

Abstract
BACKGROUND AND PURPOSE
Endocannabinoids have both anti-inflammatory and neuroprotective properties against harmful stimuli. We previously demonstrated that the endocannabinoid 2-arachidonoylglycerol (2-AG) protects hippocampal neurons by limiting the inflammatory response via a CB1 receptor-dependent MAPK/NF-κB signalling pathway. The purpose of the present study was to determine whether PPARγ, an important nuclear receptor, mediates 2-AG-induced inhibition of NF-κB phosphorylation and COX-2 expression, and COX-2-enhanced miniature spontaneous excitatory postsynaptic currents (mEPSCs).
EXPERIMENTAL APPROACH
By using a whole-cell patch clamp electrophysiological recording technique and immunoblot analysis, we determined mEPSCs, expression of COX-2 and PPARγ, and phosphorylation of NF-kB in mouse hippocampal neurons in culture.
KEY RESULTS
Exogenous and endogenous 2-AG-produced suppressions of NF-κB-p65 phosphorylation, COX-2 expression and excitatory synaptic transmission in response to pro-inflammatory interleukin-1β (IL-1β) and LPS were inhibited by GW9662, a selective PPARγ antagonist, in hippocampal neurons in culture. PPARγ agonists 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and rosiglitazone mimicked the effects of 2-AG on NF-κB-p65 phosphorylation, COX-2 expression and mEPSCs, and these effects were eliminated by antagonism of PPARγ. Moreover, exogenous application of 2-AG or elevation of endogenous 2-AG by inhibiting its hydrolysis with URB602 or JZL184, selective inhibitors of monoacylglycerol lipase (MAGL), prevented the IL-1β- and LPS-induced reduction of PPARγ expression. The 2-AG restoration of the reduced PPARγ expression was blocked or attenuated by pharmacological or genetic inhibition of the CB1 receptor.
CONCLUSIONS AND IMPLICATIONS
Our results suggest that CB1 receptor-dependent PPARγ expression is an important and novel signalling pathway in endocannabinoid 2-AG-produced resolution of neuroinflammation in response to pro-inflammatory insults.
LINKED ARTICLES
This article is part of a themed issue on Cannabinoids in Biology and Medicine. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.163.issue-7
Keywords: neuroinflammation, endocannabinoids, monoacylglycerol lipase, peroxisome proliferator-activated receptor-γ, cyclooxygenase-2, nuclear factor κ-B
Introduction
Endocannabinoids (eCBs) are endogenous signalling mediators involved in a variety of physiological, pharmacological and pathological processes (Freund et al., 2003; Kano et al., 2009; Marrs et al., 2010; Pertwee et al., 2010). The eCB system consists of eCBs, cannabinoid receptors (CB1 and CB2), enzymes synthesizing eCBs and metabolizing eCBs and cannabinoid transporters. Emerging evidence now suggests that eCBs possess anti-inflammatory and neuroprotective properties against harmful insults (Marsicano et al., 2003; Walter and Stella, 2004; Panikashvili et al., 2001; 2005, 2006; Eljaschewitsch et al., 2006; Mackie, 2006; Centonze et al., 2007; Zhang and Chen, 2008; Chen et al., 2011). Neuroinflammation is the immune system's response to infection and injury in the CNS and has been implicated in the pathogeneses and neuropathology of many prevalent neurological and neurodegenerative diseases, such as multiple sclerosis, Alzheimer's and Parkinson's diseases (Chen, 2010). However, there are currently no effective medications to prevent chronic inflammation and alleviate or treat chronic inflammation-induced brain disorders. As eCBs have the ability to restrict inflammation, they may have the potential to prevent neuropathology and treat neuroinflammation-induced brain disorders. While eCB modulation of both GABAergic and glutamatergic synaptic transmission and plasticity via a CB1 receptor-dependent mechanism has been extensively investigated (Alger, 2002; Freund et al., 2003; Piomelli, 2003; Chevaleyre et al., 2006; Mackie, 2006; Kano et al., 2009), the mechanisms underlying the ability of eCB to limit neuroinflammation and provide neuronal protection have not been elucidated (Sarne and Mechoulam, 2005; Van der Stelt and Di Marzo, 2005; Fowler et al., 2010).
PPARs are members of the nuclear receptors/transcription factors super-family. Three different PPAR genes (PPARα, PPARδ– also called PPARβ and PPARγ) have been identified. These nuclear receptors regulate expression of the genes involved in metabolism, cell differentiation and inflammation (Daynes and Jones, 2002; Kozak et al., 2002; Luna-Medina et al., 2005; Drew et al., 2006; Bensinger and Tontonoz, 2008; Bright et al., 2008; Necela et al., 2008). Growing evidence suggests that PPARs are likely a target for eCBs (Lenman and Fowler, 2007; O'Sullivan, 2007; Sun et al., 2007; O'Sullivan and Kendall, 2010; Pertwee et al., 2010; Pistis and Melis, 2010). However, little information is available as to whether PPARs are involved in this inhibitory effect of eCBs on neuroinflammation. 2-Arachidonoyl glycerol (2-AG), the most abundant eCB and a full agonist for CB1 and CB2 receptors (Sugiura et al., 2006; Kano et al., 2009), has been shown to be an important endogenous signalling mediator protecting neurons from pro-inflammatory, excitotoxic stimuli and other harmful insults (Gopez et al., 2005; Panikashvili et al., 2001; 2005, 2006; Melis et al., 2006; Kreutz et al., 2007). Rockwell et al. (2006) demonstrated that 2-AG-induced suppression of IL-2 in Jurkat T cells is mediated by activation of PPARγ through a CB1/2 receptor-independent mechanism. We demonstrated previously that direct application of 2-AG or elevation of endogenous 2-AG by inhibiting its hydrolysis protects hippocampal neurons from the effects of pro-inflammatory IL-1β and LPS, excitotoxic glutamate and kanaic acid and β-amyloid stimuli by limiting COX-2 expression via a CB1 receptor-dependent NF-κB signalling pathway (Zhang and Chen, 2008; Chen et al., 2011). In this report, we show that 2-AG-induced suppression of NF-κB phosphorylation, COX-2 expression and COX-2 elevation-enhanced excitatory synaptic transmission in hippocampal neurons in culture in response to pro-inflammatory insults is mediated by the CB1 receptor-dependent expression of PPARγ.
Methods
Cell culture
Primary hippocampal neurons from mouse pups (P0 to P1) were cultured as described previously (Sang et al., 2005; 2007, 2010; Zhang and Chen, 2008; Chen et al., 2011), according to the guidelines approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center in New Orleans. Briefly, hippocampi were dissected out under a microscope and triturated in serum-free culture medium after the meninges had been removed. Tissue was incubated in oxygenated trypsin for 10 min at 37°C and then mechanically triturated. Cells were spun down and resuspended in Neurobasal/B27 medium (Invitrogen Corp., Carlsbad, CA, USA) supplemented with 0.5 mM L-glutamine, penicillin/streptomycin and 25 µM glutamate. Cells (1 × 106) were loaded into poly-d-lysine-coated six-well plates for Western blot analysis and 35 mm culture dishes for electrophysiological recordings. Medium was changed every 3 days with the same medium without glutamate until use. The percentage of astroglial cells in the culture was ∼5% at 10 days in vitro (DIV), as estimated by staining with a neuronal marker NeuN, astrocytic marker GFAP and microglial marker OX-42 in conjunction with the 4′,6-diamidino-2-phenylindole staining as described previously (Sang et al., 2005; Zhang and Chen, 2008). Cultures were used between 10 and 21 DIV.
Electrophysiological recordings
Miniature spontaneous excitatory postsynaptic currents (mEPSCs) were recorded in hippocampal neurons in culture under voltage clamp using an Axopatch-200B amplifier (Molecular Devices, Palo Alto, CA) as described previously (Sang et al., 2005; 2006, 2007, 2010; Zhang and Chen, 2008). Recording pipettes (4–5 MΩ) were pulled from borosilicate glass with a micropipette puller (Sutter Instrument, Novato, CA). The internal pipette solution contained (in mM) 115.0 Cs gluconate, 15.0 CsCl, 4.0 NaCl, 10.0 HEPES, 0.5 EGTA, 4.0 Mg2ATP and 0.5 Na2GTP (pH 7.25 with CsOH). The membrane potential was held at −70 mV. The external solution contained (in mM): 130.0 NaCl, 2.5 KCl, 1.0 MgCl2, 10.0 HEPES, 1.25 NaH2PO4, 2.0 CaCl2 and 25.0 glucose (pH 7.4 with NaOH). To isolate mEPSCs, tetrodotoxin (TTX, 0.5 to 1 µM), a voltage-gated Na+ channel blocker and bicuculline (10 µM), a GABAA receptor blocker, were included in the external solution. All experiments were performed at room temperature (22–24°C). The frequency, amplitude and kinetics of mEPSCs were analysed using the MiniAnalysis program (Synaptosoft, Fort Lee, NJ).
Immunoblot
Hippocampal neurons in cultures were extracted and immediately homogenized in a one-to-one volume of modified radioimmunoprecipitation assay lysis buffer consisting of a number of protease inhibitors. Supernatants were fractionated on 10% SDS-PAGE gels (Bio-Rad Lab, Hercules, CA, USA) and transferred onto PVDF membranes (Bio-Rad). The membrane was incubated with anti-COX-2 polyclonal antibodies (dilution of 1:1000; Cayman Chemical, Ann Arbor, MI), anti-NF-κB-p65 and phospho-NF-κB antibodies (1:1000; Cell Signaling, Danvers, MA) and anti-PPARγ (1:1000; Abcam, Cambridge,MA) at 4°C overnight. The blot was washed and incubated with a secondary antibody (goat anti-rabbit, 1:10 000; Vector Laboratories, Burlingame, CA) at room temperature for 1 h. Proteins were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences, UK). The densities of specific bands were quantified by densitometry using FUJIFILM Multi Gauge software (version 3.0) (Fujifilm USA Inc., Valhalla, NY, USA). Band densities were normalized to the total amount of protein loaded in each well as determined by mouse anti-β-actin (1:4000; Sigma, St. Louis, MO) as described previously (Zhang and Chen, 2008; Sang et al., 2005; 2010;).
Chemicals and drugs
2-AG, AEA, URB602, JZL184, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), GW9662 and rosiglitazone were purchased from Cayman Chemical. T0070907 was purchased from Tocris Bioscience (Ellisville, MO, USA). These chemicals were dissolved in DMSO to make stock solutions at concentrations of 20 to 50 mM, distributed in small vials and then diluted with the external solution or culture medium to the desired concentrations just before experiments. Rimonabant (provided by Chemical Synthesis and Drug Supply Program, the National Institute of Mental Health) was dissolved in DMSO to make up stock solutions at concentrations of 50 to 100 mM. All other drugs and chemicals were obtained from Sigma, unless stated otherwise. To rule out potentially nonspecific effects of the solvents, the same amount of ethanol or DMSO was included in the control external solution and culture medium. Nomenclature of cannabinoid receptors follows Alexander et al. (2009).
Data analysis
Data are presented as mean ± SEM. Unless stated otherwise, anova with Fisher's PLSD test or Student–Newman–Keuls test was used for statistical comparison when appropriate. The Kolmogorov–Smirnov test was used for comparisons of mEPSCs distribution. Differences were considered significant when P < 0.05.
Results
Inhibition of COX-2 and NF-kB phosphorylation by 2-AG is mediated by PPARγ
To determine whether 2-AG-inudced anti-inflammatory effects involve PPARs, we targeted PPARγ since it represses the expression of inflammatory genes in response to pro-inflammatory stimuli (Bensinger and Tontonoz, 2008). We used phosphorylation of NF-κB-p65 and expression of COX-2 as biomarkers for neuroinflammation in cultured hippocampal neurons as described previously (Zhang and Chen, 2008). As shown in Figure 1A,B, phosphorylation of NF-κB-p65 and expression of COX-2 were significantly elevated in hippocampal neurons in culture treated with IL-1β (10 ng·mL−1) or LPS (1 µg·mL−1), commonly used pro-inflammatory stimuli. This elevation was inhibited or attenuated by exogenous application of 2-AG (3 µM), consistent with our previous observations (Zhang and Chen, 2008). However, the suppressive effect of 2-AG on NF-κB-p65 phosphorylation and COX-2 expression induced by IL-1β or LPS was blocked by GW9662 (5 µM), a selective PPARγ antagonist. This suggests that the 2-AG-produced suppression of IL-1β- and LPS-induced NF-κB-p65 phosphorylation and COX-2 expression is mediated by PPARγ. This was further confirmed by the results showing that IL-1β or LPS down-regulated the expression of PPARγ, and this down-regulation was prevented or restored by 2-AG (Figure 1C,D).
Figure 1.
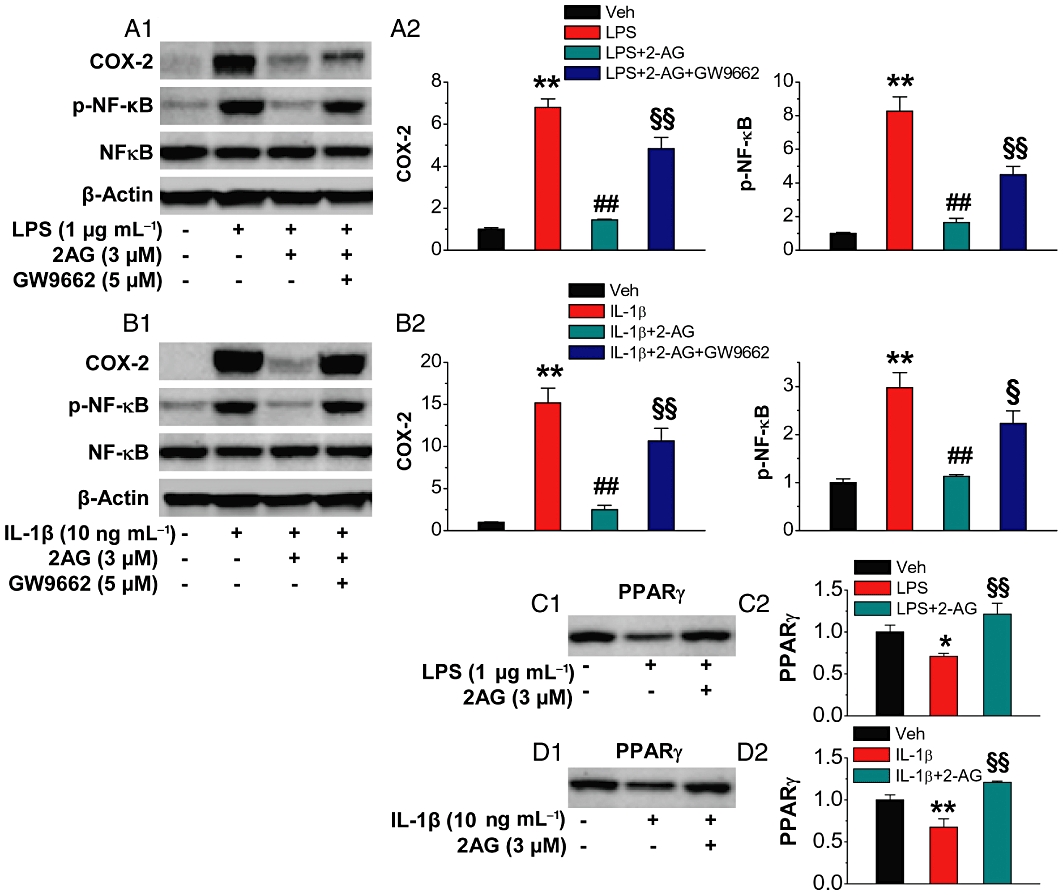
Exogenous application of 2-AG suppresses NF-κB phosphorylation and COX-2 expression and elevates PPARγ expression in response to pro-inflammatory IL-1β and LPS insults. Hippocampal neurons in culture were treated with IL-1β (10 ng·mL−1) for 6 h or LPS (1 µg·mL−1) for 16 h in the absence and presence of 2-AG or GW9662 (5 µM). The different time points used for the treatments of IL-1β and LPS were based on our previous studies where we identified that IL-1β- or LPS-induced COX-2 expression and NF-kB phosphorylation reached the peaks at these time points (Zhang and Chen, 2008). 2-AG or GW9662 was added 30 min before IL-1β or LPS application. (A1–A2) Immunoblot analysis of 2-AG suppression of LPS-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (B1–B2) Immunoblot analysis of 2-AG suppression of IL-1β-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (C1–C2) 2-AG restores LPS-induced down-regulation of PPARγ (n = 3). (D1–D2) 2-AG restores IL-1β-induced down-regulation of PPARγ (n = 3). **P < 0.01, compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §P < 0.05, §§P < 0.01 compared with IL-1β+2-AG or LPS+2-AG.
PPARγ is involved in 2-AG-induced suppression of COX-2-enhanced excitatory synaptic transmission
An elevation of COX-2 expression by pro-inflammatory IL-1β or LPS enhances mEPSCs (Sang et al., 2005; Zhang and Chen, 2008). We thus treated cultures with IL-1β or LPS for 16 and 24 h, respectively, and recorded mEPSCs in hippocampal neurons in the absence and presence of 2-AG. As expected, IL-1β or LPS, which elevates COX-2 expression, significantly augmented the frequency but not the amplitude of mEPSCs (Figure 2). This enhancement was suppressed in the presence of 2-AG (1 µM). However, application of GW9662 reversed this 2-AG-induced suppression, suggesting a role for PPARγ in the 2-AG-induced suppression of COX-2-enhanced mEPSCs. To further ascertain the effect of PPARγ inhibition on 2-AG-induced suppression of mEPSCs in IL-1β- or LPS-treated cultures, we used another selective PPARγ antagonist, T0070907 (T007) (Lee et al., 2002). Similar to GW9662, T007 (1 µM) also blocked the effect of 2-AG (Figure S1). We also measured the kinetics of mEPSCs in IL-1β- or LPS-treated neurons in the absence and presence of 2-AG or GW9662. There were no significant differences in the rise or decay time constants between the vehicle controls and the treated neurons (data not shown).
Figure 2.
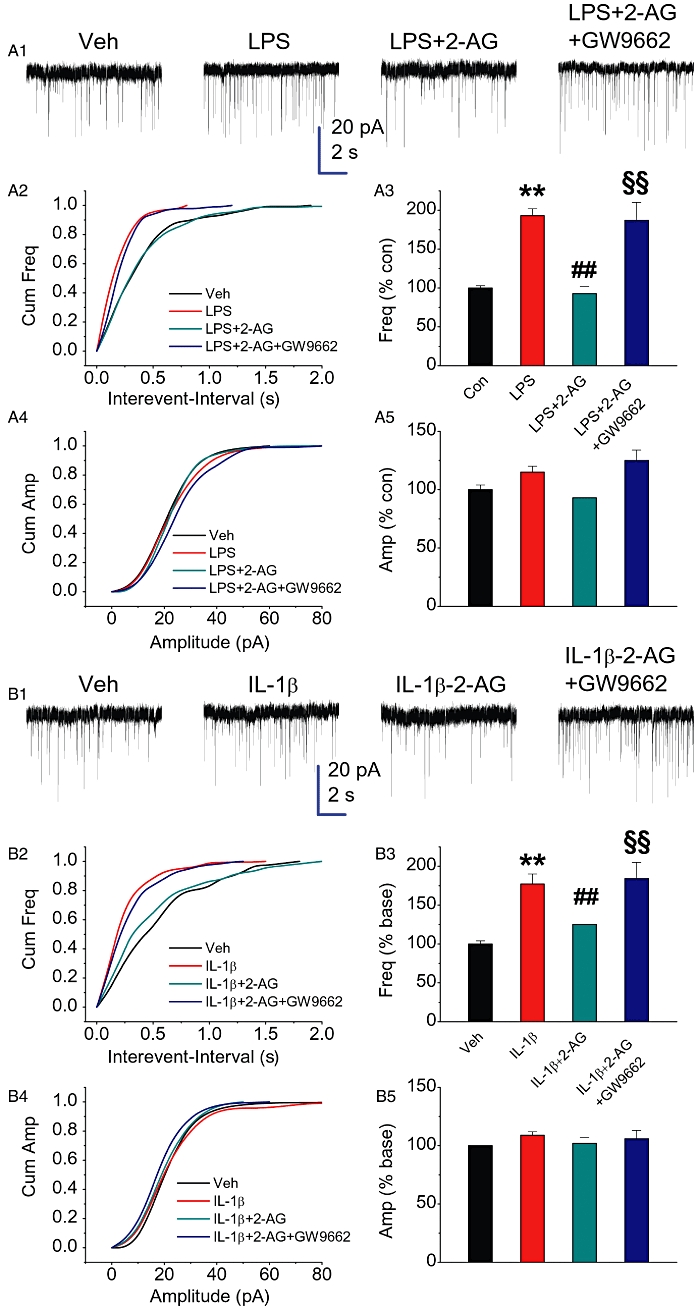
2-AG inhibits COX-2 elevation-induced enhancement of mEPSCs. Hippocampal neurons in culture were treated with IL-1β (10 ng·mL−1) for 16 h or LPS (2 µg·mL−1) for 24 h in the absence and presence of 2-AG (1 µM) or GW9662 (5 µM). The different time points used for the treatment of IL-1β and LPS were based on our previous studies where we identified that IL-1β or LPS significantly enhanced synaptic activity at these time points (Sang et al., 2005; Zhang and Chen, 2008). (A1) Representative sweeps of mEPSCs recorded in vehicle control-, LPS-, LPS + 2-AG- and LPS + 2-AG+GW9662-treated neurons. Scale bar: 20 pA/2 s. (A2) Cumulative probability of mEPSCs frequency recorded in neurons with different treatments. (A3) Mean percentage changes in the frequency of mEPSCs in neurons with different treatments. (A4) Cumulative probability of mEPSCs amplitude. (A5) Mean percentage changes in the amplitude of mEPSCs. (B1) Representative sweeps of mEPSCs recorded in vehicle control-, IL-1β-, IL-1β+2-AG (1 µM)- and IL-1β+2-AG+GW9662 (5 µM)-treated neurons. (B2) Cumulative probability of mEPSCs frequency recorded in neurons with different treatments. (B3) Mean percentage changes in the frequency of mEPSCs. (B4) Cumulative probability of mEPSCs amplitude. (B5) Mean percentage changes in the amplitude of mEPSCs. **P < 0.01 compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §§P < 0.01 compared with IL-1β+2-AG or LPS+2-AG (n = 24–32).
Endogenous 2-AG-induced suppression of neuroinflammation is mediated by PPARγ
To raise the levels of endogenous 2-AG, we used two selective MAGL inhibitors, URB 602 and JZL184. Since JZL184 displays higher selectivity and potency than URB602 for MAGL over FAAH (Hohmann et al., 2005; Long et al., 2009a,b;), we used URB 602 at 10 µM or JZL184 at 1 µM. As seen in Figure 3A–D, administration of URB602 or JZL184 significantly reduced IL-1β- or LPS-induced phosphorylation of NF-κB-p65 and expression of COX-2. URB602- and JZL184-induced suppression of IL-1β- or LPS-induced NF-κB-p65 phosphorylation and COX-2 expression was blocked by GW9662. To determine whether an elevation of endogenous 2-AG is also capable of restoring the IL-1β- or LPS-reduced expression of PPARγ, we detected the expression of PPARγ in the absence and presence of URB602 or JZL184. As shown in Figure 3E–H, URB602 (10 µM) or JZL184 (1 µM) restored the IL-1β- or LPS-induced down-regulation of PPARγ. From these results, it appears that both exogenous and endogenous 2-AG-induced inhibition of COX-2 expression and NF-κB phosphorylation are mediated by PPARγ.
Figure 3.
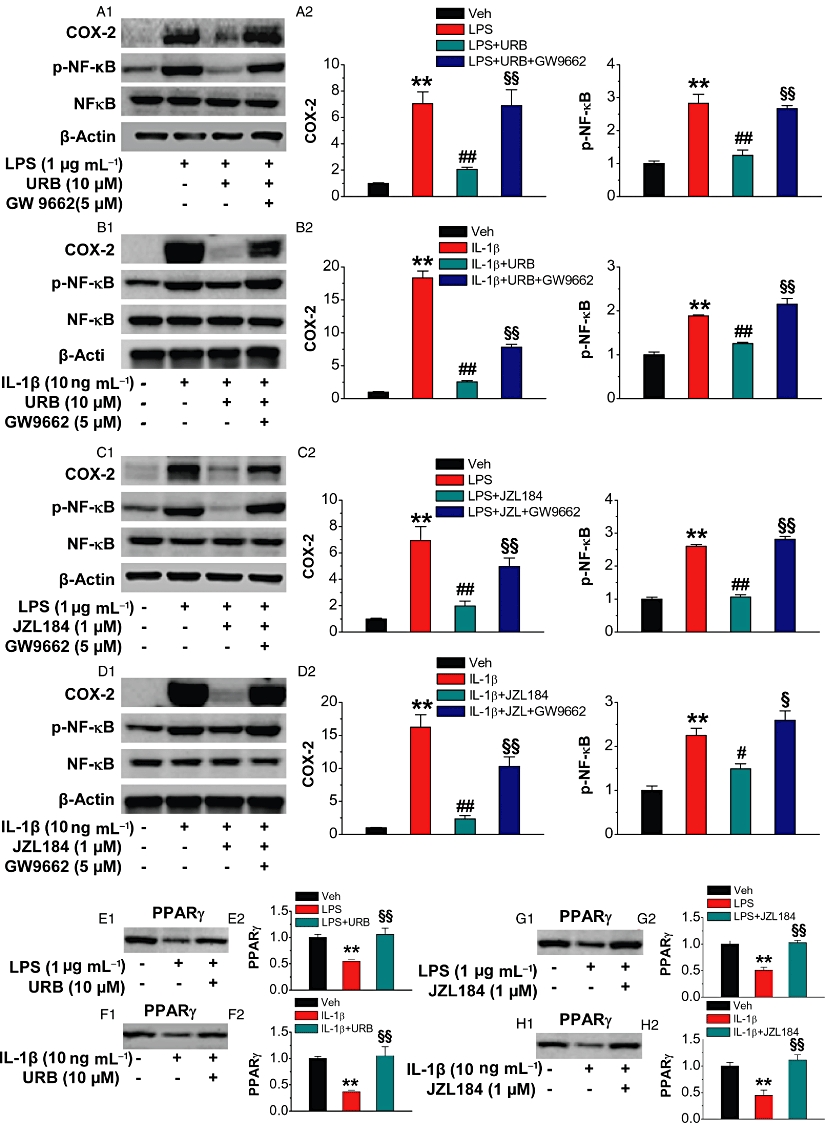
Endogenous 2-AG suppresses NF-κB phosphorylation and COX-2 expression and prevents down-regulation of PPARγ expression induced by IL-1β and LPS. Hippocampal neurons in culture treated with IL-1β (10 ng·mL−1) or LPS (1 µg·mL−1) were the same as described in Figure 1. Selective MAGL inhibitors URB602 (URB, 10 µM) and JZL184 (1 µM) were added to the culture 30 min before IL-1β or LPS application in order to elevate endogenous 2-AG. (A1–A2) Immunoblot analysis of URB suppression of LPS-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (B1–B2) Immunoblot analysis of URB suppression of IL-1β-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (C1–C2) Immunoblot analysis of JZL184 suppression of LPS-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (D1–D2) Immunoblot analysis of JZL184 suppression of IL-1β-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (E1–H2). Elevation of endogenous 2-AG by inhibiting MAGL with URB602 or JZL184 restores LPS- or IL-1β-induced down-regulation of PPARγ (n = 3 per group). **P < 0.01, compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §P < 0.05, §§P < 0.01 compared with IL-1β+ URB or JZL184, LPS+URB or JZL184.
PPARγ is involved in endogenous 2-AG-induced suppression of COX-2-enhanced excitatory synaptic transmission
If the exogenous application of 2-AG is capable of suppressing the COX-2-enhanced mEPSCs, then elevating the level of endogenous 2-AG by inhibiting its hydrolysis, as described above, should also be able to inhibit the enhanced frequency of mEPSCs in IL-1β- or LPS-treated cultures. Moreover, if PPARγ mediates this endogenous 2-AG-induced suppression of NF-κB phosphorylation and COX-2 expression, then blockade of PPARγ should be able to reverse the endogenous 2-AG-induced suppression of COX-2-enhanced mEPSCs. As seen in Figures 4 and 5, treating the culture with URB602 or JZL184 significantly reduced IL-1β- or LPS-induced enhancement of mEPSCs frequency, suggesting that an elevation of endogenous 2-AG also is capable of preventing the increase in excitatory synaptic transmission induced by COX-2. The URB602- or JZL184-produced suppression was blocked by GW9662. This suggests that, similar to the effects of exogenously applied 2-AG, the elevation of endogenous 2-AG produced by inhibiting MAGL is sufficient to reduce the release of excitatory neurotransmitter glutamates induced by COX-2 and PPARγ mediates this inhibitory effect of endogenous 2-AG on excitatory synaptic transmission.
Figure 4.
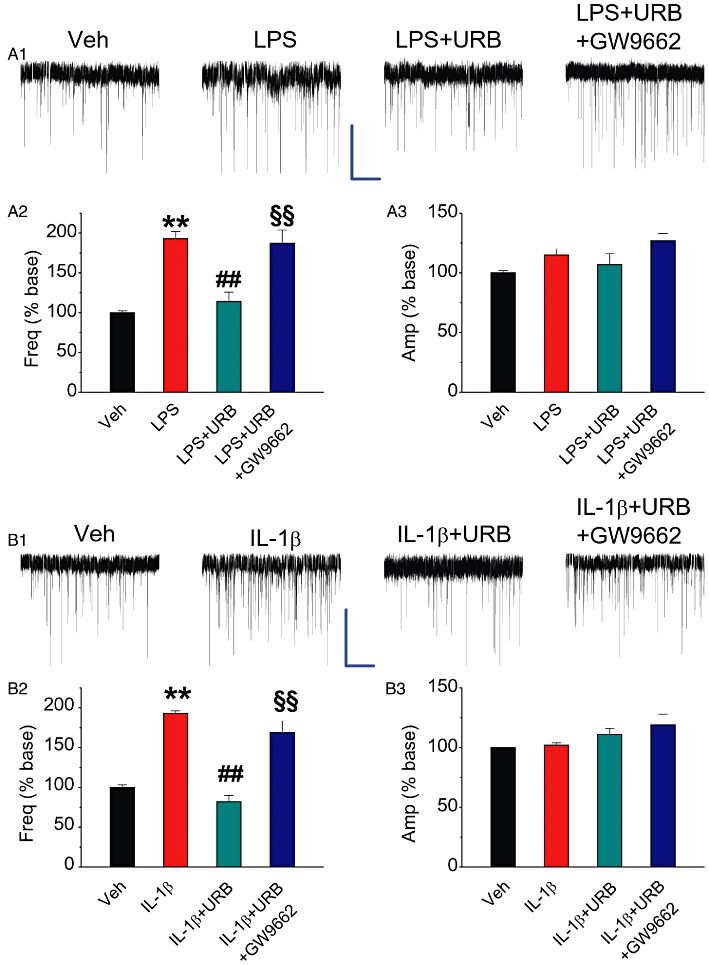
MAGL inhibitor URB602 attenuates COX-2 elevation-induced enhancement of mEPSCs. Hippocampal neurons in culture treated with IL-1β or LPS were the same as described in Figure 2. (A1) Representative sweeps of mEPSCs recorded in vehicle control-, LPS-, LPS+URB (10 µM)- and LPS+URB+GW9662 (5 µM)-treated neurons. Scale bar: 20 pA/2 s. (A2) Mean percentage changes in the frequency of mEPSCs in neurons with different treatments. (A3). Mean percentage changes in the amplitude of mEPSCs. (B1) Representative sweeps of mEPSCs recorded in vehicle control-, IL-1β-, IL-1β+ URB (10 µM)- and IL-1β+ URB+GW9662 (5 µM)-treated neurons. (B2)Mean percentage changes in the frequency of mEPSCs. (B3) Mean percentage changes in the amplitude of mEPSCs. **P < 0.01 compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §§P < 0.01 compared with IL-1β+ URB or LPS+URB (n = 20–34).
Figure 5.
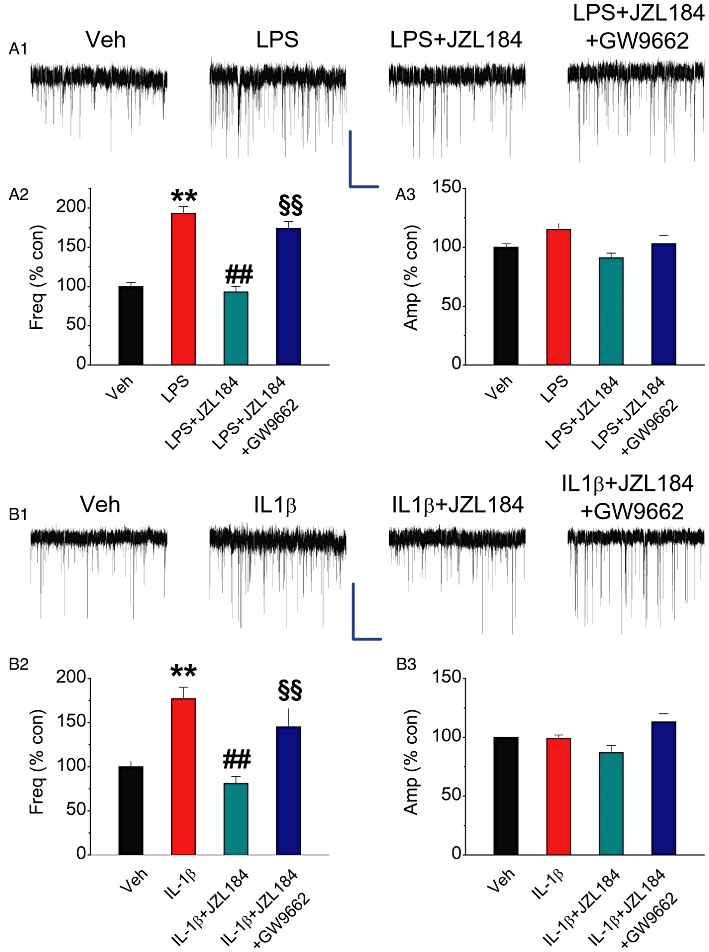
MAGL inhibitor JZL184 attenuates COX-2 elevation-induced enhancement of mEPSCs. Hippocampal neurons in culture treated with IL-1β or LPS were the same as described in Figure 2. (A1) Representative sweeps of mEPSCs recorded in vehicle control-, LPS-, LPS+JZL184 (1 µM)- and LPS+JZL184+GW9662 (5 µM)-treated neurons. Scale bar: 20 pA/2 s. (A2) Mean percentage changes in the frequency of mEPSCs in neurons with different treatments. (A3). Mean percentage changes in the amplitude of mEPSCs. (B1) Representative sweeps of mEPSCs recorded in vehicle control-, IL-1β-, IL-1β+ JZL184 (1 µM)- and IL-1β+ JZL184+GW9662 (5 µM)-treated neurons. (B2) Mean percentage changes in the frequency of mEPSCs. (B3) Mean percentage changes in the amplitude of mEPSCs. **P < 0.01 compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §§P < 0.01 compared with IL-1β+ JZL184 or LPS+JZL184 (n = 23–30).
PPARγ agonists suppress phosphorylation of NF-κB, expression of COX-2 and enhancement of mEPSCs induced by IL-1β and LPS
To determine whether PPARγ agonists or activators mimic the actions of 2-AG in resolving IL-1β- or LPS-induced NF-κB-p65 phosphorylation, COX-2 expression and enhanced mEPSCs, we used 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and rosiglitazone (Ros), both PPARγ agonists (Park et al., 2003). As seen in Figure 6, administration of 15d-PGJ2 (2 µM) or Ros (1 µM) significantly reduced IL-1β- or LPS-induced phosphorylation of NF-κB-p65 and expression of COX-2; these effects of 15d-PGJ2 and Ros were blocked by GW9662. Similarly, 15d-PGJ2 and Ros also suppressed IL-1β- or LPS-induced enhancement of mEPSCs, and again this suppression was blocked by antagonism of PPARγ (Figures 7 and 8).
Figure 6.

PPARγ agonists inhibit phosphorylation of NF-κB and expression of COX-2 induced by IL-1β-and LPS. Hippocampal neurons in culture-treated with IL-1β (10 ng·mL−1) or LPS (1 µg·mL−1) were the same as described in Figure 1. PPARγ agonists 15d-PGJ2 (2 µM) or rosiglitazone (Ros, 1 µM) were added to the culture 30 min before IL-1β or LPS. (A1–A2) Immunoblot analysis of 15d-PGJ2 suppression of LPS-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (B1-–B) Immunoblot analysis of 15d-PGJ2 inhibition of IL-1β-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (C1–C2) Immunoblot analysis of Ros suppression of LPS-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW9662 (n = 3). (D1–D2) Immunoblot analysis of Ros suppression of IL-1β-induced NF-κB-p65 phosphorylation and COX-2 expression in the absence and presence of GW (n = 3). **P < 0.01, compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §P < 0.05, §§P < 0.01 compared with IL-1β+15d-PGJ2 or Ros, LPS+15d-PGJ2 or Ros.
Figure 7.
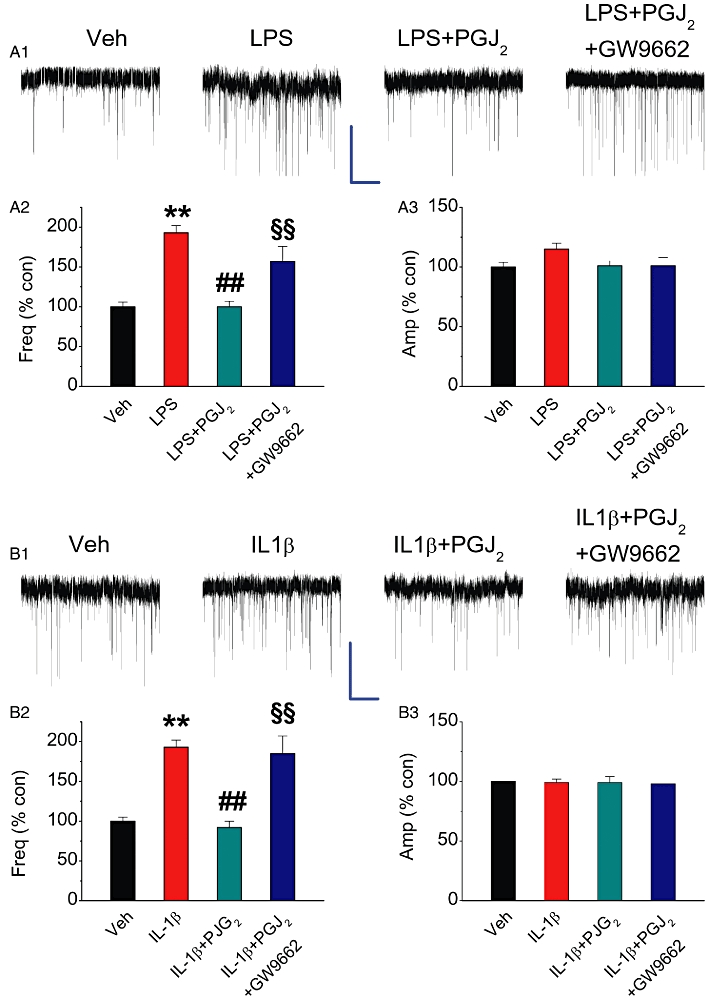
PPARγ agonist 15d-PGJ2 reduces COX-2 elevation-induced enhancement of mEPSCs. Hippocampal neurons in culture-treated with IL-1β or LPS were the same as described in Figure 2. (A1) Representative sweeps of mEPSCs recorded in vehicle control-, LPS-, LPS+15d-PGJ2 (2 µM)- and LPS+15d-PGJ2+GW9662 (5 µM)-treated neurons. Scale bar: 20 pA/2 s. (A2) Mean percentage changes in the frequency of mEPSCs in neurons with different treatments. (A3). Mean percentage changes in the amplitude of mEPSCs. (B1) Representative sweeps of mEPSCs recorded in vehicle control-, IL-1β-, IL-1β+15d-PGJ2 (2 µM)- and IL-1β+15d-PGJ2+GW9662 (5 µM)-treated neurons. (B2) Mean percentage changes in the frequency of mEPSCs. (B3) Mean percentage changes in the amplitude of mEPSCs. **P < 0.01 compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §§P < 0.01 compared with IL-1β+15d-PGJ2 or LPS+15d-PGJ2 (n = 23–30).
Figure 8.
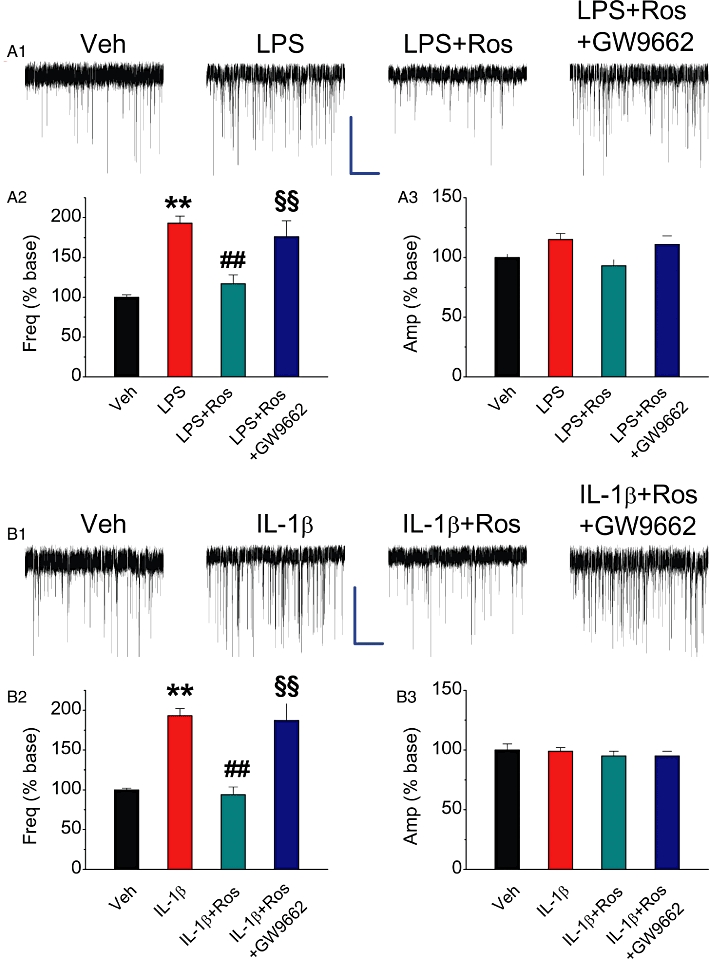
PPARγ activator rosiglitazone reduces COX-2 elevation-induced enhancement of mEPSCs. Hippocampal neurons in culture treated with IL-1β or LPS were the same as described in Figure 2. (A1) Representative sweeps of mEPSCs recorded in vehicle control-, LPS-, LPS + Ros (1 µM)- and LPS + Ros + GW9662 (5 µM)-treated neurons. Scale bar: 20 pA/2 s. (A2) Mean percentage changes in the frequency of mEPSCs in neurons with different treatments. (A3) Mean percentage changes in the amplitude of mEPSCs. (B1) Representative sweeps of mEPSCs recorded in vehicle control-, IL-1β-, IL-1β+ Ros (1 µM)- and IL-1β+ Ros+GW9662 (5 µM)-treated neurons. (B2) Mean percentage changes in the frequency of mEPSCs. (B3) Mean percentage changes in the amplitude of mEPSCs. **P < 0.01 compared with the vehicle control; ##P < 0.01 compared with IL-1β or LPS; §§P < 0.01 compared with IL-1β+ Ros or LPS + Ros (n = 23–30).
CB1 receptor mediates 2-AG-produced restoration of reduced PPARγ expression by LPS
To determine whether 2-AG-induced increase in PPAR expression is dependent on CB1 receptors, we conducted another set of experiments where the culture was treated with rimonabant (RIM), a selective CB1 receptor antagonist, in the presence of LPS and 2-AG, URB602 or JZL184. As shown in Figure 9A–C, LPS significantly decreased the expression of PPARγ, and this decrease was restored by 2-AG (3 µM), URB602 (10 µM) or JZL184 (1 µM). However, the action of 2-AG, URB602 or JZJ184 on PPARγ expression was blocked by RIM (1 µM), suggesting a CB1 receptor-mediated effect. To further confirm that restoration of the LPS-suppressed PPARγ expression by direct application of 2-AG or enhancement of endogenous 2-AG is mediated via a CB1 receptor, we treated cultured hippocampal neurons from mice deficient in the CB1 receptor with LPS in the presence of 2-AG, URB602 or JZl184. As shown in Figure 9D, 2-AG, URB602 or JZl184 failed to restore the LPS-induced suppression of PPARγ, confirming that the action of 2-AG on PPARγ expression is CB1 receptor-dependent. To determine whether endogenous 2-AG-produced suppression of LPS- or IL-1β-induced enhancement of mEPSCs is also mediated via a CB1 receptor, we recorded mEPSCs in culture treated with RIM in the presence of LPS or IL-1β and URB602. As illustrated in Figure S2, URB602-induced suppression of the increase in mEPSCs induced by LPS- or IL-1β was blocked by RIM (1 µM). These results suggest that 2-AG-induced increase in PPARγ expression is mediated primarily via the CB1 receptor.
Figure 9.

Exogenous and endogenous 2-AG prevents IL-1β-and LPS-induced decrease in PPARγ expression via a CB1 receptor-dependent mechanism. Hippocampal neurons in culture were treated with LPS (1 µg·mL−1) for 6 h. 2-AG (A1–A2), URB602 (B1–B2), JZL184 (C1–C2) or rimonabant (RIM, 1 µM) were administered 30 min before LPS. Expression of PPARγ was detected using the immunoblot analysis. **P < 0.01 compared with the vehicle control; #P < 0.05, ##P < 0.01 compared with LPS; §P < 0.05 compared with LPS+2-AG, + URB or +JZL184 (n = 3 per group). Hippocampal neurons in culture from mice deficient in the CB1 receptor were treated with LPS (1 µg·mL−1) for 6 h in the absence and presence of 2-AG, URB602 or JZL184 (D1–D2). **P < 0.01 compared with the vehicle control. There are no statistically significant differences between LPS and LPS+2-AG, LPS+URB602 or LPS+JZl184 (n = 3 per group).
We need to mention here that 2-AG or URB602 alone did not alter the basal expression of COX-2 or the basal activity of mEPSCs. This is consistent with previously described observations (Zhang and Chen, 2008). In addition, we demonstrated that treating the culture with JZL184, rosiglitazone, 15d-PGJ2 or GW alone did not alter the basal activity of mEPSCs (data not shown). In particular, we provide evidence that JZL184, Ros, 15d-PGJ2 or GW9662 alone did not significantly alter the basal expression of COX-2 (Figure S3). This suggests that 2-AG probably functions as an important signalling mediator maintaining the homeostasis of brain function.
Discussion and conclusion
In the present study, we provide evidence that exogenous application of 2-AG or the elevation of endogenous 2-AG, produced by inhibiting its hydrolysis with selective MAGL inhibitors URB602 and JZl184, is capable of suppressing NF-kB-p65 phosphorylation and COX-2 expression. This expands upon our previous work where we discovered that 2-AG protects neurons against harmful insults by limiting the inflammatory response (Zhang and Chen, 2008; Chen et al., 2011). In particular, we demonstrate here that the 2-AG-produced suppression NF-κB-p65 phosphorylation, COX-2 expression and mEPSC enhancement by pro-inflammatory IL-1β- or LPS is mediated via PPARγ. This suggests PPARγ is a target for 2-AG in protecting neurons against pro-inflammatory insults. Since the anti-neuroinflammatory effects and restoration of the LPS-reduced PPARγ expression by exogenous and endogenous 2-AG are largely blocked by pharmacological or genetic inhibition of the CB1 receptor, the actions of 2-AG were not through direct interaction with nuclear PPARγ. More likely, they are mediated primarily by CB1 receptor-dependent changes in PPARγ expression.
Arachidonoyl ethanolamide (AEA or anandamide) and 2-AG are the two most studied eCBs. However, despite their similar chemical structure, 2-AG and AEA display distinct profiles in their synthesis, metabolism, cannabinoid receptor binding affinity and synaptic modulation (Freund et al., 2003; Sugiura et al., 2006; Kano et al., 2009). In particular, AEA and 2-AG exhibit differences in their ability to limit neuroinflammation and protect neurons from harmful insults. For instance, 2-AG has been shown to protect neurons from brain ischaemia, traumatic brain injury and pro-inflammatory stimuli (Panikashvili et al., 2001; 2005, 2006; Melis et al., 2006; Kreutz et al., 2007; Zhang and Chen, 2008). However, AEA shows a paradoxical phenomenon in its neuroprotective effects against inflammatory and excitotoxic stimuli and even induces neurotoxicity per se (Cernak et al., 2004; Movsesyan et al., 2004; Sarne and Mechoulam, 2005; Van der Stelt and Di Marzo, 2005; Fowler et al., 2010). This implies that 2-AG is probably an endogenously intrinsic signalling mediator protecting neurons against harmful insults and maintaining tissue homeostasis (Yang and Chen, 2008). Since the exogenous application or endogenous elevation of 2-AG suppressed the pro-inflammatory IL-1β- or LPS-induced phosphorylation of NF-κB-p65 and expression of COX-2 (two key markers in inflammation), strengthening the 2-AG signalling pathway by inhibition of MAGL will be beneficial in resolving neuroinflammation, which is the root of many neurological disorders and neurodegenerative diseases (Chen, 2010). MAGL is the enzyme that hydrolyzes 85% of 2-AG in the brain (Blankman et al., 2007). Thus, inhibiting MAGL with selective MAGL inhibitors will raise the levels of endogenous 2-AG (Hohmann et al., 2005; Comelli et al., 2007; King et al., 2007; Long et al., 2009a; 2009b; Pan et al., 2009). As such, this may lead to potential interventions for preventing, alleviating and treating brain disorders associated with neuroinflammation.
Although PPARγ was originally shown to regulate lipid metabolism and adipocyte differentiation, there is accumulating evidence indicating that PPARγ possesses anti-inflammatory and neuroprotective properties, induced by regulating the transcription of genes involved in inflammation (Jiang et al., 1998; Ricote et al., 1998; Daynes and Jones, 2002; Luna-Medina et al., 2005; Drew et al., 2006; Bensinger and Tontonoz, 2008; Bright et al., 2008; Necela et al., 2008; Racke and Drew, 2008). PPARγ regulates gene transcription by binding to conserved DNA sequences termed peroxisome proliferator response elements (PPRE) as heterodimers with retinoic X receptor (Bensinger and Tontonoz, 2008). Increasing evidence suggests that eCBs are probably PPARγ activators (O'Sullivan, 2007; Sun et al., 2007; O'Sullivan and Kendall, 2010; Pertwee et al., 2010; Pistis and Melis, 2010). It has been shown that 2-AG suppression of the expression of IL-2, an autocrine/paracrine T-cell growth factor, is mediated via a CB1/2 receptor-independent activation of PPARγ (Rockwell et al., 2006), suggesting that 2-AG may be able to directly activate nuclear PPARγ by crossing both the plasma and nuclear membranes (O'Sullivan, 2007; Sun et al., 2007; O'Sullivan and Kendall, 2010; Pistis and Melis, 2010). However, 2-AG may also be able to activate PPARγ and restore neuroinflammation-induced down-regulation of PPARγ expression through a CB1 receptor-dependent pathway. In the present study, we observed that exogenous and endogenous 2-AG-produced suppression of NF-κB-p65 phosphorylation and COX-2 expression in response to pro-inflammatory IL-1β or LPS are blocked by antagonism of PPARγ with a selective PPARγ inhibitor. Moreover, 2-AG prevented the IL-1β- or LPS-induced down-regulation of PPARγ. It has been shown previously that COX-2 participates in synaptic transmission and plasticity via PGE2, which facilitates the synaptic release of the excitatory neurotransmitter glutamate (Chen et al., 2002; Chen and Bazan, 2005; Sang et al., 2005; Slanina and Schweitzer, 2005; Akaneya and Tsumoto, 2006; Yang et al., 2008). We demonstrated that the frequency of the mEPSCs in cultured hippocampal neurons is significantly elevated by an increase in COX-2 expression, and this enhancement is suppressed by 2-AG (Zhang and Chen, 2008). However, 2-AG-produced suppression of COX-2-enhance mEPSCs was blocked or attenuated by antagonism of PPARγ. Our results provide convincing evidence that PPARγ probably mediates 2-AG-produced inhibition of COX-2 expression and NF-kB phosphorylation.
Based on our previous and present results, PPARγ may not be directly targeted by 2-AG because inhibition of the CB1 receptor eliminates or decreases the effects 2-AG on neuroinflammation and neuroprotection. For instance, we demonstrated previously that 2-AG-produced neuroprotection and suppression of COX-2 expression in response to pro-inflammatory and other harmful insults are mediated via CB1 receptor-dependent inhibition of MAPK/NF-κB phosphorylation (Zhang and Chen, 2008; Chen et al., 2011). In addition, inhibition of the CB1 receptor blocked URB602-produced suppression of IL-1β- or LPS-enhanced mEPSCs (Figure S2). In particular, pharmacological and genetic inhibition of the CB1 receptor significantly attenuated restorative effect of 2-AG on IL-1β- or LPS-induced down-regulation of PPARγ (Figure 9). If the actions of 2-AG on NF-κB phosphorylation and COX-2 expression are through crossing both the plasma and nuclear membranes to directly activate PPARγ, then inhibition of the CB1 receptor, which is expressed on the surface plasma membrane, should fail to inhibit 2-AG-induced expression of PPARγ that regulates the expression of genes involved inflammation. In addition, antagonism of the CB1 receptor should fail to block 2-AG suppression of COX-2-induced increase in mEPSCs. Apparently, this is not the case. This indicates that the signalling mechanisms mediating 2-AG-induced activation/expression of PPARγ in our study are different from those implicated by Rockwell et al., (2006). The exact mechanism for this discrepancy between the two studies is still not clear, but it is probably due to the different preparations used.
A recent study shows that the suppression of IL-2 in T cells produced by AEA or 2-AG is COX-2-dependent (Rockwell et al., 2008). 2-AG and AEA are substrates for COX-2 and can be oxidatively metabolized by COX-2 to prostaglandin glycerol esters or ethanolamides (Kozak et al., 2004; Sang and Chen, 2006; Yang and Chen, 2008). However, it is not clear which COX-2 metabolites mediate the 2-AG- or AEA-induced IL-2 suppression. We demonstrated previously that the effects of COX-2 metabolites of 2-AG on synaptic transmission and neurodegeneration are opposite to that of their precursor 2-AG (Sang et al., 2006, 2007; Yang et al., 2008; Zhang and Chen, 2008). For instance, acute application of PGE2-G, a major COX-2 metabolite of 2-AG, enhances excitatory synaptic transmission, while 2-AG inhibits it in cultured hippocampal neurons. Moreover, treatment of neurons in culture with PGE2-G induces neurodegeneration and apoptosis and increases NF-κB phosphorylation, while 2-AG protects neurons against neurodegeneration and apoptosis and inhibits NF-κB phosphorylation. Therefore, in our study it is unlikely that 2-AG-produced suppression of COX-2 expression and NF-kB-p65 phosphorylation and increase of PPARγ expression are mediated through COX-2 metabolites of 2-AG.
It has been proposed that there is a reciprocally negative feedback loop between NF-κB and PPARγ, suggesting that there is cross-talk between these two transcription factors (Bensinger and Tontonoz, 2008; Necela et al., 2008). We speculate that under physiological conditions, inflammatory genes are tonically repressed by co-repressor complexes. However, exposure to pro-inflammatory stimuli such as IL-1β or LPS activates astroglial toll-like receptor 4 (TLR4) or IL-1 receptors, which elevate phosphorylation of MAPK and NF-κB. Increased expression and activity of NF-κB not only suppresses PPARγ expression and activity but also directly triggers inflammatory gene transcription. The CB1 receptor is expressed both in hippocampal neurons and astroglial cells (Sinha et al., 1998; Waksman et al., 1999; Stella, 2004; Eljaschewitsch et al., 2006; Zhang and Chen, 2008). Thus, 2-AG binds to Gi-coupled CB1 receptors and/or CB2 receptors. Activation of CB1/2 receptors suppresses phosphorylation of NF-κB through ERK/p38MAPK (Zhang and Chen, 2008) and increases the expression of PPARγ, which represses inflammatory gene transcription (Bensinger and Tontonoz, 2008; Necela et al., 2008). Nevertheless, the possibility that 2-AG could cross the plasma and nuclear membranes to directly induce PPARγ activation and expression, leading to repression of NF-κB and inflammatory gene transcription, cannot be excluded. It is likely that 2-AG-initiated signalling events prevent or inhibit inflammatory gene transcription, resulting in resolution of inflammation and neuroprotection.
In this study, we provide evidence for the first time that PPARγ mediates exogenous and endogenous 2-AG-produced suppression of pro-inflammatory IL-1β- or LPS-induced NF-κB-p65 phosphorylation and COX-2 expression, the two key inflammatory markers. We also provide evidence that PPARγ mediates exogenous and endogenous 2-AG-produced inhibition of enhanced mEPSCs resulting from COX-2 elevation in hippocampal neurons in culture. 2-AG-induced PPARγ expression appears to be dependent on the CB1 receptor and interplay between NF-κB-p65 and PPARγ, indicating that this would be a previously unrevealed signalling pathway mediating 2-AG-produced anti-inflammatory and neuroprotective effects. However, it is still not clear how the interaction or cross-talk between NF-κB and PPARγ occurs when 2-AG activates the CB1 receptor. More work is needed to elucidate this intriguing signalling pathway. Resolution of neuroinflammation is believed to be an efficacious therapeutic approach for prevention and treatment of neurodegenerative diseases such as Alzheimer's disease (Walker and Lue, 2007; Chen, 2010; Glass et al., 2010). Therefore, the results obtained in the present study suggest that endogenous 2-AG plays an important role in regulation of innate and adaptive immune systems in maintaining tissue homeostasis, and that approaches strengthening endogenous 2-AG signalling by inhibiting its hydrolysis or facilitating its synthesis will be potentially efficacious therapeutic interventions for preventing, relieving and treating chronic neuroinflammation-induced brain disorders.
Acknowledgments
The authors thank NIH Mental Health Institute Chemical Synthesis and Drug Supply Program for providing rimonabant (SR141716) and MIH Mental Health Institute transgenic core for providing CB1 knockout mice. This work was supported by National Institutes of Health grants NS054886 and AG039669.
Glossary
Abbreviations
- 2-AG
2-arachidonoylglycerol
- eCBs
endocannabinoids
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- RIM
rimonabant
- MAGL
monoacylglycerol lipase
- mEPSCs
miniature spontaneous excitatory postsynaptic currents
- URB
URB602
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Teaching Materials; Figs 1–9 as PowerPoint slide.
Additional Supporting Information may be found in the online version of this article:
Figure S1 Inhibition of PPARγ blocks 2-AG-produced suppression of COX-2 elevation-induced enhancement of mEPSCs. T007: T0070907 (1 μM).
Figure S2 Inhibition of the CB1 receptor eliminates URB602 suppression of COX-2 elevation-induced enhancement of mEPSCs. RIM: rimonabant (1 μM).
Figure S3 JZL184, rosiglitazone (Ros), 15d-PGJ2, or GW9662 alone do not significantly alter basal expression of COX-2 in cultured hippocampal neurons. The treatment of JZL184, Ros, 15d-PGJ2 and GW9662 was the same as that described in other figures. COX-2 expression was detected using Western blot analysis.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Akaneya Y, Tsumoto T. Bidirectional trafficking of prostaglandin E2 receptors involved in long-term potentiation in visual cortex. J Neurosci. 2006;26:10209–10221. doi: 10.1523/JNEUROSCI.3028-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC). 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Bensinger ST, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravavtt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res. 2008;2008:1–12. doi: 10.1155/2008/658520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Finazzi-Agro A, Bernardi G Maccarrone M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol Sci. 2007;28:180–187. doi: 10.1016/j.tips.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Cernak I, Vink R, Natale J, Stoica B, Lea PM, 4th, Movsesyan V, et al. The ‘dark side’ of endocannabinoids: a neurotoxic role for anandamide. J Cereb Blood Flow Metab. 2004;24:564–578. doi: 10.1097/00004647-200405000-00011. [DOI] [PubMed] [Google Scholar]
- Chen C. COX-2's new role in inflammation. Nat Chem Biol. 2010;6:402–402. doi: 10.1038/nchembio.375. [DOI] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neurosci. 2011;178:159–168. doi: 10.1016/j.neuroscience.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-Mediated Synaptic Plasticity in the CNS. Ann Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol. 2007;152:787–794. doi: 10.1038/sj.bjp.0707425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49:183–189. doi: 10.1016/j.neuint.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Rojo ML, Rodriguez-Gaztelumendi A. Modulation of the endocannabinoid system: Neuroprotection or neurotoxicity? Exp Neurol. 2010;224:37–47. doi: 10.1016/j.expneurol.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MA, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- King AR, Duranti A, Tontini A, Rivara S, Rosengarth A, Clapper JR, et al. RB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem Biol. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Gupta RA, Moody JS, Ji C, Boeglin WE, DuBois RN, et al. 15-Lipoxygenase metabolism of 2-arachidonylglycerol. Generation of a peroxisome proliferator-activated receptor alpha agonist. J Biol Chem. 2002;277:23278–23286. doi: 10.1074/jbc.M201084200. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JL, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Curr Pharmaceut Design. 2004;10:659–667. doi: 10.2174/1381612043453081. [DOI] [PubMed] [Google Scholar]
- Kreutz S, Koch M, Ghadban C, Korf HW, Dehghani F. Cannabinoids and neuronal damage: differential effects of THC, AEA and 2-AG on activated microglial cells and degenerating neurons in excitotoxically lesioned rat organotypic hippocampal slice cultures. Exp Neurol. 2007;203:246–257. doi: 10.1016/j.expneurol.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- Lenman A, Fowler CJ. Interaction of ligands for the peroxisome proliferator-activated receptor gamma with the endocannabinoid system. Br J Pharmacol. 2007;151:1343–1351. doi: 10.1038/sj.bjp.0707352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Normura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009a;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis producs cannabinoid behavioral effects. Nat Chem Biol. 2009b;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Medina R, Cortes-Canteli M, Alonso M, Santos A, Martı'nez A, Perez-Castillo A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferatoractivated receptor γ activation. J Biol Chem. 2005;280:21453–21462. doi: 10.1074/jbc.M414390200. [DOI] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Melis M, Pillolla G, Bisogno T, Minassi A, Petrosino S, Perra S, et al. Protective activation of the endocannabinoid system during ischemia in dopamine neurons. Neurobiol Dis. 2006;24:15–27. doi: 10.1016/j.nbd.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Movsesyan VA, Stoica BA, Yakovlev AG, Knoblach SM, Lea PM, 4th, Cernak I, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121–1132. doi: 10.1038/sj.cdd.4401442. [DOI] [PubMed] [Google Scholar]
- Necela BM, Su W, Thomson EA. Toll-like receptor 4 mediates cross-talk between peroxisom proliferator-activated receptor γ and nuclear factor-κB in macrophages. Immunol. 2008;125:344–358. doi: 10.1111/j.1365-2567.2008.02849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA. Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiol. 2010;215:611–616. doi: 10.1016/j.imbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, et al. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:427–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Shein NA, Mechoulam R, Trembovler V, Kohen R, Alexandrovich A, et al. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis. 2006;22:257–264. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem. 2003;278:14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signaling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pistis M, Melis M. From surface to nuclear receptors: the endocannabinoid family extends its assets. Curr Med Chem. 2010;17:1450–1467. doi: 10.2174/092986710790980014. [DOI] [PubMed] [Google Scholar]
- Racke MK, Drew PD. PPARs in neuroinflammation. PPAR Res. 2008;2008:638356. doi: 10.1155/2008/638356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor- is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE. Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor gamma independently of cannabinoid receptors 1 and 2. Mol Pharmacol. 2006;70:101–111. doi: 10.1124/mol.105.019117. [DOI] [PubMed] [Google Scholar]
- Rockwell CE, Raman P, Kaplan BL, Kaminski NE. A COX-2 metabolite of the endogenous cannabinoid, 2-arachidonyl glycerol, mediates suppression of IL-2 secretion in activated Jurkat T cells. Biochem Pharmacol. 2008;76:353–361. doi: 10.1016/j.bcp.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;12:425–434. doi: 10.1177/1073858406290794. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Chen C. PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol (Lond) 2006;572:735–745. doi: 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J Neurochem. 2007;103:1966–1977. doi: 10.1111/j.1471-4159.2007.04668.x. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Chen C. Anandamide potentiation of miniature spontaneous excitatory synaptic transmission is mediated via IP3 pathway. Neurochem Intl. 2010;56:590–596. doi: 10.1016/j.neuint.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarne Y, Mechoulam R. Cannabinoids: between neuroprotection and neurotoxicity. Curr Drug Target- CNS Neurol Dis. 2005;4:677–684. doi: 10.2174/156800705774933005. [DOI] [PubMed] [Google Scholar]
- Sinha D, Bonner TI, Bhat NR, Matsuda LA. Expression of the CB1 cannabinoid receptor in macrophage-like cells from brain tissue: immunochemical characterization by fusion protein antibodies. J Neuroimmunol. 1998;82:13–21. doi: 10.1016/S0165-5728(97)00181-1. [DOI] [PubMed] [Google Scholar]
- Slanina KA, Schweitzer P. Inhibition of cyclooxygenase-2 elicits a CB1-mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacol. 2005;49:653–659. doi: 10.1016/j.neuropharm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Stella N. Cannabinoid signaling in glial cells. Glia. 2004;48:267–277. doi: 10.1002/glia.20084. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45:405–446. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, et al. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol. 2007;152:734–743. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Stelt M, Di Marzo V. Cannabinoid receptors and their role in neuroprotection. NeuroMol Med. 2005;7:37–50. doi: 10.1385/NMM:7:1-2:037. [DOI] [PubMed] [Google Scholar]
- Waksman Y, Olson JM, Carlisle SJ, Cabral GA. The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J Pharmacol Exp Ther. 1999;288:1357–1366. [PubMed] [Google Scholar]
- Walker D, Lue L-F. Anti-inflammatory and immune therapy for Alzheimer's disease: current status and future directions. Curr Neuropharmacol. 2007;5:232–243. doi: 10.2174/157015907782793667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chen C. COX-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhang J, Andreasson K, Chen C. COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol Cell Neurosci. 2008;37:682–695. doi: 10.1016/j.mcn.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem. 2008;283:22601–22611. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.