

Abstract
IGF-I plays an important role in smooth muscle cell proliferation and migration. In vascular smooth muscle cells cultured in 25 mm glucose, IGF-I stimulated a significant increase in Src homology 2 domain containing protein tyrosine phosphatase substrate-1 (SHPS-1) phosphorylation compared with 5 mm glucose and this increase was required for smooth muscle cell proliferation. A proteome-wide screen revealed that carboxyl-terminal SRC kinase homologous kinase (CTK) bound directly to phosphotyrosines in the SHPS-1 cytoplasmic domain. Because the kinase(s) that phosphorylates these tyrosines in response to IGF-I is unknown, we determined the roles of IGF-I receptor (IGF-IR) and CTK in mediating SHPS-1 phosphorylation. After IGF-I stimulation, CTK was recruited to IGF-IR and subsequently to phospho-SHPS-1. Expression of an IGF-IR mutant that eliminated CTK binding reduced CTK transfer to SHPS-1, SHPS-1 phosphorylation, and cell proliferation. IGF-IR phosphorylated SHPS-1, which provided a binding site for CTK. CTK recruitment to SHPS-1 resulted in a further enhancement of SHPS-1 phosphorylation. CTK knockdown also impaired IGF-I-stimulated SHPS-1 phosphorylation and downstream signaling. Analysis of specific tyrosines showed that mutation of tyrosines 428/452 in SHPS-1 to phenylalanine reduced SHPS-1 phosphorylation but allowed CTK binding. In contrast, the mutation of tyrosines 469/495 inhibited IGF-IR-mediated the phosphorylation of SHPS-1 and CTK binding, suggesting that IGF-IR phosphorylated Y469/495, allowing CTK binding, and that CTK subsequently phosphorylated Y428/452. Based on the above findings, we conclude that after IGF-I stimulation, CTK is recruited to IGF-IR and its recruitment facilitates CTK's subsequent association with phospho-SHPS-1. This results in the enhanced CTK transfer to SHPS-1, and the two kinases then fully phosphorylate SHPS-1, which is necessary for IGF-I stimulated cellular proliferation.
IGF-I plays an important role in proliferation, migration, and hypertrophy of vascular smooth muscle cells (VSMC) (1). IGF-I binds to the type 1 IGF receptor (IGF-IR), which then undergoes a conformational change that activates the intrinsic tyrosine kinase, which allows recruitment of downstream signaling molecules such as insulin receptor substrate-1 (IRS-1) (2). However, in response to hyperglycemia, vascular cells down-regulate IRS-1 (3). IGF-I signaling is required for VSMC to survive hyperglycemic stress, and these cells undergo an adaptive response wherein an alternative signaling pathway is activated. This response is dependent on a complex of signaling proteins that are recruited to the transmembrane scaffold Src homology 2 domain containing protein tyrosine phosphatase substrate-1 (SHPS-1) (4). We have shown that the cytoplasmic domain (CD) of SHPS-1, which contains four tyrosines, is phosphorylated in response to IGF-I under hyperglycemic conditions in cells and in vivo (5, 6). Assembly of this complex on SHPS-1 facilitates both phosphatidylinositol 3-kinase and MAPK pathway activation and subsequently enhances VSMC cell migration and proliferation (7, 8).
SHPS-1 is a substrate for several receptor tyrosine kinases (3, 9–12), but the identities of the kinases that phosphorylate each of the four tyrosine residues have not been clearly defined. Recent studies from our laboratory and the group of Ishiura and colleagues (13) have shown that the protein tyrosine kinase Csk-homologous kinase (CTK) binds directly to phosphorylated SHPS-1 (4) and therefore has the potential to function as SHPS-1 kinase (13).
CTK contains SH3, SH2, and kinase domains in addition to a SH3-SH2 connector and SH2-kinase linker. This is similar to the structure of other Src family kinases (SFK). CTK has been shown to inactivate SFK by phosphorylating their consensus C-terminal tail tyrosine and by noncatalytic binding (14–19). However, unlike SFK, CTK lacks the autophosphorylation site. The SH2 domain of CTK has been shown to target it to transmembrane receptor tyrosine kinases such as epithelial growth factor receptor family tyrosine, kinase ErbB2 (20), nerve growth factor (NGF) receptor TrkA (21, 22), and stem cell factor receptor c-kit (23). Recruitment of CTK to plasma membrane has been associated with enhanced cell growth responsiveness.
We previously used an unbiased proteome-wide screen, which identified CTK as a protein that bound directly to tyrosine phosphorylated SHPS-1 (4). Because the IGF-I receptor phosphorylates SHPS-1 in vitro, we hypothesized that during hyperglycemic stress IGF-I receptor (IGF-IR) and CTK function coordinately to regulate SHPS-1 phosphorylation. These studies were undertaken to determine whether IGF-IR and CTK could phosphorylate SHPS-1 in VSMC to identify the specific tyrosines in the SHPS-1 CD that are phosphorylated by IGF-IR and CTK and to determine the mechanism by which the SHPS-1/CTK/IGF-IR complex functions coordinately to regulate the assembly and activation of its associated signaling components in response to hyperglycemic stress and IGF-I.
Results
Effects of high glucose on IGF-I-stimulated SHPS-1 phosphorylation
VSMC maintained in 25 mm glucose (DMEM-HG) showed a significant increase (4.6 ± 0.6 fold, n =3, P < 0.001) in SHPS-1 tyrosyl phosphorylation in response to IGF-I compared with cells that were maintained in 5 mm glucose (DMEM-NG), which is consistent with our previous in vivo observations (24) (Fig. 1A). Maintaining cells in 25 vs. 5 mm glucose had no effect on total SHPS-1 protein levels. Because we had previously demonstrated that purified IGF-IR could phosphorylate SHPS-1 in vitro, we determined whether IGF-IR associated with SHPS-1 in cells and whether this association was altered in response to hyperglycemia. In response to IGF-I, the SHPS-1/IGF-IR association increased 3.0 ± 0.2-fold (n = 3, P < 0.001) in cells exposed to DMEM-HG, compared with the cells exposed to DMEM-NG (Fig. 1A). Taken together with our previous observation, the findings suggest that IGF-I stimulated IGF-IR/SHPS-1 association in response to hyperglycemia is important for the increase in IGF-I stimulated SHPS-1 tyrosyl phosphorylation.
Fig. 1.

Effects of high glucose on IGF-I-stimulated SHPS-1 phosphorylation and IGF-IR/SHPS-1 association. A, Quiescent VSMC cultures were incubated in SFM containing 5 (DMEM-NG) or 25 (DMEM-HG) mm glucose overnight. Cells were stimulated with IGF-I (50 ng/ml) for 5 min. The extent of SHPS-1 tyrosine phosphorylation was determined by immunoprecipitating (IP) cell lysates with an anti-SHPS-1 antibody and then immunoblotting (IB) with an antiphosphotyrosine (pY) antibody (upper panel). Similarly the extent of the SHPS-1 association with IGF-IR was determined by IP using an anti-SHPS-1 antibody and then IB with an anti IGF-IR antibody (middle panel). As a loading control, the cell lysates were immunoprecipitated and immunoblotted with an anti-SHPS-1 antibody (lower panel). The bar graphs show pooled results from at least three independent experiments. The error bars represent mean ± se. ***, P < 0.001 when amount of SHPS-1 phosphorylation or its association with IGF-IR at 5 min in response to IGF-I is compared between DMEM-NG and DMEM-HG cells. B, Quiescent VSMC cultured and maintained in DMEM-NG or DMEM-HG were serum starved and then incubated with or without the IGF-IR tyrosine kinase inhibitor, PQ401 (10 μm) for 1–2 h. IGF-I was added for 5 min. Cell lysates were immunoprecipitated with an anti-IGF-IR antibody (first panel) or with an anti-SHPS-1 antibody (third panel) and immunoblotted for pY. For loading controls, IP and IB of cell lysates were performed with anti-IGF-IR (second panel) or anti-SHPS-1 antibody (fourth panel). The bar graphs show pooled results from at least three independent experiments. Error bars represent mean ± se. **, P < 0.01 when amount of SHPS-1 phosphorylation at 5 min in response to IGF-I is compared between DMEM-NG and DMEM-HG cells. C, VSMC expressing WT SHPS-1 (SHPS1-WT), VSMC expressing Y428F/Y452F mutant (Y12F), and VSMC expressing Y469F/Y495F mutant (Y34F) were serum starved in DMEM-HG and analyzed for recombinant protein expression. Cell lysates were immunoblotted using an anti-HA antibody (top panel), anti-SHPS-1 antibody (middle panel), and anti-β-actin antibodies (bottom panel). D, Confluent cells expressing SHPS1-WT or the SHPS-1-mutants (Y12F and Y34F) were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for 5 min. The extent of HA-tagged SHPS-1 and IGF-IR phosphorylation was determined by IP HA (first panel) or IGF-IR (third panel) and then immunoblotted with an anti-pY antibody. An equal amount of protein from cell lysates from the same experiment was used to immunoblot for total HA (second panel) and total IGF-IR (fourth panel).
To determine whether activation of IGF-IR was required for the increase in SHPS-1 tyrosyl phosphorylation in response to IGF-I, we used an IGF-IR kinase inhibitor, PQ401. PQ401 significantly inhibited IGF-I stimulated tyrosyl phosphorylation of IGF-IR (57 ± 4.8% decrease, n = 3, P < 0.01), and it significantly inhibited SHPS-1 tyrosyl phosphorylation in response to IGF-I in high glucose (HG) conditions (53 ± 4.9% decrease, n = 3, P < 0.001; Fig. 1B). There was no difference in the IGF-IR tyrosyl phosphorylation in 5 mm glucose (3.9 ± 0.5-fold) compared with 25 mm glucose (4.9 ± 0.3-fold) (n = 3, P = NS). To determine which of the four tyrosines in the SHPS-1 cytoplasmic domain (SHPS-1/CD) were phosphorylated in response to IGF-I stimulation, two mutants were prepared wherein tyrosines 428 and 452 (Y12F) or tyrosines 469 and 495 (Y34F) were substituted with phenylalanine. The amount of hemagglutinin (HA) tagged SHPS-1 mutant and wild-type (WT) SHPS-1 expression was similar when cells express the WT and mutant forms of SHPS-1 were compared (Fig. 1C). IGF-I stimulated a significant increase in tyrosyl phosphorylation of WT SHPS-1 (3.9 ± 0.4-fold, n = 3 P < 0.001) and in the phosphorylation of the Y12F mutant (2.1 ± 0.3-fold; n =3, P < 0.001). However it had no effect on tyrosyl phosphorylation of the Y34F mutant (Fig. 1D). This suggests that phosphorylation of tyrosines 469 and 495 is mediated by IGF-IR.
CTK phosphorylates SHPS-1 in vitro and its binding to SHPS-1 is stimulated by IGF-I in VSMC
Because both tyrosines 469 and 495 are contained in YXXL/V motifs, they could provide a binding site for SH2 domain-containing proteins. We have previously shown that two proteins, viz. the tyrosine phosphatase SHP-2 and the tyrosine kinase CTK, bind directly to tyrosine phosphorylated SHPS-1 (4). Therefore, we investigated whether CTK could phosphorylate SHPS-1. In vitro phosphorylation assays showed that CTK directly phosphorylated the CD of SHPS-1 (6.1 ± 0.5-fold increase n = 3, P < 0.001; Fig. 2A). Because IRS-1 inhibits SHPS-1 phosphorylation in smooth muscle cells (3), IRS-1 was added to the in vitro assays. The addition of IRS-1 inhibited CTK-mediated SHPS-1 phosphorylation (76.5 ± 3.6% decrease, P < 0.01; Fig. 2A). To determine whether CTK had the potential to phosphorylate SHPS-1 in VSMC, we analyzed the CTK/SHPS-1 association. In cells exposed to 25 mm glucose, CTK binding to SHPS-1 was increased (4.4 ± 0.1-fold n = 3, P < 0.001) after 5 min of IGF-I stimulation (Fig. 2B), whereas in 5 mm glucose there were no changes (Fig. 2C). Interestingly in VSMC maintained in HG, CTK binding to IGF-IR was increased 3.2 ± 0.3-fold (n = 3, P < 0.01) in 1 min (Fig. 2B), whereas after 5 min there was only a 1.4± 0.1-fold increase. Cells maintained in normal glucose (NG) showed no increase (Fig. 2C). To confirm the result, we immunoprecipitated SHPS-1 or IGF-IR and immunoblotted for CTK. In cells maintained in 25 mm glucose, IGF-I had a minimal effect on CTK association with SHPS-1 at 1 min, but their association was significantly increased (4.5 ± 0.4-fold, P < 0.05) at 5 min. The CTK/IGF-IR association was significantly increased (10 ± 1.6-fold, P < 0.01) at 1 min, but it was reduced at 5 min (Supplemental Fig. 1A, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). The difference in the temporal sequence of CTK binding to IGF-IR compared with its binding to SHPS-1 suggests that CTK is recruited initially to IGF-IR.
Fig. 2.

In vitro phosphorylation of SHPS-1 CD by CTK and CTK association with SHPS-1 and IGF-IR A, The cytoplasmic domain of the SHPS-1 (SHPS-1/CD) generated by in vitro translation using wheat germ extract was incubated in vitro (denoted by the + sign) with and without known amount of CTK and IRS-1 as described in Materials and Methods. The reaction mix was analyzed by immunoblotting (IB) for phosphotyrosine (upper panel) or SHPS-1 (middle panel) and CTK (bottom panel). The relative fold increase in the SHPS-1/CD phosphorylation and suppression by IRS-1 is shown in the bar graph. The error bars represent mean ± se. **, P < 0.01. B and C, Quiescent VSMC cultures were incubated in SFM containing DMEM-HG (B) or DMEM-NG (C) overnight. Cells were stimulated with IGF-I (50 ng/ml) for the periods of time indicated. The extent of the CTK association with SHPS-1 and IGF-IR was determined by immunoprecipitating (IP) using an anti-CTK antibody and then immunoblotted with an anti SHPS-1(first panel) or anti-IGF-IR antibody (second panel). Cell lysates from the same experiment were used to immunoprecipitate and immunoblot with anti-CTK antibody (third panel). The bar graphs show pooled results from at least three independent experiments. Error bars represent mean ± se. **, P < 0.01 when the amount of CTK associated with SHPS-1 at 5 min or IGF-IR at 1 min in response to IGF-I is compared with no stimulation or when the amount at 1 min is compared with 5 min. *, P < 0.05 when the amount of CTK associated with IGF-IR at 5 min is compared with no stimulation.
Silencing CTK impairs IGF-I-induced SHPS-1 phosphorylation
To definitively determine whether CTK was phosphorylating SHPS-1 in cells, we prepared a stable cell line using CTK short hairpin RNA (shRNA). CTK expression was reduced by 66 ± 6% (n =5, P < 0.001; Fig. 3A). This resulted in significant 85 ± 2% (n = 3, P < 0.01) reduction in the degree of increase in SHPS-1 tyrosyl phosphorylation after IGF-I stimulation compared with control cells (Fig. 3B). Recruitment of SHP-2 to phosphorylated SHPS-1 has been demonstrated to be essential for the assembly and activation of downstream signaling elements that lead to MAPK activation and stimulation of cellular proliferation (25). Analysis of the components of the SHPS-1 complex revealed a significant decrease in the amount of SHP-2 transferred to SHPS-1 in the CTK silenced cells compared with control cells in response to IGF-I (65 ± 6.7%, P < 0.001; Fig. 3B). Src homology 2 domain-containing protein (Shc) recruitment to SHPS-1 and its subsequent phosphorylation have been shown to be SHP-2 dependent (5); therefore, inhibition of SHP-2 transfer resulted in a significant decrease in Shc phosphorylation (53 ± 4.1%, P < 0.01; Fig. 3B) and phosphorylation of MAPK (38 ± 3.7%, P < 0.001) when CTK shRNA cells were compared with control cells (Fig. 3C). CTK knockdown also resulted in reduction in cellular proliferation in response to IGF-I (2.5 ± 0.04-fold increase compared with a 1.2 ± 0.02-fold increase, n = 3, P < 0.01; Fig. 3D). Taken together, these results demonstrate that CTK is required for maximal IGF-I stimulated SHPS-1 phosphorylation, which in turn is required for IGF-I stimulated downstream signaling events and cell prolifration.
Fig. 3.

Silencing CTK impairs IGF-I induced SHPS-1 phosphorylation. A, Confluent VSMC expressing an empty control vector (EVC) or CTK shRNA maintained in DMEM-HG and serum starved for 18 h in HG were analyzed for CTK protein expression (top panel) or for β-actin (lower panel). B, Confluent EVC and CTK shRNA cell cultures were serum starved for 18 h and IGF-I (50 ng/ml) was added for 5 min. Cell lysates were immunoprecipitated (IP) with an anti-SHPS-1 antibody and then immunoblotted (IB) for pY (first panel) or SHP-2 (second panel) or with an anti-SHPS-1 antibody (third panel). Cell lysates from similar experiments were immunoprecipitated with an anti-pY antibody and immunoblotted with an anti-Shc antibody (fourth panel). Twenty micrograms of protein from the same whole cell lysate were directly immunoblotted with anti-Shc antibody (fifth panel). The bar graphs show pooled results from at least three independent experiments. Error bars represent mean ± se. **, P < 0.01 when amount of SHPS-1 phosphorylation at 5 min in response to IGF-I is compared between EVC and CTK shRNA cells. C, Lysates containing 20 μg of protein from the same or similar experiment were directly immunoblotted with an anti-pMAPK antibody or an anti β-actin antibody. D, EVC and CTK shRNA cells were plated (3 × 104) in DMEM-HG with 2% FBS before exposure to IGF-I (50 ng/ml) in DMEM-HG with 0.2% platelet-poor plasma. Forty-eight hours after the addition of IGF-I, cell number was determined by tryptan blue staining and counting. The bar graphs show pooled results from at least three independent experiments. The error bars represent mean ± se. ***, P < 0.001 when cell number in response to IGF-I is compared between EVC and CTK shRNA cells.
IGF-IR and CTK phosphorylate specific tyrosines on SHPS-1
The results suggest that CTK phosphorylates SHPS-1 and that IGF-IR may regulate a CTK/SHPS-1 association. To determine whether specific tyrosines in SHPS-1 were phosphorylated by each kinase, we used the SHPS-1 mutants. Exposure to PQ401 (an IGF-IR kinase inhibitor) inhibited IGF-IR autophosphorylation in WT cells as well as cells expressing the SHPS-1 Y12F and Y34F mutants (Fig. 4, A–C). This was associated with loss of CTK transfer to IGF-IR in each cell type (Fig. 4, A–C). PQ401 treatment also inhibited SHPS-1 phosphorylation in the WT and Y12F cells, and this was associated with inhibition of IGF-I-stimulated CTK transfer to SHPS-1 (Fig. 4, A and B). In contrast, cells expressing the SHPS-1 Y34F mutant showed complete inhibition of SHPS-1 phosphorylation and CTK transfer in response to IGF-I. This loss of phosphorylation was not due to a conformational change induced by mutagenesis because CTK could phosphorylate the Y34F mutant in vitro (Fig. 4D). In addition, an in vitro assay performed using the IGF-IR kinase showed that IGF-IR had almost no ability to phosphorylate SHPS-1 Y429/453 and that it phosphorylated SHPS-1 Y469/495 to much greater extent (Supplemental Fig. 2A). Taken together with the results shown in Fig. 1D, these results suggest that IGF-IR phosphorylates Y469 and Y495 and that this is required for CTK transfer. Because the Y34F mutant cannot be phosphorylated, there is no CTK binding site available. This inhibition of CTK transfer results in failure to phosphorylate tyrosines 428 and 452.
Fig. 4.
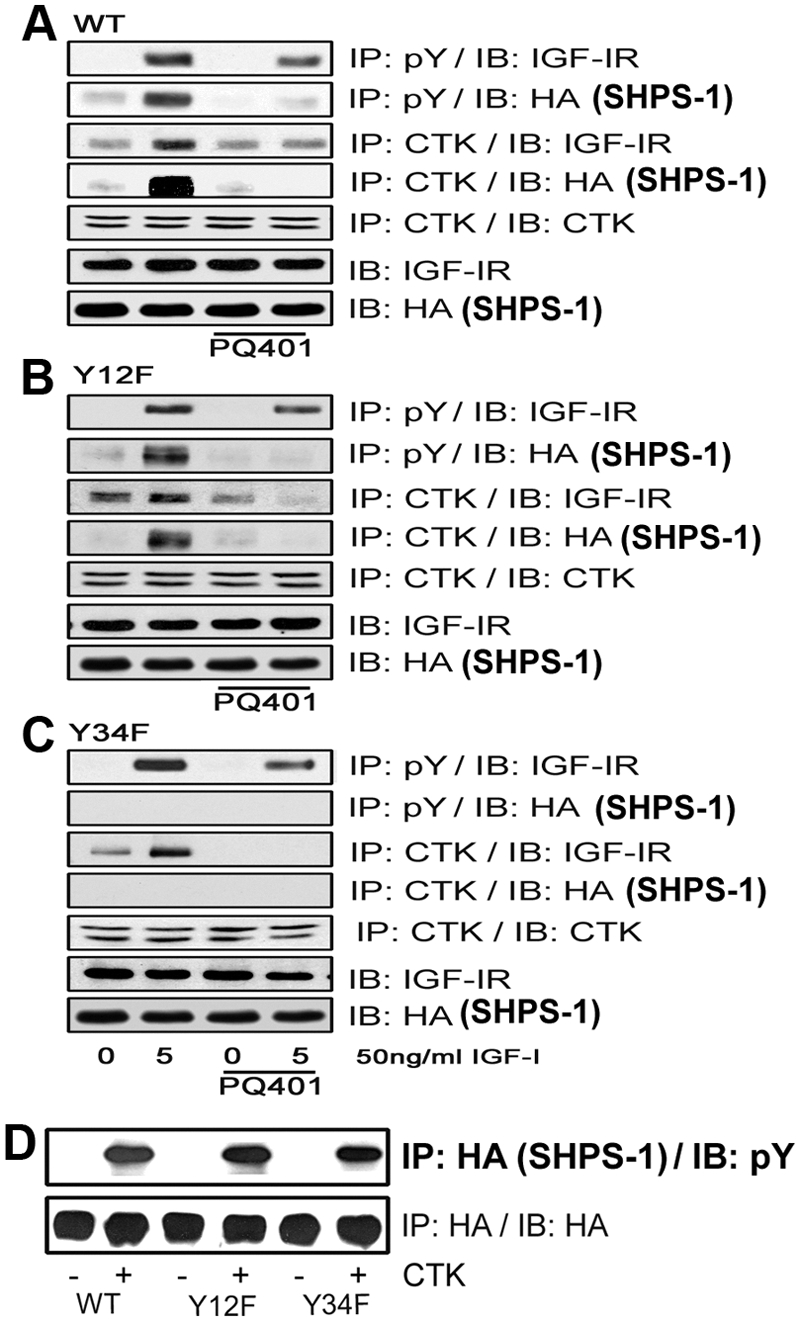
IGF-IR and CTK phosphorylate specific tyrosines on SHPS-1. A–C, Quiescent VSMC expressing SHPS1-WT (A), Y12F (B), and Y34F (C) were cultured and maintained in DMEM-HG and then serum starved overnight before being incubated with or without the IGF-IR tyrosine kinase inhibitor, PQ401 (10 μm), for 1–2 h. IGF-I was added for 5 min. Cell lysates were immunoprecipitated (IP) with an anti-pY antibody and then immunoblotted (IB) for IGF-IR (first panel) or HA (second panel). Similarly, cell lysates from the same experiments were immunoprecipitated with an anti-CTK antibody and then immunoblotted with anti-IGF-IR (third panel), anti-HA (fourth panel), or anti-CTK antibodies (fifth panel). Twenty micrograms of protein from the same whole-cell lysate were directly immunoblotted with an anti-IGF-IR antibody (sixth panel) or with an anti-HA antibody (seventh panel). D, Confluent SHPS-WT, Y12F, and Y34F cells were serum starved for 16 h in DMEM-HG, and the cell lysates were immunoprecipitated with anti-HA antiserum. The immunoprecipitates were incubated with or without CTK to determine HA-tagged SHPS-1 phosphorylation in vitro. After centrifugation the resultant supernatants were immunoblotted using a pY antibody (upper panel) or an HA antibody as a loading control (lower panel).
Functional consequences of loss of CTK transfer and CTK-mediated SHPS-1 phosphorylation
To further analyze the effects of altering the specific SHPS-1 tyrosine residues on CTK transfer and CTK-mediated SHPS-1 phosphorylation, we used the Y12F and Y34F mutants. IGF-I stimulated a 3.6 ± 0.2-fold increase in CTK transfer to SHPS-1 in WT cells, which was reduced to a 2.1 ± 0.1-fold increase in Y12F cells (n = 3, P < 0.01) (Fig. 5A). In contrast, cells expressing the Y34F mutant had no detectible CTK/SHPS-1 association (Fig. 5A). IGF-I stimulated a 3.3 ± 0.2-fold increase in MAPK phosphorylation in cells expressing WT SHPS-1 compared with a 1.8 ± 0.1-fold increase in Y12F mutant cells, (n = 3, P < 0.01; Fig. 5B). IGF-I stimulated cellular proliferation 3.8 ± 0.3-fold in WT compared with 2.1 ± 0.3-fold in Y12F mutant cells (n = 3, P < 0.01; Fig. 5C), and it stimulated migration 3.3 ± 0.3-fold in WT compared with 1.9 ± 0.1-fold in Y12F mutant cells (n = 3, P < 0.001; Fig. 5D). Cells expressing the Y34F mutant showed a minimal increase in MAPK activation and no significant changes in IGF-I-stimulated proliferation or migration. These results clearly indicate that CTK transfer to SHPS-1 and CTK-mediated SHPS-1 phosphorylation is required for optimal stimulation of cell proliferation and migration in response to IGF-I.
Fig. 5.

Substitution for specific tyrosines on SHPS-1 alters CTK association and IGF-I signaling-stimulated actions. A, Confluent VSMC expressing SHPS1-WT and SHPS1 (Y12F and Y34F) mutant cells were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for 5 min. Cell lysates were immunoprecipitated (IP) using anti-CTK antiserum then immunoblotted (IB) for HA-tagged SHPS-1. An equal amount of protein from cell lysates was used for IP/IB to estimate total CTK. B, Twenty micrograms of protein from the same cell lysates were directly immunoblotted with anti-pMAPK or anti-β-actin. Error bars represent mean ± se. *, P < 0.05 when phosphorylation of MAPK at 5 min in response to IGF-I is compared between WT and Y12F; **, P < 0.01 when Y12F and Y34F are compared. C, SHPS-1-WT, Y12F, and Y34F cells were plated (3 × 104) in DMEM-HG with 2% FBS before exposure to IGF-I (50 ng/ml) in DMEM-HG with 0.2% platelet-poor plasma. Forty-eight hours after the addition of IGF-I, the cell number was determined by tryptan blue staining and counting. The bar graphs show pooled results from at least three independent experiments. Error bars represent mean ± se. ***, P < 0.001 when the increase in cell number cell number in response to IGF-I in WT cells is compared with control; **, P < 0.01 when increase in cell number in response to IGF-I in Y12F cells is compared with control; **, P < 0.01 when the increases in cell number between WT and Y12F cells or between Y12F and Y34F cells are compared. D, SHPS-1 WT, Y12F, and Y34F cells were grown to confluent density in six-well plates in DMEM-HG containing 10% FBS. After wounding, they were allowed to migrate with or without IGF-I in medium containing 0.2% FBS for 48 h. The total number of cells migrating past the wound edge in five predetermined areas was quantified. ***, P < 0.001 when the number of WT cell migration in response to IGF-I is compared with control or with the number of migration of Y12F or Y34F cells.
Inhibition of CTK binding to IGF-IR impairs SHPS-1 tyrosyl phosphorylation
Because the CTK-IGF-IR association preceded CTK-SHPS-1 association (Fig. 2B), we hypothesized that activated IGF-IR may function to recruit additional CTK to the membrane fraction and that this may facilitate CTK recruitment to SHPS-1. CTK binds to the phosphotyrosine in YXXL/I/V motifs through its SH2 domain. Therefore, we used a cell-permeable synthetic peptide (peptide 298) that contained the 968NGVLYASV 976 sequence that is contained within IGF-IR because it had a high probability of binding to the SH2 domain of CTK. Exposure of nontransfected VSMC maintained in high glucose to this peptide resulted in significant inhibition of CTK binding to IGF-IR (53 ± 4.5% reduction, n = 3, P < 0.01; Fig. 6A). Importantly, exposure to peptide 298 inhibited SHPS-1 phosphorylation (77 ± 1% reduction, n = 3, P < 0.01) and SHP-2 recruitment to phosphorylated SHPS-1 (54 ± 7% reduction, n = 3, P < 0.05) compared with cells not exposed to the peptide in response to IGF-I. The peptide also resulted in impairment of IGF-I stimulated MAPK phosphorylation (52 ± 4.7% reduction, n = 3, P < 0.01; Fig. 6A). The peptide had no effect on IGF-IR phosphorylation (data not shown).
Fig. 6.

CTK transfer to IGF-IR facilitates SHPS-1 phosphorylation. A, VSMC maintained in DMEM-HG were serum starved and then incubated with or without cell-permeable peptide 298 at a concentration of 10 μg/ml for 2 h. IGF-I was added for 5 min with and without the peptide. Cell lysates were immunoprecipitated (IP) with an anti-CTK antibody and immunoblotted (IB) with anti-IGF-IR (fourth panel), anti-SHPS1 (fifth panel), or anti-CTK antibodies (sixth panel). The cell lysates were also immunoprecipitated with anti-SHPS-1 antibody and immunoblotted for pY (first panel) or SHP-2 (second panel). Twenty micrograms of protein from the same or similar cell lysates were directly immunoblotted with anti-SHPS-1 (third panel), pMAPK (seventh panel) or anti-β-actin (eighth panel) antibodies. The bar graph shows the relative decrease in SHPS-1 phosphorylation for at least three independent experiments. The error bars represent mean ± se. **, P < 0.001 when the increase in SHPS-1 phosphorylation in response to IGF-I is compared with or without peptide 298 (Pep298). Ctrl, Control. B, Confluent VSMC expressing IGF-IR-WT and IGF-IR-Y4F mutant cells were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for 5 or 10 min. Cell lysates were immunoprecipitated with an anti-IGF-IR antibody and immunoblotted for pY (first panel) or for total IGF-IR (second panel). Cell lysates from the same experiments were immunoprecipitated with an anti-CTK antibody and immunoblotted using an anti-IGF-IR antibody (third panel), an anti-SHPS1 antibody (fifth panel), or an anti-CTK antibody (fourth and sixth panels). C, Lysates from an experiment similar to that shown in B were immunoprecipitated with an anti-SHPS-1 antibody and immunoblotted for pY (first panel) or total SHPS-1 (second panel). Twenty micrograms of total protein from the above cell lysates were directly immunoblotted with anti-pMAPK (third panel) or anti-β-actin (fourth panel). The bar graph represents the pooled results from three independent experiments and shows the relative decrease in SHPS-1 phosphorylation. The error bars represent mean ± se. **, P < 0.01; * P < 0.05 indicate the significant differences between two treatments or two cell types. D, IGF-I-R-WT and IGF-I-R-Y4F cells were plated (3 × 104) in DMEM-HG with 2% FBS before exposure to IGF-I (50 ng/ml) in DMEM-HG with 0.2% platelet-poor plasma. Forty-eight hours after the addition of IGF-I, cell number was determined by tryptan blue staining and counting. The bar graphs show pooled results from at least three independent experiments. Error bars represent mean ± se. ***, P < 0.001 when number of proliferating cells is compared between WT and Y4F cells in response to IGF-I. P, NS indicates no significant difference between two treatments. E, Confluent VSMC expressing IGF-IR-WT and IGF-IR-Y4F mutant cells were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for 1 min. As indicated, cell lysates were precleared with protein A beads and then immunoprecipitated with an anti-IGF-IR antibody. Immunoprecipitates were used in an in vitro kinase reaction as described in Materials and Methods. The relative kinase activity is depicted in the bar graph. The error bars represent mean ± se. P = NS. **, P < 0.01 indicates a significant difference between two treatments. P, NS indicates no significant difference.
Inhibition of the IGF-IR-CTK association by the peptide did not completely abolish IGF-IR-CTK binding. This might be due to the fact that the IGF-IR β-subunit contains four tyrosines (973, 1014, 1192, and 1213), all of which are contained within YXXV/L motifs that are phosphorylated in response to IGF-I (26). Therefore, we prepared an IGF-IR mutant in which all four tyrosines were mutated to phenylalanine (IGF-IR Y4F mutant) (Supplemental Fig. 1B). When the cells expressing this mutant were analyzed, IGF-IR phosphorylation was minimally decreased (25 ± 6.6% reduction, n = 3, P < 0.05) compared with WT (Fig. 6B). In vitro kinase assays showed that the mutant receptor had normal tyrosine kinase activity (Fig. 6E), and there was no significant difference in IRS-1 phosphorylation when compared with cells expressing the WT/SHPS-1 (data not shown). CTK binding to IGF-IR was markedly inhibited (71 ± 2.8% reduction n = 3, P < 0.001; Fig. 6B). Furthermore, CTK transfer to SHPS-1 was also reduced in response to IGF-I in cells expressing the IGF-IR Y4F mutant compared with the WT cells (55 ± 13% reduction, n = 3, P < 0.01; Fig. 6B). Importantly, IGF-I-stimulated SHPS-1 tyrosine phosphorylation was significantly impaired (63 ± 8% reduction, n = 3, P < 0.05) as was MAPK phosphorylation (70 ± 2.9% reduction, n = 3, P < 0.001) (Fig. 6C). Therefore, it appears that CTK binding to IGF-IR facilitates its transfer to SHPS-1. The proliferation of the cells expressing WT IGF-IR was significantly increased by IGF-I (3.8 ± 0.2-fold, n = 3, P < 0.001; Fig. 6D), whereas there was no significant change in cells expressing the IGF-IR Y4F mutant.
IGF-IR/SHPS-1 association is required for IGF-IR-stimulated SHPS-1 tyrosyl phosphorylation and is mediated in part by CTK
To determine whether the IGF-IR/SHPS-1 association was required for IGF-IR to phosphorylate SHPS-1, we expressed an SHPS-1 mutant with a truncated extracellular domain and showed that it had reduced IGF-IR/SHPS-1 association in response to IGF-I (69 ± 6.7% reduction, n = 3, P < 0.01; Fig. 7A). Cells expressing this mutant showed reduced IGF-I-stimulated SHPS-1 phosphorylation (78 ± 4.4-fold reduction, n = 3, P < 0.001; Fig. 7A). Under basal HG conditions, a small amount of CTK is bound to SHPS-1 (Fig. 2B). To determine the functional significance of this small amount of CTK in mediating IGF-IR/SHPS-1 association, we used cells expressing several different constructs that had a reduced CTK/SHPS-1 association. The SHPS-1/IGF-IR association is attenuated in CTK shRNA-expressing cells (52 ± 5.7% reduction, n = 3, P < 0.01; Fig. 7B) and in cells expressing the SHPS-1 Y34F mutant (69 ± 9.4% decrease, n = 3, P < 0.05; Fig. 7C). To confirm the significance of CTK binding to IGF-IR for IGF-IR/SHPS-1 association, we analyzed the cells expressing the IGF-IR Y4F mutant that has reduced CTK/IGF-IR association. After stimulation by IGF-I, the SHPS-1/IGF-IR association was decreased significantly (59 ± 9.1% decrease, n = 3, P < 0.01; Fig. 7D). To confirm that a complex that contains all three components is formed in response to HG plus IGF-I, we undertook a double-coimmunoprecipitation experiment in which the IGF-IR-containing immunoprecipitate was further immunoprecipitated with anti-CTK and immunoblotted for SHPS-1. After IGF-I stimulation there was a significant increase in complex formation (Supplemental Fig. 1C).
Fig. 7.

IGF-IR/SHPS-1 association and IGF-IR-stimulated SHPS-1 phosphorylation is mediated in part by CTK. A, Confluent VSMC expressing SHPS-1WT or SHPS-1 IgA deletion mutant were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for the times indicated. Cell lysates were immunoprecipitated (IP) with an anti-SHPS-1 antibody and then immunoblotted (IB) for IGF-IR (top panel) or with an anti-pY antibody (middle panel). Twenty micrograms of protein from the same or similar whole-cell lysate were directly IB with an anti-SHPS-1 antibody (lower panel). B, Confluent empty control vector (EVC), and CTK shRNA cell cultures were serum starved for 16 h and IGF-I (50 ng/ml) was added for times indicated. The cell lysates were immunoprecipitated with an anti-SHPS-1 antibody and then immunoblotted for IGF-IR or using an anti-SHPS-1 antibody. C, Confluent VSMC-expressing SHPS-1-WT and SHPS-1 (Y12F and Y34F) mutant cells were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for 5 min. The extent of HA-tagged SHPS-1 association with IGF-IR was determined by immunoprecipitating HA and immunoblotting for IGF-IR or with an anti-HA antibody as a loading control. D, Confluent VSMC-expressing IGF-IR-WT and IGF-IR-Y4F mutant cells were serum starved for 16 h in DMEM-HG and then exposed to IGF-I for the indicated period of time. Cell lysates were immunoprecipitated with an anti-SHPS-1 antibody and immunoblotted using an anti-IGF-IR antibody or an anti-SHPS-1 antibody.
Discussion
SHPS-1 has been proposed to be a substrate for several receptor tyrosine kinases, Src kinase, and SFK (3, 9–12). Recently the protein kinase, CTK, was shown to phosphorylate SHPS-1 in neutrite cells (13). Using an unbiased proteome-wide screen for SHPS-1 CD interacting proteins in VSMC, we found that CTK was the only kinase that bound directly to phosphorylated SHPS-1 (4). We recently reported that activated IGF-IR phosphorylated the SHPS-1/CD in vitro (3), but the specific tyrosine residues within the SHPS-1/CD that are phosphorylated by these kinases in VSMC have not been defined. The novel finding of this study is that IGF-I stimulates a cooperative interaction between IGF-IR and CTK that leads to CTK recruitment to IGF-IR, which is required for an optimal increase in SHPS-1 phosphorylation in response to IGF-I.
After IGF-I binding, the IGF-IR tyrosine kinase is activated, which autophosphorylates specific residues within seconds and maximal stimulation is reached at 1 min (27). We have previously shown that a robust SHPS-1 phosphorylation response requires 5 min in VSMC in response to IGF-I (28), and this finding was confirmed in these studies (Supplemental Fig. 2B). Data from our previous study showed that the increase in SHPS-1 phosphorylation was associated with an increase in IGF-IR/SHPS-1 association in hyperglycemic conditions (3), which is supported by our current observations. Our current results show that VSMC expressing the mutant form of SHPS-1 that lacks the IgA domain has a decreased association with IGF-IR and decreased IGF-I-stimulated SHPS-1 phosphorylation. Taken together, these results suggest that IGF-IR and SHPS-1 have to associate for IGF-IR to phosphorylate SHPS-1 and support our previous observation that the extracellular region of SHPS-1 is essential for its phosphorylation (24, 29). This important function of IGF-IR cannot be rescued by the insulin receptor because VSMC have preponderance of IGF-IR and are insulin resistant due to the low fraction of available insulin receptors (30). Those investigators showed that the signal transduction and the biological responses in smooth muscle cells are primarily due to IGF-IR, not IR activation. Further detailed studies will be required to identify the regions of IGF-IR and SHPS-1 that mediate this interaction.
Because CTK was shown to be recruited to IGF-IR at 1 min and this occurred before the increase in IGF-I-stimulated CTK binding to SHPS-1 and IGF-I-stimulated SHPS-1 phosphorylation, this raised the question as to whether basal CTK binding to SHPS-1 was facilitating an IGF-IR/SHPS-1 association. Using several models, we investigated whether the interaction between IGF-IR and SHPS-1 is mediated through CTK. Although a major increase in SHPS-1 phosphorylation is required to detect a substantial increase in CTK binding to SHPS-1, a small amount of CTK binding to SHPS-1 is detectible under basal, hyperglycemic conditions. Our studies clearly show that if this binding is eliminated by mutation the CTK binding site on SHPS-1 or by using CTK knockdown, an IGF-IR/SHPS-1 association is inhibited as is IGF-IR-mediated SHPS-1 phosphorylation. After IGF-I stimulation, IGF-IR recruits CTK to phosphorylated tyrosines on IGF-IR through an SH2 domain/phosphotyrosine interaction and disruption of the IGF-IR/CTK binding by either the peptide that contains that YASV sequence or specific mutation of the four YXXL/V motifs in IGF-IR significantly impaired CTK association with IGF-IR. This result is in agreement with the prior studies that show that CTK preferentially binds to tyrosine kinases such as Erb2, c-kit receptor tyrosine kinase, paxillin, TrkA receptor, and PYK2 via a YXX/L/I/V motif (20, 21, 23) and supports the proposal of Ayrapetov et al. (31) that the CTK SH2 domain preferentially binds to this sequence. The studies by Mikkola and Bergman (32) suggest that the binding of specific proteins to the SH2 domain of CTK can regulate its activation. The four tyrosine residues that were mutated in the IGF-IR β-subunit are phosphorylated in response to IGF-I (26) and play an important role in IGF-IR-mediated pathways including phosphatidylinositol 3-kinase/AKT activation (33) and control of apoptosis (34). Taken together, these results suggest that basal CTK binding to SHPS-1 is required for the initiation of IGF-I stimulated IGF-IR/SHPS-1 association, which subsequently results in IGF-IR-stimulated SHPS-1 phosphorylation and further increase in SHPS-1/CTK association.
Similar to the changes in IGF-IR autophosphorylation, CTK recruitment to IGF-IR occurred at 1 min, a time point at which there was no change or minimal increase in IGF-I-stimulated SHPS-1 phosphorylation (Supplemental Fig. 2B) and blocking CTK recruitment to IGF-IR inhibited the IGF-I-induced increase in SHPS-1 phosphorylation. These findings strongly suggest that IGF-IR activation is required to recruit additional CTK to the cell membrane fraction. This finding raises the question of why additional CTK recruitment to IGF-IR has to occur before an increase in IGF-IR-mediated SHPS-1 phosphorylation. CTK has only one SH2 domain; therefore, for CTK to bind to IGF-IR and SHPS-1 simultaneously through SH2 domain-YXXL/V interactions would require CTK dimerization. Although CTK dimerization has not been reported, a homologous kinase carboxyl-terminal src kinase (CSK) does form stable dimers through its SH3 domain (35). Importantly, substitution for Asn at position 64 (contained in the SH3 domain) with Ala enhances CSK dimerization. CTK contains a sequence that is nearly identical with CSK in this region of its SH3 domain, but unlike CSK the Ala residue is present in the unmodified protein. Although in normoglycemic conditions CTK exists predominantly as monomer, under hyperglycemic conditions in response to IGF-I, we observed increased CTK dimerization (Radhakrishnan, Y., and D. Clemmons, unpublished observations). This suggests that in the presence of IGF-I and hyperglycemia, CTK can dimerize and that its recruitment and activation in the submembrane fraction (36, 37) leads to the facilitation of dimerization, which allows IGF-IR/SHPS-1/CTK complex formation (Supplemental Fig. 1C).
After CTK recruitment to IGF-IR, there is a major increase in the SHPS-1/CTK association and in SHPS-1 phosphorylation. Our results obtained using the SHPS-1 mutants suggest that IGF-IR rapidly phosphorylates Y469 and 495 in the SHPS-1/CD, which provides a binding site for the additional CTK to be recruited to SHPS-1 and that CTK subsequently phosphorylates Y428/452 leading to SHP-2 recruitment. This initial phosphorylation of SHPS-1 by IGF-IR leads to enhanced CTK transfer and could facilitate SHPS-1/IGF-IR association and IGF-IR-mediated SHPS-1 phosphorylation. Subsequently when sufficient CTK has been transferred, it becomes the predominate kinase for completing SHPS-1 phosphorylation. This hypothesis is further strengthened by our observation using the Y12F mutant cells, which showed that when tyrosines Y469/495 of SHPS-1 are phosphorylated in response to IGF-I, there is a detectable increase in an IGF-IR/CTK association. However, cells expressing this mutant showed reduced SHPS-1 phosphorylation compared with WT cells, suggesting that after CTK transfer it phosphorylates 428/452, thus allowing an optimal increase in SHPS-1 phosphorylation. When tyrosines 469/495 were mutated to phenylalanine, there was no detectable SHPS-1 phosphorylation, no CTK transfer, and no increase in an IGF-IR/SHPS-1 association. Our interpretation is that in the absence of CTK binding to SHPS-1, the initial IGF-IR-mediated phosphorylation of SHPS-1/CD does not occur, and hence, there is no detectable increase in phosphorylation. Our findings differ from the observations of Mitsuhashi (13) et al. in which they observed the residues Y469/495 to be substrates of CTK. This finding might be explained by the fact that their study did not measure tyrosine phosphorylation directly and that CTK-associated SHPS-1 function was determined using HIS3 yeast and coexpressing SHPS-1 mutants with CTK. Our in vitro studies show that these residues can be phosphorylated by CTK, but our results in intact cells support the conclusion that the primary role of pY469/495 is to provide a binding site for CTK. SHP2 also binds to 469/495 tyrosines (data not shown), and this is supported by the previous observation that mutation of these two tyrosine impairs SHP-2 binding (38).
The relative role of CTK as an activator of other signaling pathways, which have been linked to growth regulation, was not addressed directly in this study. However, overexpression of CTK has been shown to increase proliferation and invasion in MCF7 cells, neurite outgrowth in PC12 cells, and multinucleation of myeloid cells (20–22, 39, 40). In sharp contrast to animal model knockdown of CTK, in cultured cells RNA interference of CTK led to a decrease in phosphorylation of MAPK in response to NGF in PC12 cells (39). Our results support these observations because IGF-I-induced phosphorylation of MAPK was significantly impaired in CTK shRNA cells, and this was associated with a reduction in cell proliferation. Therefore, the decrease in VSMC proliferation in response to IGF-I in CTK shRNA cells and in the cells expressing the Y469/495F mutant is consistent with the previous observation that CTK kinase dead cells have decreased SHPS-1 phosphorylation and neurite outgrowth in response to NGF stimulation (13). Taken together, these results demonstrate a physiologically significant growth regulatory role for CTK in controlling IGF-I-stimulated SHPS-1 phosphorylation, and this interaction constitutes an important growth regulatory function by which vascular cells alter their response to hyperglycemic stress.
Our prior studies have shown that the ability of vascular cells to survive hyperglycemic stress is dependent on the assembly of the SHPS-1 signaling complex, and complex assembly requires SHPS-1 phosphorylation. The findings in this study define the roles of two tyrosine kinases, IGF-IR and CTK, in regulating this process. The cooperative interaction that is described is unique for IGF-IR and suggests that has the potential to regulate the recruitment and activation of other kinases and that this may be an important determinant of IGF-I's anabolic and antiapoptotic actions.
Materials and Methods
Human IGF-I was a gift from Genentech (South San Francisco, CA). Immobilon-P membranes were purchased from Millipore Corp. (Bedford, MA). DMEM containing 4500 mg glucose per liter (DMEM-HG), DMEM containing 900 mg glucose per liter (DMEM-NG), streptomycin, penicillin, and recombinant human CTK expressed in Escherichia coli with His-tag were purchased from Invitrogen (Carlsbad, CA). Antiphosphotyrosine (pY99), hereafter referred to as pY, the HA epitope, anti-CTK, anti-IGF-IR, and anti-SHP2 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal antibodies for SHPS-1 and Shc were obtained from Upstate Biotechnology (Lake Placid, NY). SHPS-1, HA, and IGF-IR polyclonal antiserum were also prepared in our laboratory. Antibodies against phospho-MAPK or total MAPK and β-actin were purchased from Cell Signaling (Beverly, MA). The IGF-IR tyrosine kinase inhibitor, PQ401, was purchased from Tocris Bioscience (Ellisville, MO). Recombinant rat IRS-1 expressed in Sf9 cells with His-tag was purchased from Millipore. A synthetic peptide containing cell permeability sequence of protein transduction domain 4 and the sequence from IGF-IR YARAAARQARA938GNGVLYASV946 (underlined and hereafter referred to as the peptide 298) was synthesized by the Protein Chemistry Core Facility at the University of North Carolina at Chapel Hill. The peptide sequence was confirmed by mass spectrometry.
Construction, expression, purification, and phosphorylation of the SHPS-1/CD and IGF-IR
The SHPS-1/CD, containing amino acids 394–503 was PCR amplified with SP6 promoter sequence followed by reverse transcription, which was performed to obtain mRNA (4). Using this template, SHPS-1/CD was generated, using in vitro translation using wheat germ extract as per the manufacturer's recommendation (Promega, Madison, WI). Mouse fibroblasts (NWTb3) that overexpress human IGF-IR were a kind gift from Dr. Derek LeRoith (Mount Sinai School of Medicine, New York, NY) (41).
IGF-IR was purified from the NWTb3 cell lysate and was activated with IGF-I as described previously (3). The activated IGF-IR or recombinant CTK was used to phosphorylate the CD of SHPS-1 in a buffer containing 50 mm HEPES-NaOH (pH 7.6), 3 mm MnCl2, 10 mm MgCl2, 0.1 mm EGTA, 1 mm dithiothreitol, 0.1 mm Na3O4, and 0.2 mm ATP and incubating at 30 C for 30 min. To detect tyrosine phosphorylation of SHPS-1/CD, the final product was immunoblotted using an anti-pY antibody.
Cell culture
Porcine VSMC were isolated from the porcine aortic explants and maintained as previously described (7). The cells were maintained in DMEM-HG growth medium or DMEM-NG growth medium with 10% fetal bovine serum (Hyclone, Logan, UT) and streptomycin (100 ng/ml) and penicillin (100 U/ml).
Generation of pLenti-expression vectors
WT IGF-IR in pBluescript II KS was a kind gift from Dr. Derek LeRoith. Using this as a template, IGF-IR was PCR amplified and cloned into pENTR/D-TOPO and transferred into pLentiCMV DEST vector as previously described (3). Using the IGF-IR-WT as a template, an IGF-IR mutant was generated wherein the tyrosines at positions 973, 1014, 1192, and 1213 were mutated to phenylalanine (IGF-IR-Y4F). PCR amplifications were carried out using following the complementary primer 5′-GGAATGGAGTGCTGTtTGCCTCTGTGAACCC-3′ and the reverse primer 5′-GGGTTCACAGAGGCAaACAGCACTCCATTCC-3′ to generate Y973F; the complementary primer 5′-CGTTTGGGATGGTCTtTGAAGGAGTTGCCAA-3′ and the reverse primer 5′-TTGGCAACTCCTTCAaAGACCATCCCAAACG-3′ to generate Y1014F; the complementary primer 5′-GGAGTCTTCACCACTTtCTCGGACGTCTGGTCC-3′ and the reverse primer 5′-ACCAGACGTCCGAGaAAGTGGTGAAGACTCC-3′ to generate Y1192F; and the complementary primer 5′-CTGGCCGAGCAGCCCTtCCAGGGCTTGTCCAAC-3′ and the reverse primer 5′-GTTGGACAAGCCCTGGaAGGGCTGCTCGGCCAG-3′ to generate Y1213F in which lower-case bases indicate the substitutions.
The full-length SHPS-1 sequence was PCR amplified with HA-containing primers using previously generated pcDNA-SHPS-1 as a template and cloned into the expression vector pLenti6/V5-D-TOPO and then transferred into pLentiCMV DEST vector as previously reported (4). Using the pENTR SHPS1-WT as a template, two SHPS-1 mutants were generated. In the first mutant, the first two tyrosines in the CD at positions 428 and 452 were mutated to phenylalanine (Y12F). In the second mutant, the third and fourth tyrosines at positions 469 and 495 were also substituted with phenylalanine (Y34F). The first PCR amplifications were carried out using complementary primer 5′-aggacacaaatgatatcacatTtgcagacctgaacctgcccaa-3′ and reverse primer 5′-ttgggcaggttcaggtctgcaAatgtgatatcatttgtgtcct-3′ to generate Y428F. After selection of the correct clone based on DNA sequencing, the Y428F DNA was used to generate Y452F using complementary primer 5′-cccaacaaccacacggagtTtgccagcattcagaccagc-3′ and reverse primer 5′-gctggtctgaatgctggcaAactccgtgtggttgttggg-3′ in which capitalized bases indicate the substitution that resulted in what is hereafter termed the Y12F mutant. Similarly, for generation of what is hereafter termed the Y34F mutant, PCR amplifications were carried out using complementary primer 5′-tcggaggacaccctcaccttTgctgacctggacatggtcca-3′ and reverse primer 5′-tggaccatgtccagg tcagcaaAggtgagggtgtcctccga-3′ to generate Y469F. After selection of the correct clone based on DNA sequencing, the Y469F DNA was used to generate Y495F using complementary primer 5′-gagccgtccttctcagagtTcgccagcgtccaggtcccg-3′ and reverse primer 5′-cgggacctggacgctggcgAactctgagaaggacggctc-3′ in which capitalized bases indicate the substitution. The IgA domain in the extracellular region of SHPS-1 was deleted using SHPS-1 WT and PCR amplification. The reaction was carried out using a forward primer 5′-gcgtcctgcgcctggactgtgacctccctgatc-3′ and a reverse primer 5′-gatcagggaggtcacagttgaccaggcgcaggacgc-3′. The products were sequenced (University of North Carolina Genome Analysis Facility, Chapel Hill, NC) to confirm the correct incorporation of the changes. After the sequencing of the cDNA's encoding, the WT and mutant forms of the protein were transferred from the entry vector into the pLentiCMV DEST vector using the LR clonase reaction following the manufacturer's instructions (Invitrogen).
Construction of a plasmid containing shRNA for CTK silencing
Based on Invitrogen web site design tools, three sequences containing 21 oligonucleotides, GGTGGAGCATTACAGCAAGGA, GGATATGGTGGAGCATTACAG, and GGAGCATTACAGCAAGGACAA, located within porcine CTK (4), were used to construct the shRNA template plasmid. The oligonucleotides were synthesized by the Nucleic Acids Core Facility at the University of North Carolina, annealed, and ligated into BLOCK-iT U6 RNAi entry vector (Invitrogen) following the manufacturer's instructions. The complete sequence was verified by DNA sequencing. The expression vector was generated using the Gateway LR recombination reaction between the entry vector and BLOCK-iT Lentiviral RNAi gateway vector (Invitrogen). The expression vector was used as a control. After confirmation of the sequence, plasmid DNA was prepared by a plasmid minikit (Promega).
Generation of virus stocks
The 293FT cells (Invitrogen) were prepared for generation of virus stocks of each individual pLenti construct. All the transfections and viral stock generations were carried out as previously described (5).
Establishment of porcine VSMC-expressing pLenti constructs
Early passage VSMC (three to five) were seeded at 3 × 105/well in each of two wells of a six-well plate the day before transduction. The viral stocks were thawed and the viral complexes precipitated as described previously (5). All the cells expressing pLenti or shRNA constructs were maintained in DMEM-HG. The expression of the HA-tagged WT and mutants for each construct were detected by immunoblotting with a 1:1000 dilution of anti-HA antibody using 20 μg of protein.
Immunoprecipitation and immunoblotting
Cells were seeded at 5 × 105 cells per 10-cm plate (Beckton Dickinson Labware, Franklin Lakes, NJ) and grown for 5–7 d to reach confluency. The cultures were incubated in serum-free DMEM-HG or DMEM-NG for 14–18 h before the addition of IGF-I (as indicated). In some experiments, nontransduced VSMC were preincubated with or without the synthetic peptide or the PQ401 inhibitor for 1–2 h before IGF-I was added. After cell lysis all immunoprecipitations and immunoblotting were carried out as described previously (5). The proteins were detected using enhanced chemiluminescence (Pierce Chemical Co., Rockford, IL), and the images were captured using autoradiographic film (Denville Scientific Inc., South Plainfield, NJ). For some experiments, multiple exposures were taken so that the signals that were used for quantification were in the linear range of the film. The images obtained were scanned using an HP Scanjet 8300 (Hewlett Packard, Palo Alto, CA). Densitometric analyses of the images were determined using ImageJ, version 1.37v, software (National Institutes of Health, Bethesda, MD). To determine differences in MAPK phosphorylation, arbitrary scanning units obtained for phospho-MAPK band intensities were divided by arbitrary scanning units obtained for total β-actin protein band intensities for each time point analyzed. The fold values or average ratio values obtained from at least three independent experiments for each lane were pooled.
In vitro kinase assays
The IGF-IR in vitro kinase assays were performed using a modified protocol of Buckley et al. (42). In short, cell lysates were precleared with protein A beads and immunoprecipitated with IGF-IR antibody and then washed three times with radioimmunoprecipitation assay and once with kinase buffer and then resuspended in 25 μl of kinase reaction mixture containing ATP (final concentration 0.03 mm), 2 μl of 32pATP (5 μCi/μl), and 2 μl of poly(Glu-Tyr) (kind gift from Dr. Lee Graves, University of North Carolina, Chapel Hill, NC). The reaction was carried out at 30 C for 20 min and stopped with 20 μl of 40% trichloroacetic acid. Twenty-five microliters of the reacted mix were spotted on phosphocellulose and subsequently washed three times with 0.75% phosphoric acid for 15 min and finally one time with acetone for 3 min and dried. The 32p was measured by scintillation counting (Beckman, Palo Alto, CA).
Cell proliferation and migration assays
Assessment of VSMC proliferation was performed as described previously (43). IGF-I is known to stimulate smooth muscle cell chemokinesis, and cellular migration was analyzed as previously described (44). Briefly, VSMC were plated in six-well dishes and grown to confluency. The monolayer of cells was wounded with a razor blade. After wounding, cells were incubated with serum-free medium (SFM) plus 0.2% fetal bovine serum (FBS) in the presence or absence of IGF-I (50 ng/ml) at 37 C for 48 h. The wounded monolayers were then fixed and stained (Diff Quick; Dade Behring Inc., Newark, DE), and the number of cells migrating into the wound area was counted. At least five of the previously selected 1-mm areas at the edge of the wound were counted for each data point.
Statistical analysis
A Student's t test was used to compare the differences between the treatments. The results that are shown in all experiments are representative of at least three separate experiments and P ≤ 0.05 was considered statistically significant.
Acknowledgments
We thank Jane Badley-Clarke for help in preparation of IGF-IR WT, IGF-IR Y4F, and SHPS-1 deletion IgA constructs; Walker Busby, Jr. for IGF-IR purification; Kevin Kohler for technical assistance; and Laura Lindsey for assistance in preparing the manuscript.
This work was supported by National Institutes of Health Grants HL56580 and AG02331 (to D.R.C.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CD
- Cytoplasmic domain
- CSK
- carboxyl-terminal src kinase
- CTK
- CSK-homologous kinase
- EVC
- empty control vector
- FBS
- fetal bovine serum
- HA
- hemagglutinin
- HG
- high glucose
- IGF-IR
- IGF-I receptor
- IRS-1
- insulin receptor substrate-1
- NG
- normal glucose
- NGF
- nerve growth factor
- SFK
- Src family kinase
- SFM
- serum-free medium
- Shc
- Src homology 2 domain-containing protein
- SHPS-1
- Src homology 2 domain containing protein tyrosine phosphatase substrate-1
- shRNA
- short hairpin RNA
- VSMC
- vascular smooth muscle cell
- WT
- wild type.
References
- 1. Arnqvist HJ, Bornfeldt KE, Chen Y, Lindström T. 1995. The insulin-like growth factor system in vascular smooth muscle: interaction with insulin and growth factors. Metabolism 44:58–66 [DOI] [PubMed] [Google Scholar]
- 2. Joslin E, Kahn CR. 2005. Joslin's diabetes mellitus. 14th ed. Boston: Lippincott Williams, Wilkins [Google Scholar]
- 3. Radhakrishnan Y, Busby WH, Jr, Shen X, Maile LA, Clemmons DR. 2010. Insulin-like growth factor-I-stimulated insulin receptor substrate-1 negatively regulates Src homology 2 domain-containing protein-tyrosine phosphatase substrate-1 function in vascular smooth muscle cells. J Biol Chem 285:15682–15695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen X, Xi G, Radhakrishnan Y, Clemmons DR. 2009. Identification of novel SHPS-1-associated proteins and their roles in regulation of insulin-like growth factor-dependent responses in vascular smooth muscle cells. Mol Cell Proteomics 8:1539–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ling Y, Maile LA, Lieskovska J, Badley-Clarke J, Clemmons DR. 2005. Role of SHPS-1 in the regulation of insulin-like growth factor I-stimulated Shc and mitogen-activated protein kinase activation in vascular smooth muscle cells. Mol Biol Cell 16:3353–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maile LA, Capps BE, Miller EC, Allen LB, Veluvolu U, Aday AW, Clemmons DR. 2008. Glucose regulation of integrin-associated protein cleavage controls the response of vascular smooth muscle cells to insulin-like growth factor-I. Mol Endocrinol 22:1226–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maile LA, Capps BE, Ling Y, Xi G, Clemmons DR. 2007. Hyperglycemia alters the responsiveness of smooth muscle cells to insulin-like growth factor-I. Endocrinology 148:2435–2443 [DOI] [PubMed] [Google Scholar]
- 8. Radhakrishnan Y, Maile LA, Ling Y, Graves LM, Clemmons DR. 2008. Insulin-like growth factor-I stimulates Shc-dependent phosphatidylinositol 3-kinase activation via Grb2-associated p85 in vascular smooth muscle cells. J Biol Chem 283:16320–16331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elbaz N, Bedecs K, Masson M, Sutren M, Strosberg AD, Nahmias C. 2000. Functional trans-inactivation of insulin receptor kinase by growth-inhibitory angiotensin II AT2 receptor. Mol Endocrinol 14:795–804 [DOI] [PubMed] [Google Scholar]
- 10. Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. 1997. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386:181–186 [DOI] [PubMed] [Google Scholar]
- 11. Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, Tsuda M, Takada T, Kasuga M. 1996. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol 16:6887–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stofega MR, Wang H, Ullrich A, Carter-Su C. 1998. Growth hormone regulation of SIRP and SHP-2 tyrosyl phosphorylation and association. J Biol Chem 273:7112–7117 [DOI] [PubMed] [Google Scholar]
- 13. Mitsuhashi H, Futai E, Sasagawa N, Hayashi Y, Nishino I, Ishiura S. 2008. Csk-homologous kinase interacts with SHPS-1 and enhances neurite outgrowth of PC12 cells. J Neurochem 105:101–112 [DOI] [PubMed] [Google Scholar]
- 14. Chong YP, Mulhern TD, Zhu HJ, Fujita DJ, Bjorge JD, Tantiongco JP, Sotirellis N, Lio DS, Scholz G, Cheng HC. 2004. A novel non-catalytic mechanism employed by the C-terminal Src-homologous kinase to inhibit Src-family kinase activity. J Biol Chem 279:20752–20766 [DOI] [PubMed] [Google Scholar]
- 15. Klages S, Adam D, Class K, Fargnoli J, Bolen JB, Penhallow RC. 1994. Ctk: a protein-tyrosine kinase related to Csk that defines an enzyme family. Proc Natl Acad Sci USA 91:2597–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuo SS, Moran P, Gripp J, Armanini M, Phillips HS, Goddard A, Caras IW. 1994. Identification and characterization of Batk, a predominantly brain-specific non-receptor protein tyrosine kinase related to Csk. J Neurosci Res 38:705–715 [DOI] [PubMed] [Google Scholar]
- 17. Bennett BD, Cowley S, Jiang S, London R, Deng B, Grabarek J, Groopman JE, Goeddel DV, Avraham H. 1994. Identification and characterization of a novel tyrosine kinase from megakaryocytes. J Biol Chem 269:1068–1074 [PubMed] [Google Scholar]
- 18. McVicar DW, Lal BK, Lloyd A, Kawamura M, Chen YQ, Zhang X, Staples JE, Ortaldo JR, O'Shea JJ. 1994. Molecular cloning of lsk, a carboxyl-terminal src kinase (csk) related gene, expressed in leukocytes. Oncogene 9:2037–2044 [PubMed] [Google Scholar]
- 19. Sakano S, Iwama A, Inazawa J, Ariyama T, Ohno M, Suda T. 1994. Molecular cloning of a novel non-receptor tyrosine kinase, HYL (hematopoietic consensus tyrosine-lacking kinase). Oncogene 9:1155–1161 [PubMed] [Google Scholar]
- 20. Kim S, Zagozdzon R, Meisler A, Baleja JD, Fu Y, Avraham S, Avraham H. 2002. Csk homologous kinase (CHK) and ErbB-2 interactions are directly coupled with CHK negative growth regulatory function in breast cancer. J Biol Chem 277:36465–36470 [DOI] [PubMed] [Google Scholar]
- 21. Kuo SS, Armanini MP, Phillips HS, Caras IW. 1997. Csk and BatK show opposite temporal expression in the rat CNS: consistent with its late expression in development, BatK induces differentiation of PC12 cells. Eur J Neurosci 9:2383–2393 [DOI] [PubMed] [Google Scholar]
- 22. Yamashita H, Avraham S, Jiang S, Dikic I, Avraham H. 1999. The Csk homologous kinase associates with TrkA receptors and is involved in neurite outgrowth of PC12 cells. J Biol Chem 274:15059–15065 [DOI] [PubMed] [Google Scholar]
- 23. Price DJ, Rivnay B, Fu Y, Jiang S, Avraham S, Avraham H. 1997. Direct association of Csk homologous kinase (CHK) with the diphosphorylated site Tyr568/570 of the activated c-KIT in megakaryocytes. J Biol Chem 272:5915–5920 [DOI] [PubMed] [Google Scholar]
- 24. Maile LA, Capps BE, Miller EC, Aday AW, Clemmons DR. 2008. Integrin-associated protein association with SRC homology 2 domain containing tyrosine phosphatase substrate 1 regulates IGF-I signaling in vivo. Diabetes 57:2637–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lieskovska J, Ling Y, Badley-Clarke J, Clemmons DR. 2006. The role of Src kinase in insulin-like growth factor-dependent mitogenic signaling in vascular smooth muscle cells. J Biol Chem 281:25041–25053 [DOI] [PubMed] [Google Scholar]
- 26. Li M, He Z, Ermakova S, Zheng D, Tang F, Cho YY, Zhu F, Ma WY, Sham Y, Rogozin EA, Bode AM, Cao Y, Dong Z. 2007. Direct inhibition of insulin-like growth factor-I receptor kinase activity by (-)-epigallocatechin-3-gallate regulates cell transformation. Cancer Epidemiol Biomarkers Prev 16:598–605 [DOI] [PubMed] [Google Scholar]
- 27. Shemer J, Adamo M, Raizada MK, Heffez D, Zick Y, LeRoith D. 1989. Insulin and IGF-I stimulate phosphorylation of their respective receptors in intact neuronal and glial cells in primary culture. J Mol Neurosci 1:3–8 [DOI] [PubMed] [Google Scholar]
- 28. Maile LA, Clemmons DR. 2002. Regulation of insulin-like growth factor I receptor dephosphorylation by SHPS-1 and the tyrosine phosphatase SHP-2. J Biol Chem 277:8955–8960 [DOI] [PubMed] [Google Scholar]
- 29. Maile LA, Badley-Clarke J, Clemmons DR. 2003. The association between integrin-associated protein and SHPS-1 regulates insulin-like growth factor-I receptor signaling in vascular smooth muscle cells. Mol Biol Cell 14:3519–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chisalita SI, Johansson GS, Liefvendahl E, Bäck K, Arnqvist HJ. 2009. Human aortic smooth muscle cells are insulin resistant at the receptor level but sensitive to IGF1 and IGF2. J Mol Endocrinol 43:231–239 [DOI] [PubMed] [Google Scholar]
- 31. Ayrapetov MK, Nam NH, Ye G, Kumar A, Parang K, Sun G. 2005. Functional diversity of Csk, Chk, and Src SH2 domains due to a single residue variation. J Biol Chem 280:25780–25787 [DOI] [PubMed] [Google Scholar]
- 32. Mikkola ET, Bergman M. 2003. Conserved hydrophobicity in the SH2-kinase linker is required for catalytic activity of Csk and CHK. FEBS Lett 544:11–14 [DOI] [PubMed] [Google Scholar]
- 33. Vasilcanu D, Girnita A, Girnita L, Vasilcanu R, Axelson M, Larsson O. 2004. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene 23:7854–7862 [DOI] [PubMed] [Google Scholar]
- 34. Jernberg-Wiklund H, Nilsson K. 2007. Control of apoptosis in human multiple myeloma by insulin-like growth factor I (IGF-I). Adv Cancer Res 97:139–165 [DOI] [PubMed] [Google Scholar]
- 35. Levinson NM, Visperas PR, Kuriyan J. 2009. The tyrosine kinase Csk dimerizes through its SH3 domain. PLoS One 4:e7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chong YP, Mulhern TD, Cheng HC. 2005. C-terminal Src kinase (CSK) and CSK-homologous kinase (CHK)—endogenous negative regulators of Src-family protein kinases. Growth Factors 23:233–244 [DOI] [PubMed] [Google Scholar]
- 37. Chong YP, Chan AS, Chan KC, Williamson NA, Lerner EC, Smithgall TE, Bjorge JD, Fujita DJ, Purcell AW, Scholz G, Mulhern TD, Cheng HC. 2006. C-terminal Src kinase-homologous kinase (CHK), a unique inhibitor inactivating multiple active conformations of Src family tyrosine kinases. J Biol Chem 281:32988–32999 [DOI] [PubMed] [Google Scholar]
- 38. Takada T, Matozaki T, Takeda H, Fukunaga K, Noguchi T, Fujioka Y, Okazaki I, Tsuda M, Yamao T, Ochi F, Kasuga M. 1998. Roles of the complex formation of SHPS-1 with SHP-2 in insulin-stimulated mitogen-activated protein kinase activation. J Biol Chem 273:9234–9242 [DOI] [PubMed] [Google Scholar]
- 39. Zagozdzon R, Kaminski R, Fu Y, Fu W, Bougeret C, Avraham HK. 2006. Csk homologous kinase (CHK), unlike Csk, enhances MAPK activation via Ras-mediated signaling in a Src-independent manner. Cell Signal 18:871–881 [DOI] [PubMed] [Google Scholar]
- 40. Zagozdzon R, Bougeret C, Fu Y, Avraham HK. 2002. Overexpression of the Csk homologous kinase facilitates phosphorylation of Akt/PKB in MCF-7 cells. Int J Oncol 21:1347–1352 [PubMed] [Google Scholar]
- 41. Blakesley VA, Kato H, Roberts CT, Jr, LeRoith D. 1995. Mutation of a conserved amino acid residue (tryptophan 1173) in the tyrosine kinase domain of the IGF-I receptor abolishes autophosphorylation but does not eliminate biologic function. J Biol Chem 270:2764–2769 [DOI] [PubMed] [Google Scholar]
- 42. Buckley DA, Cheng A, Kiely PA, Tremblay ML, O'Connor R. 2002. Regulation of insulin-like growth factor type I (IGF-I) receptor kinase activity by protein tyrosine phosphatase 1B (PTP-1B) and enhanced IGF-I-mediated suppression of apoptosis and motility in PTP-1B-deficient fibroblasts. Mol Cell Biol 22:1998–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nam TJ, Busby WH, Jr, Rees C, Clemmons DR. 2000. Thrombospondin and osteopontin bind to insulin-like growth factor (IGF)-binding protein-5 leading to an alteration in IGF-I-stimulated cell growth. Endocrinology 141:1100–1106 [DOI] [PubMed] [Google Scholar]
- 44. Jones JI, Prevette T, Gockerman A, Clemmons DR. 1996. Ligand occupancy of the α-V-β3 integrin is necessary for smooth muscle cells to migrate in response to insulin-like growth factor. Proc Natl Acad Sci USA 93:2482–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]