

Abstract
Double-strand DNA break repair and homologous recombination in Escherichia coli proceed by the RecBCD pathway, which is regulated by cis-acting elements known as χ sites. A crucial feature of this regulation is the RecBCD enzyme-directed loading of RecA protein specifically onto the 3′-terminal, χ-containing DNA strand. Here we show that RecBC enzyme (lacking the RecD subunit) loads RecA protein constitutively onto the 3′-terminal DNA strand, with no requirement for χ. This strand is preferentially utilized in homologous pairing reactions. We propose that RecA protein loading is a latent property of the RecBCD holoenzyme, which is normally blocked by the RecD subunit and is revealed following interaction with χ.
Keywords: RecBC, RecA, helicase, χ, recombination, DNA repair
Genetic recombination is an important cellular process, providing for both genomic maintenance and genetic diversity. In Escherichia coli the main pathway of recombination is the RecBCD pathway (Kowalczykowski et al. 1994). This pathway is responsible for homologous recombination accompanying conjugation and transduction, and for repair of double-stranded DNA (dsDNA) breaks through recombination between the damaged DNA and an intact homologous chromosome (for review, see Kogoma 1996). These potentially fatal breaks can arise during DNA replication (Kuzminov 1995; Michel et al. 1997) or as a direct result of high energy irradiation (for review, see Friedberg et al. 1995). RecA protein and RecBCD enzyme are key enzymes in this recombinational repair process. RecA protein is responsible for the recognition of homology between DNA molecules and for mediating the process of DNA strand exchange that leads to recombination intermediates (for review, see Roca and Cox 1997; Bianco et al. 1998). RecBCD enzyme is a multifunctional, heterotrimeric enzyme possessing DNA helicase, DNA-dependent ATPase, ATP-dependent exonuclease, and ATP-stimulated endonuclease activities. It acts as an initiator of homologous recombination at double-strand breaks by producing a suitable single-stranded DNA (ssDNA) substrate for RecA protein.
RecBCD enzyme-mediated recombination is controlled by DNA sequences known as χ sites (5′-GCTGGTGG-3′), which stimulate recombination by 5- to 10-fold (Lam et al. 1974; Stahl et al. 1975; Smith et al. 1981), and which are recognized by the translocating enzyme (Bianco and Kowalczykowski 1997). Stimulation is detected as far as 10 kb downstream of the χ site (Stahl et al. 1980; Ennis et al. 1987; Cheng and Smith 1989; Myers et al. 1995a).
The χ sequence acts as a regulatory switch by modifying the activity of RecBCD enzyme. This χ-dependent regulation involves both an attenuation and a switch in polarity of the enzyme’s nuclease activity (see Fig. 1). Prior to interaction with χ, the DNA strand that terminates 3′ at the entry site is degraded to a much greater extent than the 5′-terminated strand (Dixon and Kowalczykowski 1991, 1993). Following interaction with the χ sequence, however, the nuclease activity acting on the 3′-terminated strand is attenuated at χ, and a weaker nuclease activity acting on the 5′-terminated strand is upregulated (Anderson and Kowalczykowski 1997a). As the enzyme continues to unwind and degrade the 5′-terminated strand downstream of χ, it produces a ssDNA species with χ at the 3′ end (Ponticelli et al. 1985); this fragment is designated the ‘top-strand, downstream χ-specific fragment’ (see Fig. 1).
Figure 1.

The processing of dsDNA containing χ by RecBCD and RecBC enzymes generates characteristic ssDNA fragments. The two strands of DNA are defined relative to the double-stranded end at which RecBCD (or RecBC) enzyme enters. The strand of DNA that terminates 3′ at the entry site is termed the top strand; the complementary strand is termed the bottom strand. The region of dsDNA between χ and the entry site of RecBCD enzyme is referred to as the upstream region, and the region between χ and the opposite end is termed the downstream region. (↼ above the χ site) The direction that RecBCD enzyme must travel to recognize χ. For RecBCD enzyme, both an attenuation in nuclease activity and a switch in the polarity of exonuclease degradation occur upon χ recognition: The 3′ → 5′ nuclease activity is attenuated downstream of χ; a weaker 5′ → 3′ exonuclease activity is up-regulated. The consequence is production of both a bottom-strand, upstream χ-specific fragment and a top-strand, downstream χ-specific fragment. A third major unwinding product of RecBCD enzyme, not shown in the figure, is full-length, ssDNA. Full-length ssDNA is generated by RecBCD enzyme from the bottom strand if the enzyme fails to recognize χ. RecBC enzyme, which is essentially devoid of dsDNA nuclease activity, generates only full-length ssDNA from both strands. (Adapted from Anderson and Kowalczykowski 1997a).
The initial events in homologous recombination can be reconstituted in vitro using RecBCD enzyme to unwind dsDNA in the presence of RecA protein and the single-stranded DNA binding (SSB) protein (Roman and Kowalczykowski 1989a; Roman et al. 1991). In theseRecABCD pairing reactions, RecA protein preferentially incorporates the top-strand, downstream χ-specific fragment into joint molecules over all other nonspecific unwinding products (Dixon and Kowalczykowski 1991, 1995). Until recently, it was unclear why these χ-specific fragments are preferred substrates for RecABCD-dependent homologous pairing. However, it was shown that RecBCD enzyme, when activated by χ, directs the loading of RecA protein preferentially onto the top-strand, downstream χ-specific ssDNA (Anderson and Kowalczykowski 1997b). Efficient pairing of this unwinding product is dependent upon the concerted action of RecBCD enzyme and RecA protein; if their action is uncoupled, by adding RecA protein to the DNA after unwinding is complete, the efficient pairing of this χ-specific ssDNA is lost. Loss of efficient pairing in this case results from the inability of RecA protein to compete effectively with SSB protein for binding to ssDNA produced by RecBCD enzyme.
Study of the RecBC enzyme (which lacks the RecD subunit) provides additional insight into the manner by which χ modifies the RecBCD enzyme. Null mutations in either the recA, recB, or recC genes virtually eliminate dsDNA break repair (Sargentini and Smith 1986) and reduce homologous recombination to, at most, 1% of wild-type levels (Clark and Margulies 1965; Howard-Flanders and Theriot 1966; Emmerson 1968). In contrast, mutations in the recD gene do not have this phenotype. In both conjugation and transduction, recD null mutants exhibit levels of recombination that are comparable to or elevated relative to wild-type strains (Chaudhury and Smith 1984a; Lovett et al. 1988). For recombination of λ red gam phage, recD mutants are not responsive to χ, but are nevertheless hyper-recombinogenic (Chaudhury and Smith 1984a; Thaler et al. 1989). It was also found that for recD null mutants, recombinational exchanges in replication-blocked phage λ crosses are focused to the site of dsDNA breaks, resembling the distribution obtained with recBCD+ cells when χ is placed near a dsDNA end (Thaler et al. 1988, 1989). Together, these findings establish that the RecBC enzyme is fully proficient in homologous recombination, in the absence of an interaction with χ.
A number of biochemical observations support this hypothesis: RecBC enzyme is a processive helicase (Palas and Kushner 1990; Masterson et al. 1992; Korangy and Julin 1993); it does not respond to χ by producing χ-specific fragments (Yu et al. 1998; J.J. Churchill and S.C. Kowalczykowski, unpubl.); and it cannot unwind DNA under conditions in which RecBCD enzyme helicase activity is known to be reversibly inactivated by χ (Dixon et al. 1994). A particularly striking feature of RecBC enzyme is the absence of significant dsDNA exonuclease activity (Palas and Kushner 1990; Korangy and Julin 1993; Anderson et al. 1997), a property that could explain the elevated recombination proficiency of recD null mutants. However, whether RecBC enzyme can load RecA protein was completely unknown.
Here we demonstrate that RecBC enzyme constitutively loads RecA protein onto ssDNA during unwinding, without any requirement for χ. RecA protein is loaded only onto the top strand, defined as the strand that terminates 3′ at the dsDNA entry site for the enzyme (see Fig. 1). When DNA is unwound by RecBC enzyme in the presence of RecA protein, this strand is preferentially incorporated into joint molecules in homologous pairing assays. The constitutive loading of RecA protein by RecBC enzyme, coupled with this enzyme’s virtual absence of dsDNA exonuclease activity, accounts for the recombination proficiency of recD mutants. The strand-specific loading of RecA protein by RecBC enzyme is yet another feature that this enzyme, absent the RecD subunit, shares with the χ-modified holoenzyme. We propose that prior to interaction with χ, the RecD subunit blocks the loading of RecA protein by the holoenzyme; χ-induced modifications to the holoenzyme (which also result in the switch in nuclease polarity) clear this block and expose surfaces that are involved in the recruitment of RecA protein to the incipient ssDNA.
Results
DNA unwound by RecBC enzyme is efficiently incorporated by RecA protein into joint molecules, without a requirement for χ
We tested whether RecBC enzyme can serve as an initiator of RecA protein-mediated joint molecule formation (RecABC reactions), to see whether RecBC enzyme mimics the behavior of χ-modified RecBCD enzyme. In the coupled reactions reported below, RecBC enzyme unwound DNA that does not contain χ, in the presence of saturating amounts of RecA and SSB proteins. In the uncoupled reactions, pairing of ssDNA was uncoupled from RecBC enzyme action by substituting heat-denatured DNA, omitting RecBC enzyme, and initiating the reaction with the addition of a RecA and SSB protein mixture. Therefore, in the uncoupled reactions, RecA protein must compete, unassisted by RecBC enzyme, with SSB protein for binding to ssDNA.
The results from these two reactions are shown in Fig. 2, and demonstrate that RecBC enzyme can initiate RecA protein-promoted pairing between two fully duplex and homologous DNA substrates (linear dsDNA and covalently closed circular dsDNA). Interestingly, however, in the absence of unwinding by RecBC enzyme, the pairing of heat-denatured full-length ssDNA by RecA protein is extremely inefficient. In the coupled reaction with RecBC enzyme and RecA protein, 35% of the available ssDNA is incorporated into joint molecules after 2 min, but only 1% is incorporated in the uncoupled reaction. Maximal pairing is achieved in <10 min, with a 13.5-fold higher extent of pairing in the coupled process with RecBC enzyme than in the uncoupled process (Fig. 2B). Therefore, we conclude that efficient joint molecule formation is dependent on the concerted action of RecBC enzyme and RecA protein. For comparison, a coupled reaction with RecBCD holoenzyme, using χ-containing DNA, is shown to illustrate the preferential incorporation of top-strand, downstream χ-specific fragments into joint molecules (74% incorporated at 10 min) over full-length ssDNA (8% incorporated at 10 min). As reported previously (Anderson and Kowalczykowski 1997b), the full-length ssDNA is incorporated poorly into joint molecules in the RecABCD-coupled reactions—a failing that is characteristic for bottom-strand, full-length ssDNA produced by the RecBCD holoenzyme and that is in marked contrast to the RecABC reaction shown here. Furthermore, unwinding by RecBC enzyme produces two full-length strands of DNA but, as will be established below, only one of these is targeted for RecA pairing, arguing that the pairing efficiency for top-strand DNA in the RecABC pairing reaction (∼35% × 2 = 70%) rivals that for top-strand, downstream χ-specific ssDNA in the RecABCD reaction.
Figure 2.


Efficient formation of joint molecules in RecABC reactions requires the concerted action of RecA protein and RecBC enzyme. Linearized dsDNA was labeled at the 5′ end with 32P. The positions of labeled dsDNA substrate, ssDNA unwinding products, and joint molecules are indicated. Reaction conditions are described in Materials and Methods. (A) The coupled reactions were initiated by addition of either RecBC enzyme or RecBCD enzyme, in the presence of both RecA protein and SSB. In the uncoupled reaction, RecBC and RecBCD enzymes were omitted, heat-denatured DNA was substituted for dsDNA, and the reaction was initiated by the addition of a mixture of RecA and SSB proteins. The DNA substrates were XbaI-restricted χ0 pSKPB10, and HindIII-restricted χ+F (for the reaction with RecBCD). The sizes (in nucleotides) of fragments generated by RecBCD enzyme are indicated. (B) The efficiency of joint molecule formation is compared graphically for coupled vs. uncoupled reactions. (█) RecBC, coupled; (□) uncoupled (denatured DNA); (●) RecBCD, coupled χ-specific; (○) RecBCD, coupled full length. The efficiency of joint molecule formation is defined as the percentage of the specific ssDNA generated that appears in the respective joint molecule. The data for the coupled RecBC enzyme and uncoupled reactions represent an average of duplicates; there was no significant variation between replicates. Joint molecule formation from χ-specific fragments and full-length ssDNA produced by RecBCD enzyme is also shown for comparison.
RecBC enzyme loads RecA protein onto the unwound ssDNA
The preferential pairing of the top-strand, downstream χ-specific fragment, in homologous pairing reactions with RecA protein and RecBCD enzyme, results from the specific targeting of RecA protein onto this fragment. Because RecA protein polymerizes cooperatively in a 5′ → 3′ direction (Register and Griffith 1985), the loading of RecA protein onto the ssDNA at any point downstream of χ results in the formation of a nucleoprotein filament that extends to the 3′ terminus of this χ-specific fragment. In the case of RecBCD enzyme, the specificity of RecA protein loading onto this fragment was established by showing that only the top-strand, downstream χ-specific fragment is protected from degradation by exonuclease I (Exo I), a 3′-specific nuclease; by contrast, neither the full-length ssDNA nor bottom-strand, upstream χ-specific fragments are protected (Anderson and Kowalczykowski 1997b).
We tested the proposition that RecBC enzyme might also load RecA protein onto ssDNA during unwinding, which would be manifested as the protection of DNA from degradation by Exo I. As before, the fate of ssDNA produced by RecBC enzyme in a coupled unwinding reaction was compared with the fate of ssDNA in an uncoupled reaction. The results, depicted in Figure 3, show that ssDNA in the uncoupled reaction is quickly and completely degraded following addition of Exo I. In contrast, in the coupled reaction, where RecBC enzyme unwinds dsDNA in the presence of RecA protein, two species of full-length ssDNA, with different degrees of susceptibility to Exo I, are clearly distinguished. One species is quickly degraded by Exo I; however, the other species, representing ∼35% of the ssDNA produced, is protected by RecA protein even after 20 min of exposure to Exo I. For comparison, a standard coupled reaction with RecA protein, RecBCD holoenzyme, and HindIII linearized χ-containing DNA is also shown in Figure 3; as reported previously (Anderson and Kowalczykowski 1997b), the top-strand, downstream χ-specific fragment is protected from Exo I (74% remaining after 20 min), whereas the other major products of unwinding (full-length ssDNA and bottom-strand, upstream χ-specific fragment) are quickly degraded.
Figure 3.

RecBC enzyme loads RecA protein onto ssDNA during unwinding, ensuring that 3′ ends are coated. The protocol described in the legend to Fig. 2 was followed, except that supercoiled DNA was omitted and, after 3 min of reaction, excess M13 ssDNA and ATPγ-S were added as described in Materials and Methods. After a further 2 min incubation, Exo I was added. The zero time point was taken just prior to addition of Exo I. (dsDNA) The starting dsDNA substrate; (denat. DNA) heat-denatured substrate.
We hypothesized that the two different species of full-length ssDNA produced by RecBC enzyme in the presence of RecA protein represent the top and bottom strands of dsDNA, respectively, and that only top-strand DNA is protected from Exo I because RecA protein has been specifically recruited onto this strand. Such behavior would be analogous to the asymmetric loading of RecA protein onto only the top-strand, downstream χ-specific fragment by RecBCD holoenzyme.
RecBC enzyme targets RecA protein asymmetrically, directing it preferentially onto the strand that terminates with a 3′ end at the enzyme’s entry site
To test for strand-specific loading of RecA protein by RecBC enzyme, it was necessary to distinguish between each of the two full-length strands that are produced by RecBC enzyme. Furthermore, as either strand of duplex DNA may serve as the top or bottom strand (depending upon which of the two dsDNA ends RecBC enzyme enters from), it was necessary to generate a dsDNA-unwinding substrate that restricts access of the enzyme to only one of the two ends. RecBCD holoenzyme is unable to enter dsDNA at ends with ssDNA tails longer than ∼25 nucleotides (Taylor and Smith 1985). Initially, assuming that such tails would also block access to the DNA by RecBC enzyme, our strategy was to resect the DNA at one end with Exo III, perform Exo I protection assays, and use Southern hybridization with strand-specific oligonucleotide probes to distinguish between the top and the bottom strands.
When both ends of NdeI-cut pBR322 χ0 dsDNA were resected with Exo III to produce tails ∼90 nucleotides long, we found that RecBC enzyme, like RecBCD holoenzyme, cannot unwind DNA with these modified ends (data not shown). The resected DNA was then cut into two fragments with the restriction enzyme AlwNI. This treatment produces fragments of 3.7 and 0.6 kb, respectively, each bearing a 5′-ssDNA tail at one end because of Exo III resection, and a nearly blunt AlwNI 3′-overhanging end at the other. Access of RecBC enzyme to this DNA is therefore restricted to the AlwNI-cleaved end.
Exo I protection assays were conducted and followed by Southern hybridization as described in Materials and Methods. The results (Fig. 4) show that the ssDNA unwinding product that is preserved from degradation by Exo I corresponds to the top strand (Fig. 4A). In contrast, ssDNA corresponding to the bottom strand is quickly degraded (Fig. 4B). A control reaction without RecA protein shows that protection of the top strand is RecA protein-dependent (Fig. 4A); in another control reaction without Exo I, no ssDNA is lost (Fig. 4B). These results demonstrate that RecBC enzyme, like χ-modified RecBCD enzyme, directs RecA protein only onto one strand of ssDNA, that is, the top strand.
Figure 4.
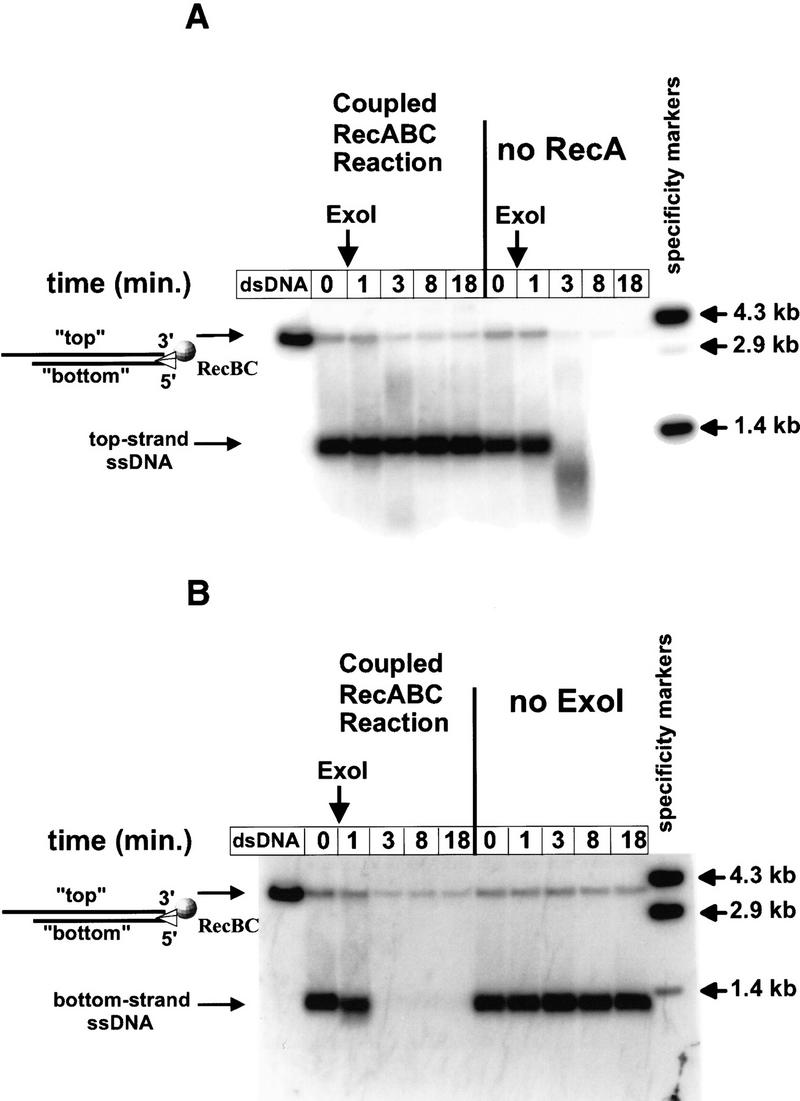
RecBC enzyme loads RecA protein only onto the DNA strand that terminates 3′ at the entry site. The starting dsDNA substrate consisted of two fragments, 3.7 and 0.6 kb, described in Materials and Methods. Each fragment has one end that was resected by Exo III and is inaccessible to RecBC enzyme and another AlwNI-restricted end at which the enzyme can enter. Exo I protection assays were performed. Control reactions were also performed in which either RecA protein or Exo I was omitted. After electrophoresis and Southern transfer, the DNA was probed with one of two strand-specific probes. (A) Top strand (probed with PB19); (B) bottom strand (probed with PB27). (The 0.6-kb fragment hybridizes to neither probe and is not shown.) The positions of 3.7-kb dsDNA starting material and ssDNA unwinding product are indicated. (Specificity marker) Lanes demonstrate the specificity of the two probes. These lanes contain a mixture of NdeI-restricted pBR322 χ0 dsDNA (the 4.3-kb fragment, which hybridizes to both probes) and pBR322 χ0 DNA restricted with EcoRI and AvaI (which produces fragments of 1.4 and 2.9 kb; probe PB19 hybridizes only to the smaller fragment, whereas PB27 hybridizes only to the larger fragment).
In RecABC pairing reactions, joint molecules are formed by invasion only of ssDNA produced from the top strand of the unwound DNA.
The previous experiment confirmed that in the coupled reaction with RecBC enzyme, only the top strand is protected by RecA protein from degradation by Exo I; the bottom strand is not protected. Because the loading of RecA protein onto top-strand, downstream χ-specific fragments by RecBCD holoenzyme results in the preferential incorporation of this fragment into joint molecules (Anderson and Kowalczykowski 1997b), it was logical to ask whether the top strand of DNA produced by RecBC enzyme unwinding might also be preferentially incorporated into joint molecules. To address this question, Exo III-resected DNA was again used to limit entry of RecBC enzyme to one end; this time, however, the resected DNA was labeled with 32P at either the 5′ or 3′ end, as described in the legend to Figure 5. After labeling, the DNA was cut with AlwNI to yield fragments of 3.7 and 0.6 kb, each with one Exo III-resected end and one AlwNI-cleaved end. For each fragment, the strand that is labeled with 32P at the 3′ end corresponds to the bottom strand during unwinding; 5′-labeled ssDNA corresponds to the top strand. It is thus possible to track the fate of each strand separately, depending on which labeled substrate is used.
Figure 5.

Only the DNA strand that terminates 3′ at the entry site is incorporated into joint molecules in RecABC pairing reactions. Plasmid DNA (pBR322 χ0) was linearized with NdeI, resected with Exo III, and then labeled either at the 5′ ends with [γ-32P]ATP or at the 3′ ends using Klenow fragment, [α-32P]ATP, and two of the other dNTPs (dCTP was omitted to prevent the polymerase from filling in the Exo III-resected ends). After labeling, the DNA was cut with AlwNI to produce the 3.7- and 0.6-kb fragments described in Fig. 4. Reactions conditions were as described in Fig. 3, except that dsDNA was preincubated with SSB protein in the absence of RecA protein for 2 min prior to initiation of unwinding to prevent RecA protein from binding to the 5′ tails (generated by Exo III resection) on the top strand. Unwinding was initiated with a mixture of RecBC enzyme and RecA protein. Joint molecules from the two different fragments are indicated.
Pairing reactions are shown in Figure 5, comparing the 5′- versus the 3′-labeled substrates. Each of the two fragments is unwound within 2 min. Joint molecules incorporating ssDNA from the 3.7-kb fragment migrate more slowly on the gel and can be distinguished from joints incorporating the smaller 0.6-kb fragment, as indicated.
In the reaction with 5′-labeled starting material, the labeled ssDNA product (corresponding to the top strand) is incorporated into joint molecules with high efficiency. At 2 min, 43% of the available ssDNA (3.7 kb) is incorporated into joint molecules, whereas in the reaction with 3′-labeled DNA, only 4% of the labeled ssDNA (corresponding in this case to the bottom strand) is present in joint molecules. A similar pattern is seen when comparing the behavior of 5′- versus 3′-labeled DNA for the 0.6-kb fragment. In both cases, it is clear that only DNA from the 5′-labeled top strand is utilized by RecA protein in the formation of joint molecules. This result is consistent with the earlier observation that only DNA from the top strand is protected from degradation by Exo I, because of the targeted loading of RecA protein onto this strand as the DNA is unwound by RecBC enzyme.
Discussion
It is important to know the molecular basis for recombination hot spot activity. In E. coli, specifically, it is important to understand what makes χ a recombination hot spot, and how this DNA sequence coordinates the activity of RecBCD enzyme and other proteins to both stimulate and focus recombination in its immediate vicinity. Two specific consequences of the interaction between χ and RecBCD enzyme are (1) the modulation of the enzyme’s nuclease activity (see Fig. 1), and (2) the asymmetric loading of RecA protein, onto the ssDNA containing χ (the top-strand, downstream χ-specific ssDNA in Fig. 1).
In contrast to the RecBCD holoenzyme, the RecBC enzyme, which lacks the RecD subunit, lacks significant dsDNA exonuclease activity. Nevertheless, here we demonstrate that it can serve as an initiator for RecA protein-dependent homologous pairing. But, perhaps most importantly, we find that the RecBC enzyme loads RecA protein onto ssDNA, in a manner similar to the χ-stimulated RecBCD enzyme (see model in Fig. 6). Just as with the holoenzyme, the loading of RecA protein by RecBC enzyme is both coupled to DNA unwinding and asymmetric, being confined to the top strand. However, unlike the holoenzyme, RecBC enzyme loads RecA protein constitutively, without the requirement for χ.
Figure 6.
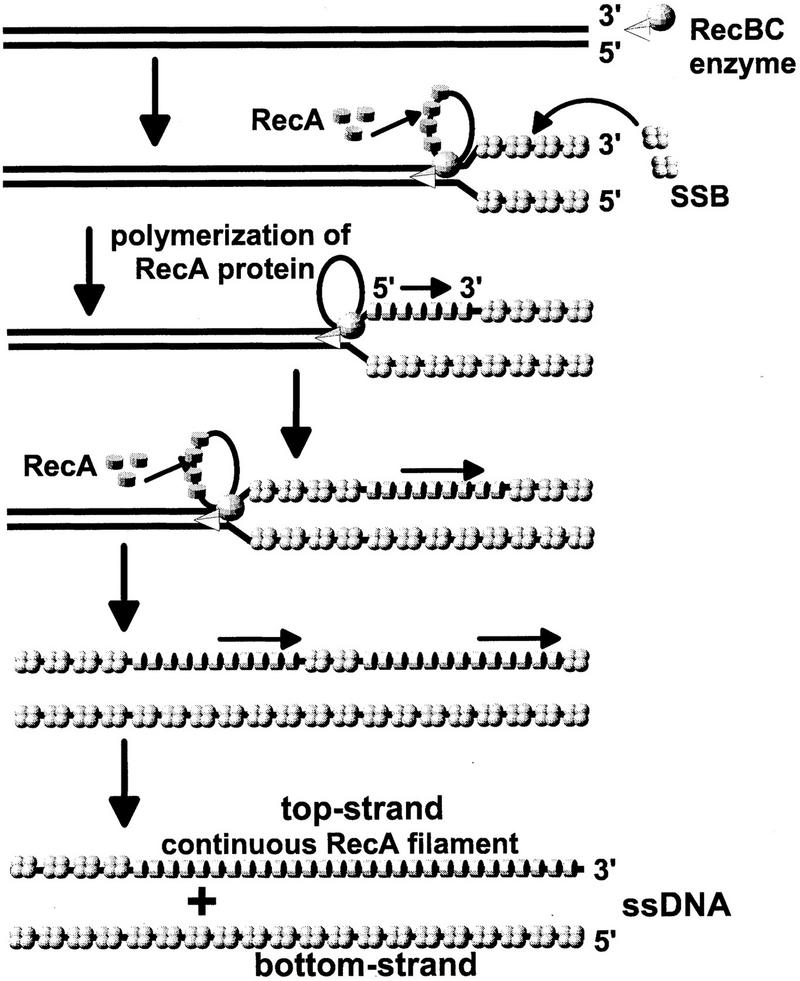
Model for helicase-facilitated loading of RecA protein onto the 3′-terminated top strand, during unwinding by RecBC/RecBCD enzyme. The model reconciles the 3′ → 5′ translocation of RecBC (or RecBCD) enzyme, relative to the top strand, with the requirement for 5′ → 3′ polymerization of RecA protein onto the same strand during unwinding. RecBC enzyme initiates unwinding at an intact duplex DNA end at the top. Unwinding progresses with time from the top to the bottom. Looped unwinding structures were demonstrated for RecBCD holoenzyme in electron microscope studies (Taylor and Smith 1980; Telander-Muskavitch and Linn 1980; Braedt and Smith 1989), but it is not known whether RecBC enzyme produces such loops. Therefore, hypothetical loops are shown for RecBC enzyme. As ssDNA is released during unwinding, SSB protein binds to the 5′-terminated bottom strand. RecBC enzyme periodically promotes the nucleation of RecA protein onto the 3′-terminated top strand, so that successive tracts of RecA protein are interspersed initially with SSB protein. Polymerization of RecA protein in the 5′ → 3′ direction extends these tracts, displacing SSB, and ultimately forming a continuous RecA nucleoprotein filament on the top strand only.
In the RecABC pairing reactions, only the top strand, the strand to which RecA protein is specifically targeted, is incorporated into joint molecules (Fig. 5). This result corresponds to the preferential incorporation of the top-strand, downstream χ-specific fragment into joint molecules in RecABCD pairing reactions (Dixon and Kowalczykowski 1991; Anderson and Kowalczykowski 1997b). Considering that unwinding by RecBC enzyme produces two full-length strands of DNA, but only one of these is targeted for RecA pairing, Figure 2B shows that the pairing efficiency for top-strand DNA in the RecABC pairing reaction (∼35% × 2 = 70%) rivals that for top-strand, downstream χ-specific fragments in the RecABCD reaction (74%).
With respect to the asymmetric loading of RecA protein, RecBC enzyme thus behaves as an analog of χ-modified RecBCD holoenzyme. This result concurs with the interpretation that has emerged consistently from both genetic and biochemical characterization of recD deficient variants (see introductory section for details). The fact that functional RecD subunit is not required for recombination in vivo suggests that its function is chiefly a regulatory one. Two striking characteristics of RecBC enzyme are its virtual absence of dsDNA exonuclease activity and its constitutive capacity to load RecA protein, indicating that RecD protein is involved in controlling these specific functions. Although many similarities exist between RecBC enzyme and χ-modified RecBCD enzyme, we note that RecBC enzyme lacks the 5′ → 3′ exonuclease activity that is up-regulated in the holoenzyme following interaction with χ (Anderson et al. 1997).
The constitutive loading of RecA protein by RecBC enzyme argues that the capacity to load RecA protein onto the top strand of DNA is an intrinsic property of the RecBCD holoenzyme; however, this latent activity is masked until interaction with χ. We can imagine two different means by which such masking might occur. The first possibility is that the RecBCD holoenzyme might itself also load RecA protein onto the top strand constitutively, even upstream of χ, but that this function is either obscured or simply not productive because of degradation of top-strand DNA by the 3′ → 5′ exonuclease activity. According to this view, χ activates the recombinogenic potential of a constitutive RecA protein-loading function, by attenuating this nuclease activity. The second possibility is that the RecBCD holoenzyme is physically incapable of interaction with RecA protein until the enzyme has undergone the structural changes that are elicited by interaction with χ; after a productive interaction, this χ-modified RecBCD enzyme reveals a new subunit surface that permits interaction with RecA protein. Based on preliminary experiments involving a truncated form of RecBC enzyme (Yu et al. 1998) we presently favor the latter explanation (J.J. Churchill, D. Anderson, and S.C. Kowalczykowski, unpubl.). Thus, we advocate the idea that RecA protein-loading requires more than simply helicase activity coupled with the absence of nuclease activity, but rather that loading is a specialized function specific to the RecBCD (and RecBC) helicase.
The facilitated loading of RecA protein might involve a direct interaction between RecBCD (or RecBC) enzyme and RecA protein. Interestingly, interspecies complementation in vivo demonstrates that optimal recombination is achieved when both RecBCD enzyme and RecA protein are from the same species (Rinken et al. 1991; de Vries and Wackernagel 1992). To date, no direct interaction between these enzymes has been detected using either coimmunoprecipitation or biosensor experiments (D.G. Anderson and S.C. Kowalczykowski, unpubl.).
An interesting feature of RecBC/RecBCD enzyme-assisted loading of RecA protein is the puzzling strand asymmetry of this process. This loading of RecA protein is confined to the strand that terminates with a 3′ end at the entry site of the helicase. During unwinding, the helicase is translocating in a 3′ → 5′ direction relative to this strand. This presents something of a paradox, as the assembly of RecA protein onto ssDNA proceeds in the opposite direction, that is, 5′ → 3′. The conflicting polarities of unwinding and RecA protein filamentation can be reconciled by assuming that RecBCD (or RecBC) enzyme promotes the nucleation of RecA protein at various intervals along the 3′-terminated strand as ssDNA is generated during unwinding. Each nucleation site is extended by RecA protein polymerization in the 5′ → 3′ direction, until contact is made with a preceding tract of RecA protein, and a continuous filament is formed (see Fig. 6). Discontinuities at the junctions of these tracts can be corrected by successive rounds of disassembly and reassembly of RecA protein monomers within the filament, accompanied by successive rounds of ATP hydrolysis (Menetski et al. 1990; Rehrauer and Kowalczykowski 1993; Kowalczykowski and Krupp 1995; Shan and Cox 1997). Thus, the facilitated loading of RecA protein can extend for a considerable distance beyond χ, explaining how recombination can be stimulated downstream of χ past heterologies over 9 kb in length (Myers et al. 1995a).
The facilitated loading of RecA protein onto ssDNA during unwinding, together with the absence of destructive dsDNA exonuclease activity in RecBC enzyme, accounts for the recombination proficiency of recD null mutants. RecA protein-loading also accounts for the observation that recombination in these mutants is focused at the site of dsDNA breaks (Thaler et al. 1989), as is recombination in wild-type strains following exposure of RecBCD enzyme to χ (Koppen et al. 1995; Myers et al. 1995b). Furthermore, RecA protein-loading may explain why recombination in recD mutants is not dependent on recF or recO function (Lovett et al. 1988; Lloyd and Buckman 1995), whereas all three genes, recF, recO, and recR, are required in a recB or recC mutant background (Clark and Sandler 1994; Kowalczykowski et al. 1994). Genetic and biochemical evidence suggest that together, the products of these genes allow RecA protein to compete more effectively with SSB protein for binding to ssDNA (Umezu et al. 1993; Umezu and Kolodner 1994; Webb et al. 1997). Thus, the χ-activated RecBCD enzyme provides the equivalent of recFOR function by directing the loading of RecA protein onto ssDNA downstream of χ while disfavoring the binding of SSB protein. In agreement, the RecBC enzyme loads RecA protein constitutively, explaining the lack of dependence on recF and recO for recombination in recD null mutants.
The novel modes by which χ regulates the activity of a multifunctional helicase/nuclease reveal fundamental features by which homologous recombination and dsDNA break repair are controlled in E. coli, as well as other prokaryotes (Chedin et al. 1998; el Karoui et al. 1998). The χ-dependent loading of RecA protein by RecBCD enzyme is an important aspect of this interplay. Understanding how this phenomenon occurs will reveal much about the basis for χ recombination hot spot activity.
Materials and methods
Enzymes
RecBC enzyme was purified as a heterodimer from the recBCD deletion strain V186 (Chaudhury and Smith 1984b) bearing three compatible plasmids: pPB700 (Boehmer and Emmerson 1991), expressing RecB protein and conferring ampicillin resistance; pPB520 (Boehmer and Emmerson 1991), expressing RecC protein and conferring chloramphenicol resistance; and pMS421 (Heath and Weinstock 1991) expressing lacIq and conferring streptomycin resistance. Cells were grown at 37°C in Terrific Broth containing 50 μg/ml thymine, 25 μg/ml chloramphenicol, 60 μg/ml ampicillin, and 50 μg/ml streptomycin.
RecBC enzyme was purified following the method previously described for the purification of RecBCD holoenzyme (Roman and Kowalczykowski 1989b), with an additional chromatographic column (Sephacryl S-200) added to separate RecBC heterodimer from any free RecB and RecC subunits. RecBC heterodimer eluted in the void volume. No peaks corresponding to RecB or RecC proteins eluted from the column, indicating that the preparation was essentially free of unassociated subunits.
RecBC enzyme concentration was determined using an extinction coefficient of 3.6 × 105/m per cm at 280 nm, derived by adding the respective extinction coefficients (Korangy and Julin 1993) for the individual subunits. No contaminating protein bands were detected when 1 μg of protein was loaded on an SDS–polyacrylamide gel and stained with Coomassie Brilliant blue. The percentage of functional enzyme in the RecBC preparation was determined as described below (see Determination of the functional RecBC enyzme concentration).
RecBCD enzyme was purified according to the method of Roman and Kowalczykowski (1989b), with further purification by an additional FPLC Mono-Q step as described by Anderson and Kowalczykowski (1997b). RecA protein was purified using a procedure based on spermidine precipitation (Griffith and Shores 1985; S.C. Kowalczykowski, unpubl.). RecA protein concentration was determined using an extinction coefficient of 2.7 × 104/m per cm at 280 nm. SSB protein was isolated from strain RLM727 and purified according to LeBowitz (1985). SSB protein concentration was determined using an extinction coefficient of 3.0 × 104/m per cm (Ruyechan and Wetmur 1975).
Exo III was purchased from Promega. All other DNA-modifying enzymes and restriction endonucleases were purchased from New England Biolabs. The enzymes were used according to Sambrook et al. (1989) or as indicated by the specific vendor.
Determination of the functional RecBC enzyme concentration
The activity of RecBC enzyme was measured by complementation with a saturating amount of hexahistidine-tagged RecD protein (Chen et al. 1997). A fixed amount of purified RecBC enzyme was incubated with increasing amounts of tagged RecD protein on ice for 30 min in a 132-μl reconstitution mixture consisting of 20 mm potassium phosphate at pH 7.0, 100 mm NaCl, 1 mg/ml bovine serum albumin, and 10% (vol/vol) glycerol. The reconstitution mixture was then assayed for ATP-dependent nuclease activity (Eichler and Lehman 1977). The nuclease activity of the reconstituted RecBC enzyme, when fully saturated with RecD protein, was compared to that of a known quantity of wild-type RecBCD enzyme to determine the percentage of functional, reconstitution-competent RecBC enzyme. The RecBC enzyme was estimated by this method to be ∼20% active.
DNA substrates
The plasmid χ0 pSKPB10 was provided by Piero Bianco of this laboratory and was derived from pBluescript II SK(−) phagemid (Stratagene) by the insertion of a fragment containing the RAD52 gene from S. cerevisiae to generate a plasmid of 4263 bp. All other plasmid DNA substrates are previously described (Anderson and Kowalczykowski 1997b). Purification and restriction of all plasmid DNA, and 5′-end labeling with [γ-32P]ATP, were as described previously (Anderson and Kowalczykowski 1997b); 3′-end labeling of Exo III resected pBR322 χ0 was performed with the Klenow fragment (Exo−) of DNA polymerase I in the presence of dGTP, dTTP, and [γ-32P]dATP (NEN).
Resection of DNA by Exo III was performed as follows: 100 μg/ml NdeI-cut pBR322 χ0 DNA was treated for 1 min at 25°C with 17500 U/ml Exo III in 66 mm Tris-HCl at pH 8.0, 0.66 mm MgCl2. After Exo III was inactivated by heating at 70°C for 15 min, the mixture was extracted with phenol/chloroform, precipitated with sodium acetate, and resuspended in TE buffer as described in Sambrook et al. (1989).
Joint molecule formation assay
Homologous pairing reactions contained 25 mm Tris-acetate at pH 7.5, 8 mm magnesium acetate, 5 mm ATP, 1 mm dithiothreitol, 1 mm phosphoenolpyruvate, 4 U/ml pyruvate kinase, 40 μm nucleotide linear dsDNA, 80 μm nucleotides supercoiled DNA, 20 μm RecA protein, 8 μm SSB protein, and either 5.4 nm functional RecBC or 0.8 nm functional RecBCD enzyme. These conditions were essentially those of Dixon and Kowalczykowski (1991) except for a twofold higher concentration of both supercoiled DNA and SSB protein. Assays were performed at 37°C. Coupled reactions were started by the addition of RecBC or RecBCD enzyme after preincubation of all other components for 2 min. For uncoupled reactions, heat-denatured DNA was substituted for dsDNA, helicase was omitted, and the reactions were initiated by the addition of a mixture of RecA and SSB proteins. Samples were taken at the indicated time points and deproteinized; both gel electrophoresis and quantitation were performed as described previously (Anderson and Kowalczykowski 1997b).
Exo I protection assay (non-strand-specific)
Conditions were the same as for joint molecule formation assays, except for the omission of supercoiled DNA. After 3 min of reaction with RecBC or RecBCD enzyme, a mixture of ATPγ-S and nonhomologous M13mp7 ssDNA was added to final concentrations of 5 mm and 200 μm, respectively. (ATPγ-S induces a high-affinity state and stabilizes RecA protein that is bound to ssDNA. Addition of nonhomologous ssDNA insures that any free RecA protein remaining in solution is sequestered away from the labeled ssDNA unwinding product.) After 2 min of further incubation, Exo I was added to a concentration of 100 U/ml. Uncoupled reactions contained heat-denatured DNA instead of dsDNA and were initiated by the addition of a mixture of RecA and SSB proteins in the absence of any helicase. Samples were taken at the indicated times and processed as described above for joint molecule formation assays, except that electrophoresis was at 2 V/cm for 15 hr in 1.2% agarose gels. The assay was used to generate the data in Figure 3.
Strand-specific Exo I protection assay
The DNA substrate was χ0 pBR322 that had been restricted with NdeI, resected with Exo III, and then restricted again with AlwNI as described in Results. Reaction conditions were as described above for non-strand-specific Exo I protection assays, except that SSB protein was at 4 μm, and was added to the 5′-tailed dsDNA substrate and incubated for 2 min prior to addition of RecA protein (to prevent RecA protein from binding to the 5′ tails generated by Exo III resection). Addition of RecA protein was immediately followed by initiation of the reaction with RecBC enzyme. After 3 min of reaction, a mixture of ATPγS and nonhomologous ssDNA [poly(dT)] was added to concentrations of 5 mm and 200 μm, respectively. After 2 min of further incubation, Exo I was added to 100 U/ml. Samples taken at the indicated time points were stopped, split, and loaded onto two separate 1.2% agarose gels. Following electrophoresis (2 V/cm for 15 hr), the DNA in each of these gels was transferred to charged nylon and probed with one of two different labeled oligonucleotides as described below. The assay was used to generate the data in Figure 4.
Southern transfer and strand-specific probing with oligonucleotide probes
The oligonucleotide probes were PB19 (5′-GGCATGGCGGCCGACGCGCT-3′) and PB27 (5′-GGCCAGGACCCAACGCTGCC-3′). For the pBR322 χ0 substrate that was linearized with NdeI restriction enzyme, treated with Exo III, and restricted again with AlwNI enzyme, PB19 hybridizes specifically to the top-strand (i.e., the strand which terminates 3′ at the AlwNI overhanging end that is the entry site for RecBC enzyme), and PB27 hybridizes specifically to the complementary, or bottom strand. Before Southern transfer, the agarose gels were soaked twice in 0.25 m HCl for 10 min; twice in 0.5 m NaOH, 1 m NaCl for 15 min; and then twice in 0.5 m Tris-HCl at pH 7.4, 3 m NaCl for 30 min. DNA was transferred by capillary action in 5× SSC (0.75 m NaCl, 75 mm sodium citrate at pH 7.0) to a charged nylon membrane (Hybond-N+, Amersham) and covalently linked to the membrane by UV irradiation. The oligonucleotide probes were labeled with 0.67 unit of polynucleotide kinase in a 30-μl reaction volume, using 8 pmoles of oligonucleotide and 80 μCi of [γ-32P]ATP. Unincorporated nucleotides were removed by a G-25 MicroSpin column (Pharmacia); 4 pmoles of labeled probe were used in each hybridization. Prehybridization (20 min) and hybridization (1 hr) were at 42°C in Rapid-hyb buffer (Amersham). Following hybridization, membranes were washed for 20 min in 5× SSC, 0.1% SDS, at room temperature, and the signal was quantified using a Storm 840 PhosphorImager (Molecular Dynamics) with ImageQuaNT software.
Acknowledgments
We are especially grateful to Doug Julin and Misook Yu for sharing their results regarding the nuclease domain of RecB subunit before publication. In addition, we thank Peter Emmerson of the University of Newcastle and Paul Boehmer of the New Jersey Medical School for the gift of plasmids that overexpress RecB and RecC proteins. We also thank the many members of the Kowalczykowski laboratory for their critical reading of this manuscript: Deana Arnold, Piero Bianco, Frederic Chedin, Frank Harmon, Noriko Kantake, Julie Kleiman, Jim New, and Tomohiko Sugiyama. This work was supported by a Molecular and Cellular Biology Training Grant (T32-GM-07377) to J.J.C. from the National Institutes of Health (NIH) and by a grant from NIH, (GM-41347).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sckowalczykowski@ucdavis.edu; FAX (530) 752-5939.
References
- Anderson DG, Kowalczykowski SC. The recombination hot spot χ is a regulatory element that switches the polarity of DNA degradation by the RecBCD enzyme. Genes & Dev. 1997a;11:571–581. doi: 10.1101/gad.11.5.571. [DOI] [PubMed] [Google Scholar]
- ————— The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell. 1997b;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- Anderson DG, Churchill JJ, Kowalczykowski SC. Chi-activated RecBCD enzyme possesses 5′ → 3′ nucleolytic activity, but RecBC enzyme does not: Evidence suggesting that the alteration induced by Chi is not simply ejection of the RecD subunit. Genes Cells. 1997;2:117–128. doi: 10.1046/j.1365-2443.1997.1130311.x. [DOI] [PubMed] [Google Scholar]
- Bianco PR, Kowalczykowski SC. The recombination hot spot Chi is recognized by the translocating RecBCD enzyme as the single strand of DNA containing the sequence 5′-GCTGGTGG-3′. Proc Natl Acad Sci. 1997;94:6706–6711. doi: 10.1073/pnas.94.13.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco PR, Tracy RB, Kowalczykowski SC. DNA strand exchange proteins: A biochemical and physical comparison. Front Biosci (online) 1998;3:D570–D603. doi: 10.2741/a304. [DOI] [PubMed] [Google Scholar]
- Boehmer PE, Emmerson PT. Escherichia coli RecBCD enzyme: Inducible overproduction and reconstitution of the ATP-dependent deoxyribonuclease from purified subunits. Gene. 1991;102:1–6. doi: 10.1016/0378-1119(91)90529-k. [DOI] [PubMed] [Google Scholar]
- Braedt G, Smith GR. Strand specificity of DNA unwinding by recBCD enzyme. Proc Natl Acad Sci. 1989;86:871–875. doi: 10.1073/pnas.86.3.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury AM, Smith GR. A new class of Escherichia coli recBC mutants: Implications for the role of recBC enzyme in homologous recombination. Proc Natl Acad Sci. 1984a;81:7850–7854. doi: 10.1073/pnas.81.24.7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Escherichia coli recBC deletion mutants. J Bacteriol. 1984b;160:788–791. doi: 10.1128/jb.160.2.788-791.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedin F, Noirot P, Biaudet V, Ehrlich SD. A 5-nucleotide sequence protects DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of Bacillus subtilis. Mol Microbiol. 1998;29:1369–1378. doi: 10.1046/j.1365-2958.1998.01018.x. [DOI] [PubMed] [Google Scholar]
- Chen HW, Ruan B, Yu M, Wang J, Julin DA. The RecD subunit of the RecBCD enzyme from Escherichia coli is a single-stranded DNA-dependent ATPase. J Biol Chem. 1997;272:10072–10079. doi: 10.1074/jbc.272.15.10072. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Smith GR. Distribution of Chi-stimulated recombinational exchanges and heteroduplex endpoints in phage lambda. Genetics. 1989;123:5–17. doi: 10.1093/genetics/123.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Margulies AD. Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc Natl Acad Sci. 1965;53:451–459. doi: 10.1073/pnas.53.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ. Homologous genetic recombination: The pieces begin to fall into place. Crit Rev Microbiol. 1994;20:125–142. doi: 10.3109/10408419409113552. [DOI] [PubMed] [Google Scholar]
- de Vries J, Wackernagel W. Recombination and UV resistance of Escherichia coli with the cloned recA and recBCD genes of Serratia marcescens and Proteus mirabilis: Evidence for an advantage of intraspecies combination of P. mirabilis RecA protein and RecBCD enzyme. J Gen Microbiol. 1992;138:31–38. doi: 10.1099/00221287-138-1-31. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kowalczykowski SC. Homologous pairing in vitro stimulated by the recombination hot spot, Chi. Cell. 1991;66:361–371. doi: 10.1016/0092-8674(91)90625-9. [DOI] [PubMed] [Google Scholar]
- ————— The recombination hot spot χ is a regulatory sequence that acts by attenuating the nuclease activity of the E. coli RecBCD enzyme. Cell. 1993;73:87–96. doi: 10.1016/0092-8674(93)90162-j. [DOI] [PubMed] [Google Scholar]
- ————— Role of the Escherichia coli recombination hotspot, χ, in RecABCD-dependent homologous pairing. J Biol Chem. 1995;270:16360–16370. doi: 10.1074/jbc.270.27.16360. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Churchill JJ, Kowalczykowski SC. Reversible inactivation of the Escherichia coli RecBCD enzyme by the recombination hotspot χ in vitro: Evidence for functional inactivation or loss of the RecD subunit. Proc Natl Acad Sci. 1994;91:2980–2984. doi: 10.1073/pnas.91.8.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler DC, Lehman IR. On the role of ATP in phosphodiester bond hydrolysis catalyzed by the recBC deoxyribonuclease of Escherichia coli. J Biol Chem. 1977;252:499–503. [PubMed] [Google Scholar]
- el Karoui M, Ehrlich D, Gruss A. Identification of the lactococcal exonuclease/recombinase and its modulation by the putative Chi sequence. Proc Natl Acad Sci. 1998;95:626–631. doi: 10.1073/pnas.95.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson PT. Recombination deficient mutants of Escherichia coli K12 that map between thyA and argA. Genetics. 1968;60:19–30. doi: 10.1093/genetics/60.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis DG, Amundsen SK, Smith GR. Genetic functions promoting homologous recombination in Escherichia coli: A study of inversions in phage lambda. Genetics. 1987;115:11–24. doi: 10.1093/genetics/115.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- Griffith J, Shores CG. RecA protein rapidly crystallizes in the presence of spermidine: A valuable step in its purification and physical characterization. Biochemistry. 1985;24:158–162. doi: 10.1021/bi00322a022. [DOI] [PubMed] [Google Scholar]
- Heath JD, Weinstock GM. Tandem duplications of the lac region of the Escherichia coli chromosome. Biochimie. 1991;73:343–352. doi: 10.1016/0300-9084(91)90099-m. [DOI] [PubMed] [Google Scholar]
- Howard-Flanders P, Theriot L. Mutants of Escherichia coli K-12 defective in DNA repair and in genetic recombination. Genetics. 1966;53:1137–1150. doi: 10.1093/genetics/53.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T. Recombination by replication. Cell. 1996;85:625–627. doi: 10.1016/s0092-8674(00)81229-5. [DOI] [PubMed] [Google Scholar]
- Köppen A, Krobitsch S, Thoms B, Wackernagel W. Interaction with the recombination hot spot χ in vivo converts the RecBCD enzyme of Escherichia coli into a c-independent recombinase by inactivation of the RecD subunit. Proc Natl Acad Sci. 1995;92:6249–6253. doi: 10.1073/pnas.92.14.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korangy F, Julin DA. Kinetics and processivity of ATP hydrolysis and DNA unwinding by the RecBC enzyme from Escherichia coli. Biochemistry. 1993;32:4873–4880. doi: 10.1021/bi00069a024. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC, Krupp RA. DNA-strand exchange promoted by RecA protein in the absence of ATP: Implications for the mechanism of energy transduction in protein-promoted nucleic acid transactions. Proc Natl Acad Sci. 1995;92:3478–3482. doi: 10.1073/pnas.92.8.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC, Dixon SA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- Lam ST, Stahl MM, McMilin KD, Stahl FW. Rec-mediated recombinational hot spot activity in bacteriophage lambda. II. A mutation which causes hot spot activity. Genetics. 1974;77:425–433. doi: 10.1093/genetics/77.3.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz J. ‘Biochemical mechanism of strand initiation in bacteriophage lambda DNA replication’. Ph.D. thesis. Baltimore, MD: Johns Hopkins University; 1985. [Google Scholar]
- Lloyd RG, Buckman C. Conjugational recombination in Escherichia coli: Genetic analysis of recombinant formation in Hfr × F− crosses. Genetics. 1995;139:1123–1148. doi: 10.1093/genetics/139.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett ST, Luisi-DeLuca C, Kolodner RD. The genetic dependence of recombination in recD mutants of Escherichia coli. Genetics. 1988;120:37–45. doi: 10.1093/genetics/120.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson C, Boehmer PE, McDonald F, Chaudhuri S, Hickson ID, Emmerson PT. Reconstitution of the activities of the RecBCD holoenzyme of Escherichia coli from the purified subunits. J Biol Chem. 1992;267:13564–13572. [PubMed] [Google Scholar]
- Menetski JP, Bear DG, Kowalczykowski SC. Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc Natl Acad Sci. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RS, Stahl MM, Stahl FW. Chi recombination activity in phage lambda decays as a function of genetic distance. Genetics. 1995a;141:805–812. doi: 10.1093/genetics/141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RS, Kuzminov A, Stahl FW. The recombination hot spot c activates RecBCD recombination by converting Escherichia coli to a recD mutant phenocopy. Proc Natl Acad Sci. 1995b;92:6244–6248. doi: 10.1073/pnas.92.14.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palas KM, Kushner SR. Biochemical and physical characterization of exonuclease V from Escherichia coli. Comparison of the catalytic activities of the RecBC and RecBCD enzymes. J Biol Chem. 1990;265:3447–3454. [PubMed] [Google Scholar]
- Ponticelli AS, Schultz DW, Taylor AF, Smith GR. Chi-dependent DNA strand cleavage by recBC enzyme. Cell. 1985;41:145–151. doi: 10.1016/0092-8674(85)90069-8. [DOI] [PubMed] [Google Scholar]
- Register JC, III, Griffith J. The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J Biol Chem. 1985;260:12308–12312. [PubMed] [Google Scholar]
- Rehrauer WM, Kowalczykowski SC. Alteration of the nucleoside triphosphate (NTP) catalytic domain within Escherichia coli RecA protein attenuates NTP hydrolysis but not joint molecule formation. J Biol Chem. 1993;268:1292–1297. [PubMed] [Google Scholar]
- Rinken R, de Vries J, Weichenhan D, Wackernagel W. The recA-recBCD dependent recombination pathways of Serratia marcescens and Proteus mirabilis in Escherichia coli: Functions of hybrid enzymes and hybrid pathways. Biochimie. 1991;73:375–384. doi: 10.1016/0300-9084(91)90104-9. [DOI] [PubMed] [Google Scholar]
- Roca AI, Cox MM. RecA protein: Structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Kowalczykowski SC. Formation of heteroduplex DNA promoted by the combined activities of Escherichia coli RecA and RecBCD proteins. J Biol Chem. 1989a;264:18340–18348. [PubMed] [Google Scholar]
- ————— Characterization of the helicase activity of the Escherichia coli RecBCD enzyme using a novel helicase assay. Biochemistry. 1989b;28:2863–2873. doi: 10.1021/bi00433a018. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Dixon DA, Kowalczykowski SC. RecBCD-dependent joint molecule formation promoted by the Escherichia coli RecA and SSB proteins. Proc Natl Acad Sci. 1991;88:3367–3371. doi: 10.1073/pnas.88.8.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruyechan WT, Wetmur JG. Studies on the cooperative binding of the Escherichia coli DNA unwinding protein to single-stranded DNA. Biochemistry. 1975;14:5529–5534. doi: 10.1021/bi00696a023. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sargentini NJ, Smith KC. Quantitation of the involvement of the recA, recB, recC, recF, recJ, recN, lexA, radA, radB, uvrD, and umuC genes in the repair of X-ray-induced DNA double-strand breaks in Escherichia coli. Radiat Res. 1986;107:58–72. [PubMed] [Google Scholar]
- Shan Q, Cox MM. RecA filament dynamics during DNA strand exchange reactions. J Biol Chem. 1997;272:11063–11073. doi: 10.1074/jbc.272.17.11063. [DOI] [PubMed] [Google Scholar]
- Smith GR, Kunes SM, Schultz DW, Taylor A, Triman KL. Structure of Chi hotspots of generalized recombination. Cell. 1981;24:429–436. doi: 10.1016/0092-8674(81)90333-0. [DOI] [PubMed] [Google Scholar]
- Stahl FW, Crasemann JM, Stahl MM. Rec-mediated recombinational hot spot activity in bacteriophage lambda. III. Chi mutations are site-mutations stimulating rec-mediated recombination. J Mol Biol. 1975;94:203–212. doi: 10.1016/0022-2836(75)90078-9. [DOI] [PubMed] [Google Scholar]
- Stahl FW, Stahl MM, Malone RE, Crasemann JM. Directionality and nonreciprocality of Chi-stimulated recombination in phage lambda. Genetics. 1980;94:235–248. doi: 10.1093/genetics/94.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Smith GR. Unwinding and rewinding of DNA by the recBC enzyme. Cell. 1980;22:447–457. doi: 10.1016/0092-8674(80)90355-4. [DOI] [PubMed] [Google Scholar]
- ————— Substrate specificity of the DNA unwinding activity of the RecBC enzyme of Escherichia coli. J Mol Biol. 1985;185:431–443. doi: 10.1016/0022-2836(85)90414-0. [DOI] [PubMed] [Google Scholar]
- Telander-Muskavitch KM, Linn S. Electron microscopy of E. coli recBC enzyme reaction intermediates. In: Alberts B, editor. Mechanistic studies of DNA replication and genetic recombination. New York, NY: Academic Press; 1980. pp. 901–918. [Google Scholar]
- Thaler DS, Sampson E, Siddiqi I, Rosenberg SM, Stahl FW, Stahl M. A hypothesis: Chi-activation of recBCD enzyme involves removal of the recD subunit. In: Friedberg E, Hanawalt P, editors. Mechanisms and consequences of DNA damage processing. New York, NY: Alan R. Liss, Inc.; 1988. pp. 413–422. [Google Scholar]
- Thaler DS, Sampson E, Siddiqi I, Rosenberg SM, Thomason LC, Stahl FW, Stahl MM. Recombination of bacteriophage lambda in recD mutants of Escherichia coli. Genome. 1989;31:53–67. doi: 10.1139/g89-013. [DOI] [PubMed] [Google Scholar]
- Umezu K, Kolodner RD. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem. 1994;269:30005–30013. [PubMed] [Google Scholar]
- Umezu K, Chi NW, Kolodner RD. Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci. 1993;90:3875–3879. doi: 10.1073/pnas.90.9.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb BL, Cox MM, Inman RB. Recombinational DNA repair: The RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell. 1997;91:347–356. doi: 10.1016/s0092-8674(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Yu M, Souaya J, Julin DA. The 30-kDa C-terminal domain of the RecB protein is critical for the nuclease activity, but not the helicase activity, of the RecBCD enzyme from Escherichia coli. Proc Natl Acad Sci. 1998;95:981–986. doi: 10.1073/pnas.95.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]