains of the Gcn5 family. (A) Sequence comparison between the putative HAT domains of yeast Gcn5, human P/CAF, human Gcn5 and Tetrahymena p55. The protein sequence within the putative HAT domains are shown by single amino acid letter-code. Numbers at left indicate amino acid residues. The highly conserved regions I–IV (Brownell et al. 1996) are indicated at the top. Solid boxes indicate amino acid identity and shaded boxes indicate similarity. (B) Yeast Gcn5 substitution mutants created in the study. The substitution mutants are named according to the group of amino acids mutated to alanine, each of which is indicated by brackets.
To identify critical amino acids for HAT enzymatic activity and to determine whether HAT activity is crucial for Gcn5’s role in transcription and growth, an alanine-scan mutagenesis was performed. Three or four adjacent amino acids were grouped (indicated as three or four amino acid letter code) and mutagenized to alanine (Fig. 1B) using site-directed mutagenesis. The mutagenic oligonucleotides (Table 1) contained a PstI restriction site allowing initial positive identification of each mutant, which was confirmed by DNA sequencing. Thirty-one substitution mutants were created and their phenotypes studied.
Table 1.
Sequences of mutagenic oligonucleotides
| Namea
|
Amino acidb
|
Sequence of oligonucleotides
|
|---|---|---|
| LKN | 120 | GATGGTCCTAACTGGAGCTGCAGCCATTTTTCAAAAGCAATTACC |
| IFQ | 123 | CTAACTGGATTAAAAAACGCTGCAGCAAAGCAATTACCAAAAATG |
| KQL | 126 | GGATTAAAAAACATTTTTCAAGCTGCAGCACCAAAAATGCCC |
| PKM | 129 | GGATTAAAAAACATTTTTCAAAAGCAATTAGCTGCAGCGCCCAAAGAATACATTGCC |
| PKE | 132 | CAATTACCAAAAATGGCTGCAGCATACATTGCCAGGTTAG |
| RLV | 135 | ATGCCCAAAGAAGCTGCAGCCAGGTTAGTCTATG |
| YIA | 138 | CAAAGAATACATTGCCGCTGCAGCCTATGATCGAAGTCATC |
| YDR | 141 | GCCAGGTTAGTCGCTGCAGCAAGTCATCTTTCCATG |
| SHL | 144 | GTTAGTCTATGATCGAGCTGCAGCTTCCATGGCTGTCATTAGG |
| GGI | 159 | CGATAAGAGAGAAGCCGCAGCAATTGTTTTCTGTGCC |
| FAE | 171 | CCATTGACTGTCGTAGCTGCAGCAACATATCGACCTTTCG |
| IVF | 174 | GAATTCGCAGAAGCTGCAGCCTGTGCCATCAGTTCG |
| CAI | 177 | CGCAGAAATTGTTTTCGCTGCAGCCAGTTCGACGGAACAG |
| EQV | 183 | GCCATCAGTTCGACGGCTGCAGCACGCGGTTATGGTGCG |
| RGY | 186 | GGAACAGGTAGCTGCAGCTGGTGCGCATC |
| GAHL | 189 | GGTACGCGGTTATGCTGCAGCTGCAATGAATCACTTAAAAGAC |
| MNHL | 193 | GGTTATGGTGCGCATCTAGCGGCTGCAGCAAAAGACTATGTTAG |
| KDY196 | 196 | GAATCACTTAGCTGCAGCTGTTAGAAATACC |
| NIK | 205 | GTTAGAAATACCTCGGCTGCAGCATATTTTTTGACATATGC |
| YFL | 208 | CCTCGAACATAAAAGCTGCAGCGACATATGCAGATAATTAC |
| TYA | 211 | CGAACATAAAATATTTTTTGGCTGCAGCAGATAATTACGC |
| DNY | 214 | TATTTTTTGACATATGCAGCTGCAGCCGCTATTGGATACTTT |
| AIGY | 217 | GCAGATAATTACGCTGCTGCAGCCTTTAAAAAGCAAGGC |
| FKK | 221 | TACGCTATTGGATACGCTGCAGCGCAAGGCTTCACTAA |
| QGF | 224 | CGCTATTGGATACTTTAAAAAGGCTGCAGCCACTAAAGAAATCACGTTGG |
| TKEI | 227 | AAAAAGCAAGGCTTCGCTGCAGCAGCCACGTTGGATAAAAG |
| GYI | 239 | GTATATGGATGGCTGCAGCTAAAGATTATGAAGG |
| KDY242 | 242 | GGATGGGATATATTGCTGCAGCTGAAGGTGGT |
| EGG | 245 | GGATATATTAAAGATTATGCTGCAGCTACGCTGATGCAATG |
| TLM | 248 | GATTATGAAGGTGGTGCTGCAGCGCAATGTTCTATGTTACC |
| PRI | 256 | GCAATGTTCTATGTTAGCTGCAGCACGATATTTGGACGC |
The name of each mutant represents the amino acids mutagenized.
The position of the first amino acid mutagenized in each mutant is indicated.
Growth complementation by Gcn5 substitution mutants in the gcn5− strain
The ability of the mutant GCN5 genes to complement phenotypes caused by GCN5 disruption in yeast was tested. Yeast lacking GCN5 grow poorly on minimal synthetic media, and transformation of wild-type GCN5 completely complements the growth defect (Georgakopoulos and Thireos 1992; Marcus et al. 1994). Complementation by the Gcn5 substitution mutants was tested by transforming plasmids bearing each of the mutant genes into the gcn5− strain and assessing growth using colony size (Fig. 2A) and doubling time (Fig. 2B). The mutant genes were expressed using the natural promoter and terminator of GCN5 to obtain normal levels of protein expression. The majority of Gcn5 substitution mutants (19 of the 30 mutants shown, as well as GGI located between regions I and II) complemented growth as well as wild-type Gcn5, as the yeast transformants yielded colony sizes that were indistinguishable from wild type (Fig. 2A). Several mutants (YIA, RGY, DNY, KDY242, and TLM) displayed variable intermediate colony sizes, ranging between that achieved by yeast bearing wild-type GCN5 or a disruption of the gene (Fig. 2A). Six mutants (KQL, PKM, FAE, KDY196, AIGY, and FKK) completely failed to complement the colony growth defect in the gcn5− strain (Fig. 2A).
Figure 2.
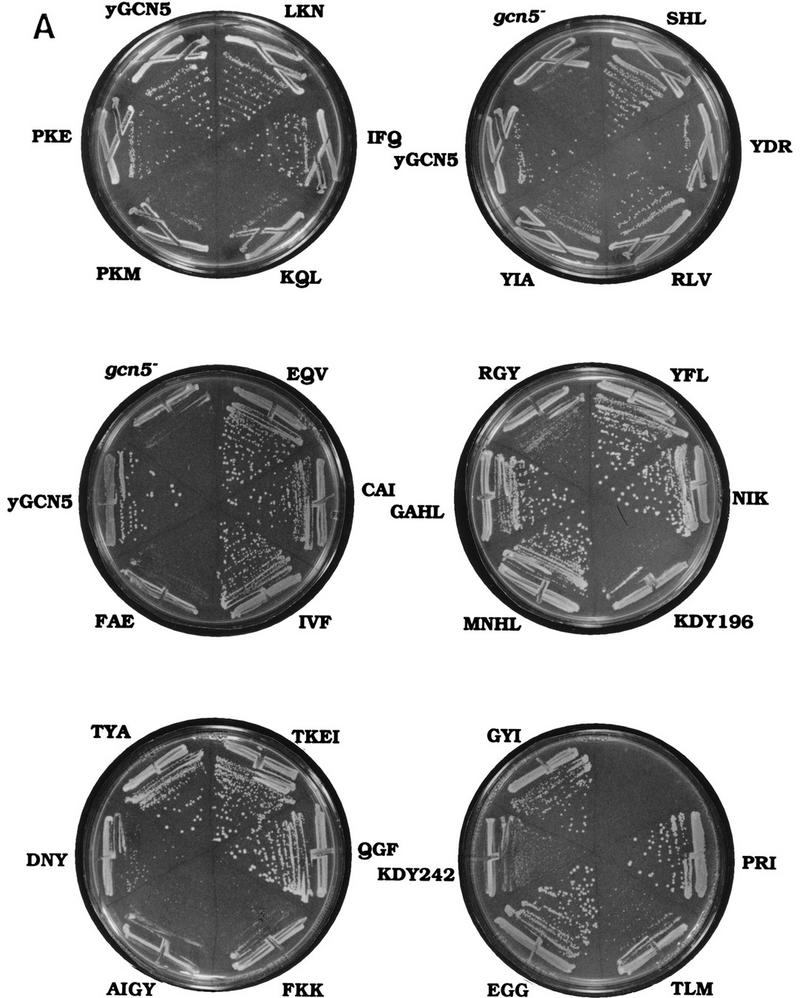

Growth phenotypes of GCN5 substitution mutants in the gcn5− strain. Wild-type GCN5, each of the GCN5 mutants, or vector alone were introduced into gcn5− strain. (A) Colony growth assay. Transformants were streaked onto synthetic minimal medium. Colony growth was assessed after 2 days of growth at 30°C. (B) Doubling time of the Gcn5 substitution mutants. Aliquots of cultures in liquid synthetic minimal medium were taken every 2 hr and the A600 of the cultures was measured. The generation time was calculated as the OD doubling time during exponential growth.
Relative growth phenotypes were quantitated by measuring doubling times in liquid minimal growth media. The Gcn5 mutants that exhibited the poorest growth in the colony assay were analyzed, as well as several representative mutants that exhibited wild-type growth. The results paralleled the colony assays because the most defective mutants (KQL, PKM, FAE, KDY196, AIGY, and FKK) grew at essentially the rate of the gcn5− strain, and the wild-type mutants grew comparably with GCN5+ (Fig. 2B). Furthermore, the RGY mutant that showed intermediate-sized colonies was also intermediate in its rate of doubling (Fig. 2B). Based on the two growth complementation assays, we tentatively grouped the mutants into three classes—those that were essentially wild type, those that exhibited significant loss of growth complementation, and those that were completely defective.
The failure of Gcn5 mutants to complement growth may be caused by defective function of the Gcn5 substitution mutants, or alternatively, by structural defects in the protein causing either instability or inability to form requisite protein–protein interactions in vivo. To address the question of protein stability, the expression levels of the mutant Gcn5 proteins in yeast were determined relative to wild-type Gcn5. Gcn5 was expressed under control of the galactose-inducible GAL1-10 promoter and intracellular protein expression levels were determined by Western analysis (Fig. 3, top). Although there was variation in protein levels, all Gcn5 mutants were present at levels comparable with wild-type Gcn5, and, in particular, all mutants displaying partial or complete defects in the growth assay were detectable in vivo (Fig. 3, top).
Figure 3.

Stability and Ada2 interaction of Gcn5 substitution mutants in yeast. Wild-type GCN5, mutant GCN5, or vector alone were cotransformed with GST–ADA2, and proteins were induced using the GAL1-10 promoter. (Top) Gcn5 protein levels in the yeast extracts were determined by Western blot analysis using Gcn5 antisera. (Bottom left) The yeast extracts were incubated with GST beads to allow GST–Ada2 to bind. Wild-type or mutant Gcn5 bound to GST–Ada2 was determined by Western analysis using Gcn5 antiserum. (Bottom right) Co-expression of wild-type Gcn5 with GST alone was used as a negative control.
To determine the structural integrity of the mutant proteins in vivo, we tested interaction with Ada2, as our previous results demonstrated that Ada2 interaction is critical for Gcn5 function in vivo (Candau et al. 1997). Yeast ADA2 was fused to glutathione-S-transferase (GST) followed by cotransformation into yeast of expression plasmids bearing GST–ADA2 and GCN5. Yeast extracts were incubated with GST beads and the binding of wild-type Gcn5 or Gcn5 mutants to GST–Ada2 was determined by immunoblot. The Gcn5 mutants were able to bind to Ada2 and importantly, mutants defective for growth complementation also interacted with Ada2 (Fig. 3, bottom). Overall, the yeast expression experiments and Ada2-interaction results indicate that the defective phenotypes of the substitution mutants were not due to global structural defects.
Transcriptional activation in the presence of Gcn5 mutants in the gcn5− strain
The ability of Gcn5 substitution mutants to support transcriptional activation in the gcn5− strain was tested. Full transcriptional potency by certain transcriptional activators requires Gcn5 (Georgakopoulos and Thireos 1992; Marcus et al. 1994). Two chimeric activators were tested. The first was the yeast Gal4 DNA-binding domain fused to the potent activation domain of herpes simplex virus activator VP16 (Gal4–VP16), and the second was the bacterial LexA DNA binding domain fused to the activation domain from the yeast activator Gcn4 (LexA–Gcn4). The expression reporters were bacterial LacZ driven by yeast Gal4 DNA-binding sites or bacterial LexA DNA-binding sites, and transcriptional potency was determined by quantitative β-galactosidase assays.
Both Gal4–VP16 and LexA–Gcn4 transcriptional activity were reduced in the GCN5 deletion strain to ∼9% of wild type (Fig. 4). The Gcn5 substitution mutants divided into three classes. Most Gcn5 mutants were capable of potentiating Gal4–VP16 or LexA–Gcn4 activity to approximately the level observed in the GCN5+ strain (Fig. 4). Several mutants that displayed partial complementation in the growth assay also showed an intermediate level of activity in potentiating transcriptional activation by both activators (Fig. 4). The six mutants (KQL, PKM, FAE, KDY196, AIGY, and FKK) that were unable to complement the growth defect of gcn5−, also failed to complement gcn5− function in the transcription assay for either activator (Fig. 4).
Figure 4.

Transcriptional activation in the presence of Gcn5 substitution mutants. GAL4–VP16 (left panel) and LexA–GCN4 (right panel) were cotransformed with wild-type GCN5, each of the GCN5 mutants or vector alone, along with the appropriate LacZ reporters. β-Gal activities of the mutants are shown as a percentage of wild-type Gcn5, which was set at 100%. (Solid bars) Mutants that were defective in the growth and transcription assays; (dark gray bars) mutants having intermediate phenotypes; (light gray bars) mutants having activity similar to wild-type Gcn5. Error bars represent the standard error about the mean from three independent experiments.
Therefore, the majority of the Gcn5 substitution mutants had similar phenotypes exhibited by wild-type Gcn5 in growth and transcription assays (hereafter referred to as wild-type mutants; light gray bars in Fig. 4). The assays identified six Gcn5 mutants (KQL, PKM, FAE, KDY196, AIGY, and FKK) that profoundly reduced Gcn5 function in vivo (“defective” mutants; black bars in Fig. 4). In addition, several mutants (YIA, RGY, DNY, KDY242, and TLM) displayed various degrees of partial function and are likely to represent important, but not crucial, residues for Gcn5 activity (“intermediate” mutants; dark gray bars in Fig. 4).
Dominant-negative effect of Gcn5 mutants
Mutations in catalytic site residues may cause mutant proteins to exert dominant-negative effects when overexpressed in vivo. Dominant-negative phenotypes are likely caused by the mutant enzyme competing with the wild-type counterpart for association with other proteins or assembly into protein complexes. To determine whether the phenotypically defective Gcn5 mutants exert a dominant defective growth effect in vivo, each was overexpressed in yeast containing normal levels of wild-type Gcn5.
Overexpression of wild-type Gcn5 itself caused yeast to grow slowly compared with transformed vector (Fig. 5), perhaps attributable to sequestration of associated proteins (Gill and Ptashne 1988). Overexpression of the wild-type Gcn5 mutants showed similar phenotypes as wild-type Gcn5 (data not shown) and were therefore judged not to be dominant-negative. Overexpression of most defective Gcn5 mutants caused even stronger growth inhibition relative to wild-type Gcn5 (Fig. 5). This was true both of mutants that were completely (KQL, PKM, FAE, AIGY, and FKK) or partially (YIA, RGY, and KDY242) defective (Fig. 5). Two defective mutants (completely defective KDY196 and partially defective TLM) exhibited a similar phenotype as wild-type Gcn5 (Fig. 5) and therefore were not dominant-negative. The mutants are likely to exert their dominant-negative effects by incorporation into normal protein complexes (Grant et al. 1997) because first, they are vastly overexpressed relative to wild-type Gcn5, and second, they interact normally with Ada2 (Fig. 3) and within native complexes (Fig. 6B,C) in the gcn5− strain where wild-type Gcn5 is not present.
Figure 5.

Dominant-negative effects of Gcn5 HAT mutants. Wild-type GCN5, the GCN5 mutants, or vector alone were transformed into yeast strain bearing wild-type GCN5. The transformants were restreaked onto galactose plates to induce expression of transformed Gcn5 proteins and grown at 25°C for 6 days.
Figure 6.



Free histone HAT activity of Gcn5 substitution mutants in SAGA and Ada complexes. (A) SAGA and Ada complexes prepared from Gcn5 mutants displaying wild-type activity in functional assays (light gray bars in Fig. 4) and Gcn5-defective mutants (solid bars in Fig. 4) were tested for HAT activity. SAGA and Ada complexes of each mutants containing peak Gcn5 and Ada2 proteins detected by Western blots (B,C) were used. Complexes were incubated with free core histones and [3H]acetyl CoA. Reaction mixtures were then subjected to liquid scintillation accounting (top) as well as SDS-PAGE and fluorography (middle and bottom). The activity from SAGA complexes (middle) and Ada complexes (bottom) are indicated by arrows. Quantitation of corresponding scintillation assays for SAGA is shown as a percentage of wild-type Gcn5. Error bars represent the standard error about the mean from three independent experiments. Western blots of Gcn5 and Ada2 proteins from SAGA (B) and Ada (C) complexes. Peak fractions from SAGA and Ada complexes were detected for the presence of Gcn5 and Ada2 using immunoblot analysis using Gcn5 and Ada2 antisera.
Therefore, two lines of evidence indicate that the Gcn5 mutants displaying defects in vivo are functional. First, all of the defective mutants possess structural integrity as manifested by their ability to interact with Ada2 (Fig. 4), and second, the majority of the defective mutants exhibit dominant negativity in vivo (Fig. 5).
Histone acetylation by Gcn5 substitution mutants within Ada and SAGA complexes
Previous studies have tested HAT activity of recombinant proteins. The HATs and HDACs are active within high-molecular-weight complexes, however, which influences their specificity (Rundlett and Grunstein 1996; Grant et al. 1997). To directly assay whether the defective phenotypes of the substitution mutants were caused by loss of ability to acetylate histones, HAT activity of the Gcn5 substitution mutants was tested in the context of high-molecular-weight native HAT complexes (Grant et al. 1997). Two high-molecular-weight native yeast complexes have been identified. Both contain Gcn5 as the HAT catalytic subunit, and specifically acetylate histone H3 and, to a lesser extent, histone H2B. The smaller complex (800 kD) was named Ada and the larger one (2 MD) SAGA, based on the identity of their constituent proteins (Grant et al. 1997). We partially purified Ada and SAGA complexes from yeast expressing the Gcn5 substitution mutants, to determine whether the mutations affected the HAT activity within these physiological HAT complexes.
Wild-type Gcn5 or the substitution mutants were transformed into the gcn5− strain using vectors to express normal levels of protein. Whole-cell extracts were prepared from 26 out of the 31 mutants created and were fractionated through Ni2+ agarose and MonoQ ion exchange resins (Grant et al. 1997). HAT activities were determined on free histone substrates for 22 out of these 26 extracts, which were determined by Western analysis to have nearly normal Gcn5/Ada2 protein levels in both the Ada and SAGA complexes (see below). These included 11 mutants that were partially or completely defective in growth (Fig. 2) and transcription (Fig. 4), as well as 11 wild-type mutants.
The HAT activity of the peak SAGA fraction from wild-type or mutant Gcn5 was then determined by liquid HAT assay quantitation of [3H]acetyl group incorporation into histones using scintillation counting and visualization of histones by PAGE and fluorography. SAGA fractions derived from strains bearing wild-type mutant Gcn5 were able to acetylate histones, as shown by the examples of LKN, PKE, YDR, GAHL, and GYI (Fig. 6A). Four of these had levels as high as, or nearly as high as, wild-type Gcn5 (LKN, PKE, YDR, and GYI) as quantitated by scintillation counting (Fig. 6A, top) and confirmed by visualization of [3H]acetyl incorporation into histone H3 (Fig. 6A, middle). These mutant complexes displayed Gcn5 and Ada2 protein levels that were comparable with wild-type Gcn5 (Fig. 6B, top). The remaining wild-type mutant (GAHL) possessed >50% of wild-type activity (Fig. 6A) and was found to be somewhat reduced in protein level compared with wild-type Gcn5 and the other wild-type mutants (Fig. 6B, top; data not shown). In addition, visualization of [3H]acetyl incorporation into histone H3 catalyzed by SAGA bearing GAHL indicated clearly higher activity than the defective Gcn5 mutants (Fig. 6A, middle).
The six severely defective mutants (KQL, PKM, FAE, KDY196, AIGY, and FKK), as well as partially defective RGY, displayed greatly reduced ability to acetylate histones, using both the quantitative assay as well as histone visualization (Fig. 6A, top and middle). Five of the extracts showed background levels of HAT activity (∼25% of wild-type level). The level of acetylation by SAGA prepared from the defective FKK mutant was 45% of wild type in the quantitative assay, but was nearly as low as background levels by [3H]-histone fluorography. (As is obvious in Fig. 6, we consistently observe that fluorography is a more sensitive measure of acetylation than scintillation quantitation.) The protein levels of both Ada2 and the defective mutant Gcn5 proteins were variable, but the levels were similar to those in the strain expressing wild-type Gcn5 (Fig. 6B, bottom). Therefore, consistent with the stability assay (Fig. 3), the defects of these mutants were not attributable to lack of protein expression, or importantly, inability to be incorporated into the SAGA complex.
We then determined the HAT activity of the Ada complexes using the identical set of mutant Gcn5 proteins as used for assay of the SAGA complexes. In the case of the Ada complex only [3H]histone-H3 fluorography was used to determine HAT activity, as a strong overlapping Gcn5-independent histone H4 acetylation activity (Grant et al. 1997; see Fig. 7) obscured quantitation by scintillation counting of the Gcn5-dependent histone H3 activity. As observed for the SAGA complex, each of the wild-type mutants displayed activities comparable with wild-type Gcn5, whereas HAT activity by the defective mutants was not detectable (Fig. 6A, bottom). Western analysis of Ada2 and Gcn5 in the Ada complex (Fig. 6C) of the wild-type (Fig. 6C, top) and defective (Fig. 6C, bottom) Gcn5 mutants indicated comparable levels of protein with wild-type Gcn5. Note that the Ada complex containing the GAHL mutant eluted primarily in fraction 20 from the MonoQ column (Fig. 6C, top left), whereas the remainder of the wild-type mutants eluted in a broader peak covering fractions from 20 (Fig. 6C, top left) to 24 (Fig. 6C, top right).
Figure 7.

Nucleosomal HAT activities of Gcn5 substitution mutants in Ada complexes. The same wild-type mutants and defective mutants used in the free histone assays (Fig. 6) were subjected to nucleosomal HAT assays using Ada complexes. Even-numbered Mono Q column fractions from Ada complex of each mutant were incubated with oligonucleosome cores and [3H]acetyl CoA. Reactions were loaded onto SDS-PAGE and the gels were analyzed by fluorography. (Left) Wild-type Gcn5, vector alone, and wild-type Gcn5 substitution mutants; (right) defective Gcn5 mutants. Positions of core histones on the gel are indicated by arrows. Numbers at the top are the Mono Q column fractions. The H3/H2B nucleosomal activity is Gcn5-dependent, and the H4/H2A nucleosomal HAT activity is Gcn5-independent (Grant et al. 1997).
Overall, the results demonstrate a clear correlation between complementation of function by the Gcn5 substitution mutants in the gcn5− strain (Figs. 2 and 4) and acetylation of histones (Fig. 6). Importantly, HAT activity by the mutant Gcn5 proteins was similar in both the Ada and SAGA complexes, such that mutants that possessed wild-type levels of activity in the Ada complex were also competent in the SAGA complex and mutants that were defective in Ada were also defective in SAGA.
Acetylation of nucleosomal histones by Gcn5 substitution mutants
Both the SAGA and Ada complexes acetylate nucleosomal histones. Because the level of nucleosomal acetylation compared with free histone acetylation is markedly inhibited in the SAGA complex (D. Sterner, P. Grant, J. Workman, and S.L. Berger, unpubl.), we tested the ability of the Gcn5 substitution mutants to acetylate nucleosomal histones in the context of the Ada complex. In addition, the Gcn5-dependent H3/H2B HAT activity in the Ada complex overlaps a Gcn5-independent H4/H2A HAT activity (Grant et al. 1997; Fig. 7), which provides an internal control for variation in extract preparation.
Fractions between 18 and 30 were tested for HAT activity on nucleosomal substrates using a liquid HAT assay and incorporation of the [3H]acetyl group into histones was visualized by PAGE and fluorography. The peak of Gcn5-dependent H3/H2B activity in the wild-type extract eluted between fractions 20–24, whereas the H4/H2A activity peaked at fraction 24–26 (Fig. 7, top left). As observed previously (Grant et al. 1997), in extracts prepared from the gcn5− strain, the H4/H2A HAT activity remained intact, whereas the H3/H2B activity disappeared (Fig. 7, left).
Strikingly, five mutants that failed to complement loss of Gcn5 in vivo (KQL, PKM, FAE, AIGY, and FKK) and were defective in acetylation of free histones in the SAGA and Ada complexes also were completely defective in acetylation of nucleosomal histones in the Ada complex (Fig 7, right). The remaining defective mutant (KDY196) had severely reduced activity, which was barely above background levels present in the gcn5− extract (Fig. 7, right). In addition, one of the partially defective mutants in the in vivo assays (RGY) showed severely impaired activity in the nucleosome acetylation assay (Fig. 7, right), as also observed in the free histone assay (Fig. 6A). In contrast, the Gcn5-independent H4/H2A acetylation activity was not affected by these Gcn5 substitution mutations (Fig. 7, right). As described in the previous section, the presence of Gcn5 and Ada2 in the Ada complex was confirmed for all mutants by Western analysis (Fig 6C).
In contrast, mutants that displayed wild-type phenotypes in complementation of growth and transcription in the gcn5− strain (LKN, PKE, YDR, GAHL, and GYI), were competent for acetylation of nucleosomal H3/H2B with varying efficiencies relative to wild-type Gcn5 (Fig. 7, left). Significantly, all wild-type Gcn5 mutants were relatively strong in nucleosomal H3/H2B acetylation compared with all defective mutants. Therefore, the data demonstrate that mutations that are most deleterious for in vivo function of Gcn5 similarly completely impair its histone acetylation function as a component of native SAGA and Ada complexes.
Discussion
Several transcriptional cofactors have been revealed recently to possess histone acetyltransferase activity in vitro. These include members of the Gcn5 family as well as the unrelated coactivators p300/CBP and TAFII250. The emergence of this new class of transcription coactivators functioning as HATs has raised important questions. First, do these proteins function as acetyltransferases in vivo, second, are nucleosomal histones their natural substrates, and, finally, is acetyltransferase activity required for transcription function? To address these issues, we systematically created substitution mutants in the putative HAT domain of yeast Gcn5, and compared their effects on growth and transcriptional activation in vivo with histone acetylation in vitro. If acetylation of histones by Gcn5 is critical, then there should be strong correlation between HAT activity and in vivo function.
Correlation between in vivo function of Gcn5 and in vitro histone acetylation
We identified six substitution mutations (KQL, PKM, FAE, KDY196, AIGY, and FKK) in Gcn5 that were highly deleterious to Gcn5 function in vivo. Strikingly, five were inert for acetylation of histones, either as free or nucleosomal substrates, and the sixth mutant (KDY196) had very low activity. Importantly, other mutants that were intermediate in function in vivo (YIA, RGY, DNY, KDY242, and TLM) were also impaired significantly in both free histone and nucleosomal histone acetylation (RGY acetylation is shown in Figs. 6 and 7; others not shown). Finally, substitution mutants that displayed wild-type function in vivo, displayed relatively high levels of acetylation in vitro (five examples are shown in Figs. 6 and 7; complexes from six others were prepared and were found to possess significant levels of HAT activity). Therefore, these data (summarized in Fig. 8) demonstrate a clear correlation between residues within Gcn5 that are critical for acetylation of histone substrates, and residues that are absolutely required for growth and transcriptional activity of Gcn5.
Figure 8.

Comparison of mutant Gcn5 phenotypes in acetylation vs. transcription. Quantitative data are taken from Fig. 6A (top) and Fig. 4 (the Gal4–VP16 and LexA–Gcn4 data were averaged). Acetylation of free histones by the SAGA complex is shown at left and transcriptional activation by the chimeric activators is shown at right.
It is important to stress that we analyzed histone acetylation by Gcn5 within the context of native yeast SAGA and Ada complexes. Gcn5, as well as other HATs (Chen et al. 1997) and HDACs (Rundlett and Grunstein 1996), exists in protein complexes containing multiple components that, in the case of Gcn5, alter substrate specificity in two ways. Recombinant yeast Gcn5 acetylates only free histones and primarily histone H3 (histone H4 is acetylated to a far lesser degree) (Brownell et al. 1996). Gcn5 in native yeast SAGA and Ada complexes acetylates both free and nucleosomal histones and targets both H3 and H2B (Grant et al. 1997). The altered specificity of Gcn5 in complexes, as opposed to recombinant form, suggests that physiologically relevant acetylation activity is best evaluated in the context of these native HAT complexes, as we have done in the current study.
Mutations that impair transcription similarly impair growth
Gcn5 mutants were tested for their effects on transcription using two heterologous model activators, Gal4–VP16 and LexA–Gcn4. We showed previously that Ada2, which is present in the Ada and SAGA complexes and is required for Gcn5 function in vivo, binds to both of these activation domains (Barlev et al. 1995). The Gcn5 substitution mutants showed very similar phenotypes in the transcription assays, such that the wild-type, intermediate, and deleterious mutants displayed parallel effects for function by each activator. In addition, the natural HIS3 and PHO5 promoters also display dependency on the defective mutants, and have normal function in the presence of wild-type mutants (R. Bolotserkovskaya, N. Barlev, and S.L. Berger, unpubl.). Therefore, the effects of the substitution mutations that reduce histone acetylation have similar effects on both synthetic model promoters and natural yeast promoters, arguing that the effects are generally consistent for activators that require Gcn5 for function.
Moreover, we found a striking correlation between the effect of the substitution mutations on Gcn5 function in growth and in transcriptional activation assays, such that colony size and doubling time of the wild-type, intermediate, and deleterious mutants mirrored transcription function. The consistency between growth and transcription suggests that results obtained using the model activators Gal4–VP16 and LexA–Gcn4 reflect natural dependency of yeast genes transcription on Gcn5.
Taken together with previous data regarding function and biochemical interactions of the Ada, Gcn5, and Spt proteins (Grant et al. 1997; Roberts and Winston 1997), the data support the model that Gcn5 functions within Ada and/or SAGA complexes to acetylate nucleosomal histones in promoter regions of dependent genes. Ada and SAGA may be recruited to promoters via interactions of Ada2 or Gcn5 with activation domains (Silverman et al. 1994; Barlev et al. 1995; Chiang et al. 1996), which may position the HAT domain to acetylate nucleosomes proximal to the TATA box. Spt proteins may have distinct roles in the SAGA complex, either for complex integrity (Spt7 and Spt20/Ada5; Grant et al. 1997) or recruitment of TBP and associated proteins (Spt7 and Spt8; Eisenmann et al. 1992, 1994). Based on the strong correlation we observe between residues within Gcn5 required for HAT activity and function, we conclude that acetylation by Ada and/or SAGA appears to be a critical in vivo function.
Residues important for function within the Gen5 HAT domain
We identified critical residues for function in all four conserved subdomains of Gcn5. Mutations in regions I (KQL and PKM), II (FAE and RGY), and III (AIGY and FKK) were most debilitating for histone acetylation within the Ada and SAGA complexes. Previous evolutionary comparisons have identified conserved sequences in the putative HAT domain between the Gcn5 subfamily of acetyltransferases and many additional proteins, leading to speculation that these proteins form an extensive superfamily (Reifsnyder et al. 1996; Neuwald and Lansman 1997). In particular, there is very strong conservation of conserved Gcn5 regions II and III and members of the superfamily (Neuwald and Lansman 1997). Because the apparent commonality within the superfamily is acetyl-CoA binding, this region may bind acetyl-CoA. In particular, RGY, AIGY, and FKK show very strong conservation across the superfamily. Therefore, our results that these mutations are extremely deleterious to histone acetylation are consistent with a role in acetyl-CoA binding. Whereas AIGY and FKK are completely debilitating to function of Gcn5 in vivo, it was surprising that RGY mutant was not affected as dramatically. Because the two glycines in RGYG show the most strong conservation in this short sequence (Neuwald and Lansman 1997), it may be that the alanine substitution preserves some function. The dominant negativity of all three of these mutants RGY, AIGY, and FKK supports the interpretation of a defect in acetyl-CoA binding, but retention of other critical protein interactions.
Interestingly, the FAE mutant, which occurs at the amino-terminal border of region II, is one of the most dysfunctional mutants, but is not strongly conserved in the superfamily. The biochemical function of the FAE residues will be important to determine. In addition, another region of conservation in the superfamily, located between regions I and II, contains the conserved GGI (Neuwald and Lansman 1997). In our study, the GGI mutant was not affected in vivo, but, as is true for the RGYG mutant, the alanine substitutions may not be profound sequence changes for these particular residues. Overall, our data support previous speculation that regions II and III are involved in acetyl-CoA binding.
Regions I and IV of the Gcn5 subfamily display no significant homology to the other members of the superfamily (Neuwald and Lansman 1997). The KQL and PKM mutations in region I, however, result in similar phenotypes as the mutations in the putative acetyl-CoA-binding site in regions II and III. These phenotypes include loss of histone acetyltransferase activity, and loss of growth and transcription function in vivo, as well as strong dominant negativity. One possibility is that region I is involved in binding of the particular substrates acetylated by Gcn5. If this is true, then the lack of homology in region I within the superfamily would imply that the other family members acetylate substrates other than nucleosomal histones H3 and H2B. These alternate substrates could include free histones, histones H4 or H2A, or completely dissimilar acetylation targets.
The analysis of mutants KDY196 and KDY242 suggests an important role for these residues in processes other than direct nucleosome acetylation. Both mutants had extremely debilitated function in vivo, but retained low acetylation function (see Fig. 7 for KDY196; data not shown for KDY242). These phenotypes suggest a possible regulatory function for these residues or interaction with other proteins that are not involved in acetylation. In particular, because region IV is not well conserved with other acetyltransferases (Neuwald and Lansman 1997), and KDY242 was the only profoundly defective mutant in this region, this is consistent with a function of region IV specific to regulation of Gcn5.
Overall, the data demonstrate that all four highly conserved subdomains contain important elements for function of Gcn5. Regions I, II, and III are likely to be involved in enzymatic activity and substrate specificity, whereas region IV may have a regulatory role.
Are histones bona fide physiological substrates of Gcn5?
As noted above, the HAT domains of Gcn5 subfamily members, although remarkably conserved with respect to each other, display no obvious sequence conservation with the putative HAT domains of either CBP/p300 or TAFII250 (nor are the latter two HAT domains mutually conserved). These observations raise the possibility that the known HATs share yet unidentified structural motifs. A second, and not exclusive explanation is that significant differences exist in the substrates of acetylation by the Gcn5 subfamily compared with the other putative acetyltransferases. Notably, p300 has been shown recently to acetylate certain lysines in the carboxyl terminus of the tumor suppressor and activator p53 (Gu and Roeder 1997).
Our data are consistent with the hypothesis that nucleosomal histones are critical physiological substrates of the Gcn5 family. We scanned the conserved residues of the entire HAT domain, and observed a strong consistency between the effect of the most deleterious Gcn5 substitution mutants on function in vivo and their effect on histone acetylation in vitro. Moreover, recent data indicate that normal remodeling of nucleosomes at the PHO5 promoter during transcriptional activation requires Gcn5, and in particular, the remodeling mirrors the pattern of dependency on the HAT domain mutations depicted here (P. Gregory, A. Schmid, M. Zavari, L. Liu, S.L. Berger, and W. Hörz, unpubl.). Taken together the data strongly suggest that histones are among the key substrates of Gcn5 in vivo.
Materials and methods
Yeast strains
The yeast strain PSY316 (MATα ade-101 Δhis3-200 leu2-3,112 lys2 ura3-52) and its derivatives PSY316Δgcn5 (Candau and Berger 1996) were used for transformation of HAT domain substitution mutations in conserved regions II, III, and IV. The TRP1 disruption of IPY3 Δgcn5 (Roberts and Winston 1997) was constructed using the hisgURA3 cassette as described (Alani et al. 1987), and was used for integration of the HAT domain substitution mutations in conserved region I.
Plasmids
pRS414–GCN5 was made by amplifying GCN5 including its natural promoter and terminator from genomic DNA using PCR with primers containing 5′-XhoI → 3′-BglII sites and then cloning GCN5 into the low-copy yeast expression vector pRS414 at XhoI and BamHI sites. GCN5 with BglII ends was ligated into high-copy yeast galactose-inducible expression vector pRS424(PGal) to produce pRS424(PGal)-GCN5. GCN5 was mutagenized in pRSET, pRS414 or pRS424(PGal)backbone and then subcloned into other vectors by exchanging BamHI–EcoRI (for subregion I) or NcoI (subregion II–IV) fragments. GCN5 subregion I mutants were subcloned into pRS306 for integration. The yeast-inducible expression vector containing GST was made by inserting GST fragment (amplified by PCR from pGEX plasmid with BamHI and KpnI sites) into the yeast galactose inducible expression vector pYES. GST–ADA2 was made by inserting ADA2 at BglII in frame with GST.
Site-directed mutagenesis
The Chameleon double-stranded site-directed mutagenesis kit (Stratagene) was used to construct the alanine-scan mutants of Gcn5 using mutagenic oligonucleotides shown in Table 1. Briefly, the GCN5 plasmid was heat-denatured and hybridized with two oligonucleotide primers. The selection primer changed one nonessential unique restriction site to a new restriction site on the plasmid backbone. The second primer encoded the specific mutations, in which three or four amino acids in Gcn5 were changed into three or four alanine residues, and a new PstI restriction site was created simultaneously. Unmutagenized parental plasmids were selected against by digestion using the mutagenized restriction site in the vector backbone, and mutagenized progeny were identified using the introduced PstI site and were confirmed by sequencing.
Growth and dominant-negative assays
Growth phenotypes of Gcn5 substitution mutations were analyzed after transformation of IPY37Δgcn5Δtrp1 (conserved region I mutations) PSY316Δgcn5Δtrp1 (conserved regions II–IV mutations). Single colonies were streaked on minimal synthetic medium, and were incubated at 30°C for 2 days. For the doubling time assay, single colonies were inoculated into liquid fully supplemented synthetic media and rotated overnight at 30°C. The cultures were then diluted to an A600 of 0.02, and the OD was checked every 2 hr up to a total of 28 hr.
Dominant-negative assays were carried out by transforming high-copy expression plasmids pRS425(PGal) or pRS424(PGal) containing GCN5 mutants into IPY36 or PSY316 strains bearing wild-type GCN5. Transformants were plated on SD (2% glucose) plates, incubated at 30°C for 3 days. Single colonies were restreaked on SD (2% galactose) medium, and incubated at room temperature for 3 days.
Transcription assays
Transcription assays were performed in IPY37Δgcn5Δtrp1 (conserved region I mutations) and PSY316Δgcn5Δtrp1 (conserved regions II–IV mutations). Plasmids expressing Gal4–VP16FA (Berger et al. 1990) and LexA–Gcn4 (Marcus et al. 1994) and the corresponding reporters were cotransformed. β-Gal activity was determined as units per milligram of protein. The LacZ reporters used were pLGSD5 (Guarente et al. 1982) for Gal4–VP16 activation, and LexA–8x (eight LexA-binding sites) (Candau et al. 1996) for LexA–Gcn4 activation.
Yeast protein overexpression and in vivo GST-binding assay
Yeast PSY316Δgcn5 strain transformed with GST–ADA2 and pRS424(PGal)–GCN5 or GCN5 substitution mutants were inoculated into 5 ml of synthetic glucose media and grown overnight. The cultures were diluted in 100 ml of synthetic media containing 2% lactate, and grown for 5–6 hr. Galactose was added to 2% final concentration to induce protein expression. Cells were harvested when cultures reached A600 = 1. Cell pellets were washed and resuspended in 400 μl of storage buffer (20 mm HEPES at pH 8.0, 5 nm EDTA, 20% glycerol, 1 mm β-mercaptoethanol, 1 μm PMSF). Cells were disrupted by vortexing with glass beads and supernatants were collected. This extract (200 μl) was incubated with 100 μl of GST bead slurry (50%) for 1 hr in PBS buffer–0.1% Triton-100, and beads were washed extensively with PBS buffer. The proteins bound to GST beads were analyzed by SDS-PAGE and immunoblotted using Gcn5 antiserum (Candau and Berger 1996).
Yeast protein complex preparation
Strains transformed with GCN5 substitution mutants were grown selectively in 200 ml of synthetic media, and then transferred to 4 liters of YPD media and grown to A600 = 2. Cells were harvested and resuspended in 30 ml of extraction buffer (40 mm HEPES at pH 7.3, 350 mm NaCl, 0.1% Tween, 10% glycerol, protease inhibitors). Cells were lysed by glass beads using a bead beater, and extracts were clarified by centrifugation for 1 hr at 40,000g. Next, extracts were incubated with 10 ml Ni–NTA–agarose slurry (50%) at 4°C for 2.5 hr. Resins were poured into a column and washed subsequently with 10 ml of extraction buffer, 10 ml of buffer A (20 mm Imidazole at pH 7.0, 100 mm NaCl, 0.1% Tween 20, 10% glycerol, protease inhibitors). Bound proteins were eluted with 10 ml of buffer A with 300 mm imidazole. The 300 mm imidazole eluate was directly loaded onto a Mono Q HR5/5 column equilibrated in buffer B (50 mm Tris at pH 8.0, 100 mm NaCl, 0.1% Tween 20, 10% glycerol). After a 10-ml wash with buffer B, bound proteins were eluted with a 25-ml linear gradient of 100 mm to 500 mm NaCl in buffer B. Fractions were collected and subjected to HAT assay.
Western blot analysis
The substitution mutations in the HAT domain of Gcn5 unpredictably altered the elution position of the SAGA complex from the Mono Q column, and therefore the peak fraction of SAGA elution for each mutant was determined by Western blot analysis using both Ada2 and Gcn5 antisera (data not shown). Then final Western blot analyses were performed with this peak fraction from the MonoQ column (Fig. 6B). For the Ada complex, peak fractions for protein analysis were fractions 20 or 24; fraction 22 could not be used because of a strong nonspecific cross-reacting protein (data not shown). Western blot analyses were performed with Gcn5 and Ada2 antisera (Barlev et al. 1995; Candau and Berger 1996).
HAT assays for free and nucleosomal histones
Different amount of fractions (2 and 6 μl) were used for the nucleosomal HAT assay and similar histone acetylation patterns (H3/H2B relative to H4/H2A) were observed (data not shown), indicating that the amount of extracts used in the HAT assay was in the linear range of HAT enzymatic activity. For nucleosomal HAT assays, MonoQ fractions from Ada complex of each Gcn5 mutant were used. Three microliters of each even numbered fraction (from 18 to 30) were incubated with 1 μg of oligonucleosome cores and [3H]acetyl CoA in HAT buffer (Grant et al. 1997) at 30°C for 30 min. The reactions were subjected to SDS-PAGE, and gels were then treated with Intensify Buffer (DuPont NEN) and fluorographed. For free histone HAT assays, the same fractions of SAGA and Ada complexes used in Western analysis were incubated with 1 μg core histones and [3H]acetyl CoA in HAT buffer. Reactions and fluorograph were carried out as indicated above.
Acknowledgments
We are grateful to R. Marmorstein for preparation of Figure 1A. We thank C.D. Allis and M.H. Kuo for communication of unpublished results. We thank J. Workman for oligonucleosomes and D. Sterner and F. Winston for yeast strains. We thank P. Lieberman, G. Moore, F. Rauscher, G. Rovera, and J. Workmen for support and valuable discussions; N. Barlev and G. Moore for critical reading of the manuscript; and A. Kulak for expert editorial help. The work was supported by grants from the National Science Foundation and The Council for Tobacco Research to S.L.B; S.L.B is the recipient of an American Cancer Society Junior Faculty Research Award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL berger@wista.wistar.upenn.edu; FAX (215) 898-0063.
References
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alland L, Muhle R, Hou Jr H, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Bannister A, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Barlev N, Candau R, Wang L, Darpino P, Silverman N, Berger S. Characterization of physical interactions of the putative transcriptional adaptor, ADA2, with acidic activation domains and TATA-binding protein. J Biol Chem. 1995;270:19337–19344. doi: 10.1074/jbc.270.33.19337. [DOI] [PubMed] [Google Scholar]
- Berger SL, Cress WD, Cress A, Triezenberg SJ, Guarente L. Selective inhibition of activated but not basal transcription by the acidic activation domain of VP16: Evidence for transcriptional adaptors. Cell. 1990;61:1199–1208. doi: 10.1016/0092-8674(90)90684-7. [DOI] [PubMed] [Google Scholar]
- Berger SL, Pina B, Silverman N, Marcus GA, Agapite J, Regier JL, Triezenberg SJ, Guarente L. Genetic isolation of ADA2: A potential transcriptional adaptor required for function of certain acidic activation domains. Cell. 1992;70:251–265. doi: 10.1016/0092-8674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- Bradbury EM. Reversible histone modifications and the chromosome cell cycle. BioEssays. 1992;14:9–16. doi: 10.1002/bies.950140103. [DOI] [PubMed] [Google Scholar]
- Brownell J, Zhou J, Ranalli T, Kobayashi R, Edmondson D, Roth S, Allis C D. Tetrahymena histone acetyltransferase A: A transcriptional co-activator linking gene expression to histone acetylation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- Candau R, Berger SL. Structural and functional analysis of yeast putative adaptors: Evidence for an adaptor complex in vivo. J Biol Chem. 1996;271:5237–5345. doi: 10.1074/jbc.271.9.5237. [DOI] [PubMed] [Google Scholar]
- Candau R, Moore P, Wang L, Barlev N, Ying C, Rosen C, Berger S. Identification of functionally conserved human homologues of the yeast adaptors ADA2 and GCN5. Mol Cell Biol. 1996;16:593–602. doi: 10.1128/mcb.16.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candau R, Zhou J, Allis CD, Berger SL. Histone acetyltransferase activity and interaction with ADA2 are critical for GCN5 function in vivo. EMBO J. 1997;16:555–565. doi: 10.1093/emboj/16.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin R, Schiltz R, Chakravarti D, Nash A, Nagy L, Privalsky M, Nakatani Y, Evans R. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with PCAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chiang Y, Komarnitsky P, Chase D, Denis C. ADR1 activation domains contact the histone acetyltransferase GCN5 and the core transcriptional factor TFIIB. J Biol Chem. 1996;271:32359–32365. doi: 10.1074/jbc.271.50.32359. [DOI] [PubMed] [Google Scholar]
- Cote J, Quinn J, Workman JL, Peterson CL. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science. 1994;265:53–60. doi: 10.1126/science.8016655. [DOI] [PubMed] [Google Scholar]
- Eisenmann D, Arndt K, Ricupero S, Rooney J, Winston F. SPT3 interacts with TFIID to allow normal transcription in Saccharomyces cerevisiae. Genes & Dev. 1992;6:1319–1331. doi: 10.1101/gad.6.7.1319. [DOI] [PubMed] [Google Scholar]
- Eisenmann D, Chapon C, Roberts S, Dollard C, Winston F. The Saccharomyces cerevisiae SPT8 gene encodes a very acidic protein that is functionally related to SPT3 and TATA-binding protein. Genetics. 1994;137:647–657. doi: 10.1093/genetics/137.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fascher K, Schmitz J, Horz W. Structural and functional requirements for the chromatin transition at the PHO5 promoter in Saccharomyces cerevisiae upon PHO5 activation. J Mol Biol. 1993;231:658–667. doi: 10.1006/jmbi.1993.1317. [DOI] [PubMed] [Google Scholar]
- Gansheroff L, Dollard C, Tan P, Winston F. The S. cerevisiase SPT7 gene encodes a very acidic protein important for transcription in vivo. Genetics. 1995;139:523–536. doi: 10.1093/genetics/139.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos T, Thireos G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J. 1992;11:4145–4152. doi: 10.1002/j.1460-2075.1992.tb05507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill G, Ptashne M. Negative effect of the transcriptional activator GAL4. Nature. 1988;334:4721–4724. doi: 10.1038/334721a0. [DOI] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell J, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, Berger SL, Workman JL. yGCN5 function within multisubunit ADA and SPT/ADA adaptor complexes to acetylate nucleosomal histones. Genes & Dev. 1997;11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Nucleosomes: regulators of transcription. Trends Genet. 1990;6:395–400. doi: 10.1016/0168-9525(90)90299-l. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder R. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Guarante L. Transcriptional coactivators in yeast and beyond. Trends Biol Sci. 1995;20:517–521. doi: 10.1016/s0968-0004(00)89120-3. [DOI] [PubMed] [Google Scholar]
- Guarente L, Yocum RR, Gifford P. A GAL10-CYC1 hybrid yeast promoter identifies the GAL4 regulatory region as an upstream site. Proc Natl Acad Sci. 1982;79:7410–7414. doi: 10.1073/pnas.79.23.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression [see comments] Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Kadosh D, Struhl K. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell. 1997;89:365–371. doi: 10.1016/s0092-8674(00)80217-2. [DOI] [PubMed] [Google Scholar]
- Kingston RE, Bunker CA, Imbalzano AN. Repression and activation by multiprotein complexes that alter chromatin structure. Genes & Dev. 1996;10:905–920. doi: 10.1101/gad.10.8.905. [DOI] [PubMed] [Google Scholar]
- Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- Lee D, Hayes J, Pruss D, Wolffe A. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Loidl P. Histone acetylation: Facts and questions. Chromosoma. 1994;103:441–449. doi: 10.1007/BF00337382. [DOI] [PubMed] [Google Scholar]
- Marcus G, Silverman N, Berger S, Horiuchi J, Guarente L. Functional similarity and physical association between GCN5 and ADA2 - putative transcriptional adaptors. EMBO J. 1994;13:4807–4815. doi: 10.1002/j.1460-2075.1994.tb06806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus G, Horiuchi J, Silverman N, Guarente L. ADA5/SPT20 links the ADA and SPT genes, which are involved in yeast transcription. Mol Cell Biol. 1996;16:3197–3205. doi: 10.1128/mcb.16.6.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizzen C, Yang X-J, Kokubo T, Brownell J, Bannister A, Owen-Hughes T, Workman J, Wang L, Berger SL, Kouzarides T, Nakatani Y, Allis CD. The TAF250 Subunit of TFIID Has Histone Acetyltransferase Activity. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Lansman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- Ogryzko V, Schlitz R, Russanova V, Howard B, Nakatani Y. The Transcriptional Coactivators p300 and CBP are Histone Acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Owen-Hughes T, Workman JL. Experimental analysis of chromatin function in transcription control. Crit Rev Eukary Gene Exp. 1994;4:403–441. [PubMed] [Google Scholar]
- Paranjape SM, Kamakaka RT, Kadonaga JT. Role of chromatin structure in the regulation of transcription by RNA polymerase II. Annu Rev Biochem. 1994;63:265–297. doi: 10.1146/annurev.bi.63.070194.001405. [DOI] [PubMed] [Google Scholar]
- Pazin M, Kadonaga J. What’s up and down with histone deacetylation and transcription? Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Dingwall A, Scott MP. Five SWI/SNF gene products are components of a large multisubunit complex required for transcriptional enhancement [see comments] Proc Natl Acad Sci. 1994;91:2905–2908. doi: 10.1073/pnas.91.8.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piña B, Berger S, Marcus GA, Silverman N, Agapite J, Guarente L. ADA3: A gene, identified by resistance to GAL4-VP16, with properties similar to and different from those of ADA2. Mol Cell Biol. 1993;13:5981–5989. doi: 10.1128/mcb.13.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifsnyder C, Lowell J, Clarke A, Pillus L. Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nature Genet. 1996;14:44–49. doi: 10.1038/ng0996-42. [DOI] [PubMed] [Google Scholar]
- Roberts S, Winston F. SPT20/ADA5 encodes a novel protein functionally related to the TATA-binding protein and important for transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:3206–3213. doi: 10.1128/mcb.16.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Essential functional interactions of SAGA, a Saccaromyces cerevisiae complex of Spt, Ada and GCN5 proteins, with the Snf/Swi and Srt/mediator complexes. Genetics. 1997;147:451–465. doi: 10.1093/genetics/147.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundlett SE, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci. 1996;93:14503–14508. doi: 10.1073/pnas.93.25.14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Lang V, Cook R, Brand C. Identification of native complexes containing the yeast coactivator-repressor proteins NGG1-ADA3 and ADA2. J Biol Chem. 1997;272:5571–5578. doi: 10.1074/jbc.272.9.5571. [DOI] [PubMed] [Google Scholar]
- Silverman N, Agapite J, Guarente L. Yeast ADA2 protein binds to the VP16 protein activation domain and activates transcription. Proc Natl Acad Sci. 1994;91:11665–11668. doi: 10.1073/pnas.91.24.11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. Yeast transcriptional regulatory mechanisms. Annu Rev Genet. 1995;29:651–674. doi: 10.1146/annurev.ge.29.120195.003251. [DOI] [PubMed] [Google Scholar]
- Svaren J, Schmitz J, Horz W. The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J. 1994;13:4856–4862. doi: 10.1002/j.1460-2075.1994.tb06812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcription regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ. Structure and function of transcriptional activation domains. Curr Opin Genet Dev. 1995;5:190–196. doi: 10.1016/0959-437x(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Turner BM, O’Neill LP. Histone acetylation in chromatin and chromosomes. Sem Cell Biol. 1995;6:229–236. doi: 10.1006/scel.1995.0031. [DOI] [PubMed] [Google Scholar]
- Vettese-Dadey M, Grant PA, Hebbes TR, Crane-Robinson C, Allis CD, Workman JL. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Gaber R. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in S. cerevisiae. Mol Cell Biol. 1991;11:6317–6327. doi: 10.1128/mcb.11.12.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade P, Wolffe A. Histone acetyltransferases in control. Curr Biol. 1997;7:82–84. doi: 10.1016/s0960-9822(06)00042-x. [DOI] [PubMed] [Google Scholar]
- Wang L, Mizzen C, Ying C, Candau R, Barlev N, Brownell J, Allis CD, Berger S. Histone Acetyltransferase activity is conserved between yeast and human GCN5 and required for complementation of growth and transcriptional activation. Mol Cell Biol. 1997;17:519–527. doi: 10.1128/mcb.17.1.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe A. Chromatin: Structure and function. London, UK: Academic Press; 1992. [Google Scholar]
- Yang X, Ogryzko V, Nishikawa J, Howard B, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral E1A oncoprotein. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Zawel L, Reinberg D. Common themes in assembly and function of eukaryotic transcription complexes. Annu Rev Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]