

Abstract
BACKGROUND AND PURPOSE
The endocannabinoid 2-arachidonoylglycerol (2-AG) is degraded primarily by monoacylglycerol lipase (MGL). We compared peripheral antinociceptive effects of JZL184, a novel irreversible MGL inhibitor, with the reversible MGL-preferring inhibitor URB602 and exogenous 2-AG in rats.
EXPERIMENTAL APPROACH
Nociception in the formalin test was assessed in groups receiving dorsal paw injections of vehicle, JZL184 (0.001–300 µg), URB602 (0.001–600 µg), 2-AG (ED50), 2-AG + JZL184 (at their ED50), 2-AG + URB602 (at their ED50), AM251 (80 µg), AM251 + JZL184 (10 µg), AM630 (25 µg) or AM630 + JZL184 (10 µg). Effects of MGL inhibitors on endocannabinoid accumulation and on activities of endocannabinoid-metabolizing enzymes were assessed.
KEY RESULTS
Intra-paw administration of JZL184, URB602 and 2-AG suppressed early and late phases of formalin pain. JZL184 and URB602 acted through a common mechanism. JZL184 (ED50 Phase 1: 0.06 ± 0.028; Phase 2: 0.03 ± 0.011 µg) produced greater antinociception than URB602 (ED50 Phase 1: 120 ± 51.3; Phase 2: 66 ± 23.9 µg) or 2-AG. Both MGL inhibitors produced additive antinociceptive effects when combined with 2-AG. Antinociceptive effects of JZL184, like those of URB602, were blocked by cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2) antagonists. JZL184 suppressed MGL but not fatty-acid amide hydrolase or N-arachidonoyl-phosphatidylethanolamine phospholipase D activities ex vivo. URB602 increased hind paw 2-AG without altering anandamide levels.
CONCLUSIONS AND IMPLICATIONS
MGL inhibitors suppressed formalin-induced pain through peripheral CB1 and CB2 receptor mechanisms. MGL inhibition increased paw skin 2-AG accumulation to mediate these effects. MGL represents a target for the treatment of inflammatory pain.
LINKED ARTICLES
This article is part of a themed issue on Cannabinoids in Biology and Medicine. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-7
Keywords: JZL184, URB602, 2-arachidonoylglycerol, anandamide, cannabinoid antagonists, endocannabinoid levels, formalin, hind paw, inflammatory pain, MGL
Introduction
The endocannabinoid system represents an emerging target for pharmacotherapies aimed at alleviating pathological pain. Endocannabinoids are endogenous lipid-signalling molecules that mimic the pharmacological actions of the principal psychoactive component of marijuana, Δ9-tetrahydrocannabinol (Δ9-THC) (Gaoni and Mechoulam, 1964). The two best-studied endocannabinoids are arachidonoylethanolamide (AEA; anandamide) (Devane et al., 1992) and 2-arachidonoyl glycerol (2-AG) (Mechoulam et al., 1995). Endocannabinoids are produced in the cell membrane from phospholipid precursors and possess cannabimimetic properties because they bind to and activate cannabinoid receptor 1 (CB1) (Devane et al., 1988) and/or cannabinoid receptor 2 (CB2) (Munro et al., 1993) subtypes (receptor nomenclature follows Alexander et al., 2009). AEA is an agonist with a fourfold selectivity for CB1 (Ki = 89 nM) over CB2 (Ki = 371 nM) receptors (Pertwee et al., 1995). 2-AG is an agonist with a threefold selectivity for CB1 (Ki = 472 nM) over CB2 (Ki = 1400 nM) receptors (Mechoulam et al., 1995). The existence of additional endocannabinoids (Di Marzo, 2006) as well as cannabinoid receptor subtypes (Begg et al., 2005; Kreitzer and Stella, 2009) is also supported. AEA is mainly hydrolysed by the enzyme fatty-acid amide hydrolase (FAAH) (Cravatt et al., 1996), whereas 2-AG is mainly, although not exclusively, hydrolysed by the enzyme monoacylglycerol lipase (MGL) (Goparaju et al., 1999; Wang and Ueda, 2009).
Peripheral antinociceptive actions of cannabinoids have been demonstrated in many animal models of persistent pain (see Guindon and Hohmann, 2009). Peripheral mechanisms of analgesic action offer the potential to separate the therapeutic effects of cannabinoids from the unwanted CNS side effects. Local administration of exogenous AEA in the paw reduces pain behaviour produced by formalin injection (Calignano et al., 1998; Russo et al., 2007). However, one limitation of studies evaluating antinociceptive effects of exogenous endocannabinoids is their failure to demonstrate that their endogenously derived counterparts produce similar effects under physiologically relevant doses and conditions. The development of pharmacological agents that inhibit the enzymatic degradation of endocannabinoids (i.e. through inhibition of FAAH or MGL) provides means for increasing endocannabinoid levels at sites where they are mobilized and degraded under physiological conditions. This strategy offers the potential to improve current understanding of the functional roles of endogenous AEA and 2-AG in modulating pain under different physiological and pathological conditions.
Suppressing the enzymatic degradation of AEA or 2-AG reduces pain behaviour in inflammatory pain models (see Jhaveri et al., 2007; Guindon and Hohmann, 2009). Antinociception produced by pharmacological inhibitors of FAAH, including URB937, URB597, PF-3845, N-arachidonoyl 5-HT (AA-5-HT) and ibuprofen (see Guindon and Hohmann, 2009), in models of inflammatory pain are well characterized (Maione et al., 2007; Jhaveri et al., 2008; Ahn et al., 2009; Clapper et al., 2010; Costa et al., 2010). Fewer studies have evaluated the effect of inhibiting the degradation of 2-AG (Hohmann et al., 2005; Guindon et al., 2007a; Bisogno et al., 2009; Long et al., 2009a), possibly due to the relative lack of selective pharmacological tools. A first generation of MGL inhibitors included the reversible N-biphenyl carbamate URB602 (Hohmann et al., 2005) (Figure 1). Following site-specific injection in the brain, this MGL-preferring inhibitor elevated 2-AG accumulation, without altering levels of AEA, under conditions that were biochemically validated (Hohmann et al., 2005). However, URB602 cannot be used systemically as a selective MGL inhibitor and can suppress FAAH in vitro (Hohmann et al., 2005 in peri-aqueductal gray; King et al., 2007 in intact brain slices; but see Saario et al. 2005 and Vandevoorde et al., 2007). More recently, a new generation of MGL inhibitors have been described that includes OMDM169 (Bisogno et al., 2009) and JZL184 (Long et al., 2009a). JZL184 is a piperidine carbamate and an irreversible MGL inhibitor that suppresses formalin-induced pain behaviour following systemic administration in mice (Long et al., 2009a) (Figure 1). URB602 and JZL184 have recently been examined for peripheral antinociceptive effects in rats (Guindon et al., 2007a; Spradley et al., 2010).
Figure 1.

Chemical structures of the monoacylglycerol lipase inhibitors URB602 and JZL184, the endocannabinoid 2-arachidonoyl glycerol (2-AG), the CB1 receptor antagonist AM251 and the CB2 receptor antagonist AM630.
However, it remains unclear whether the effects of URB602 or JZL184 in these latter studies can be attributed solely to inhibition of MGL. Consequently, the role of 2-AG in controlling peripheral nociception remains poorly understood. The present study was designed to evaluate the role of MGL inhibition in regulating peripheral antinociception in rats. First, structurally distinct compounds (JZL184 and URB602) were used to investigate the peripheral antinociceptive effects of MGL inhibition in an animal model of tonic pain, the formalin test. Second, we confirmed that JZL184 and URB602, administered to the dorsal hind paw surface, acted through a local site of action. Third, we examined whether the combination of JZL184 or URB602, at their ED50 doses, with exogenous 2-AG would produce an additive antinociceptive effect. Fourth, we examined the receptor mechanism by which JZL184 produces peripheral antinociceptive effects, using selective antagonists for cannabinoid CB1 (AM251) and CB2 (AM630) receptors. Fifth, we measured the impact of intra-paw injection of the irreversible MGL inhibitor JZL184 on the activity of enzymes implicated in hydrolysis (MGL, FAAH) and synthesis [N-arachidonoyl-phosphatidylethanolamine phospholipase D (NAPE-PLD)] of endocannabinoids in paw skin. Finally, we measured levels of AEA and 2-AG in hind paw skin of rats that received JZL184 and URB602 prior to formalin administration. Our studies suggest that pharmacological inhibitors of MGL produce peripheral antinociception through MGL inhibition-induced increases in 2-AG accumulation, and subsequent activation of CB1 and CB2 receptors. These studies further validate JZL184 and URB602 as research tools for studying antinociceptive effects produced by inhibition of 2-AG hydrolysis in rats.
Methods
Animals
All animal care and experimental protocols were approved by the University of Georgia Animal Care and Use Committee, and followed the guidelines for the treatment of animals conformed to the International Association for the Study of Pain (Zimmermann, 1983). Three hundred and seven adult male Sprague–Dawley rats (Harlan, Indianapolis, IN, USA) weighing 275–350 g, at the time of testing, were used in these experiments. Animals were housed two per cage in standard plastic cages with sawdust bedding in a climate-controlled room, under a 12 h light/dark cycle. The rats received free access to standard rodent chow and water.
Formalin test
The formalin test is a well-established model of persistent pain characterized by a transient, biphasic pattern of pain behaviour. The early phase is characterized by acute activation of C and Aδ fibres. The late phase involves an inflammatory reaction in peripheral tissue (Tjölsen et al., 1992), the development of CNS sensitization (Coderre and Melzack, 1992) and additionally involves activation of primary afferent nociceptors (Puig and Sorkin, 1996). Rats were acclimatized to the testing environment (clear Plexiglass box 20 × 40 × 20 cm) for 15 min or until cessation of exploratory behaviour. Rats were injected subcutaneously in the dorsal surface of the right hind paw (i.paw) with either JZL184 (0.001, 0.01, 0.03, 0.1, 1, 3, 10, 30, 100 or 300 µg), URB602 (0.001, 0.1, 1, 10, 30, 66, 100, 300 or 600 µg), 2-AG (1 µg), AM251 (80 µg) or AM630 (25 µg). Drug or vehicle was administered, in a 50 µL volume, 15 min before a dorsal paw injection of 2.5% formalin (50 µL), delivered to the same site. Following each injection, the rat was immediately placed back in the observation chamber. Nociceptive behaviour was observed with the help of a mirror angled at 45° below the observation chamber. Observation of the animal's behaviour was performed in consecutive 5-min periods for 60 min, following formalin administration. In each 5-min bin, the total time the animal spent in three different behavioural categories was recorded: (0) the injected paw has little or no weight placed on it; (1) the injected paw is raised; (2) the injected paw is licked, shaken or bitten. Nociceptive behaviour was quantified using the composite pain score-weighted scores technique (CPS-WST0,1,2) (Watson et al., 1997), where each pain behaviour is weighted by the amount of time spent in each category (0,1,2). The area under the curve (AUC), which corresponds to CPS-WST0,1,2 × time (min), was calculated for the acute phase (0–15 min; Phase 1) and the inflammatory phase (15–60 min; Phase 2) using the trapezoidal rule.
Protocol
All experiments were conducted in a randomized manner and without knowledge of the treatments by the same experimenter. In a first study, the dose–response curves for JZL184 and URB602 were determined using the AUC of Phase 1 or Phase 2 pain behaviour. In a second study, the antinociceptive effects of JZL184 (300 µg) and URB602 (600 µg) were evaluated following injection in the paw, ipsilateral or contralateral to formalin, to exclude the possibility that systemic leakage contributed to the pattern of results obtained. In a third study, antinociceptive effects of ED50 doses of JZL184 (0.03 µg i.paw) or URB602 (66 µg i.paw), in combination with 2-AG [ED50 dose of 1 µg i.paw (Guindon et al., 2007a)], were quantified to evaluate the presence of additive or synergic effects of these drugs. In a fourth study, antinociceptive effects of JZL184 (at 10 µg i.paw, an analgesic dose) were studied in the presence or absence of either AM251 or AM630 to determine whether these effects were mediated through CB1 and/or CB2 receptors. The CB1 receptor antagonist AM251 exhibits 306-fold selectivity for CB1 over CB2 receptors (Gatley et al., 1996), whereas the CB2 receptor antagonist AM630 exhibits 70–165-fold selectivity for CB2 over CB1 receptors (Pertwee et al., 1995; Ross et al., 1999). The doses employed (AM251 at 80 µg i.paw and AM630 at 25 µg i.paw) were those which blocked peripheral antinociceptive effects of URB602 in Wistar rats (Guindon et al., 2007a). For the first study (n = 4–6 per group for URB602 and n = 6–8 per group for JZL184) and for all the other behavioural studies (n = 6 per group), drugs, administered either alone or in combination, were dissolved in the same total volume (50 µL) and injected into the right hind paw. Preliminary experiments (n = 8 per group; data not shown) confirmed that formalin-induced pain behaviour did not change following intra-paw administration of either vehicle [polyethylene glycol 300 (PEG 300): Tween 80 in a 4:1 ratio or dimethylsulfoxide (DMSO): ethanol: cremophor: 0.9% saline in a 1:1:1:17 ratio were all purchased from Fisher (Pittsburgh, PA, USA)].
Peripheral oedema
At the end of the formalin test, maximal paw thickness was measured at the base of the right hind paw using a digital micrometer (Mitutoyo Corporation, Aurora, IL, USA) with a resolution of 0.001 mm (Petricevic et al., 1978; Guindon et al., 2007a).
Sample generation for biochemical studies
Formalin-injected rats receiving either JZL184 (100 µg, n = 10), URB602 (300 µg, n = 10) or their respective vehicles [4:1 of PEG 300: Tween 80, n = 6 and 1:1:1:17 DMSO: ethanol: cremophor: normal saline (0.9% NaCl in water), n = 6, respectively] were killed at the peak of Phase 1 (5 min post-formalin) or Phase 2 (35 min post-formalin) pain behaviour. Paw skin was dissected from the entire dorsal paw surface (excluding the toes) (Figure 9I) and used for evaluation of endocannabinoid content. Different groups of rats receiving these same drug treatments were killed at the peak of Phase 2 pain behaviour. The irreversible MGL inhibitor JZL184 was used to determine whether enzyme activity (MGL, FAAH and NAPE-PLD) was suppressed ex vivo in the entire dorsal paw skin surface (excluding the toes). The reversible MGL inhibitor URB602 was used to determine whether regional differences in endocannabinoid accumulation could be unmasked when tissue was further dissected into distal, middle and proximal zones of paw skin (Figure 10G). Paw skin was fast frozen in isopentane, pre-cooled on dry ice. The frozen sample was subsequently dissected into distal, middle and proximal zones of skin on dry ice. Dissection was performed on an inverted glass Petri dish filled with dry ice, surrounded by a container of dry ice. The paw skin was cut in three pieces (distal, middle and proximal; see Figure 10G) of similar length. Each segment was weighed separately and then further dissected into two or three smaller pieces to facilitate homogenization. All pieces from the same zone were homogenized together. The tissue remained frozen until it was placed in cold methanol containing the standards. The weight range for these samples were as follows: 78.60 ± 7.78 mg (proximal), 122.46 ± 11.43 mg (middle) and 75.33 ± 9.08 mg (distal) for vehicle-treated groups and 73.0 ± 8.44 mg (proximal), 103.15 ± 5.70 mg (middle), 68.56 ± 6.16 mg (distal) for URB602-treated groups respectively. Distal, middle and proximal skin segments were dissected into zones of qualitatively similar lengths. Thus, it is probable that differences in the degree of skin inflammation within each zone contributed to observed differences in tissue weights between the zones. The middle zone of skin weighed more than the distal or proximal zones, consistent with a greater inflammatory response in the skin corresponding to the centre of the injection site. All samples exceeded 50 mg and, consequently, limitations in assay sensitivity associated with proximity to detection thresholds or small pieces of tissue could not contribute to the pattern of results obtained.
Figure 9.

(A–I) Endocannabinoid levels in ‘whole’ dorsal hind paw skin following administration of JZL184 (100 µg i.paw) and URB602 (300 µg i.paw) in (A–D) Phase 1 (5 min) and (E–H) Phase 2 (35 min) of the formalin test. URB602 did not alter either (A,E) 2-arachidonoylglycerol (2-AG) or (B,F) anandamide (AEA) during either (A,B) Phase 1 or (E,F) Phase 2 of the formalin test. JZL184 decreased (D) AEA levels during Phase 1 but not (H) Phase 2 of the formalin test. JZL184 did not alter 2-AG levels during either (C) Phase 1 or (G) Phase 2 of the formalin test. The vehicle for JZL184 was associated with 10-fold higher levels of Phase 1 AEA levels compared with the vehicle for URB602 (B,D). Data are expressed as mean ± SEM (n = 6–10 per group). *P < 0.003 versus vehicle group.
Figure 10.
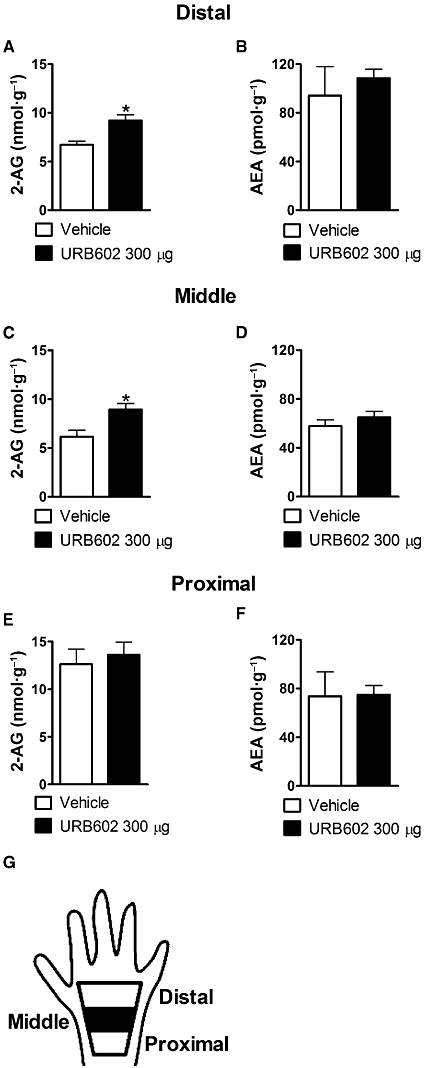
(A–G) Effects of URB602 on 2-arachidonoylglycerol (2-AG) and anandamide (AEA) levels in proximal, middle and distal hind paw skin segments during Phase 2 (35 min) of the formalin test. URB602 selectively increased 2-AG levels in the middle and distal hind paw skin (A,C) without altering AEA levels (B,D). URB602 did not alter 2-AG or AEA levels in the proximal hind paw skin (E,F). Data are expressed as mean ± SEM (n = 5–9 per group). *P < 0.01 versus vehicle group.
Drugs were dissolved in the same total volume (50 µL) of their respective vehicles and injected into the right hind paw for evaluation of enzyme activity (n = 4–9 per group) and endocannabinoid content (n = 5–10 per group).
Lipid extraction
Dissected frozen paw tissues were weighed and homogenized in methanol (1 mL/100 mg of tissue) containing [2H8]-2-AG and [2H4]-AEA (prepared as described previously in Fu et al., 2007; Schwartz et al., 2008) as internal standards. Lipids were extracted using two volumes of chloroform and washed with one volume of water. The organic extract obtained after centrifugation at 2400×g for 15 min at 4°C was fractionated by open-bed silica gel column chromatography, as described (Cadas et al., 1997). Briefly, the extract was dissolved in 2 mL of chloroform and loaded onto small glass columns packed with Silica Gel G (60-Å 230–400 Mesh ASTM; Whatman, Clifton, NJ, USA). 2-AG and AEA were eluted with 2 mL of chloroform/methanol (9:1, v/v). The lipids were collected and dried under nitrogen, and the lipid pellet was reconstituted in 60 µL of methanol.
Liquid chromatography/mass spectrometry (LC/MS)
Tissue levels of 2-AG and AEA were quantified by LC/MS (Giuffrida et al., 2000; Fu et al., 2007; Schwartz et al., 2008). An 1100-LC system coupled with a 1946A-MS detector (Agilent Technologies, Inc., Palo Alto, CA, USA) equipped with an electrospray ionization interface was used. 2-AG and AEA were eluted on a XDB Eclipse C18 column (50 × 4.6 mm inner diameter, 1.8 µm, Zorbax, Agilent Technologies, Inc.) using a linear gradient of methanol in water (from 85% to 90% methanol in 2.5 min), at a flow rate of 1 mL·min−1. Column temperature was kept at 40°C. MS detection was in the positive ionization mode, capillary voltage was set at 3000 V and fragmentor voltage varied from 120 to 140 V. Nitrogen was used as drying gas at a flow rate of 13 L·min−1 and a temperature of 350°C. Nebulizer pressure was set at 60 psig. For quantification purposes, we monitored, in the selective ion-monitoring mode, the Na+ adducts ([M + Na+]) of [2H8]-2-AG (mass-to-charge ratio, m/z, 409), 2-AG (m/z, 401), [2H4]-AEA (m/z, 374) and AEA (m/z, 370).
MGL activity
Tissues were homogenized in ice-cold Tris-HCl (50 mM, pH 7.5, 10 vol) containing 0.32 M sucrose. Homogenates were centrifuged at 1000×g for 10 min. Supernatants were incubated at 37°C for 30 min in Tris-HCl buffer (50 mM, pH 8, 0.5 mL) containing fatty acid-free bovine serum albumin (0.05%), protein (50 µg) and 2-mono-oleoyl glycerol, rac [3H-glycerol] (10 000 dpm, specific activity 20–40 Ci·mmol−1). The reactions were stopped by adding chloroform/methanol (2/1, v/v, 1 mL). Radioactivity was measured in the aqueous layers by liquid scintillation counting.
FAAH activity
Tissues were homogenized in ice-cold Tris-HCl (50 mM, pH 7.5, 10 vol) containing 0.32 M sucrose. Homogenates were centrifuged at 1000×g for 10 min, and the supernatants were kept. Reactions were conducted at 37°C for 30 min in Tris-HCl buffer (50 mM, pH 7.5, 0.5 mL) containing fatty acid-free bovine serum albumin (0.05%), protein (50 µg) and [3H-ethanolamine]anandamide (10 000 dpm, specific activity 20 Ci·mmol−1). After stopping the reactions with chloroform/methanol (2/1, v/v, 1 mL), radioactivity was measured in the aqueous layers by liquid scintillation counting.
NAPE-PLD activity
Tissues were homogenized in ice-cold Tris-HCl (50 mM, pH 7.4, 10 vol) containing 0.32 M sucrose. Homogenates were centrifuged at 1000×g for 10 min. NAPE-PLD activity was measured at 37°C for 30 min in Tris-HCl buffer (50 mM, pH 7.4, 0.2 mL) containing 0.1% Triton X-100, phenylmethylsulphonylfluoride (1 mM), protein (100 µg) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-heptadecenoyl (100 µM) as substrate. The reactions were stopped by adding chloroform/methanol (2/1, v/v) containing [2H4]-heptadecenoylethanolamide as internal standard. After centrifugation at 1500×g at 4°C for 15 min, the organic layers were collected and dried under nitrogen. The pellets were suspended in 50 µL of methanol and analysed by LC/MS. For NAPE-PLD assay, 2[H4]-heptadecenoylethanolamide and N-heptadecenoylethanolamide were eluted on a XDB Eclipse C18 column (50 × 4.6 mm inner diameter, 1.8 µm, Zorbax, Agilent Technologies, Inc.) using a linear gradient of 85–90% of A in B over 5 min at a flow rate of 1.5 mL·min−1. Mobile phase A consisted of methanol and mobile phase B consisted of water. Column temperature was kept at 40°C. MS detection was in the positive ionization mode, capillary voltage was set at 4000 V and fragmentor voltage varied from 120 to 140 V. Nitrogen was used as drying gas at a flow rate of 13 L·min−1 and a temperature of 350°C. Nebulizer pressure was set at 60 psig. For quantification purposes, we monitored the ions of 2[H4]-heptadecenoylethanolamide (m/z, 338) and N-heptadecenoylethanolamide (m/z, 334).
Statistical analysis
Pain behaviour for each treatment group was expressed as mean ± SEM. The dose–response curve for JZL184 and URB602 were determined using ALLFIT software (Montreal, QC, Canada) (De Léan et al., 1978). A repeated-measures analysis of variance (anova) was performed to evaluate the time course of drug effects on the formalin-induced composite pain score. The Greenhouse–Geisser correction was applied to all repeated factors; degrees of freedom reported for significant interactions are the uncorrected values. Significant interactions were further explained by performing separate one-way anovas on each individual time point, followed by Bonferroni post hoc tests. anova adapted for factorial experimental design (Winer, 1971) was used to examine differences in the AUC of pain behaviour between groups. The different components of the total variation were settled a priori using multiple regression analysis (Draper and Smith, 1998). Effects of MGL inhibitors on enzyme activity and endocannabinoid content were evaluated using independent samples t-tests (one or two tailed, as appropriate). Analyses were performed using SPSS statistical software (version 17.0; SPSS Incorporated, Chicago, IL, USA). P < 0.05 was considered significant.
Materials
JZL184 and URB602 were purchased from Cayman Chemical (Ann Arbor, MI, USA) (King et al., 2007; Long et al., 2009a). JZL184 was dissolved in a vehicle of 4:1 PEG 300 (PEG 300: Tween 80) (Long et al., 2009a). URB602 was dissolved in 1:1:1:17 mixture of DMSO: ethanol: cremophor: normal saline (0.9% NaCl in water). For higher doses of URB602 (300 µg and 600 µg), the vehicle contained 15 and 30% DMSO, respectively, as described previously (Guindon et al., 2007a). 2-AG was purchased from Tocris (Ellisville, MO, USA) and further dissolved in a 1:1:1:17 ratio of DMSO: ethanol: cremophor: normal saline (0.9% NaCl in water) (Guindon et al., 2007a). AM251 and AM630 were purchased from Cayman Chemical and dissolved in a vehicle of 1:1:1:17 DMSO: ethanol: cremophor: normal saline (0.9% NaCl in water) (Guindon et al., 2007b). JZL184, 2-AG and URB602 were stored at −20°C. Care was taken to protect 2-AG and URB602 from light. [2H8]-2-AG was purchased from Cayman Chemical. [2H4]-AEA, [2H4]-heptadecenoyl acid and [2H4]-heptadecenoylethanolamide were prepared as described previously (Fu et al., 2007; Schwartz et al., 2008) using the appropriate fatty acid chlorides purchased from Nu-Chek Prep (Elysian, MN, USA). Anandamide [ethanolamine 1-3H], 2-mono-oleoyl glycerol rac [1,2,3H-glycerol] were from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA).
Results
Antinociceptive effects of JZL184 and URB602 and their comparison
JZL184 and URB602 decreased the AUC of pain behaviour during the early phase of the formalin test with an ED50 of 0.06 ± 0.028 µg for JZL184 and 120 ± 51.3 µg for URB602. Both MGL inhibitors also suppressed pain behaviour during the late phase of formalin pain, with an ED50 of 0.03 ± 0.011 µg for JZL184 and 66 ± 23.9 µg for URB602 (Figure 2A,B). The dose–response curves had the same slope, suggesting that JZL184 and URB602 acted through a common mechanism; the slope was 1.00 ± 0.057 for JZL184 and 1.00 ± 0.301 for URB602. The dose–response curve for JZL184 was shifted to the left of the dose–response curve for URB602 (Figure 2A,B).
Figure 2.

Dose-response curve for JZL184 and URB602 in (A) Phase 1 (0–15 min) and (B) Phase 2 (15–60 min) of the formalin test. Area under the curve (AUC) of pain behaviour for each phase. Data are expressed as mean ± SEM (n = 6–8 per group for JZL184 and n = 4–6 per group for URB602).
Peripheral antinociceptive effects of JZL184 and URB602
Both JZL184 and URB602 produced time-dependent suppressions of the composite pain score relative to vehicle [F22,165 = 9.19, P = 0.001; Figure 3A]; this suppression was observed at 5 min and from 25 to 50 min post-formalin injection (P < 0.05). Analysis of the AUC of pain behaviour revealed that both JZL184 (300 µg i.paw) and URB602 (600 µg i.paw) produced antinociception relative to vehicle in both Phase 1 (F1,15 = 123.37, P < 0.001) (Figure 3B) and Phase 2 (F1,15 = 67.06, P < 0.001) (Figure 3C) of the formalin test. JZL184 produced a greater antinociceptive effect relative to URB602 during the late phase of the formalin test (F1,15 = 8.83, P < 0.01) (Figure 3A–C). Composite pain scores were also lower in groups receiving JZL184 relative to URB602 from 35 to 50 min post-formalin injection (P < 0.05), suggesting that JZL184-induced antinociception outlasted that of URB602.
Figure 3.

The MGL inhibitors JZL184 and URB602 suppressed formalin-induced pain behaviour. (A) JZL184 (300 µg i.paw) and URB602 (600 µg i.paw) suppressed the time course of formalin-induced pain behaviour. Both JZL184 and URB602 suppressed the area under the curve (AUC) of (B) Phase 1 and (C) Phase 2 pain behaviour. Data are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus all groups; xP < 0.05 versus vehicle group; +P < 0.05 versus URB602.
Formalin-induced pain behaviour was assessed following injection of the compounds (JZL184, 300 µg and URB602, 600 µg) into the contralateral hind paw to confirm that drug effects observed were mediated by a local site of action. Ipsilateral (formalin-injected) paw administration of JZL184 [F22,165 = 10.25, P < 0.001; Figure 4A] and URB602 [F22,165 = 4.69, P < 0.001; Figure 4D] produced time-dependent suppressions of composite pain scores, and thereby showed lower composite pain scores than those from groups receiving the same doses of MGL inhibitors in the contralateral paw. For each phase, the AUC of pain behaviour was lower following administration of either JZL184 [F1,15 = 74.76, P < 0.001 (Phase 1); F1,15 = 162.83, P < 0.001 (Phase 2)] or URB602 [F1,15 = 49.56, P < 0.001 (Phase 1); F1,15 = 74.71, P < 0.001 (Phase 2)] into the ipsilateral relative to contralateral paw (Figure 4A–F). Phase 1 and Phase 2 pain behaviour was also similar between groups receiving vehicle and either JZL184 (300 µg i.paw) [P = 0.16 (Phase 1); P = 0.62 (Phase 2)] or URB602 (600 µg i.paw) [P = 0.78 (Phase 1); P = 0.84 (Phase 2)] in the contralateral paw (Figure 4A–F).
Figure 4.

The MGL inhibitors JZL184 and URB602 suppressed formalin-induced pain behaviour through a peripheral mechanism. (A–C) JZL184 (300 µg i.paw) and (D–F) URB602 (600 µg i.paw) suppressed the time-course and AUC of formalin-induced pain behaviour following local administration to the ipsilateral but not the contralateral paw. Data are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus contralateral groups.
Antinociceptive effects of JZL184, URB602 and 2-AG
The MGL inhibitors (JZL184 and URB602) or the endocannabinoid 2-AG, injected at their ED50 doses for Phase 2, produced time-dependent suppressions in the composite pain scores [F33,220 = 3.16, P < 0.007]. This suppression was maximal at 5 and 30–50 min post-formalin (P < 0.05) compared with the vehicle group (Figure 5A). At 30 min post-formalin, the peak of the Phase 2 pain response, JZL184 (ED50 dose) lowered the composite pain score relative to either 2-AG or URB602 (P < 0.05). Local administration of JZL184, URB602 and 2-AG at their ED50 doses produced antinociception relative to vehicle during both Phase 1 (F1,20 = 38.92, P < 0.001) and Phase 2 (F1,20 = 53.31, P < 0.001) of the formalin test (Figure 5B,C). The antinociceptive effect of JZL184 was greater than that of either URB602 or 2-AG for both phases [F1,20 = 7.68, P < 0.025 (Phase 1); F1,20 = 29.22, P < 0.001 (Phase 2)] (Figure 5B,C). Furthermore, antinociceptive effects of URB602 did not differ from that of 2-AG for either phase of the formalin test [P = 0.74 (Phase 1); P = 0.89 (Phase 2)] (Figure 5B,C).
Figure 5.

Comparison of peripheral antinociceptive effects of JZL184, URB602 and exogenous 2-arachidonoylglycerol (2-AG) in the formalin test. (A–C) Antinociceptive effects of ED50 doses for Phase 2 of JZL184 (0.03 µg i.paw), URB602 (66 µg i.paw) and 2-AG (1 µg i.paw). Data are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus all groups; xP < 0.05 versus vehicle group; +P < 0.05 versus URB602 or 2-AG.
Antinociceptive effects of JZL184, URB602, 2-AG and their combination at ED50 doses
JZL184, 2-AG and their combination (at ED50 doses for Phase 2) produced time-dependent suppressions of the composite pain score in the formalin test [F33,220 = 5.32, P < 0.001; Figure 6A]. The combination of JZL184 and 2-AG lowered composite pain scores relative to 2-AG (P < 0.05 at 5 and 25–45 min post-formalin). Moreover, at 30 min post-formalin, JZL184 produced a greater suppression of the composite pain score than 2-AG (P < 0.05). Analysis of the AUC of pain behaviour revealed that JZL184, 2-AG and their combination, at their ED50 doses, produced antinociception relative to vehicle [F1,20 = 69.80 P < 0.001 (Phase 1); F1,20 = 115.76 (Phase 2), P < 0.001] for both phases of the formalin test (Figure 6B,C). Moreover, the combination of JZL184 with 2-AG (at ED50 doses) produced greater antinociception than either drug given alone for both the acute and inflammatory phases [F1,20 = 26.94, P < 0.001 (Phase 1); F1,20 = 42.56, P < 0.001 (Phase 2)], thereby revealing an additive antinociceptive effect of the combination of the MGL inhibitor with 2-AG (Figure 6B,C). Finally, the antinociceptive effect of JZL184 was greater than that of 2-AG for both phases [F1,20 = 4.53 (Phase 1), P < 0.05; F1,20 = 23.94 (Phase 2), P < 0.001].
Figure 6.

Effects of local injection of JZL184, URB602, 2-arachidonoylglycerol (2-AG) at their ED50 doses and their combination in the formalin test (A–F). Additive effects of the combination of either JZL184 (0.03 µg i.paw) or URB602 (66 µg i.paw) with exogenous 2-AG (1 µg i.paw) were observed for (A,D) the composite pain score and area under the curve (AUC) of (B,E) Phase 1 and (C,F) Phase 2 pain behaviour. Data are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus all groups; xP < 0.05 versus vehicle group; +P < 0.05 versus 2-AG; #P < 0.05 for JZL184 versus 2-AG; ‡P < 0.025 versus MGL inhibitors (URB602 or JZL184) and 2-AG given alone.
URB602, 2-AG and their combination (at ED50 doses for Phase 2) also produced time-dependent suppressions of the composite pain score in the formalin test [F(33, 220) = 2.31, P < 0.04; Figure 6D]. The drugs decreased the composite pain scores (P < 0.05, at 5 and 30–45 min post-formalin) compared with the vehicle group. At 30 min post-formalin, the combination of URB602 with 2-AG (at their ED50 doses) produced a lower composite pain score compared with 2-AG alone (P < 0.05). URB602, 2-AG and their combination, at their ED50 doses, also reduced the AUC of pain behaviour, relative to the vehicle group [F1,20 = 34.59, P < 0.001 (Phase 1); F1,20 = 38.55, P < 0.001 (Phase 2)] for each phase of the formalin test (Figure 6E,F). For each phase, the combination of URB602 with 2-AG (at ED50 doses) produced greater antinociception than either drug given alone [F1,20 = 5.91, P < 0.025 (Phase 1); F1,20 = 6.46, P < 0.025 (Phase 2)], thereby revealing an additive antinociceptive effect of the combination of URB602 with 2-AG (Figure 6E,F). The antinociceptive effects of URB602 were similar to that of 2-AG alone for either phase [P = 0.76 (Phase 1); P = 0.89 (Phase 2)].
Antinociceptive effects of JZL184 and its involvement with the cannabinoid receptor antagonists
Neither the CB1 antagonist AM251 nor the CB2 antagonist AM630, administered locally in the paw, altered the time course of formalin-induced pain behaviour relative to vehicle [P = 0.34 (Figure 7A)]. The AUC of pain behaviour was similar between antagonists and vehicle groups for both phases [P = 0.81 (Phase 1); P = 0.51 (Phase 2); Figure 7B,C].
Figure 7.

The MGL inhibitor JZL184 suppressed formalin-induced pain behaviour through peripheral CB1 and CB2 receptor mechanisms. (A–C) The CB1 antagonist AM251 (80 µg i.paw) and the CB2 antagonist AM630 (25 µg i.paw) do not alter formalin-induced pain behaviour relative to vehicle (i.paw). Peripheral antinociceptive effects of JZL184 (10 µg i.paw) on (D) the composite pain score and area under the curve (AUC) of (E) Phase 1 and (F) Phase 2 of pain behaviour were blocked by either AM251 (80 µg i.paw) or AM630 (25 µg i.paw) in the formalin test. Data are expressed as mean ± SEM (n = 6 per group). *P < 0.05 versus all groups.
Both AM251 and AM630 blocked the suppression of formalin-induced pain scores produced by JZL184 (10 µg i.paw) [F33,220 = 6.54, P < 0.001; P < 0.05 at 5 and 30–60 min post-formalin; Figure 8D]. The antinociceptive effects of JZL184 were blocked by either AM251 or AM630 (Figure 7D–F); for each phase, the AUC of pain behaviour was lower in groups receiving JZL184 compared with either vehicle or JZL184 co-administered with either the CB1 or CB2 antagonist [F1,20 = 66.61, P < 0.001 (Phase 1); F1,20 = 154.16, P < 0.001 (Phase 2); Figure 7E,F]. Formalin-induced pain behaviour was similar in groups receiving either vehicle or either antagonist combined with JZL184 [P = 0.97 (Phase 1); P = 0.92 (Phase 2); Figure 7E,F]. Moreover, the AUC of pain behaviour did not differ between antagonist co-administration groups [P = 0.96 (Phase 1); P = 0.51 (Phase 2)].
Figure 8.
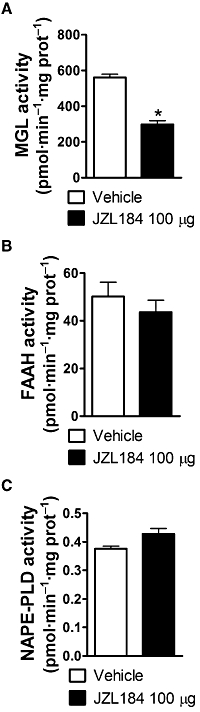
The MGL inhibitor JZL184 (100 µg i.paw) suppressed the MGL activity in ‘whole’ dorsal hind paw skin without affecting fatty-acid amide hydrolase (FAAH) and N-arachidonoyl-phosphatidylethanolamine phospholipase D (NAPE-PLD) activities. JZL184 inhibited (A) MGL activity but not (B) FAAH or (C) NAPE-PLD activity relative to vehicle. Enzyme activities were measured in animals killed at the peak of Phase 2 (35 min) of the formalin test. Data are expressed as mean ± SEM (n = 4–9 per group). *P < 0.001 versus vehicle group.
Paw oedema
Oedema of the injected paw did not differ between any of the experimental groups (Table 1).
Table 1.
Oedema in the formalin-injected paw for the different treatment groups
| Paw Oedema (thickness in mm) | Mean ± SEM |
|---|---|
| Vehicle (PEG 300:Tween 80) | 1.28 ± 0.02 |
| 2-AG 1 µg | 1.30 ± 0.03 |
| JZL184 0.03 µg | 1.31 ± 0.02 |
| URB602 66 µg | 1.29 ± 0.02 |
| JZL184 + 2-AG | 1.35 ± 0.01 |
| URB602 + 2-AG | 1.30 ± 0.03 |
| Vehicle (1:1:1:17) | 1.34 ± 0.03 |
| JZL184 0.03 µg | 1.33 ± 0.01 |
| AM251 80 µg | 1.28 ± 0.03 |
| AM630 25 µg | 1.29 ± 0.02 |
| AM251 + JZL184 | 1.34 ± 0.02 |
| AM630 + JZL184 | 1.35 ± 0.01 |
PEG 300, polyethylene glycol 300.
MGL, FAAH and NAPE-PLD activities
Both MGL inhibitors exhibited identical mechanisms of peripheral antinociception (see slopes of dose–response curves in Figure 2). However, the irreversible nature of MGL inhibition produced by JZL184 enabled us to use this compound to evaluate ex vivo whether MGL activity in rat paw skin was selectively altered by intra-paw injection of JZL184. JZL184 (100 µg) inhibited MGL activity (t10 = 6.92, P < 0.001, one-tailed t-test) relative to the vehicle (Figure 8A) in the inflammatory phase (35 min) of the formalin test. By contrast, JZL184 did not alter FAAH (t9 = 0.73, P = 0.49, two-tailed t-test) or NAPE-PLD (t10 = 1.56, P = 0.15, two-tailed t-test) activity (Figure 8B,C).
Endocannabinoid levels in the ‘whole’ hind paw skin
Neither URB602 (300 µg) nor JZL184 (100 µg) increased either 2-AG or AEA levels during Phase 1 or Phase 2 of the formalin test when endocannabinoid content was measured in the entire dorsal hind paw tissue (P≥ 0.05; Figure 9A–I). In fact, a decrease in AEA accumulation was observed following JZL184 but not URB602 administration, and this effect was restricted to Phase 1 (t13 = 3.58, P < 0.003, two-tailed t-test) (Figure 9D). The vehicle for JZL184 was associated with 10-fold higher levels of Phase 1 AEA levels compared with the vehicle for URB602 (Figure 9B,D).
AEA and 2-AG levels in the ‘divided’ hind paws
The impact of dorsal paw injection of URB602 (300 µg) on endocannabinoid accumulation was subsequently measured in proximal, middle and distal hind paw skin segments during Phase 2 (35 min) (Figure 10A–G). This study was performed using the less potent and presumably less selective MGL inhibitor that was also dissolved in a vehicle that did not alter AEA accumulation during Phase 1 (Figure 10A–G). URB602 selectively increased 2-AG accumulation in the middle (t10 = 2.77, P < 0.01, one-tailed t-test) and distal (t11 = 3.17, P < 0.004, one-tailed t-test) hind paw skin (Figure 10A,C) without altering AEA levels (Figure 10B,D). URB602 did not alter 2-AG or AEA levels in proximal hind paw skin (P > 0.66) (Figure 10E,F).
Discussion
Two structurally distinct inhibitors of MGL were employed to validate a role for 2-AG degradation inhibitors as possible analgesics. JZL184 and URB602 produced dose-dependent peripheral antinociception during both the early and late phases of the formalin test. These effects were mediated by a local site of action, and involved both cannabinoid CB1 and CB2 receptors (see also Guindon et al., 2007a). Either MGL inhibitor also produced additive antinociceptive effects in combination with 2-AG, suggesting that inhibition of MGL enhanced 2-AG antinociceptive actions by preventing 2-AG hydrolysis. JZL184 was more potent than the MGL-preferring inhibitor URB602 in producing peripheral antinociception; the dose–response curve for JZL184 was shifted to the left of that for URB602 for each phase of the formalin test. This greater potency may be influenced by the irreversible nature of JZL184-induced MGL inhibition in comparison to that of URB602 (Hohmann et al., 2005; Long et al., 2009a). Off-target effects of URB602 have also been demonstrated in vitro (Muccioli et al., 2007; Vandevoorde et al., 2007). However, studies employing intact brain slices (King et al., 2007) and local injections of URB602 (Hohmann et al., 2005) have demonstrated that URB602 suppressed MGL and selectively increased 2-AG accumulation under biochemically validated conditions.
In whole dorsal hind paw skin, JZL184 suppressed MGL activity without altering FAAH or NAPE-PLD activity, illustrating the selectivity of JZL184 for inhibiting MGL. Analogous experiments could not be performed ex vivo with URB602 due to the reversible nature of URB602-induced MGL inhibition. Nonetheless, dorsal paw injection of either JZL184 or URB602 failed to increase 2-AG levels in whole dorsal hind paw skin during either phase of the formalin test. The mismatch between the ability of JZL184 to inhibit MGL activity in the entire paw skin, but its failure to alter endocannabinoid content in identically dissected samples, is noteworthy. Due to the irreversible nature of JZL184-induced MGL inhibition, assays of MGL activity may be more sensitive than assays of endocannabinoid content, at least in peripheral paw skin. Moreover, assays of endocannabinoid content do not differentiate between bulk and signalling competent pools of endocannabinoids. Thus, regionally specific increases in paw skin 2-AG levels produced by JZL184 may be masked by the high lipid content in paw skin, especially when the entire dorsal paw skin was used for LC/MS analysis.
The JZL184 vehicle was associated with 10-fold higher levels of AEA (but not 2-AG) in hind paw skin relative to the URB602 vehicle during Phase 1 (5 min post-formalin). Vehicle-induced increases in AEA, but not 2-AG levels, were also observed using a propofol vehicle (Guindon et al., 2007c) and larger volumes (100 µL) of saline (Jhaveri et al., 2008). The JZL184 vehicle could have interfered with lipid extraction and/or the LC/MS detection threshold. In fact, polyethylene glycol, a component of the JZL184 vehicle, administered intraperitoneally, can confound certain metabolomic measurements, especially in peripheral tissues where it can accumulate and reside for extended periods (Long et al., 2009b). However, Phase 2 AEA and 2-AG levels were not altered by either JZL184 or URB602, suggesting that vehicle effects were transient.
The regional distribution of endocannabinoid accumulation in rat hind paw skin was subsequently measured in proximal, middle and distal hind paw skin during Phase 2 (35 min post-formalin). This study was performed using a single MGL inhibitor (URB602) that was dissolved in the vehicle that did not alter Phase 1 AEA accumulation. The use of the less potent and presumably less selective MGL inhibitor thus provides a more conservative test of the hypothesis that MGL inhibitors produce peripheral antinociception by selectively increasing levels of 2-AG. In our studies, URB602 selectively increased 2-AG accumulation in distal and middle hind paw skin, while leaving AEA levels unchanged. Moreover, endocannabinoid levels in proximal hind paw skin were not altered by URB602. Thus, larger samples of tissue may mask detection of regionally restricted changes in endocannabinoid accumulation produced by MGL inhibition. The regionally restricted changes in 2-AG accumulation produced by URB602 in our study may correspond to the injection spread of the MGL inhibitor in the paw, and/or the localization of the inflammatory response produced by formalin. This interpretation is also consistent with the greater observed weight of the middle zone of paw skin relative to distal and proximal segments, despite the fact that paw skin was dissected into zones of similar lengths.
The development of MGL−/− mice (Schlosburg et al., 2010) provides new opportunities to uncover the role of 2-AG in pain modulation. MGL inhibitors with different selectivity have been developed: URB602 (Hohmann et al., 2005), OMDM169 (Bisogno et al., 2009) and JZL184 (Long et al., 2009a). Antinociceptive effects of these compounds in different animal pain models have been reported (see Guindon and Hohmann, 2009). In our study, local administration of URB602 and JZL184 produced dose-dependent antinociceptive effects in both phases of the formalin test. By contrast, OMDM169, administered systemically, affected the late phase only (Bisogno et al., 2009). OMDM169 and URB602 can also inhibit FAAH at higher concentrations which should translate to a weaker selectivity of these compounds following systemic injection. By contrast, JZL184 shows high selectivity in inhibiting MGL over FAAH in mouse brain membranes following systemic administration, producing an eightfold increase in brain 2-AG levels without altering AEA levels (Long et al., 2009a). The piperidine carbamate scaffold of JZL184 irreversibly inactivates MGL in mice (Long et al., 2009b). Recent in vitro studies have nonetheless questioned whether JZL184 may be used as effectively as an MGL inhibitor in rats (Pan et al., 2009). Our findings demonstrate that JZL184 suppresses MGL activity in rat hind paw skin and is more potent than URB602 in producing peripheral antinociception. Our lab has recently demonstrated that JZL184 produces modality-specific peripheral antinociception in the capsaicin model that did not overlap with a FAAH inhibitor (URB597) administered via the same route (Spradley et al., 2010). These observations further argue for selectivity of JZL184 in suppressing MGL in vivo.
The presence of cannabinoid receptors on primary afferent neurons is supported by many experiments (Hohmann and Herkenham, 1999b; Ständer et al., 2005). Cannabinoid receptors are synthesized in dorsal root ganglion cells, which are the source of primary afferent input to the spinal cord (Hohmann and Herkenham, 1999a,b; Ständer et al., 2005; Walczak et al., 2005). Both cannabinoid receptor subtypes have been found in skin and in adnexal structures which may also contribute to peripheral antinociception (Ibrahim et al., 2005; Ständer et al., 2005).
Cannabinoids act locally through distinct CB1 and CB2 receptors to suppress both the development (Clayton et al., 2002; Nackley et al., 2003) and maintenance (Gutierrez et al., 2007) of inflammatory nociception (see Jhaveri et al., 2007). However, it remains to be determined whether antinociceptive effects of 2-AG are mediated through peripheral CB2 receptors. Antinociceptive effects of MGL inhibitors may be influenced by the compound employed, the route of administration, the animal model used and the level of endocannabinoid tone produced by the injury. Thus, involvement of either CB1 and/or CB2 receptors may differ based upon these factors. Antihyperalgesic effects of JZL184 were purely CB1 receptor-mediated in the chronic constriction injury model (Kinsey et al., 2009, systemic). JZL184 also produced CB1 receptor-mediated antinociception in the formalin test, but mediation by CB2 receptors was not assessed (Long et al., 2009a, systemic). In our study, JZL184 produced both CB1-and CB2-mediated peripheral antinociception in rats, as observed recently in the capsaicin model (Spradley et al., 2010), thereby revealing a CB2 receptor component in JZL184-induced antinociception. CB1/CB2 receptors are implicated in antinociceptive effects of URB602 (Guindon et al., 2007a, local) and OMDM169 (Bisogno et al., 2009, systemic) in formalin and partial sciatic nerve ligation models (Desroches et al., 2008, local). URB602 also produces CB2 receptor-mediated antinociception in the carrageenan model (Comelli et al., 2007, systemic). It remains to be determined whether cannabinoid receptor-independent mechanisms also contribute to the in vivo pharmacological profile of MGL inhibitors.
JZL184 inactivates MGL with good efficacy and selectivity in different peripheral tissues in mice (Long et al., 2009b) when given intraperitoneally. Our findings demonstrate that local administration of JZL184 inhibits MGL activity without affecting FAAH and NAPE-PLD activities in hind paw skin of rats. Nonetheless, antinociceptive doses of JZL184 and URB602 failed to increase levels of 2-AG in the whole hind paw skin during either phase of the formalin test. However, when the hind paw skin was further subdivided, URB602, the less potent MGL inhibitor, nonetheless increased 2-AG but not AEA levels in the middle and distal paw skin segments. Systemic OMDM169 also increased levels of 2-AG, but not AEA, in the formalin-injected paw (Bisogno et al., 2009). Moreover, FAAH inhibitors increase both AEA and 2-AG (Jhaveri et al., 2008) or AEA only (Costa et al., 2010) in carrageenan-inflamed paws. A peripherally restricted FAAH inhibitor URB937 also produces CB1-mediated antinociception in the formalin model by elevating levels of AEA and other fatty-acid amides with no change in 2-AG (Clapper et al., 2010). Differences in the mechanism of action (FAAH vs. MGL), route of administration and duration of inflammation limit the ability to make comparisons between studies. These studies nonetheless demonstrate that inhibition of endocannabinoid hydrolysis in the periphery suppresses the development of inflammatory pain.
In conclusion, MGL inhibition in the periphery produces antinociception and increases 2-AG accumulation in rat hind paw skin. The MGL inhibitors JZL184 and URB602, administered locally in the paw, acted through a common mechanism to suppress formalin-induced pain behaviour with identical patterns of pharmacological specificity, although with different potencies. The combination of JZL184 or URB602 with exogenous 2-AG also produce additive antinociceptive effects. Furthermore, JZL184 inhibited MGL activity without affecting activity of enzymes catalysing AEA hydrolysis (FAAH) or synthesis (NAPE-PLD). Finally, we showed that URB602 produced regionally restricted increases in 2-AG levels in rat hind paw skin without altering AEA levels. Our findings provide evidence that inhibition of MGL in the periphery modulates the endocannabinoid system to block the development of inflammatory pain. More work is necessary to determine whether MGL inhibitors are effective for the treatment of pathological pain in humans.
Acknowledgments
JG is supported by a Fonds de la recherche en santé du Québec (FRSQ) postdoctoral fellowship. DP is supported by DA021644 and AGH is supported by DA021644 and DA028200. The contribution of the Agilent Technologies/UCI Analytical Discovery Facility, Center for Drug Discovery is gratefully acknowledged.
Glossary
Abbreviations
- Δ9-THC
Δ9-tetrahydrocannabinol
- 2-AG
2-arachidonoyl glycerol
- AA-5-HT
N-arachidonoyl 5-HT
- AEA
anandamide
- AUC
area under the curve
- CB
cannabinoid
- CNS
central nervous system
- CPS-WST0
1,2, composite pain score-weighted scores technique
- DMSO
dimethylsulfoxide
- FAAH
fatty-acid amide hydrolase
- LC/MS
liquid chromatography-mass spectrometry
- m/z
mass-to-charge ratio
- MGL
monoacylglycerol lipase
- NAPE-PLD
N-arachidonoyl-phosphatidylethanolamine phospholipase D
- PEG
300, polyethylene glycol 300
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–10 as PowerPoint slide.
References
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th Edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, et al. Endocannabinoid Research Group. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta. 2009;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Cadas H, di Tomaso E, Piomelli D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci. 1997;17:1226–1242. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton N, Marshall FH, Bountra C, O'Shaughnessy CT. CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain. 2002;96:253–260. doi: 10.1016/S0304-3959(01)00454-7. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol. 2007;152:787–794. doi: 10.1038/sj.bjp.0707425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B, Bettoni I, Petrosino S, Comelli F, Giagnoni G, Di Marzo V. The dual fatty acid amide hydrolase/TRPV1 blocker, N-arachidonoyl-serotonin, relieves carrageenan-induced inflammation and hyperalgesia in mice. Pharmacol Res. 2010;61:537–546. doi: 10.1016/j.phrs.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- De Léan A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- Desroches J, Guindon J, Lambert C, Beaulieu P. Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br J Pharmacol. 2008;155:913–924. doi: 10.1038/bjp.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2006;160:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- Draper NR, Smith H. Applied Regression Analysis. New York: Wiley; 1998. [Google Scholar]
- Fu J, Astarita G, Gaetani S, Kim J, Cravatt BF, Mackie K, et al. Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem. 2007;282:1518–1528. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- Gatley SJ, Gifford AN, Volkow ND, Lan R, Makriyannis A. 123I-labellded AM251: a radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors. Eur J Pharmacol. 1996;307:331–338. doi: 10.1016/0014-2999(96)00279-8. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Rodriguez de Fonseca F, Piomelli D. Quantification of bioactive acylethanolamides in rat plasma by electrospray mass spectrometry. Anal Biochem. 2000;280:87–93. doi: 10.1006/abio.2000.4509. [DOI] [PubMed] [Google Scholar]
- Goparaju SK, Ueda N, Taniguchi K, Yamamoto S. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol. 1999;57:417–423. doi: 10.1016/s0006-2952(98)00314-1. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol. 2007a;150:693–701. doi: 10.1038/sj.bjp.0706990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Desroches J, Dani M, Beaulieu P. Pre-emptive antinociceptive effects of a synthetic cannabinoid in a model of neuropathic pain. Eur J Pharmacol. 2007b;568:173–176. doi: 10.1016/j.ejphar.2007.04.060. [DOI] [PubMed] [Google Scholar]
- Guindon J, Lo Verme J, Piomelli D, Beaulieu P. The antinociceptive effects of local injections of propofol in rats are mediated in part by cannabinoid CB1 and CB2 receptors. Anesth Analg. 2007c;104:1563–1569. doi: 10.1213/01.ane.0000263278.05423.a3. [DOI] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, Hohmann AG. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. Br J Pharmacol. 2007;150:153–163. doi: 10.1038/sj.bjp.0706984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999a;92:1171–1175. doi: 10.1016/s0306-4522(99)00220-1. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999b;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci USA. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol. 2007;152:624–632. doi: 10.1038/sj.bjp.0707433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- King AR, Duranti A, Tontini A, Rivara S, Rosengarth A, Clapper JR, et al. URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem Biol. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer FR, Stella N. The therapeutic potential of novel cannabinoid receptors. Pharmacol Ther. 2009;122:83–96. doi: 10.1016/j.pharmthera.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009b;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, et al. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB(2) receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;119:747–757. doi: 10.1016/s0306-4522(03)00126-x. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, et al. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R, Griffin G, Fernando S, Li X, Hill A, Makriyannis A. AM630, a competitive cannabinoid receptor antagonist. Life Sci. 1995;56:1949–1955. doi: 10.1016/0024-3205(95)00175-6. [DOI] [PubMed] [Google Scholar]
- Petricevic M, Wanek K, Denko CW. A new mechanical method for measuring rat paw edema. Pharmacology. 1978;16:153–158. doi: 10.1159/000136761. [DOI] [PubMed] [Google Scholar]
- Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1996;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo R, Lo Verme J, La Rana G, D'Agostino G, Sasso O, Calignano A, et al. Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR-α receptor agonist GW7647. Eur J Pharmacol. 2007;566:117–119. doi: 10.1016/j.ejphar.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Järvinen T, et al. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008;8:281–288. doi: 10.1016/j.cmet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradley JM, Guindon J, Hohmann AG. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res. 2010;62:249–258. doi: 10.1016/j.phrs.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ständer S, Schmelz M, Metze D, Luger T, Rukwied R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177–188. doi: 10.1016/j.jdermsci.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Tjölsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wang J, Ueda N. Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat. 2009;89:112–119. doi: 10.1016/j.prostaglandins.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Watson GS, Sufka KJ, Coderre TJ. Optimal scoring strategies and weights for the formalin test in rats. Pain. 1997;70:53–58. doi: 10.1016/s0304-3959(96)03299-x. [DOI] [PubMed] [Google Scholar]
- Winer BJ. Statistical Principles in Experimental Design. New York: McGraw-Hill; 1971. [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.