

Abstract
BACKGROUND AND PURPOSE
Previous findings have indicated that a cannabinoid, such as Δ9-THCV, which has antioxidant properties and the ability to activate CB2 receptors but to block CB1, might be a promising therapy for alleviating symptoms and delaying neurodegeneration in Parkinson's disease (PD).
EXPERIMENTAL APPROACH
The ability of Δ9-THCV to reduce motor inhibition and provide neuroprotection was investigated in rats lesioned with 6-hydroxydopamine and in mice lesioned with lipopolysaccharide (LPS).
KEY RESULTS
Acute administration of Δ9-THCV attenuated the motor inhibition caused by 6-hydroxydopamine, presumably through changes in glutamatergic transmission. Moreover, chronic administration of Δ9-THCV attenuated the loss of tyrosine hydroxylase–positive neurones caused by 6-hydroxydopamine in the substantia nigra, through an effect related to its antioxidant properties (it was reproduced by cannabidiol -enriched botanical extract). In addition, CB2 receptor–deficient mice responded to 6-hydroxydopamine in a similar manner to wild-type animals, and CB2 receptors were poorly up-regulated in the rat substantia nigra in response to 6-hydroxydopamine. By contrast, the substantia nigra of mice that had been injected with LPS exhibited a greater up-regulation of CB2 receptors. In these animals, Δ9-THCV also caused preservation of tyrosine hydroxylase–positive neurones. This effect probably involved CB2 receptors as it was also elicited by the selective CB2 receptor agonist, HU-308, and CB2 receptor–deficient mice were more vulnerable to LPS lesions.
CONCLUSIONS AND IMPLICATIONS
Given its antioxidant properties and its ability to activate CB2 but to block CB1 receptors, Δ9-THCV has a promising pharmacological profile for delaying disease progression in PD and also for ameliorating parkinsonian symptoms.
LINKED ARTICLES
This article is part of a themed issue on Cannabinoids in Biology and Medicine. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-7
Keywords: Δ9-THCV, cannabinoid receptors, Parkinson's disease, 6-hydroxydopamine-lesioned rats, LPS-lesioned mice, neuroprotection
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder; its aetiology is associated with environmental insults, genetic susceptibility or interactions between both causes (Thomas and Beal, 2007). The major clinical symptoms in PD are tremor, bradykinesia, postural instability and rigidity, symptoms that result from the severe dopaminergic denervation of the striatum caused by the progressive death of dopaminergic neurones of the substantia nigra pars compacta (Nagatsu and Sawada, 2007). Major symptoms in PD (e.g. bradykinesia) can be attenuated with dopaminergic replacement therapy (Thomas and Beal, 2007). However, this therapy does not work for all PD patients, and when used for more than 5–10 years, it provokes an irreversible dyskinetic state (Pezzoli and Zini, 2010). Therefore, the search for novel symptomatic therapies, as well as for treatments effective in delaying the progression of nigrostriatal damage in PD, is still the major challenge in PD therapy.
Cannabinoid-based compounds have recently been proposed as promising therapies in PD (García-Arencibia et al., 2009). Thus, the blockade of CB1 receptors, which are highly abundant in basal ganglia structures, may be effective in reducing the motor inhibition typical of PD patients (Fernández-Espejo et al., 2005; González et al., 2006; Kelsey et al., 2009), as well as in enhancing the therapeutic effect of moderate doses of levodopa, which are less pro-dyskinetic (Kelsey et al., 2009). Both effects are in accordance with the overactivity of the cannabinoid system observed in PD patients and in animal models of this disease (Di Marzo et al., 2000; Romero et al., 2000; Lastres-Becker et al., 2001; Gubellini et al., 2002; Pisani et al., 2005). In addition, CB1 receptor–deficient mice also showed less severe dyskinesias, when lesioned with 6-hydroxydopamine and treated with levodopa, compared with wild-type animals (Pérez-Rial et al., 2010). However, the efficacy of CB1 receptor blockade was restricted to specific circumstances, that is the use of low doses (Fernández-Espejo et al., 2005; González et al., 2006; Kelsey et al., 2009) and very extensive nigral damage (Fernández-Espejo et al., 2005), conditions that were not reproduced in the only clinical trial conducted so far with CB1 receptor blockers, which included a population of patients that were all good responders to levodopa (Mesnage et al., 2004). Therefore, this potential therapeutic strategy merits further clinical investigation, this time with PD patients that respond poorly to levodopa. On the other hand, other studies have reported that compounds that directly or indirectly activate rather than block CB1 receptors also alleviate the symptoms associated with PD (Ferrer et al., 2003; Segovia et al., 2003; Fernández-Espejo et al., 2004), thus stressing the complexity of the effects of cannabinoids in PD.
Some cannabinoids have been reported to protect nigral neurones from death caused by different cytotoxic stimuli in various experimental models of PD (Lastres-Becker et al., 2005; García-Arencibia et al., 2007; Jiménez-Del-Rio et al., 2008). These include the phytocannabinoids, Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol (CBD) (Lastres-Becker et al., 2005; García-Arencibia et al., 2007), the synthetic CB1/CB2 receptor agonist CP55 940 (Jiménez-Del-Rio et al., 2008) and the anandamide analogue AM404 (García-Arencibia et al., 2007). A priori these compounds acted through antioxidant mechanisms that seem to be independent of CB1 or CB2 receptors, although selective CB2 receptor agonists also showed efficacy in MPTP-lesioned mice (Price et al., 2009) but not in 6-hydroxydopamine-lesioned rats (García-Arencibia et al., 2007). It is also noteworthy that CB1 receptor–deficient mice display an increased vulnerability to 6-hydroxydopamine lesions (Pérez-Rial et al., 2010). However, selective CB1 receptor agonists do not seem to protect against 6-hydroxydopamine-induced damage (García-Arencibia et al., 2007), and they may even aggravate major parkinsonian symptoms, given the hypokinetic effects associated with the activation of the CB1 receptor (García-Arencibia et al., 2009).
Therefore, these previous data provide good evidence that a cannabinoid having antioxidant properties and the ability to activate CB2 receptors but to block CB1 receptors, might serve to alleviate parkinsonian symptoms and to arrest/delay neurodegeneration in PD. Δ9-THC and CBD are two phytocannabinoids that display cannabinoid receptor–independent antioxidant properties in PD (Lastres-Becker et al., 2005; García-Arencibia et al., 2007). However, Δ9-THC is an agonist of CB1 receptors and acutely aggravates motor inhibition, and CBD has negligible activity at both CB1 and CB2 receptors. In addition, although rimonabant may be efficacious as a symptom-relieving agent (Fernández-Espejo et al., 2005; González et al., 2006; Kelsey et al., 2009), it does not appear to have neuroprotective effects in PD. Indeed, enhanced damage has been reported after genetic ablation of CB1 receptors and lesion with 6-hydroxydopamine (Pérez-Rial et al., 2010). However, the phytocannabinoid Δ9-tetrahydrocannabivarin (Δ9-THCV) has a pharmacological profile that seems to be particularly appropriate for PD; it is an antioxidant and appears to act as a CB1 receptor antagonist (when used at doses lower than 3 mg·kg−1) but is a potent CB2 receptor agonist. In vitro, it has been shown to displace [3H]-CP55940 from human CB1 and CB2 receptors, antagonize CP55940-induced stimulation of [35S]-GTPγS binding to human CB1 receptors, inhibit forskolin-induced stimulation of cAMP production in human CB2-expressing Chinese hamster ovary cells and stimulate [35S]-GTPγS binding both to human CB2 receptors and to mouse spleen membranes, all at submicromolar concentrations (Thomas et al., 2005; Pertwee et al., 2007; Bolognini et al., 2010). In vivo, it has been shown to antagonize certain CB1 receptor–mediated behavioural effects of Δ9-THC in mice (Pertwee et al., 2007) and decrease carrageenan-induced mouse paw oedema in a manner that can be antagonized by the CB2 antagonist, SR144528, but not by the CB1 antagonist, rimonabant (Bolognini et al., 2010).
The aim of the present study was to demonstrate that Δ9-THCV can alleviate the symptoms associated with PD by blocking CB1 receptors at low doses (Thomas et al., 2005) and also induce neuroprotection due to its antioxidant properties and/or CB2 agonist activity. We used rats subjected to i.c.v. injection of 6-hydroxydopamine as a model to screen for evidence of amelioration and neuroprotection (Rodriguez Diaz et al., 2001; see details in González et al., 2006; García-Arencibia et al., 2008). In an initial experiment, we compared the ability of Δ9-THCV to improve the motor inhibition produced by 6-hydroxydopamine in these animals with that of rimonabant, a compound that has already been shown to have beneficial effects in this experimental model (González et al., 2006). Concentrations of neurotransmitters in the basal ganglia of these animals were determined, in particular those of glutamate as this neurotransmitter has been found to be involved in the anti-parkinsonian effect that follows the blockade of CB1 receptors (García-Arencibia et al., 2008). In a second experiment, Δ9-THCV was administered to 6-hydroxydopamine-lesioned animals for a period of 14 days and its ability to protect nigral neurones from 6-hydroxydopamine insult was evaluated by tyrosine hydroxylase immunostaining, a parameter that, although does not completely reflect neurodegeneration/neuroprotection per se, is frequently used for this purpose. OX-42 immunostaining (microglial activation) was also determined. Given that CB2 receptor agonists failed to provide neuroprotection in 6-hydroxydopamine-lesioned rats (García-Arencibia et al., 2007), effects of Δ9-THCV in these rats should be assigned a priori only to its cannabinoid receptor–independent antioxidant properties. We explored this hypothesis further: (i) by comparing the effects of Δ9-THCV with those produced by CBD, which showed positive effects in previous studies (Lastres-Becker et al., 2005; García-Arencibia et al., 2007); (ii) by determining the presence of CB2 receptors in the substantia nigra of 6-hydroxydopamine-lesioned rats; and (iii) by measuring the response to this neurotoxin in CB2 receptor–deficient mice. As our data confirmed that the neuroprotective effects of Δ9-THCV in 6-hydroxydopamine-lesioned rats do not appear to be mediated by CB2 receptors, we conducted additional experiments using mice intrastriatally injected with LPS, a model of PD with a higher inflammatory profile and in which up-regulation of CB2 receptors in response to damage was much more intense. HU-308, a selective CB2 receptor agonist, was used as a positive control. The response of CB2 receptor–deficient mice to LPS was also investigated.
Methods
6-Hydroxydopamine or LPS lesions and pharmacological treatments
Animals
Male Sprague–Dawley rats or CB2−/− mice and their respective wild-type littermates (bred in our animal facilities from mice donated by Dr Nancy Buckley, California State Polytechnic University, Pomona, CA, USA; see Buckley et al., 2000; Buckley, 2008) were housed in a room with a controlled photoperiod (0600–1800 h light) and temperature (22 ± 1°C). They had free access to standard food and water and were used at adult age (>2 months old and weighing around 350 g at the onset of experiments, for rats; and 3–4 months old; 25–30 g weight, for mice) for experimental purposes. All experiments were conducted according to Spanish and European guidelines (directive 86/609/EEC). In the case of mice, their genetic profile (CB2+/+, CB2+/− and CB2−/−) was determined by PCR analysis, as described by Buckley et al. (2000), using DNA extracted from a piece of tail taken from each mouse. Only homozygous mice (CB2+/+ or CB2−/−) were used in these experiments.
Intracerebroventricular or unilateral injection of 6-hydroxydopamine or LPS
Rats were subjected to an i.c.v. injection of 6-hydroxydopamine following the procedure previously described by Rodriguez Diaz et al., (2001). This model has been validated in our laboratory, and the details regarding the dose–response curves and the time course of the effects of 6-hydroxydopamine, as well as the behavioural characterization of motor deficits, and the histopathological and neurochemical evaluation of the animal brains have been described elsewhere (González et al., 2006). Rats were anaesthetized (ketamine 40 mg·kg−1+ xylazine 4 mg·kg−1, i.p.) 30 min after pretreatment with desipramine (25 mg·kg−1, i.p.), and then 6-hydroxydopamine free base (200 µg in a volume of 5 µL of saline containing 0.05% ascorbate to avoid oxidation) or saline (for control rats) was injected stereotaxically into the third ventricle (coordinates from bregma: −2 mm AP, 0 mm ML and 8 mm DV, according to Paxinos and Watson, 1986). The solution was injected slowly (0.5 µL·30 s−1), and the needle was left in place for 5 min before being slowly withdrawn. This avoids generating reflux and a rapid increase in intracranial pressure. After the application of 6-hydroxydopamine or saline, animals were used for pharmacological treatments as described in the following section. We also induced lesions with 6-hydroxydopamine using unilateral administration, the advantage of which is that contralateral structures serve as controls for the different analyses. This procedure was used in rats to seek out a possible up-regulation of CB2 receptors in the lesioned substantia nigra and in mice to evaluate possible differences in the response to 6-hydroxydopamine between CB2-deficient and wild-type animals. Rats (pretreated with 25 mg·kg−1 desimipramine, i.p., 30 min before) received injections of 6-hydroxydopamine free base (8 µg in a volume of 2 µL of saline containing 0.05% ascorbate to avoid oxidation) stereotaxically into the medial forebrain bundle (coordinates from bregma: −2.5 mm AP, −1.8 mm ML and −8.9 mm DV; Paxinos and Watson, 1986). Two weeks post lesion, animals were transcardially perfused with saline followed by fresh 4% paraformaldehyde [in 0.1 M phosphate-buffered saline (PBS)], and their brains were collected and post-fixed overnight at 4°C, cryoprotected with 30% sucrose in PBS and then frozen and stored at −80°C for immunohistochemical analysis of CB2 receptors. In the case of mice (also pretreated with 25 mg·kg−1 desimipramine, i.p., 30 min before), 6-hydroxydopamine (2 µL at a concentration of 2 µg·µL−1 saline in 0.2% ascorbate) was injected into the right striatum at a rate of 0.5 µL·min−1, using the following coordinates: +0.4 mm AP, ±1.8 mm ML and −3.5 mm DV, as described in Alvarez-Fischer et al. (2008). These animals were killed by rapid and careful decapitation 1 week after lesioning, and their brains were rapidly removed and frozen in 2-methylbutane cooled in dry ice and stored at −80°C for subsequent immunohistochemical analysis of the substantia nigra. On the other hand, other groups of mice received unilateral injections of S. Minnesota LPS (Sigma-Aldrich, Madrid, Spain) into two points of the right striatum following the procedure developed by Hunter et al. (2009). We used the following stereotaxic coordinates from bregma: +1.18 mm AP, −1.5 mm ML and −3.5 mm DV, as well as −0.34 mm AP, −2.5 mm ML and −3.2 mm DV (see details in Hunter et al., 2009). At each intrastriatal coordinate, 5 µg of LPS in a volume of 1 µL of saline was injected by using the same procedure as for 6-hydroxydopamine. Control animals were sham-operated and injected with 1 µL of saline using the same coordinates. This procedure leads to a loss of dopaminergic neurones caused predominantly by LPS-induced inflammatory events. After the application of LPS or saline, animals were subjected to pharmacological treatments as described in the following section, although a separate group remained untreated for 2 weeks and were perfused with paraformaldehyde following the same procedure as described above. Lastly, LPS was also administered to CB2-deficient and wild-type mice to evaluate possible differences in the response to LPS, following the same procedure described above.
Pharmacological treatments
Our initial experiment was directed at establishing whether Δ9-THCV, at a dose of 2 mg·kg−1 (based on previous dose–response analysis conducted in our laboratory; data not shown) can behave in vivo as a CB1 receptor antagonist in rats since it has previously been validated as a CB1 receptor antagonist in vivo only in mice (Pertwee et al., 2007; Bolognini et al., 2010). We investigated its ability to attenuate two effects induced by the cannabinoid agonist, CP-55,940: motor inhibition and anti-nociception in a model of acute pain, both effects that are thought to be mediated by CB1 receptors. To this end, naïve rats were administered CP55,940 (0.1 mg·kg−1, i.p.; purchased from Tocris Bioscience, Biogen Científica, Madrid, Spain) and/or Δ9-THCV (kindly provided by GW Pharmaceuticals Ltd, Cambridgeshire, UK) and effects on their motor behaviour were determined in a computer-aided actimeter, followed by the hot-plate test to determine their nociceptive sensitivity. In a subsequent experiment, Δ9-THCV was administered i.p. in a single dose (2 mg·kg−1) to 6-hydroxydopamine-lesioned animals 14 days after the lesion. Separate groups of animals received vehicle (Tween 80 : saline, 1:16) or rimonabant (0.1 mg·kg−1; kindly provided by Sanofi-Aventis, Montpellier, France), also i.p and at the same dose employed in our previous studies (González et al., 2006; García-Arencibia et al., 2008). Ten minutes later, the behaviours of vehicle-, Δ9-THCV- or rimonabant-injected 6-hydroxydopamine-lesioned animals and sham-operated controls were assessed in a computer-aided actimeter for a period of 10 min, at the end of which the animals were killed by rapid and careful decapitation. Their brains were rapidly removed and frozen in 2-methylbutane cooled in dry ice and stored at −80°C for analysis of neurotransmitters in basal ganglia. In the next experiment, Δ9-THCV (2 mg·kg−1) was administered i.p. to 6-hydroxydopamine-lesioned animals for a period of 14 days (one injection per day) starting shortly (approximately 16 h) after the lesion. Separate groups of animals received vehicle (Tween 80 : saline, 1:16) or CBD-enriched botanical extract (also provided by GW Pharmaceuticals Ltd, Cambridgeshire, UK) that contains 64.8% CBD, 2.3% Δ9-THC, 1.1% cannabigerol, 3.0% cannabichromene and 1.5% other phytocannabinoids. It was also administered i.p., at a dose of 4.63 mg·kg−1 (equivalent to 3 mg·kg−1 of pure CBD, the same dose used in previous studies; Lastres-Becker et al., 2005; García-Arencibia et al., 2007), and was used in this investigation as a positive control. Two hours after the final injection, the 6-hydroxydopamine-lesioned animals injected with vehicle, Δ9-THCV or CBD-enriched botanical extract and sham-operated controls were killed by rapid and careful decapitation. Their brains were rapidly removed and frozen in 2-methylbutane cooled in dry ice and stored at −80°C for subsequent immunohistochemical analysis of the substantia nigra. Lastly, Δ9-THCV (2 mg·kg−1) or HU-308 (5 mg·kg−1; purchased from Tocris Bioscience, Biogen Científica) were administered i.p. to LPS-lesioned mice for a period of 14 days (one injection per day) starting shortly (approximately 16 h) after the lesion. Separate groups of animals received vehicle (Tween 80 : saline, 1:16). Two hours after the final injection, the LPS-lesioned animals injected with vehicle, Δ9-THCV or HU-308 and sham-operated controls were killed by transcardial perfusion with 4% paraformaldehyde as described above. Their brains were collected, post-fixed, cryoprotected and subsequently used for immunohistochemical analysis of the substantia nigra.
Behavioural analysis
Computer-aided actimeter
Motor activity was analysed in a computer-aided actimeter (Actitrack, Panlab, Barcelona, Spain). This apparatus consisted of a 45 × 45 cm area, with infrared beams all around, spaced 2.5 cm, coupled to a computerized control unit that analyses the following parameters: (i) distance run in the actimeter (ambulation); (ii) mean velocity developed during this running; and (iii) time spent in fast (>5 cm s−1) and slow (<5 cm s−1) movements. Animals stayed in the actimeter for a period of 10 min, but measurements were only recorded during the final 5 min (first 5 min was used only for animal acclimatization).
Hot-plate test
Rats were placed on a hot-plate maintained at 52°C, and the latency to exhibit the first sign of pain (i.e. licking the hind paws or jumping) was measured for each animal. Animals not responding were removed after 30 s (cut-off time to avoid tissue damage).
Determination of dopamine and glutamate concentrations
Dissection procedure
Coronal slices (around 500 µm thick) were obtained manually from brains at the level containing the caudate-putamen, globus pallidus and substantia nigra. Subsequently, these structures were dissected and homogenized in 20–40 vol of cold 150 mM potassium phosphate buffer, pH 6.8 and the dopamine and glutamate content of each homogenate was analysed. An aliquot of each homogenate was used to determine the protein concentration (Lowry et al., 1951).
Determination of dopamine content
Dopamine content was determined using HPLC with electrochemical detection (Lastres-Becker et al., 2005; González et al., 2006). Briefly, homogenates were diluted in ice-cold 0.2 N perchloric acid containing 0.2 mM sodium disulphite and 0.45 mM EDTA, and dihydroxybenzylamine (20 ng·mg−1) was added as an internal standard. The diluted homogenates were then centrifuged, and the supernatants were injected into the HPLC system. This system consisted of a Spectra-Physics 8810 pump and an RP-18 column (Tracer Excel 120 ODSB; 150 mm, 4.6 mm, 5 µm particle size; Teknokroma, Barcelona, Spain). The mobile phase, previously filtered and degassed, was a solution of 100 mM citric acid, 100 mM sodium acetate, 1.2 mM heptane sulphonate, 1 mM EDTA and 7% methanol (pH 3.9), and the flow rate was 0.8 mL·min−1. The effluent was monitored with a coulochemical detector (Coulochem III, ESA) using a procedure of oxidation/reduction (conditioning cell: +360 mV; analytical cell #1: +50 mV; analytical cell #2: −340 mV). The signal was recorded on a Spectra-Physics 4290 integrator from the analytical cell #2, with a sensitivity of 50 nA (10 pg per sample). Dopamine levels were calculated from areas under the peaks using the comparison with the internal standard area. Values are expressed as ng·mg−1 of protein.
Determination of glutamate content
Homogenates were used for the analysis of glutamate content using a commercial Glutamate Assay Kit (#K629-100, BioVision, Mountain View, CA, USA) and following the instructions provided by the manufacturer. Values were calculated as nmol·mg−1 of protein and expressed as % of controls.
Immunohistochemical procedures
Coronal sections (20 µm thick) were obtained from the previously frozen brains with a cryostat and collected on gelatin-coated slides. Sections were fixed in fresh 4% paraformaldehyde prepared in 0.1 M PBS, pH 7.4. After being washed in PBS, sections were incubated overnight at room temperature with the primary antibodies: (i) monoclonal anti-tyrosine hydroxylase (Chemicon-Millipore, Temecula, CA, USA) used at 1:500; and (ii) monoclonal anti-rat CD11b clon OX-42 (Serotec, Bionova Científica, Madrid, Spain) used at 1:50. Next, sections were incubated with the secondary antibody, biotinylated horse anti-mouse antibody (Vector Elite, Burlingame, CA, USA), used at 1:200, followed by streptavidin incubation. We used Alexa Fluor-488 conjugated streptavidin (1:200, Molecular Probes, Eugene, OR, USA) for tyrosine hydroxylase immunostaining, and Alexa Fluor-546 conjugated streptavidin (1:200, Molecular Probes) for OX-42 immunostaining. Sections were then stained with 45 µM Hoechst 33342 and the slides were coverslipped with Vectashield mounting medium (Vector Elite, Burlingame, CA, USA). Negative control sections were obtained using the same protocol with omission of the primary antibody. All sections for each immunohistochemical procedure were processed at the same time and under the same conditions. In the case of tyrosine hydroxylase immunostaining in mouse sections, we used Mouse on Mouse Immunodetection Kit (Vector Elite), following the instructions provided by the manufacturer, to avoid secondary antibody cross-reaction. A Nikon Eclipse 90i microscope and a Nikon DXM 1200F camera were used for slide observation and photography. The magnitude of tyrosine hydroxylase or OX-42 immunostaining in the substantia nigra (both sides) for each animal was determined with NIH Image Processing and Analysis software (ImageJ; NIH, Bethesda, MD, USA) using three to six randomly selected sections, separated by 200 µm, and observed with a 5× objective. In all sections, the same area of substantia nigra pars compacta was analysed. Background signal was determined for each section in a small area near the one being examined. Analyses were always conducted by experimenters who were blinded to all animal characteristics.
Immunohistochemical analysis of CB2 receptor (also in those immunostainings for tyrosine hydroxylase carried out in LPS-lesioned mice) was performed in paraformaldehyde-perfused brains instead of frozen brain sections. Fixed brains were sliced in a cryostat (30 µm thick) and collected on gelatine-coated slides. Sections were incubated overnight at room temperature with polyclonal anti-CB2 receptor (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Next, sections were incubated with an Alexa-Fluor 594 chicken anti-goat secondary antibody conjugate (1:200; Molecular Probes). A Nikon Eclipse 90i microscope and a Nikon DXM 1200F camera were used for slide observation and photography.
Data analysis
Data were subjected to one-way anova followed by the Student–Newman–Keuls test, using Graph-Pad software (La Jolla, CA, USA) (version 4.0).
Results
Antagonism by Δ9-THCV of hypokinetic and analgesic effects of CP55,940
Figure 1 shows data obtained in experiments directed at establishing whether Δ9-THCV (2 mg·kg−1) behaves as a CB1 receptor antagonist in vivo in rats, as previously demonstrated in studies conducted in mice (Pertwee et al., 2007; Bolognini et al., 2010). As expected, the potent cannabinoid receptor agonist CP55,940 reduced ambulation in the actitrack [F(3,21) = 7.983, P < 0.005; Figure 1], altered other motor parameters in the same direction (data not shown) and increased the latency of the response by animals to a noxious stimulus in the hot-plate test [F(3,22) = 13.84, P < 0.001; Figure 1], two effects that are thought to be induced by the activation of CB1 receptors. The administration of Δ9-THCV did not produce any effect by itself, but it partially attenuated the effects of CP55,940 in both assays (Figure 1), thus supporting the notion that Δ9-THCV also behaves as a CB1 receptor antagonist in rats at the dose of 2 mg·kg−1.
Figure 1.
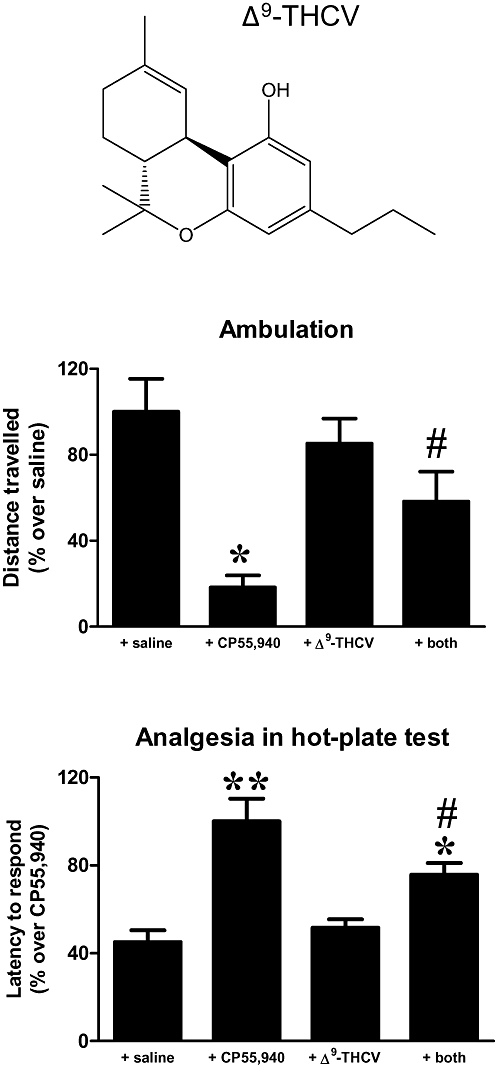
Effect of a single i.p. injection of Δ9-THCV (2 mg·kg−1; its structure is shown in the top panel) on CP55,940-induced motor inhibition and analgesia (measured in the hot-plate test) in naïve rats. Values were normalized versus the group having the maximal response and are expressed as means ± SEM (n = 5–6 rats per group). Data were subjected to one-way anova followed by the Student–Newman–Keuls test (*P < 0.05, **P < 0.005 vs. control and Δ9-THCV-treated animals; #P < 0.05 vs. CP55,940-treated animals).
Effect of Δ9-THCV on motor inhibition and dopamine and glutamate anomalies in 6-hydroxydopamine-lesioned rats
Figures 2 and 3 show the effects of an acute injection of Δ9-THCV (2 mg·kg−1) or rimonabant (0.1 mg·kg−1) on motor inhibition (lower ambulation, mean velocity and time spent in fast movements and increased time spent in slow movements) and dopamine and glutamate anomalies caused by lesioning with 6-hydroxydopamine (i.c.v). In this model, striatal dopamine content is significantly reduced (fivefold) 14 days post lesion, although this is accompanied by small motor anomalies, in accordance with human cases of parkinsonism where the appearance of motor symptoms occurs only after a loss of dopaminergic neurones greater than 50% (Nagatsu and Sawada, 2007). This was also observed in the present study (see Figures 2 and 3). Our behavioural data indicated that both Δ9-THCV and rimonabant were equally effective in enhancing ambulation [F(3,18) = 4.14, P < 0.05], mean velocity [F(3,18) = 4.22, P < 0.05] and, to a lesser extent, time spent doing fast movements [F(3,19) = 1.992, P = 0.156], reaching in all cases values similar to those exhibited by sham-operated animals (Figure 2). In addition, Δ9-THCV and rimonabant also reduced the large amount of time that these animals spent displaying slow movements [F(3,21) = 3.037, P < 0.05; Figure 2].
Figure 2.
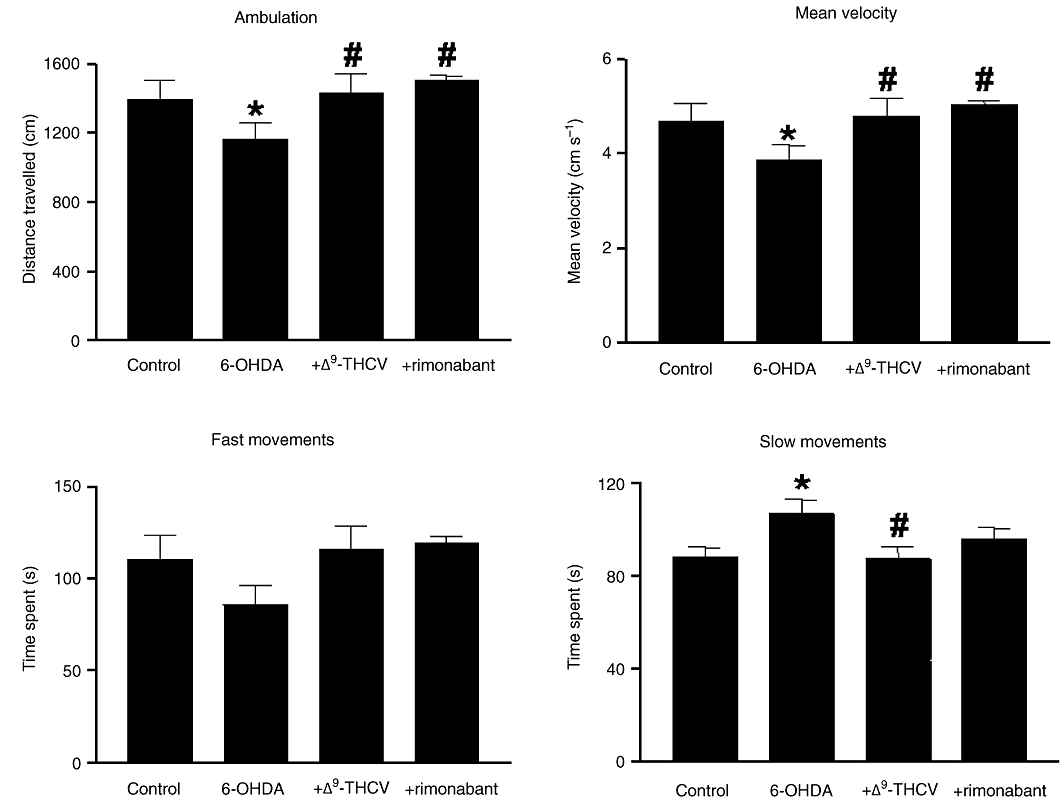
Motor parameters measured using a computer-aided actimeter. Rats were injected i.c.v. with 6-hydroxydopamine or saline (control rats) and subjected to a single i.p. injection of Δ9-THCV (2 mg·kg−1), rimonabant (0.1 mg·kg−1) or vehicle two weeks later. Values are means ± SEM (n = 5–6 rats per group), and the data were subjected to one-way anova followed by the Student–Newman–Keuls test (*P < 0.05 vs. controls; #P < 0.05 vs. 6-hydroxydopamine-lesioned animals).
Figure 3.

Dopamine content in the striatum and glutamate concentrations in different basal ganglia structures of rats injected i.c.v. with 6-hydroxydopamine or saline (control rats) and subjected to a single i.p. injection of Δ9-THCV (2 mg·kg−1) or vehicle 2 weeks later. Values are means ± SEM (n = 5–6 rats per group), and the data were subjected to one-way anova followed by the Student–Newman–Keuls test (*P < 0.005 vs. controls; #P < 0.05 vs. 6-hydroxydopamine-lesioned animals).
The effect of Δ9-THCV on behaviour was not associated with an increase in dopamine levels in the striatum, which, as mentioned above, is significantly reduced by lesioning with 6-hydroxydopamine [F(2,16) = 8.30, P < 0.005; Figure 3]. However, it was paralleled by signs of an increase in the glutamate content of the striatum, although this effect did not reach statistical significance [F(2,17) = 2.786, P = 0.094; Figure 3], as well as by a significant reduction of this chemical in the substantia nigra [F(2,17) = 3.991, P < 0.05; Figure 3]. No changes in glutamate levels were found in the globus pallidus (Figure 3). In all three structures, values of glutamate levels in control animals were as expected (striatum: 42.1 ± 5.2 nmol·mg−1 protein; globus pallidus: 50.1 ± 5.9 nmol·mg−1 protein; and substantia nigra: 36.8 ± 2.6 nmol·mg−1 protein) from previous findings (González et al., 2006). Values were normalized for graphical presentation (see Figure 3).
Effect of Δ9-THCV on tyrosine hydroxylase immunostaining in the substantia nigra of 6-hydroxydopamine-lesioned rats or LPS-lesioned mice
Figure 4 shows the effects of chronic treatment with Δ9-THCV (2 mg·kg−1; 14 days), or with a CBD-enriched botanical extract (equivalent to 3 mg·kg−1 of pure CBD; 14 days), on the damage in nigral neurones caused by 6-hydroxydopamine (i.c.v.). These data indicate that the loss of nigrostriatal dopaminergic neurones, determined by tyrosine hydroxylase immunostaining in the substantia nigra, was reduced after chronic administration of Δ9-THCV [F(3,18) = 4.869, P < 0.05; Figure 4]. Δ9-THCV also attenuated the enhanced microglial activation caused by 6-hydroxydopamine, as measured by OX-42 immunostaining in the substantia nigra [F(3,18) = 31.63, P < 0.0001; Figure 4]. Both effects were also seen, to an even greater extent, with CBD-enriched botanical extract (Figure 4), in accordance with previous data obtained with pure CBD (Lastres-Becker et al., 2005; García-Arencibia et al., 2007). This finding possibly indicates that the neuroprotective effects of Δ9-THCV in 6-hydroxydopamine-lesioned rats are due more to its antioxidant properties than to its ability to activate CB2 receptors. Previous studies have indicated that these receptors do not appear to play an important role in nigrostriatal lesions caused by 6-hydroxydopamine (García-Arencibia et al., 2007). We obtained further evidence for this by showing that immunostaining for CB2 receptors in the substantia nigra of the lesioned side was very weak (only a few cells were labelled with the anti-CB2 antibody; see Figure 5) and so not much different from that seen in the contralateral non-lesioned side of each animal (Figure 5). In addition, lesioning CB2 receptor–deficient mice with 6-hydroxydopamine led to a loss of tyrosine hydroxylase immunostaining in the substantia nigra that was of the same magnitude (approximately 50%) as that observed in wild-type animals (Figure 5). For these two last experiments, we used a unilateral lesion with 6-hydroxydopamine as this is much more appropriate for evaluating the degree of response against this neurotoxin, since this procedure allows the contralateral structures to serve as controls for the different analyses (up-regulation of CB2 receptors and loss of tyrosine hydroxylase immunostaining).
Figure 4.
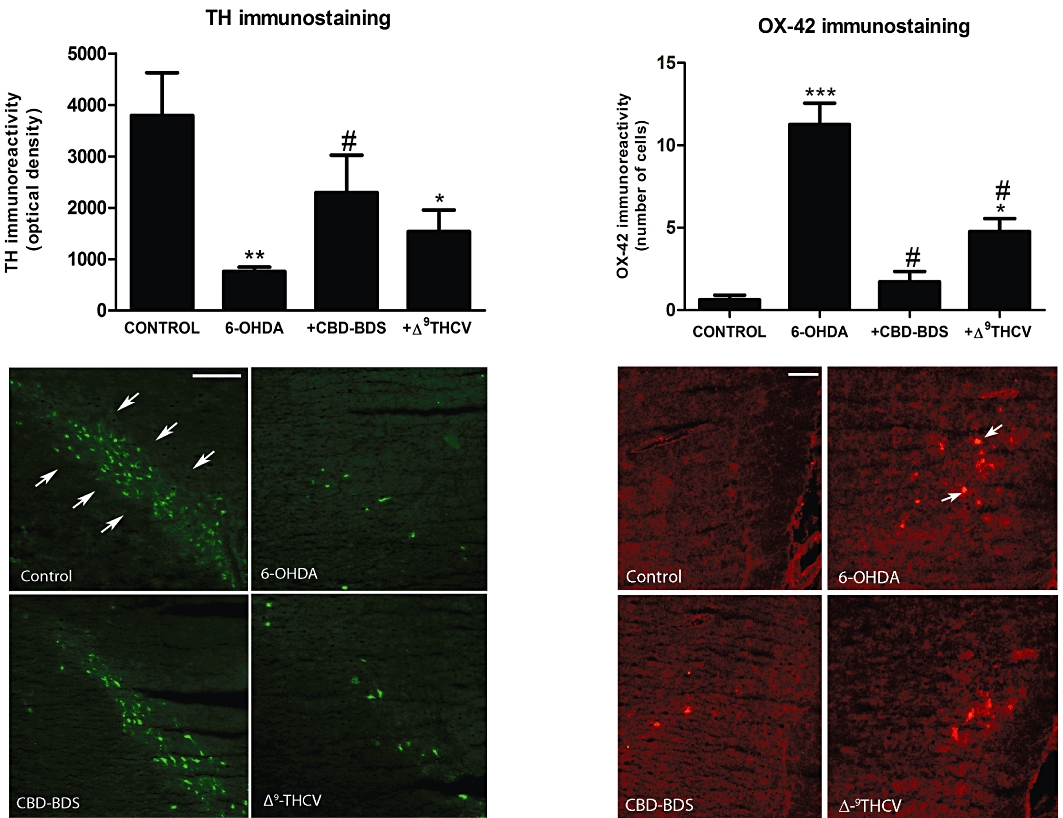
Tyrosine hydroxylase and OX-42 immunoreactivity measured in the substantia nigra of rats injected i.c.v. with 6-hydroxydopamine or saline (control rats) and subjected to chronic i.p. treatment with Δ9-THCV (2 mg·kg−1; 14 days), CBD-enriched botanical extract (4.63 mg·kg−1, equivalent to 3 mg·kg−1 of pure CBD; 14 days) or vehicle, starting 16 h after the i.c.v. injection. Values are means ± SEM (n = 5–6 rats per group), and the data were subjected to one-way anova followed by the Student–Newman–Keuls test (*P < 0.05, **P < 0.005, ***P < 0.0005 vs. controls; #P < 0.05 vs. 6-hydroxydopamine-lesioned animals). Representative tyrosine hydroxylase- and OX-42-immunostained sections of the substantia nigra are shown for the four experimental groups in the lower panels (scale bar = 100 µm for tyrosine hydroxylase and 50 µm for OX-42). Tyrosine hydroxylase– and OX-42-positive cells are indicated with arrows.
Figure 5.

Top panel: representative CB2 receptor-immunostained sections of the substantia nigra of rats unilaterally injected with 6-hydroxydopamine. Scale bar = 100 µm. CB2 receptor–positive cells are indicated with arrows. Bottom panel: tyrosine hydroxylase immunostaining measured in the substantia nigra of CB2 receptor–deficient and wild-type mice unilaterally injected with 6-hydroxydopamine. Values are means ± SEM (n = 5–6 mice per group) and are expressed as % of immunoreactivity in the lesioned side versus the non-lesioned side in the same animal. Data were subjected to Student's t-test.
Since 6-hydroxydopamine induces effects that do not appear to be counteracted by CB2 receptor activation, we conducted additional experiments using a model of PD in which CB2 receptors are expected to have a greater protective role. More specifically, mice were administered LPS by a unilateral intrastriatal injection, and it was found that immunostaining for CB2 receptors in the lesioned substantia nigra was more intense than in the contralateral non-lesioned structure (Figure 6). These were presumably CB2 receptors located in microglial cells labelled with IBA-1 (data not shown). These animals showed a significant reduction in tyrosine hydroxylase immunoreactivity [F(3,17) = 4.73, P < 0.05; see Figure 6] that reached approximately 32%, in line with data published by Hunter et al. (2009) who used different amounts of LPS. The administration of Δ9-THCV (2 mg·kg−1, i.p.) to LPS-lesioned animals was associated with the preservation of tyrosine hydroxylase–positive neurones, as indicated by immunostaining for this enzyme (Figure 6). The same effect was also observed with the selective CB2 receptor agonist HU-308 (Figure 6), thus supporting the involvement of CB2 receptors in this PD model. This was further supported by the finding that CB2 receptor–deficient mice were more vulnerable to LPS-induced lesions than wild-type animals (Figure 6).
Figure 6.

Top panel: representative CB2 receptor-immunostained sections of the substantia nigra of mice unilaterally injected with LPS. Scale bar = 100 µm. CB2 receptor–positive cells are indicated with arrows. Middle panel: tyrosine hydroxylase immunoreactivity measured in the substantia nigra of mice unilaterally injected with LPS or saline (control mice) and subjected to chronic i.p. treatment with Δ9-THCV (2 mg·kg−1), HU-308 (5 mg·kg−1) or vehicle (14 days), starting 16 h after the LPS injection. Values are means ± SEM (n = 5–6 mice per group), and the data were subjected to one-way anova followed by the Student–Newman–Keuls test (*P < 0.05 vs. the remaining groups). Bottom panel: tyrosine hydroxylase immunostaining measured in the substantia nigra of CB2 receptor–deficient and wild-type mice unilaterally injected with LPS. Values are means ± SEM (n = 5–6 mice per group) and are expressed as % of immunoreactivity in the lesioned side versus the non-lesioned side in the same animal. Data were subjected to Student's t-test (*P < 0.05 vs. wild-type animals).
Discussion
Several cannabinoids have been found to display signs of therapeutic efficacy in animal models of PD, although their effects were limited to the alleviation of specific motor symptoms [e.g. rimonabant for akinesia (Fernández-Espejo et al., 2005; González et al., 2006; Kelsey et al., 2009) or levodopa-induced dyskinesia (for review, see García-Arencibia et al., 2009), CB1 receptor agonists for tremor (Sañudo-Peña et al., 1999) and antioxidant cannabinoids (Lastres-Becker et al., 2005; García-Arencibia et al., 2007; Jiménez-Del-Rio et al., 2008) or CB2 receptor agonists (Price et al., 2009) for the progression of nigral damage]. Classical anti-parkinsonian dopaminergic-replacement therapy also possesses the same disadvantage of inducing symptom relief without delaying disease progression (Pezzoli and Zini, 2010). Therefore, the search for novel compounds that will simultaneously ameliorate motor symptoms and delay progression of nigral damage represents an important challenge for this disease. In this study, we have examined a phytocannabinoid compound, Δ9-THCV, whose structural similarity to Δ9-THC and ability to block CB1 receptors but activate CB2 receptors (Thomas et al., 2005; Pertwee et al., 2007; Bolognini et al., 2010), indicated a priori that it might meet this need both to relieve parkinsonian symptoms and to protect nigral neurones from death.
Here we have demonstrated that an acute injection of Δ9-THCV is efficacious in reducing motor inhibition in parkinsonian rats, with a potency equivalent to rimonabant. We also found that, as described previously for rimonabant (García-Arencibia et al., 2008), these effects were dopamine-independent and associated with changes in glutamatergic transmission in key structures of the basal ganglia. For example, the administration of Δ9-THCV tended to elevate glutamate levels in the striatum, an effect that may be associated with a blockade of CB1 receptors located in corticostriatal glutamatergic terminals, in accordance with the stimulant effects of rimonabant on glutamate release found previously by using in vivo microdialysis (García-Arencibia et al., 2008) and with the well-known ability of CB1 agonists to reduce the release of this transmitter (Adermark et al., 2009). Any benefits resulting from this increase in glutamate might be mediated by glutamatergic receptor subtypes (e.g. group III metabotropic receptors) located in striatopallidal neurones that have been proposed to induce anti-parkinsonian effects (Ossowska et al., 2007). It is also important to note that this stimulating effect on glutamatergic transmission is particularly marked in conditions of nigrostriatal dopaminergic denervation, being very modest in control animals, as described previously (García-Arencibia et al., 2008). This might be because dopaminergic denervation of the striatum may particularly affect CB1 receptors located in corticostriatal terminals, thus enabling a greater control of glutamate transmission by these receptors in conditions such as PD. Thus, such denervation was found some time ago, in experiments using autoradiography and in situ hybridization that do not allow cellular resolution, to be associated with an up-regulation of CB1 receptors in different neuronal subpopulations within the striatum (Mailleux and Vanderhaeghen, 1993; Lastres-Becker et al., 2001).
Glutamate levels in the substantia nigra of parkinsonian animals were reduced by Δ9-THCV. In our opinion, this was not the result of a direct action of Δ9-THCV on CB1 receptors located in subthalamonigral terminals. Instead, it was most likely an indirect effect that resulted from blockade of CB1 receptors located on striatopallidal terminals that, by enhancing GABA uptake (Maneuf et al., 1996), would be expected to cause a reduction in the activity of these neurones and a subsequent decrease in their tonic inhibitory action on GABAergic neurones projecting from the globus pallidus to the subthalamic nucleus, resulting in a greater inhibition of glutamate release from subthalamonigral neurones. This would be expected to correct the excessive subthalamonigral activity underlying parkinsonian symptoms such as tremor (Nagatsu and Sawada, 2007), which, in our 6-hydroxydopamine-lesioned animals, was only evident as a trend towards an increase in glutamate content in the substantia nigra. Such a correction is fully concordant with the type of effect expected for an anti-parkinsonian agent.
The attenuation of motor inhibition via modulation of excitatory transmission is not the only beneficial effect that Δ9-THCV may provide in PD. Thus, given its classical phytocannabinoid structure, this compound may serve as an antioxidant agent with the same efficacy as CBD or Δ9-THC, two cannabinoids that have been demonstrated to be neuroprotective in 6-hydroxydopamine-lesioned rats (Lastres-Becker et al., 2005; García-Arencibia et al., 2007). In addition, Δ9-THCV is also a potent CB2 receptor agonist that may share the ability of other agonists of this receptor to protect against nigrostriatal cell loss in MPTP-lesioned mice (Price et al., 2009). However, in our previous studies conducted in 6-hydroxydopamine-lesioned rats, we found that selective CB2 agonists did not have protective effects (García-Arencibia et al., 2007), and we have found here that CB2 receptors are poorly up-regulated in response to damage in these animals. This may be related to the fact that inflammation is a secondary event in 6-hydroxydopamine-lesioned animals, as reflected by the modest levels of OX-42 immunoreactivity found in the substantia nigra in the model used in our present study. With this idea in mind, we investigated whether Δ9-THCV induces neuroprotective effects when administered daily for 14 days to 6-hydroxydopamine-lesioned rats. Our data strongly demonstrated that Δ9-THCV partially reduced 6-hydroxydopamine-induced nigral damage as shown by measuring tyrosine hydroxylase immunostaining and by the ability of this cannabinoid to attenuate OX-42 immunoreactivity (indicative of reactive microgliosis). Both responses were also elicited by CBD-enriched botanical extract, in accordance with previous data obtained with pure CBD (Lastres-Becker et al., 2005; García-Arencibia et al., 2007). The effects of CBD, which does not bind CB2 receptors except at high concentrations, were in general of greater magnitude than the effects of Δ9-THCV, thus supporting the hypothesis that the antioxidant activity of both phytocannabinoids is possibly the key mechanism providing neuroprotection in this model of PD. This hypothesis is also supported by our observation that 6-hydroxydopamine lesions were of similar magnitude in CB2-deficient and wild-type mice, and by the above-mentioned poor up-regulation of CB2 receptors in the substantia nigra of 6-hydroxydopamine-lesioned rats. In this context, our data also suggest that the combined administration of both phytocannabinoids may enhance their neuroprotective properties in 6-hydroxydopamine-lesioned animals, while retaining the symptom-relieving effects of Δ9-THCV alone. On the other hand, although the neuroprotective effects of Δ9-THCV do not appear to be related to its ability to activate CB2 receptors in the model of 6-hydroxydopamine used here, we assume that this property may be more important in pro-inflammatory models of PD, as well as an important factor to consider when developing a Δ9-THCV-based therapy for PD patients where inflammation is also a key element in the pathogenesis. To test this hypothesis, we used the LPS-lesioned mice model of PD where dopaminergic cell death is caused predominantly by inflammatory events. The substantia nigra of these animals exhibited a more intense up-regulation of CB2 receptors compared both with the contralateral non-lesioned structures and to 6-hydroxydopamine-lesioned rats, and, accordingly, the treatment with Δ9-THCV also led to neuroprotection. We assume that this neuroprotective effect was a result of Δ9-THCV-induced activation of CB2 receptors, as it was mimicked in the same model of PD by HU-308, a selective CB2 receptor agonist, whereas mice lacking CB2 receptors appeared to be particularly vulnerable to LPS lesion.
Conclusion
In summary, given its antioxidant properties and its ability to activate CB2 but block CB1 receptors at a dose of 2 mg·kg−1, Δ9-THCV seems to have an interesting and therapeutically promising pharmacological profile. Thus, in contrast to other phytocannabinoids that have been investigated to date, it shows promise both for the treatment of disease progression in PD and for the relief of PD symptoms. This represents an important advance in the search for potential novel anti-parkinsonian agents, since Δ9-THCV administered alone or in combination with CBD may provide a much needed improved treatment for PD.
Acknowledgments
This work has been supported by grants from CIBERNED (CB06/05/0089), MICINN (SAF2006-11333 and SAF2009/11847), CAM (S-SAL-0261/2006) and GW Pharmaceuticals Ltd. These agencies had no further role in study design, the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. Authors are indebted to Yolanda García-Movellán for administrative assistance.
Glossary
Abbreviations
- CBD
cannabidiol
- CP-55940
2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl) cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- HU-308
[(1R,2R,5R)-2-[2,6-dimethoxy-4-(2-methyloctan-2-yl)phenyl]-7,7-dimethyl-4-bicyclo[3.1.1]hept-3-enyl] methanol
- PD
Parkinson's disease
- Δ9-THC
Δ9-tetrahydrocannabinol
- Δ9-THCV
Δ9-tetrahydrocannabivarin
Conflict of interest
Authors have formal links with GW Pharmaceuticals that funds some of their research.
Supporting Information
Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Adermark L, Talani G, Lovinger DM. Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur J Neurosci. 2009;29:32–41. doi: 10.1111/j.1460-9568.2008.06551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Fischer D, Henze C, Strenzke C, Westrich J, Ferger B, Höglinger GU, et al. Characterization of the striatal 6-OHDA model of Parkinson's disease in wild type and alpha-synuclein-deleted mice. Exp Neurol. 2008;210:182–193. doi: 10.1016/j.expneurol.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Bolognini D, Costa B, Maione S, Comelli F, Marini P, Di Marzo V, et al. The plant Δ9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br J Pharmacol. 2010;160:677–687. doi: 10.1111/j.1476-5381.2010.00756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE. The peripheral cannabinoid receptor knockout mice: an update. Br J Pharmacol. 2008;153:309–318. doi: 10.1038/sj.bjp.0707527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, et al. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB2 receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson's disease. FASEB J. 2000;14:1432–1438. doi: 10.1096/fj.14.10.1432. [DOI] [PubMed] [Google Scholar]
- Fernández-Espejo E, Caraballo I, Rodriguez de Fonseca F, Ferrer B, El Banoua F, Flores JA, et al. Experimental parkinsonism alters anandamide precursor synthesis, and functional deficits are improved by AM404: a modulator of endocannabinoid function. Neuropsychopharmacology. 2004;29:1134–1142. doi: 10.1038/sj.npp.1300407. [DOI] [PubMed] [Google Scholar]
- Fernández-Espejo E, Caraballo I, Rodríguez de Fonseca F, El Banoua F, Ferrer B, Flores JA, et al. Cannabinoid CB1 antagonists possess antiparkinsonian efficacy only in rats with very severe nigral lesion in experimental parkinsonism. Neurobiol Dis. 2005;18:591–601. doi: 10.1016/j.nbd.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. Eur J Neurosci. 2003;18:1607–1614. doi: 10.1046/j.1460-9568.2003.02896.x. [DOI] [PubMed] [Google Scholar]
- García-Arencibia M, González S, de Lago E, Ramos JA, Mechoulam R, Fernández-Ruiz J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson's disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007;1134:162–170. doi: 10.1016/j.brainres.2006.11.063. [DOI] [PubMed] [Google Scholar]
- García-Arencibia M, Ferraro L, Tanganelli S, Fernández-Ruiz J. Enhanced striatal glutamate release after the administration of rimonabant to 6-hydroxydopamine-lesioned rats. Neurosci Lett. 2008;438:10–13. doi: 10.1016/j.neulet.2008.04.041. [DOI] [PubMed] [Google Scholar]
- García-Arencibia M, García C, Fernández-Ruiz J. Cannabinoids and Parkinson's disease. CNS Neurol Disord Drug Targets. 2009;8:432–439. doi: 10.2174/187152709789824642. [DOI] [PubMed] [Google Scholar]
- González S, Scorticati C, García-Arencibia M, de Miguel R, Ramos JA, Fernández-Ruiz J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson's disease. Brain Res. 2006;1073–1074:209–219. doi: 10.1016/j.brainres.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, et al. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RL, Cheng B, Choi DY, Liu M, Liu S, Cass WA, et al. Intrastriatal lipopolysaccharide injection induces parkinsonism in C57/B6 mice. J Neurosci Res. 2009;87:1913–1921. doi: 10.1002/jnr.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Del-Rio M, Daza-Restrepo A, Velez-Pardo C. The cannabinoid CP55,940 prolongs survival and improves locomotor activity in Drosophila melanogaster against paraquat: implications in Parkinson's disease. Neurosci Res. 2008;61:404–411. doi: 10.1016/j.neures.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Kelsey JE, Harris O, Cassin J. The CB1 antagonist rimonabant is adjunctively therapeutic as well as monotherapeutic in an animal model of Parkinson's disease. Behav Brain Res. 2009;203:304–307. doi: 10.1016/j.bbr.2009.04.035. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Cebeira M, de Ceballos M, Zeng B-Y, Jenner P, Ramos JA, et al. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson's syndrome and of MPTP-treated marmosets. Eur J Neurosci. 2001;14:1827–1832. doi: 10.1046/j.0953-816x.2001.01812.x. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernández-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson's disease. Neurobiol Dis. 2005;19:96–107. doi: 10.1016/j.nbd.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Dopaminergic regulation of cannabinoid receptor mRNA levels in the rat caudate-putamen: an in situ hybridization study. J Neurochem. 1993;61:1705–1712. doi: 10.1111/j.1471-4159.1993.tb09807.x. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Nash JE, Crossman AR, Brotchie JM. Activation of the cannabinoid receptor by Δ9-tetrahydrocannabinol reduces gamma-aminobutyric acid uptake in the globus pallidus. Eur J Pharmacol. 1996;308:161–164. doi: 10.1016/0014-2999(96)00326-3. [DOI] [PubMed] [Google Scholar]
- Mesnage V, Houeto JL, Bonnet AM, Clavier I, Arnulf I, Cattelin F, et al. Neurokinin B, neurotensin, and cannabinoid receptor antagonists and Parkinson's disease. Clin Neuropharmacol. 2004;27:108–110. doi: 10.1097/00002826-200405000-00003. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Biochemistry of postmortem brains in Parkinson's disease: historical overview and future prospects. J Neural Transm Suppl. 2007;72:113–120. doi: 10.1007/978-3-211-73574-9_14. [DOI] [PubMed] [Google Scholar]
- Ossowska K, Konieczny J, Wardas J, Pietraszek M, Kuter K, Wolfarth S, et al. An influence of ligands of metabotropic glutamate receptor subtypes on parkinsonian-like symptoms and the striatopallidal pathway in rats. Amino Acids. 2007;32:179–188. doi: 10.1007/s00726-006-0317-y. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. London: Academic Press; 1986. [Google Scholar]
- Pérez-Rial S, García-Gutiérrez MS, Molina JA, Pérez-Nievas BG, Ledent C, Leiva C, et al. Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol Aging. 2010;32:631–645. doi: 10.1016/j.neurobiolaging.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Thomas A, Stevenson LA, Ross RA, Varvel SA, Lichtman AH, et al. The psychoactive plant cannabinoid, Δ9-tetrahydrocannabinol, is antagonized by Δ8- and Δ9-tetrahydrocannabivarin in mice in vivo. Br J Pharmacol. 2007;150:586–594. doi: 10.1038/sj.bjp.0707124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzoli G, Zini M. Levodopa in Parkinson's disease: from the past to the future. Expert Opin Pharmacother. 2010;11:627–635. doi: 10.1517/14656561003598919. [DOI] [PubMed] [Google Scholar]
- Pisani A, Fezza F, Galati S, Battista N, Napolitano S, Finazzi-Agro A, et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson's disease patients. Ann Neurol. 2005;57:777–779. doi: 10.1002/ana.20462. [DOI] [PubMed] [Google Scholar]
- Price DA, Martinez AA, Seillier A, Koek W, Acosta Y, Fernández E, et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Eur J Neurosci. 2009;29:2177–2186. doi: 10.1111/j.1460-9568.2009.06764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Diaz M, Abdala P, Barroso-Chinea P, Obeso J, González-Hernández T. Motor behavioural changes after intracerebroventricular injection of 6-hydroxydopamine in the rat: an animal model of Parkinson's disease. Behav Brain Res. 2001;122:79–92. doi: 10.1016/s0166-4328(01)00168-1. [DOI] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Pérez-Rosado A, Manzanares J, Rojo A, Fernández-Ruiz J, et al. Unilateral 6-hydroxydopamine lesions of nigrostriatal dopaminergic neurons increased CB1 receptor mRNA levels in the caudate-putamen. Life Sci. 2000;66:485–494. doi: 10.1016/s0024-3205(99)00618-9. [DOI] [PubMed] [Google Scholar]
- Sañudo-Peña MC, Tsou K, Walker JM. Motor actions of cannabinoids in the basal ganglia output nuclei. Life Sci. 1999;65:703–713. doi: 10.1016/s0024-3205(99)00293-3. [DOI] [PubMed] [Google Scholar]
- Segovia G, Mora F, Crossman AR, Brotchie JM. Effects of CB1 cannabinoid receptor modulating compounds on the hyperkinesia induced by high-dose levodopa in the reserpine-treated rat model of Parkinson's disease. Mov Disord. 2003;18:138–149. doi: 10.1002/mds.10312. [DOI] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146:917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.