

Abstract
The human immunodeficiency virus encodes the transcriptional transactivator Tat, which binds to the transactivation response (TAR) RNA stem–loop in the viral long terminal repeat (LTR) and increases rates of elongation rather than initiation of transcription by RNA polymerase II (Pol II). In this study, we demonstrate that Tat binds directly to the cyclin-dependent kinase 7 (CDK7), which leads to productive interactions between Tat and the CDK-activating kinase (CAK) complex and between Tat and TFIIH. Tat activates the phosphorylation of the carboxy-terminal domain (CTD) of Pol II by CAK in vitro. The ability of CAK to phosphorylate the CTD can be inhibited specifically by a CDK7 pseudosubstrate peptide that also inhibits transcriptional activation by Tat in vitro and in vivo. We conclude that the phosphorylation of the CTD by CAK is essential for Tat transactivation. Our data identify a cellular protein that interacts with the activation domain of Tat, demonstrate that this interaction is critical for the function of Tat, and provide a mechanism by which Tat increases the processivity of Pol II.
Keywords: HIV, transactivator Tat, phosphorylation, CTD, Pol II, CAK, CDK7
The human immunodeficiency virus (HIV) encodes a highly conserved transcriptional transactivator Tat, that is expressed early in the viral life cycle and is essential for viral replication and progression to disease (Cullen 1993; Jones and Peterlin 1994). Tat binds to the transactivation response (TAR) RNA stem–loop located from positions +1 to +60 in the viral 5′ long terminal repeat (LTR). Interactions between Tat and TAR are absolutely required for the increased processivity of RNA polymerase II (Pol II) and the production of full-length viral transcripts (Kao et al. 1987; Laspia et al. 1989; Marciniak and Sharp 1991; Kato et al. 1992). Tat is unique because it is the only eukaryotic transcription elongation factor known to function via RNA (Madore and Cullen 1995). Although the mechanism by which Tat increases transcription elongation rates is unknown, common regulatory themes must exist between viral and cellular genes because Tat can relieve Pol II pausing when artificially targeted to the c-myc promoter (Wright et al. 1994).
Tat can be divided into two functional domains. The activation domain contains 48 amino-terminal amino acids and interacts with the cellular transcriptional machinery. A 10-amino-acid basic domain is required for the binding of Tat to TAR (Jones and Peterlin 1994). Cellular proteins are clearly required for the function of Tat (Carroll et al. 1992; Madore and Cullen 1993). At least one protein, encoded by the human chromosome 12, is required for the efficient binding of Tat to TAR (Hart et al. 1989; Alonso et al. 1992). Numerous other proteins have been postulated to interact with the activation domain of Tat. These include general transcription factors (GTFs) such as the core Pol II (Mavankal et al. 1996), the TATA box-binding protein (TBP) (Kashanchi et al. 1994; Veschambre et al. 1995), the TBP-associated factor TAFII55 (Chiang and Roeder 1995) and TFIIH (Blau et al. 1996; Parada and Roeder 1996). Upstream DNA-bound activators such as Sp1 have also been identified as possible co-activators of Tat (Jeang et al. 1993). In addition, a wide variety of other proteins that interact with Tat but whose role in transcription is somewhat unclear have also been identified (Nelbock et al. 1990; Desai et al. 1991; Zhou and Sharp 1996). Recently, Tat has been demonstrated to interact with the Pol II holoenzyme (Cujec et al. 1997). This large megadalton complex consists of core Pol II, a subset of general transcription factors (TFIIE, TFIIF, TFIIH), human SRBs (suppressors of mutations in RNA polymerase B), which confer the ability of the Pol II holoenzyme to respond to activators and proteins involved in chromatin remodeling (SWI/SNF) and DNA repair (Kim et al. 1994; Ossipow et al. 1995; Chao et al. 1996; Maldonado et al. 1996; Wilson et al. 1996).
TFIIH contains nine polypeptides [ERCC3, ERCC2, p62, p54, p44, CDK7 (MO15), cyclin H, MAT 1, and p34] (Drapkin and Reinberg 1994; Hoeijmakers et al. 1996). It contains a kinase activity that can phosphorylate the carboxy-terminal domain (CTD) of Pol II (Feaver et al. 1991; Lu et al. 1992). The kinase activity resides in the cyclin-dependent kinase 7 (CDK7) subunit (Feaver et al. 1994; Roy et al. 1994; Serizawa et al. 1995; Shiekhattar et al. 1995). In association with cyclin H, CDK7 forms the CDK-activating kinase (CAK) complex that phosphorylates CDKs involved in the regulation of the cell cycle. Association of MAT 1 with the CAK dimer stabilizes the complex and allows for the activation of CAK independently of the phosphorylation of CDK7 on the threonine at position 170 (Fisher and Morgan 1994; Fisher et al. 1995). Moreover, the CAK trimer is much more efficient at phosphorylating the CTD than the CAK dimer (Rossignol et al. 1997; Yankulov and Bentley 1997). The tripartite CAK can exist in three distinct complexes in cells. The majority is present as free CAK. However, CAK can also exist as a CAK–ERCC2 complex as well as in association with the core TFIIH (ERCC3, p62, p54, p44, and p34) (Drapkin et al. 1996; Reardon et al. 1996). The association of CAK with TFIIH confers kinase activity to TFIIH and renders it transcriptionally competent. Interestingly, the yeast homolog of CDK7, Kin28, is found only in a complex with TFIIH and is devoid of CAK activity (Cismowski et al. 1995). Instead, CAK activity resides in a novel protein called Civ1 or CAK1p (Kladis et al. 1996; Thuret et al. 1996).
The eukaryotic Pol II is unique among polymerases in that it contains multiple heptapeptide repeats of the sequence YSPTSPS, which together comprise the CTD (Dahmus 1994, 1995). A large number of kinases capable of phosphorylating the CTD in vitro have been identified. However, the functional relevance of these kinases remains unclear. To date, CDK7/cyclin H (TFIIH) and CDK8/cyclin C (human homologs of the yeast SRB10/SRB11) are the major kinases associated with transcription factors that can phosphorylate the CTD (Liao et al. 1995; Serizawa et al. 1995; Shiekhattar et al. 1995).
Pol II enters into the assembling transcription complex with its CTD unphosphorylated (IIA form). However, the CTD of elongating polymerases is highly phosphorylated (IIo form) primarily on its serine and threonine residues (Laybourn and Dahmus 1989, 1990). This observation led to the suggestion that the phosphorylation of the CTD is important for promoter clearance and for the processivity of Pol II. Numerous additional observations support this contention: (1) the CTD of polymerases paused on the Drosophila hsp 70 promoter before heat shock activation are hypophosphorylated (IIa), whereas those of actively elongating polymerases are hyperphosphorylated (IIo) (O’Brien et al. 1994); (2) inhibitors of CTD kinases inhibit promoter clearance and elongation of Pol II in vitro (Yankulov et al. 1995, 1996); (3) the kinase activity of TFIIH is required for the clearance of the DHFR promoter but not for the initiation of its transcription (Akoulitchev et al. 1995); and (4) mutations in the yeast Pol II CTD, the yeast homolog of CDK7 (Kin 28), or SRB2, a subunit of the Pol II holoenzyme, each inhibit the processivity of Pol II in vivo (Akhtar et al. 1996). The identification of CTD-binding proteins with homology to serine/arginine-rich (SR) proteins suggest that the phosphorylation of the CTD might also provide a mechanism for coupling transcription and pre-mRNA processing (Yuryev et al. 1996).
Recently, we and others demonstrated that the CTD is absolutely required for the production of long transcripts from the HIV LTR in vitro and in vivo (Chun and Jeang 1996; Okamoto et al. 1996; Parada and Roeder 1996; Yang et al. 1996). In contrast, basal transcription and the production of short transcripts from the HIV LTR is independent of the CTD. Together these results suggested an important role for the CTD in the function of Tat.
Given the critical role of the CTD for the function of Tat, and the fact that TFIIH is part of the Pol II holoenzyme that interacts with Tat, we examined the possibility that Tat might interact with TFIIH. In this study, we report that Tat co-immunoprecipitates with TFIIH/CAK complexes in vivo. Furthermore, Tat binds to recombinant CAK in vitro and this association is mediated by direct interactions between Tat and CDK7. Functional relevance for this association is provided by experiments that demonstrate that Tat potentiates the ability of TFIIH and CAK to phosphorylate the CTD of Pol II. Finally, using specific pseudosubstrate peptides, which inhibit the activity of the CDK7 kinase, we demonstrate that the phosphorylation of the CTD by CDK7 is required for Tat transactivation in vitro and in vivo.
Results
Tat interacts with TFIIH in vivo
We demonstrated recently that Tat interacts with the Pol II holoenzyme (Cujec et al. 1997). Given that TFIIH is present in the Pol II holoenzyme (Ossipow et al. 1995; Maldonado et al. 1996) and the essential role that the phosphorylation of the CTD by TFIIH is believed to have in the processivity of Pol II, we examined whether Tat can associate with TFIIH independently of the Pol II holoenzyme. To determine whether Tat interacts with TFIIH in vivo, hemagglutinin (HA)-tagged wild-type and mutant Tat proteins were expressed in COS cells. Immunoprecipitations were done with an antibody that is specific for ERCC3 (αERCC3), the largest subunit of TFIIH (Drapkin et al. 1996). Because TFIIH is part of the Pol II holoenzyme, coimmunoprecipitations were done under stringent conditions (0.5 m NaCl and 1% Triton X-100), which were expected to dissociate TFIIH from the Pol II holoenzyme. To confirm that only TFIIH, and not the entire Pol II holoenzyme, was immunoprecipitated in these experiments, αERCC3 immunoprecipitates were also probed with antibodies against the largest subunit of the core Pol II (RPB1) (Thompson et al. 1989). As shown in Figure 1A, αERCC3 antibodies efficiently precipitated ERCC3 and CDK7 subunits of TFIIH, but failed to precipitate Pol II (Fig. 1A, lane 3). As a control, antibodies against the transcriptional activator CIITA (C) (Steimle et al. 1993) failed to coimmunoprecipitate any of the transcription factors (Fig. 1A, lane 2). To determine if Tat could bind to TFIIH in vivo, αERCC3 immunoprecipitates were probed with αHA antibodies. As shown in Figure 1B, although equivalent amounts of wild-type (Tat) and mutant (C30G and K41A) Tat proteins were expressed in COS cells (Fig. 1B, left three lanes), only the wild-type Tat was coimmunoprecipitated by the αERCC3 antibodies (Fig. 1B, right three lanes). Again, antibodies against the transcriptional activator CIITA (C) failed to coimmunoprecipitate Tat (Fig. 1B). Similar results were obtained when using αCDK7 antibodies (Fig. 1C). Together, these results demonstrate that Tat can interact with TFIIH in the absence of both the Pol II holoenzyme and TAR in vivo.
Figure 1.

Tat associates with TFIIH in the absence of the Pol II holoenzyme in vivo. (A) Total cell lysates (TCL) from COS cells were immunoprecipitated with αCIITA (lane 2) or αERCC3 antibodies (lane 3). One-third of the lysate was used as the input control (lane 1). Samples were separated by SDS-PAGE, transferred to membranes, and probed with the antibodies indicated on the left. (B) COS cells expressed HA-tagged wild-type (Tat) or mutant (C30G or K41A) Tat proteins. Cell lysates were immunoprecipitated with either the αCIITA (C) antibody or the αERCC3 antibody (αERCC3). Equal amounts of the lysates were loaded as input controls. Samples were processed as in A and probed with the αHA antibody. (C) Nuclear extracts (NE) or lysates from cells expressing HA-tagged wild-type Tat (Tat) or mutant Tat (mTat = K41A) were immunoprecipitated with the αCDK7 antibody. One-half of these cell lysates were loaded as input controls (lanes 1,2). Samples were processed and blots probed with αHA antibodies as described above.
Tat Binds to CDK7 in vitro
Although we could detect readily an interaction between Tat and purified TFIIH in vitro (Parada and Roeder 1996; data not shown), repeated attempts to detect specific interactions between Tat and ERCC3, ERCC2, or p62 subunits of TFIIH were unsuccessful. Of these proteins, only p62 bound to Tat and all our mutant Tat proteins (data not shown). Therefore, we tested whether Tat could bind to recombinant CAK in vitro (Fig. 2). Recombinant wild-type and mutant Tat proteins were attached to streptavidin agarose beads by virtue of streptavidin-binding peptides at their 3′ termini (Cujec et al. 1997). CDK7, cyclin H, and MAT 1 were coexpressed in insect cells by baculovirus infection. Extracts were purified initially over a HiTrap chelating column and then an anion exchange column (HiTrap Q). Relevant fractions were purified further by gel filtration and concentrated on a second HiTrap Q column (see Materials and Methods). The resulting CAK preparations were >99% pure and were subsequently used for crystallization studies (data not shown). CAK was incubated with streptavidin–agarose beads alone (C) or with streptavidin–agarose beads containing wild-type (Tat) or mutant (mTat = K41A) Tat proteins bound to them. After washing, the presence of CAK was monitored by Western blotting using αcyclin H antibodies. As shown in Figure 2, wild-type Tat (Fig. 2, lane 4), but not mutant Tat (Fig. 2, lane 5), bound to CAK very efficiently, as at least half of the input CAK was retained on Tat beads (Fig. 2, cf. lanes 2 and 4 with 5). Western blotting with αCDK7 and αMAT 1 antibodies revealed the presence of the other CAK subunits as well (data not shown). Tat proteins containing other debilitating mutations in their activation domains (C22G or C30G) (Kuppuswamy et al. 1989; Madore and Cullen 1993; Cujec et al. 1997) also failed to bind recombinant CAK in vitro (data not shown).
Figure 2.

Tat associates with recombinant CAK in vitro. Recombinant CAK trimer (50 ng) was incubated with the wild-type (Tat) or mutant (mTat = K41A) Tat proteins (1 μg) bound to streptavidin–agarose beads or with streptavidin–agarose beads alone as the control (C). Nuclear extract (10 μg, lane 1) or CAK (50 ng, lane 2) were loaded as input controls. After washing, the streptavidin beads and controls were subjected to SDS-PAGE, transferred to membranes, and probed with the αcyclin H antibody.
We next tested the ability of each of the individual CAK subunits to bind to Tat. CDK7, cyclin H, and MAT 1 were labeled with [35S]methionine by in vitro translation and CAK dimers or trimers were allowed to form. Complexes were immunoprecipitated with αCDK7 antibodies bound to protein-A–Sepharose beads. Immunoprecipitated complexes were then incubated with [γ-32P]ATP-labeled Tat and the ability of CDK7 beads to retain Tat was evaluated. As demonstrated in Figure 3, equal amounts of Tat were retained on these beads regardless of whether CDK7 was present alone (Fig. 3, lane 6), as a dimer with cyclin H (Fig. 3, lane 9), or as the CAK trimer (Fig. 3, lane 12). Although equivalent amounts of wild-type (T) or mutant (mT) Tat proteins were used in these experiments (Fig. 3, lanes 13,14), the mutant Tat did not bind to any CDK7 complexes. As expected, addition of MAT 1 to the CDK7–cyclin H dimers stabilized the CAK complex (Fisher et al. 1995) and increased the amount of CAK immunoprecipitated by αCDK7 antibodies (Fig. 3, cf. lane 7 with 10). These results suggest that the reticulocyte lysate used for in vitro translation contained minimal amounts of endogenous cyclin H or MAT 1 proteins that could associate with the introduced CDK7 monomer or CDK7–cyclin H dimer and form the tripartite CAK complex. Similar experiments failed to reveal specific interactions between Tat and cyclin H or MAT 1 in the absence of CDK7 (data not shown). Together these results demonstrate that Tat can interact with CDK7 directly and that this interaction leads to the association of Tat with the higher order CAK and TFIIH complexes.
Figure 3.

Tat associates with recombinant CDK7 in vitro. CDK7, cyclin H, and MAT 1 (p36) were labeled with 35S by translation in vitro. CDK7 alone (lanes 4–6), in combination with cyclin H (lanes 7–9), or as a mixture with cyclin H and MAT 1 (lanes 10–12), was immunoprecipitated with αCDK7 antibodies bound to protein-A–Sepharose beads. Immunoprecipitates were divided into three fractions. Whereas one-third served as a control for immunoprecipitations (lanes 4,7,10), the remaining two-thirds were incubated with wild-type (T) or mutant (mT = K41A) Tat proteins labeled at their 3′ termini with [γ-32P]ATP using the catalytic subunit of cAMP-dependent heart muscle kinase. After washing, the beads were subjected to SDS-PAGE. Gels were dried and visualized by autoradiography. One-fifth of the wild-type and mutant Tat proteins used in the binding reactions were loaded as the input controls (lanes 13,14).
Tat increases the ability of CAK to phosphorylate the CTD
Having demonstrated a strong affinity between Tat and CAK, we wished to determine whether Tat affected the ability of CAK to phosphorylate the CTD of Pol II. Wild-type or mutant CAK/TFIIH complexes were immunoprecipitated from lysates of cells that stably expressed HA-tagged wild-type CDK7 or its kinase-deficient variant (D155A). Although this mutation (D155A) abolishes the kinase activity of CDK7, it does not affect its ability to bind to cyclin H and MAT 1 (D.O. Morgan, unpubl.). Immunoprecipitates were incubated for increasing lengths of time with recombinant CTD and wild-type or mutant Tat proteins (Fig. 4A). The phosphorylation of the CTD by casein kinase (CK) was used as a marker for the electrophoretic mobility of the CTD (Fig. 4A, lane 1). Because casein kinase only phosphorylates the CTD at one site (Dahmus 1996), only the hypophosphorylated form of the CTD is apparent (Fig. 4, lane 1). Importantly, this control demonstrates that only the hypophosphorylated form of the CTD (CTDa) is present in our protein preparations. Wild-type but not mutant (mTat) Tat proteins increased the ability of immunoprecipitated CAK/TFIIH complexes to phosphorylate the hypophosphorylated (CTDa) and hyperphosphorylated (CTDo) forms of the CTD. The effect of Tat was evident after 20 min and increased throughout the duration of the experiment (Fig. 4A, cf. lanes 2–5 with 6–9). Interestingly, Tat did not affect the ability of CAK/TFIIH complexes to autophosphorylate cyclin H. As a control for the specificity of our immunoprecipitations, kinase-deficient CAK/TFIIH complexes (M) were also immunoprecipitated. As expected, these complexes failed to phosphorylate the CTD and cyclin H (Fig. 4A, lane 10).
Figure 4.

Tat increases the ability of CAK to phosphorylate the CTD of Pol II. (A) Wild-type HA-tagged CDK7 (CDK7), or a kinase-deficient mutant of CDK7 (M) were immunoprecipitated from stably expressing HeLa cells. GST–CTD protein (25 ng) was incubated with casein kinase (CK, 25 ng), or CDK7 immunoprecipitates in the presence of wild-type (Tat) or mutant (mTat = K41A) Tat proteins and γ-ATP. Kinase reaction products were subjected to SDS-PAGE, and gels were visualized by autoradiography after drying. Positions of the hypophosphorylated (CTDa) and hyperphosphorylated (CTDo) GST–CTD fusions, as well as cyclin H are indicated on the left. (B) GST–CTD was incubated with recombinant CAK (50 ng) in the presence of wild-type (Tat) or mutant (mTat = K41A) Tat proteins in a kinase reaction. Products were processed as described above. (C) Purified Pol II was incubated with CAK in the presence of wildtype (Tat) or mutant (mTat = K41A) Tat proteins in a kinase reaction. Products were processed as described above. (D) CyclinAΔ171/CDK2HA complexes bound to protein-A–Sepharose beads were incubated with CAK (50 ng) in the presence of wild-type (Tat) or mutant (mTat = K41A) Tat proteins. After washing the beads were incubated with histone H1 and kinase reactions allowed to proceed for 1 hr. Products were processed as described above. Position of labeled histone H1 (H1) is indicated.
Because approximately 10% of CAK is part of TFIIH, and the rest exists as a free complex (Drapkin and Reinberg 1994; Fisher and Morgan 1994; Fisher et al. 1995), immunoprecipitations with αCDK7 antibodies would be expected to yield both CAK and some TFIIH. To test directly whether Tat could affect the ability of CAK to phosphorylate the CTD, recombinant CAK was incubated with the CTD in the presence or absence of wild-type or mutant Tat proteins. As demonstrated in Figure 4B, wild-type Tat (Tat) was much more effective at stimulating the ability of CAK to phosphorylate the CTD than the mutant Tat protein (mTat) (Fig. 4B, cf. lanes 3 and 4 with 2, 5, and 6). The wild-type Tat also increased the ability of CAK to phosphorylate highly purified preparations of Pol II (Fig. 4C). In neither case did Tat affect the phosphorylation of cyclin H. The CDK7 monomer was incapable of phosphorylating the CTD (data not shown). By demonstrating that Tat can increase the kinase activity of CDK7, we provide functional relevance for the binding results presented in the previous section.
To test for the specificity of these effects of Tat, we next examined whether Tat affected the ability of CAK to phosphorylate cyclinAΔ171/CDK2HA complexes. CyclinAΔ171/CDK2HA complexes were bound to protein-A–Sepharose beads and incubated with CAK in the presence of wild-type or mutant Tat proteins. Because the ability of cyclinAΔ171/CDK2HA to phosphorylate histone H1 is dependent on the phosphorylation of CDK2 by CAK, the phosphorylation of histone H1 can be used as a measure of CAK activity (Fisher and Morgan 1994). As shown in Figure 4D, neither the wild-type (Tat) nor mutant Tat (mTat) affected the ability of CAK to phosphorylate cyclinAΔ171/CDK2HA. As expected, H1 was not phosphorylated if either cyclinAΔ171/CDK2HA or CAK were omitted from the reaction. Furthermore, these controls demonstrate that our Tat preparation did not contain a kinase activity capable of activating cyclinAΔ171/CDK2HA.
A CDK2 mutant peptide inhibits the phosphorylation of the CTD by CAK
The activity of some kinases can be inhibited by excess amounts of substrate peptides that contain a mutation in the amino acid phosphorylated by the kinase of interest (Poteet-Smith et al. 1997). Because CAK phosphorylates the threonine of CDK2 at position 160 (Fisher and Morgan 1994; Makela et al. 1994), we tested whether a CDK2 peptide (amino acids 149–170) having a T → A mutation (mC2p) could inhibit phosphorylation of the CTD by CAK. Increasing amounts of the CDK2 mutant peptide (mC2p) were added to kinase reactions containing recombinant CAK (0.02 μm) in the presence or absence of Tat. As demonstrated in Figure 5A, a 65-fold molar excess of mC2p (1.3 μm) inhibited the kinase activity of CAK by 50%, whereas a 650-fold excess of the peptide (13 μm) abolished almost all the activity of CAK. Interestingly, mCp inhibited both Tat-dependent and Tat-independent activity of CAK, as well as the phosphorylation of cyclin H. A randomized peptide (rC2p) having the same net charge and solubility as mC2p had no effect on the kinase activity of CAK (Fig. 5B). The mutant peptide (mC2p) did not affect the ability of CDK8/cyclin C to phosphorylate the CTD, or the activity of cyclinAΔ171/CDK2HA (data not shown).
Figure 5.

A CDK2 mutant peptide inhibits the phosphorylation of the CTD by recombinant CAK. (A) Increasing concentrations of a mutant CDK2 peptide (mC2p) were added to kinase reactions containing GST–CTD, and CAK in the presence (+) or absence (−) of Tat. Casein kinase was used as a control for the mobility of the labeled GST–CTD (lane 1). Kinase reactions were processed as described in Fig. 4. Positions of the hypophosphorylated (CTDa) and hyperphosphorylated (CTDo) GST–CTD fusions, as well as cyclin H are indicated on the left. (B) The randomized CDK2 peptide (rC2p) was added to kinase reactions as described in A. The labeled GST–CTD fusion protein is presented.
The mutant peptide inhibits Tat transactivation in vitro
Having demonstrated that Tat binds to CDK7, and that this interaction increases the ability of CAK to phosphorylate the CTD, we next examined more closely the role of CAK in Tat transactivation. We took advantage of the fact that mC2p could inhibit the CDK7-mediated kinase activity of CAK to examine the effect of this peptide on the function of Tat in vitro. As shown in Figure 6A, the addition of recombinant Tat to transcription reactions containing the wild-type HIV LTR as the template stimulated transcription 10-fold compared with basal levels (Fig. 6A, lanes 1,2). Increasing concentrations of mC2p selectively inhibited Tat transactivation. At a concentration of 65 μm, mC2p inhibited Tat transactivation to basal levels (Fig. 6A, lanes 9,10), whereas at 130 μm, transcription was abolished completely (Fig. 6A, lane 12). As a control, the randomized rC2p peptide did not affect Tat transactivation (Fig. 6B). In contrast, transcription in the absence of Tat was relatively unaffected by mC2p or rC2p, even at high concentrations of the peptide (130 μm) (Fig. 6A and B, cf. lane 1 with 11). The slight increase in basal transcription at low concentrations of mC2p or rC2p (Fig. 6, cf. lanes 5, 7, or 9 with 1 in A and B) suggests that these peptides might also have a nonspecific stabilizing effect on basal transcription. It is surprising that Tat transactivation in vitro is less sensitive to mC2p concentrations than CAK kinase activity. Perhaps high levels of CAK in nuclear extracts, or the nonspecific binding of the peptide to other proteins in these extracts, can explain the relatively high concentrations of mC2p required to affect Tat activity in the in vitro transcription reactions. Furthermore, it is also possible that CAK is less accessible to the peptide when complexed with TFIIH.
Figure 6.

A CDK2 mutant peptide inhibits Tat transactivation. (A) In vitro transcription reactions were done with linearized DNA templates containing the wild-type HIV LTR sequences in the presence (+) or absence (−) of Tat and increasing concentrations of the CDK2 mutant peptide (mC2p). Runoff transcripts (750 nucleotides) were resolved on a 5% polyacrylamide/urea sequencing gel and exposed to x-ray film after drying. (B) Increasing concentrations of the randomized CDK2 peptide (rC2p) were added to in vitro transcription reactions containing the wild-type HIV LTR sequences in the presence (+) or absence (−) of Tat. Runoff transcripts were processed as described in A. (C) Increasing concentrations of the mutant CDK2 peptide (mC2p) were added to in vitro transcription reactions containing linearized DNA templates containing the AdML promoter. Lane 7 was identical to lane 1 except that reactions were done in the presence of α-amanitin (αA = 2 μg/ml). Runoff transcripts (390 nucleotides) were processed as described in A. (D) Increasing concentrations of the mutant CDK2 peptide (mC2p) were added to in vitro transcription reactions containing linearized DNA templates containing the DHFR promoter as described in the experimental procedures. Lane 7 was identical to lane 1 except that reactions were done in the presence of α-amanitin (αA = 2 μg/ml). Runoff transcripts (390 nucleotides) were processed as described in A. The doublet corresponds to alternate transcription start sites. (E) Increasing concentrations of the randomized CDK2 peptide (rC2p) were added to in vitro transcription reactions as described in D.
To control for the possibility that mC2p might affect other transcription factors nonspecifically, we tested the effect of the peptide on transcription from adenovirus major late (AdML) and DHFR promoters. These promoters represent important controls because it has been demonstrated that transcription from the AdML promoter is independent of CDK7, whereas transcription from the DHFR promoter is absolutely dependent on the kinase activity of CDK7 (Akoulitchev et al. 1995). As shown in Figure 6C, increasing concentrations of mC2p had no effect on the transcription from the AdML promoter. In contrast, mC2p inhibited the transcription from the DHFR promoter by more than 50% at a concentration of 65 μm and completely at a concentration of 130 μm (Fig. 6D). The randomized peptide had no effect on transcription from the DHFR promoter (Fig. 6E). These results, together with studies on the inhibition of the kinase activity, suggest that the phosphorylation of the CTD by CDK7 is required for Tat transactivation in vitro.
CDK7 is required for Tat transactivation in vivo
To confirm that the kinase activity of CDK7 is required for Tat transactivation in vivo, increasing amounts of the mC2p peptide were co-electroporated into COS cells, in the presence or absence of Tat (see Materials and Methods for details). The reporter construct (pHIVΔKBCAT) consisted of the HIV LTR containing sequences encoding TAR, the initiator, the TATA box, and three Sp1-binding sites. Because transcription from the HIV LTR can be activated through NF-κB-binding sites during electroporation, the reporter construct lacked these sites (Tong-Starksen et al. 1987). Levels of specific transcripts were determined by the RNase protection assay. As documented extensively by our laboratory (Kao et al. 1987; Selby et al. 1989; Lu et al. 1993) and those of others (Ratnasabapathy et al. 1990; Sheldon et al. 1993), transcription from the HIV LTR in the absence of Tat gives rise to primarily short, nonpolyadenylated transcripts ∼55–59 nucleotides in length. On the other hand, in the presence of TAT, transcription is highly processive resulting in full-length polyadenylated transcripts that protect an 80-nucleotide-long RNA probe in our RNase protection assays. Consequently, although the levels of short transcripts are a measure of transcription initiation rates, the amount of long transcripts can be used to estimate the efficiency of transcription elongation. Importantly, because Tat does not affect transcription initiation rates, short transcripts also serve as useful internal controls for transfection efficiency and subsequent RNA manipulations. As presented in Figure 7A, the production of long transcripts from the HIV LTR was reduced dramatically in the presence of low concentrations of mC2p (5 μm), and abolished completely at a peptide concentration of 10 μm. In contrast, the production of short transcripts in the presence or absence of Tat was unaffected by mC2p, even at a peptide concentration of 20 μm. Although overall transcript levels appeared higher in the absence of the randomized peptide (Fig. 7B), the ratio of long to short transcripts remained the same regardless of the peptide concentration. Similarly, increasing concentrations of rC2p had no effect on transcription in the absence of Tat (data not shown).
Figure 7.
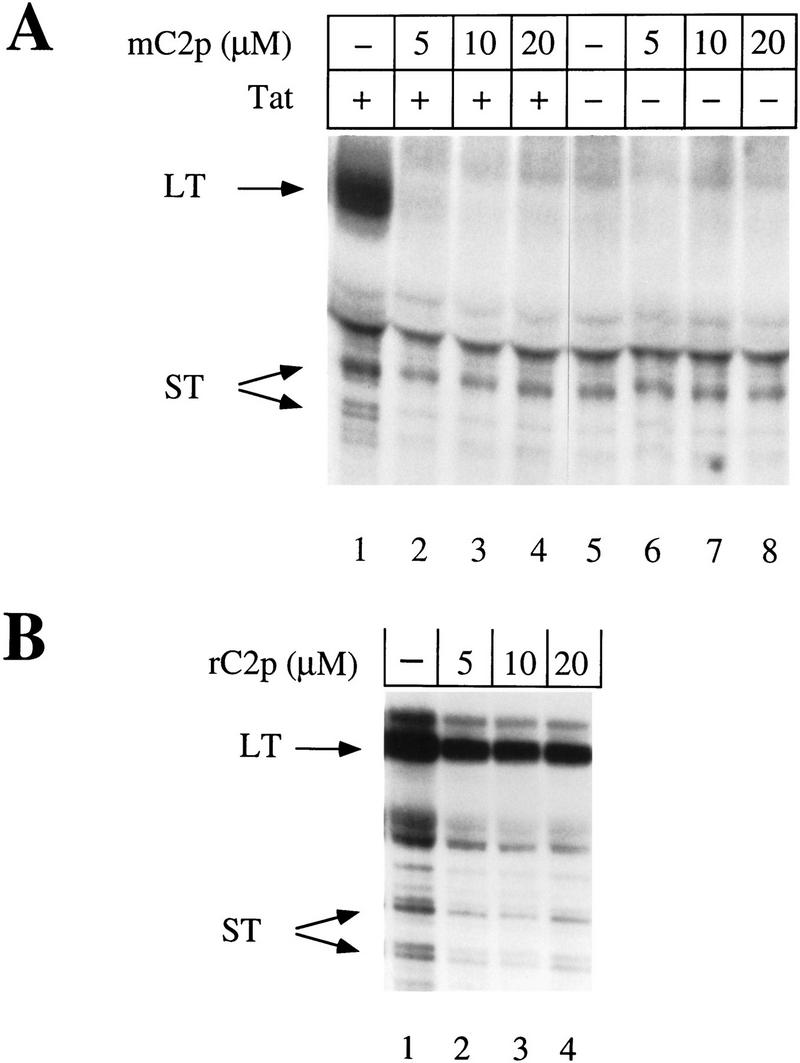
The mutant CDK2 peptide inhibits Tat transactivation in vivo. (A) Increasing concentrations of the mutant CDK2 peptide (mC2p) were cotransfected into COS cells with a reporter plasmid containing HIV LTR promoter sequences that lacked NF-κB-binding sites (pHIVΔKBCAT) and either functional (+) or nonfunctional (−) Tat. RNase protection assays were done with a probe 220 nucleotides long and hybridized to full-length transcripts of 80 nucleotides (LT) or prematurely terminated transcripts (ST) of 55–59 nucleotides (Okamoto et al. 1996). Protected fragments were resolved on a 11% polyacrylamide/urea sequencing gel and exposed to x-ray film after drying. (B) Increasing concentrations of the randomized CDK2 peptide (rC2p) were cotransfected into COS cells with a reporter plasmid containing the HIV LTR lacking NF-kB-binding sites (pHIVΔKBCAT) and functional Tat. RNase protection assays were done as in A.
To examine further the role of CDK7 on the activity of Tat in vivo, we overexpressed wild-type or a kinase-deficient mutant (D155A) of CDK7 in COS cells. RNase protection assays were used to quantify levels of transcripts from the HIV LTR or from an enhancerless promoter consisting of only a TATA box and four Sp1-binding sites (4XSp1). This promoter (4XSp1) was shown previously to be independent of the CTD (Gerber et al. 1995) and consequently should not require the kinase activity of CDK7. Transfection of HA-tagged Tat increased dramatically the ratio of long transcripts (LT) to short transcripts (ST) (Fig. 8, top panel). As expected, Tat did not affect transcription from the 4XSp1 promoter (Fig. 8, middle panel). The cotransfection of the wild-type CDK7–HA construct resulted in significant increase in the ratio of long transcripts (LT) to short transcripts (ST) compared with cells expressing only the endogenous CDK7. In contrast, the cotransfection of the kinase-deficient mutant of CDK7 (Mut) inhibited the production of long transcripts to levels below those observed in cells expressing endogenous CDK7, but had relatively little effect on the production of short transcripts. As expected, overexpression of wild-type or mutant CDK7 proteins had no effect on transcription from the 4XSp1 promoter. As additional controls, Western blotting revealed that similar levels of Tat–HA and CDK–HA proteins were expressed in these experiments (Fig. 8, bottom panels). Together with previous data, these results confirm that the transcriptional activation by Tat in vivo is dependent on the kinase activity of CDK7. In contrast, the synthesis of short transcripts from the HIV promoter is independent of the kinase activity of CDK7.
Figure 8.
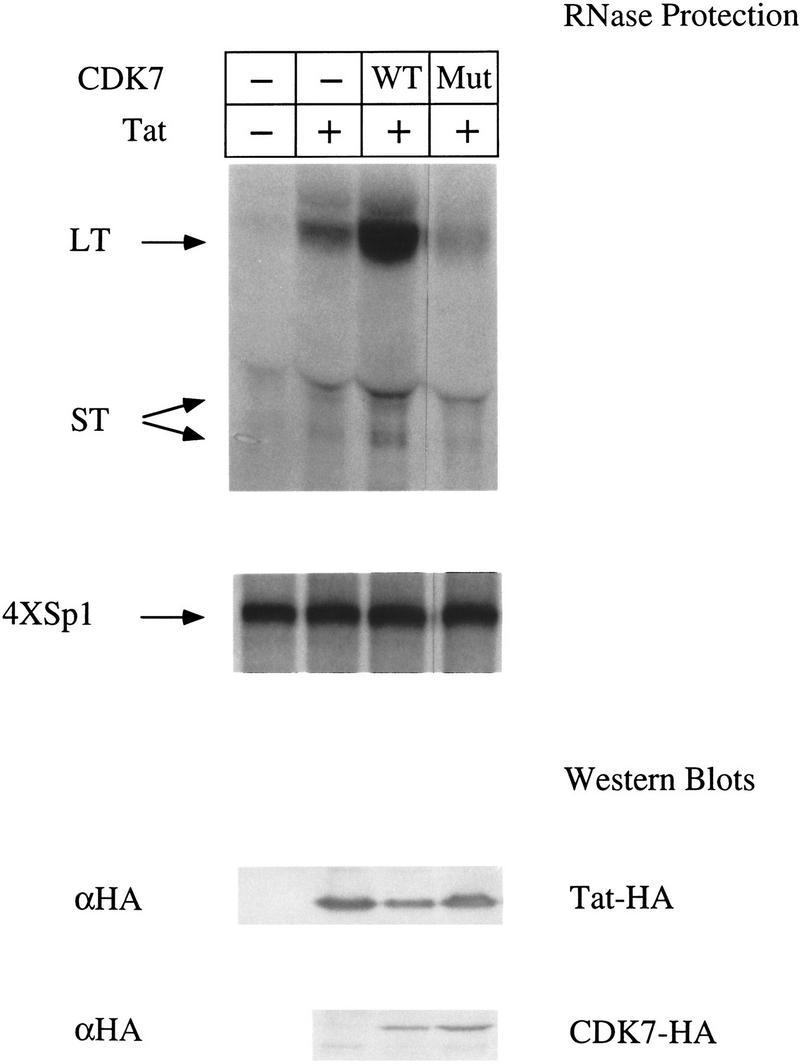
Over-expression of CDK7 affects Tat transactivation in vivo. COS cells were cotransfected with HIVSCAT and 4XSp1βglob reporter plasmids, and plasmids encoding HA-tagged functional (+) or nonfunctional (−) Tat, either alone or in combination with wild-type HA-tagged CDK7 (WT) or a kinase-deficient HA-tagged CDK7 (Mut). RNase protection assays were done using a HIV LTR probe that hybridized to full-length LTR–CAT transcripts of 80 nucleotides (LT) or prematurely terminated transcripts (ST) of 55–59 nucleotides (top panel) and a 216 nucleotide β-globin probe that hybridized to transcripts of 179 nucleotides (4XSp1) (Okamoto et al. 1996). (Bottom two panels) Lysates from transfected cells were subjected to SDS-PAGE, transferred to membranes and probed with αHA antibodies to detect Tat–HA and CDK7–HA protein levels.
Discussion
Our results indicate that Tat binds to CDK7 directly and that this interaction mediates the association between Tat and CAK or TFIIH. Furthermore, Tat stimulated the ability of recombinant CAK, as well as immunoprecipitated CAK/TFIIH complexes to phosphorylate the free CTD and purified Pol II. Finally, whereas basal transcription from the HIV LTR was independent of CDK7, the phosphorylation of the CTD by CDK7 was absolutely essential for Tat transactivation in vitro and in vivo. Together, our results reveal a cellular target of the activation domain of Tat, suggest that the interaction between Tat and its cellular counterpart is critical for the function of Tat, and suggest a model by which Tat increases the processivity of Pol II.
Tat was reported previously to bind to the p62 subunit of TFIIH (Blau et al. 1996; Parada and Roeder 1996). However in our hands, Tat mutants used in this study (C30G, K41A) bound to p62 as well as the wild-type Tat (data not shown). Instead, we found a strong and specific interaction between Tat and CDK7. This observation was supported by the finding that Tat bound to the recombinant CAK in vitro with high affinity and specificity. Despite this result, it is possible that additional contacts between Tat and other subunits of TFIIH stabilize further the binding of Tat to TFIIH in vivo. Furthermore, our finding that Tat can bind to TFIIH in vivo is consistent with our previous results demonstrating an interaction between Tat and the Pol II holoenzyme (Cujec et al. 1997).
Our initial experiments confirmed other studies that demonstrated that Tat can increase the ability of TFIIH to phosphorylate the CTD of Pol II (Parada and Roeder 1996; Garcia-Martinez et al. 1997). We extend these findings by demonstrating that Tat can also increase the ability of CAK to phosphorylate the CTD and Pol II. Although CAK was more efficient at phosphorylating free CTD than Pol II (compare the ratio of substrate phosphorylation with cyclin H phosphorylation in Figs. 4A and B), the effect of Tat was evident in both cases. It is possible that when associated with Pol II, the CTD is relatively inaccessible to CAK. Following the interaction of Pol II with TFIIH/TFIIE, SRBs, or TAR and TAR-binding proteins, the CTD of Pol II might become more accessible to phosphorylation. Although Gaynor and colleagues (Garcia-Martinez et al. 1997) reported recently that Tat has only a low affinity for CAK and that it fails to potentiate its kinase activity, these experiments were done using crude fractions from gel filtration columns and the authors themselves suggested that inhibitors in their fractions could mask potential interactions between Tat and CAK.
The CTD of Pol II has an important role in Pol II transcription (Dahmus 1995), in premRNA processing (Yuryev et al. 1996) and polyadenylation (McCracken et al. 1997). The phosphorylation of the CTD by TFIIH has an important role in the processivity of Pol II (Maldonado and Reinberg 1995). Evidence supporting this hypothesis comes from the observation that the CTDs of polymerases paused on the Drosophila hsp 70 promoter are relatively unphosphorylated, whereas those of actively elongating polymerases are high phosphorylated (O’Brien et al. 1994), and the fact that the inhibition of TFIIH abolishes transcriptional elongation (Yankulov et al. 1995; Akhtar et al. 1996). Therefore, it is remarkable that our results demonstrating that Tat potentiates the kinase activity of CAK illuminates further the pivotal role of TFIIH in phosphorylating the CTD and ensuring the processivity of Pol II. The mechanism by which phosphorylation of the CTD allows for increased processivity of Pol II is unknown. However, one might envision that the phosphorylation of the CTD results in conformational changes in Pol II, which in turn facilitate the escape of the DNA-bound polymerase from the assembled pre-initiation complex, increase the activity of its catalytic subunit, and allow for the binding of new transcription elongation factors.
Previous attempts to demonstrate that the kinase activity of TFIIH is required for Tat transactivation have centered on the use of kinase inhibitors (H8), ATP analogs (DRB), or immunodepletion experiments (Herrmann and Rice 1995; Parada and Roeder 1996). However, these approaches were limited by the absence of specific inhibitors to CDK7, and the possibility that the immunodepletion of TFIIH removed additional proteins or activities (3′ → 5′ helicase of ERCC3) required for the function of Tat. Although pseudosubstrates have been used previously to inhibit kinases such as protein kinase C (Poteet-Smith et al. 1997), we believe ours is the first report of a peptide being used to inhibit the enzymatic activity of a GTF. The use of a mutant CDK2 peptide to inhibit specifically the kinase activity of CDK7 was validated by the following: (1) the inhibition of the activity of CAK by mC2p in in vitro kinase assays; (2) the inhibition of transcription from the CDK7-dependent DHFR promoter; (3) the observation that mC2p did not affect the transcription from the CDK7-independent AdML promoter; and (4) the observation that mC2p had no effect on the kinase activity of either CDK8/cyclin C or cyclinAΔ171/CDK2HA. Consequently, we used this peptide to demonstrate that phosphorylation of the CTD by CDK7 is required for the function of Tat in vitro and in vivo. Overexpression of the wild-type and kinase-deficient CDK7 proteins in COS cells confirmed that CDK7 is essential for the production of long transcripts from the HIV LTR. Together these results extend our previous observations demonstrating that the CTD is required for Tat transactivation (Okamoto et al. 1996), and firmly establish that the HIV LTR is an enhancer-dependent promoter.
Based on these results we propose a model for how the function of Tat increases the processivity of Pol II (Fig. 9). Tat enters into the transcription complex by virtue of its association with the Pol II holoenzyme before docking of the complex onto the DNA template. Pol II then copies promoter proximal sequences and TAR is synthesized.
Figure 9.

A model of the mechanism by which Tat increases the processivity of Pol II. As depicted above, transcription from the HIV LTR can be divided into four stages: (1) assembly of the transcription complex onto the DNA template; (2) initiation of transcription; (3) promoter clearance; and (4) elongation of Pol II along the DNA template. In this schematic, the DNA template is depicted by a thick black line, the unphosphorylated form of the core Pol II (IIA) by an open oval with its unphosphorylated CTD as a thin curved line, and the nine subunits TFIIH as striped forms with the CDK7 as a solid circle. Tat is designated as an open circle and the transcription initiation site with an arrow. During assembly of the initiation complex, Tat, TFIIH and other components of the Pol II holoenzyme (omitted for the sake of simplicity) associate with the core Pol II onto the DNA template. Transcription initiates with the hydrolysis of ATP between the β and γ phosphates and the synthesis of the first phosphodiester bond. Nascent RNA (line with a large dot at its 5′ terminus) is synthesized following the promoter clearance. The CTD might be partially phosphorylated at this point and is depicted by a curved line with a partially thickened section. Following synthesis of TAR, Tat binds to the bulge region and is repositioned or modified such that it can increase the ability of TFIIH to phosphorylate the CTD. The highly phosphorylated form of Pol II (IIo), depicted as a shaded oval with its CTD as a thick curved line, is therefore rendered highly processive and can now efficiently transcribe the entire viral genome.
Following its interaction with TAR, Tat is repositioned and/or modified such that it increases the ability of CDK7 to phosphorylate the CTD. Tat might function in a catalytic manner by modifying TFIIH, it may reposition TFIIH in closer proximity of the CTD, or it might increase the length of time that TFIIH remains in contact with the CTD. In this regard it is intriguing that TFIIH remains associated with Pol II for only a relative short period of time following promoter clearance by Pol II (Zawel et al. 1995) and that core Pol II has been reported to interact with both Tat (Mavankal et al. 1996) and TAR (Wu-Baer et al. 1995). Although our data is entirely consistent with the one-step recruitment of Tat to the transcription complex via its interaction with the Pol II holoenzyme, it does not preclude the possibility that additional Tat is also recruited by nascent TAR (Keen et al. 1996). In this manner, other CTD kinases that are not part of the Pol II holoenzyme might be brought into the complex (Herrmann and Rice 1995; Yang et al. 1996). Synergistic effects of multiple kinases might ensure efficient phosphorylation of the Pol II CTD and optimal transcription elongation. The CTD of Pol II remains unphosphorylated despite the fact that TFIIH is present in the Pol II holoenzyme (Ossipow et al. 1995; Chao et al. 1996; Maldonado et al. 1996). Presumably conformational changes in TFIIH, Pol II, and perhaps other transcription factors are required before the kinase activity of TFIIH can be activated (Laybourn and Dahmus 1990; Peterson et al. 1992). The inability of TFIIH to phosphorylate the CTD before docking of the polymerase onto the DNA template would not be affected by the presence of Tat. The mechanism by which Tat increases the processivity of Pol II establishes important principles of transcriptional elongation, and suggests a mechanism by which pause sites in other promoters (c-myc) might be circumvented (Wright et al. 1994). Future work on template commitment, the ability of TAR decoys to deplete Tat at different stages of transcription, and the mapping of surfaces on CDK7 that interact with Tat will reveal further mechanistic details by which interactions between Tat and TAR stimulate the ability of TFIIH to phosphorylate the CTD of Pol II.
Materials and methods
Immunoprecipitations
The constructs pCMV–TATHA(Tat) and pCMV–TAT(C30G)HA containing wild-type or mutant Tat (C30G) fused to the influenza virus HA epitope tag (3′) were described previously (Cujec et al. 1997). A second HA-tagged mutant of Tat (K41A) was constructed by PCR-mediated mutagenesis. Briefly, PCR primers K41A (CATTGCTACGCGTGTTTCACAAGAgccGGCTTAGGC, lowercase letters denote mutation) and TAT3 (CAGTCTGAGTAGTTCGAAGAGTAG) were used to amplify a 112-bp fragment of Tat that was then cloned into the AflIII and HindIII sites of pCMV–TATHA. Both mutations (C30G, K41A) are in the activation domain of Tat and render Tat inactive without affecting its RNA-binding ability or protein expression levels (Kuppuswamy et al. 1989; Fig. 1). The Tat constructs (5 μg) were transfected into COS-7 cells by liptofectin (10 μl) according to the manufacturer’s recommendations (GIBCO BRL, Gaithersburg, MD). Approximately 36 hr after transfection the cells were lysed [50 mm HEPES–KOH at pH 7.8, 0.5 m NaCl 1% Triton X-100, 10 mm EDTA, 5 mm dithiothreitol (DTT), 0.1 mm phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml of aprotinin 10 μg/ml of leupeptin], and the supernatants immunoprecipitated with the indicated antibodies. Immunoprecipitates bound to protein-A–Sepharose beads were washed three times in CAK-binding buffer (lysis buffer + 10% glycerol). Washed beads were subjected to gradient (5%–15%) SDS-PAGE, transferred to Immobilon-NC membranes (Millipore, Bedford, MA) and reacted with the indicated antibodies. The proteins were visualized by enhanced chemiluminescence detection (Amersham, Arlington Heights, IL). The anti-HA antibody was purchased from Boehringer Mannheim (Indianapolis, IN), the CDK7 antibody from Santa Cruz Biotechnology (Santa Cruz, CA), and the cyclin H antibody from Upstate Biotechnology (Lake Placid, NY) and the Pol II antibody (αRPB 1) from Promega (Madison, WI). The CIITA antibody was from our laboratory (Steimle et al. 1993).
Purification of recombinant CAK trimer
Recombinant baculoviruses encoding human CDK7 (Fisher and Morgan 1994), MAT1 (Fisher et al. 1995), and an amino-terminally 6-Histidine-tagged version of cyclin H (Kim et al. 1996) were constructed as described. Sf9 insect cells (4 × 109) were coinfected with all three baculoviruses (multiplicity of infection for each virus: 5 PFU/cell), incubated 2 days at 28°C, and harvested by centrifugation. Cells were resuspended in 120 ml of buffer A (20 mm Na phosphate, 25 mm NaCl, 1 mm PMSF, 1 μg/ml of leupeptin, 2 μg/ml of aprotinin, 1 mm DTT at pH 7.4). NaCl concentration was raised to 300 mm and the lysate was clarified by centrifugation and loaded over tandem 5-ml Pharmacia HiTrap chelating columns loaded with CoCl2 and pre-equilibrated with buffer B (300 mm NaCl, 20 mm Na phosphate, 10% glycerol at pH 7.2). The column was washed with buffer B + 1 mm DTT and eluted with a linear gradient of 0–200 mm imidazole in buffer B + 1 mm DTT. Fractions containing the CAK trimer were pooled, diluted fourfold in buffer C (20 mm HEPES–NaOH, 1 mm EDTA, 10% glycerol, 1 mm DTT at pH 7.4), and loaded on a 5-ml Pharmacia HiTrap Q column pre-equilibrated with buffer C. The column was washed with buffer C and eluted with a linear gradient of 25–1000 mm NaCl in buffer C. Fractions containing the CAK trimer were pooled and subjected to gel filtration on a 125-ml Pharmacia Superdex 200 column pre-equilibrated in buffer C + 150 mm NaCl. Peak fractions were concentrated by ion exchange on a 1-ml Pharmacia HiTrap Q column. The pure CAK trimer (2 mg/ml; >99% homogeneous) was stored at −80°C.
In vitro binding assays
For the CAK-binding assays, wild-type Tat (STat) or mutant Tat (TatK41A) proteins containing at their 3′ ends both a phosphorylation site for the cAMP-dependent heart muscle kinase and a streptavidin binding peptide, were expressed in bacteria and bound to streptavidin–agarose beads as described previously (Cujec et al. 1997). Equilibrated Tat streptavidin-agarose beads were incubated with 250 μg of nuclear extract or 50 ng of purified CAK as described above. Pelleted beads were washed four times in CAK-binding buffer (see above), and processed as described in the immunoprecipitation protocol. For the CDK7-binding assays, Tat was eluted from the streptavidin–agarose beads with 0.8 m NaCl and 2 mm biotin and dialyzed into buffer D (25 mm HEPES–KOH at pH 7.6, 0.1 m KCl, 20% glycerol, 10 mm DTT, 0.1 mm EDTA). Approximately 50 ng of eluted Tat was labeled using 10 units of catalytic subunit of cAMP-dependent heart muscle kinase (Sigma P-2645) and 20 μCi of [γ-32P]ATP. Kinase reactions were performed in 50 μl of 0.1 m Tris at pH 7.5, 5 mm DTT, 0.5 m NaCl, 60 mm MgCl2. CDK7, cyclin H, and MAT 1 proteins were labeled with l-[35S]methionine (>1000Ci/mmole; Amersham) and the Promega TNT protein expression system. CAK complexes were formed in association buffer (20 mm HEPES–KOH at pH 7.6, 50 mm KCl, 10 mm DTT, 5 mm EDTA, and 10% glycerol) for 1 hr at 4°C and then immunoprecipitated with αCDK7 antibodies attached to protein-A–Sepharose beads. After binding, the beads were washed three times in the association buffer and then two times in CAK-binding buffer (see above). Labeled Tat (0.5 ng) was added to the CDK7 complexes and incubated for 1 hr at 25°C. Beads were washed again with binding buffer and then with PBS before loading onto SDS-PAGE (5%–20% gradient). After drying, the gels were visualized by autoradiography.
Kinase reactions
GST–CTD fusion proteins were expressed in Escherichia coli using pGCTD (a generous gift of W. Dynan) as described (Peterson et al. 1992). Fusion proteins were eluted from glutathione–Sepharose beads with 15 mm glutathione and purified by gel filtration on a S-300 column (Pharmacia, Piscataway NJ). Approximately 25 ng of the eluted GST–CTD fusion was used in each kinase reaction. Purified preparations of Pol II (a generous gift of C. Kane) were obtained as described (Hodo and Blatti 1977; Kerppola and Kane 1990) except that a Mono S column was used instead of a DEAE–5PW column in the final step of the purification. CAK/TFIIH complexes were immunoprecipitated from HeLa-cells that stably expressed HA-tagged wild-type CDK7 or a kinase-deficient mutant (D155A) under the control of a tetracycline-repressible promoter (Jin et al. 1996). Cells were lysed (50 mm HEPES–KOH at pH 7.6, 150 mm NaCl, 5 mm EDTA, 0.1% Triton X-100, 5 mm DTT, 0.2 mm PMSF, 1 mm NaF, 0.1 mm NaVO4 10 μg/ml of aprotinin, 1 μg/ml leupeptin) and immunoprecipitations done as described above. Typically, wild-type CAK/TFIIH complexes were immunoprecipitated from four 150-mm culture dishes after four days of growth in the absence of tetracycline (10 mg/ml). Immunoprecipitated beads were washed three times with lysis buffer and then two times with CTD–kinase buffer (20 mm Tris at pH 7.6, 50 mm KCl, 5 mm MgCl2, 2.5 mm MnCl2, 10 mm DTT). Reactions were supplemented with 10 μCi of [γ-32P]ATP and 50 μm of unlabeled ATP in a final reaction volume of 50 μl. Reactions were incubated for 1 hr at 30°C. In some experiments, recombinant CAK (50 ng) or casein kinase II (25 ng) (Upstate Biotechnology) was used as the source of kinase activity. Peptide concentrations were determined by the ESL Protein Assay (Boehringer Mannheim) system. The sequences of mC2p and rC2p are ARAFGVPVRTYaHEVVTLWYRA (lowercase letter denotes residue mutated from threonine) and HARTVGVWYRAEYARFVTPaVV, respectively. Histone H1 kinase assays were done as reported previously (Fisher and Morgan 1994).
In vitro transcription reactions
Run-off transcription reactions from the wild-type HIV LTR and AdML promoters linearized with NcoI and HindIII, respectively, were carried out as described (Okamoto et al. 1996; Cujec et al. 1997). Transcription reactions containing the DHFR promoter (500 ng) fused to a G-less cassette (linearized at NcoI) were supplemented with 3 mm (NH4)2SO4, 2% PEG 8000, 50 units of T1 RNase (Boehringer Mannheim), 500 units of RNase inhibitor (Boehringer Mannheim) and contained 5 μm instead of 40 μm of unlabeled UTP. Phosphocreatine, poly[d(I-C)], and poly[r(I-C)] were omitted from the reactions.
RNase protection assays
For the peptide inhibition studies 1 × 107 COS-7 cells were electroporated (500 μl) (Bio-Rad, Hercules, CA) at 210 V, 960 mF, using 2 μg of reporter DNA (pHIVΔKBCAT) containing HIV LTR sequences lacking NF-kB-binding sites, 2 μg of effector DNA (pSVTAT or pSVTATZX) and varying concentrations of mC2p or rC2p (1 μg/μl of solution). For the CDK7 overexpression studies, COS-7 cells were transfected with lipofectin using 2 μg of reporter DNA (HIVSCAT and 4XSp1), and 2 μg of effector DNA (pSVTAT or pSVTATZX) as described above. Vector alone or plasmids encoding HA-tagged wild-type CDK7 (SRα–CDK7–HA) or mutant HA-tagged CDK7 [SRα–CDK7(D155A)–HA] were cotransfected as indicated. Cells were incubated in the Opti-MEM medium for 5 hr and harvested 48 hr after transfection. Twenty micrograms of RNA was used for the RNase protection assays. To make the rabbit β-globin or the HIV LTR CAT probe, Sp6βTS or pGEMI/WT vectors were linearized with EcoRI and transcribed with Sp6 or T7 polymerases, respectively, to produce [α-32P]UTP-labeled RNA probes. Assays were performed as described (Okamoto et al. 1996), the protected fragments were separated on 11% polyacrylamide/urea sequencing gels and processed as outlined above.
Acknowledgments
We thank Michael Armanini for excellent secretarial assistance and members of our laboratories for comments on the manuscript. We thank Sasha Akoulitchev, Ron Drapkin, and Danny Reinberg for antibodies, TFIIH preparations, plasmids, and helpful suggestions. We are grateful to Ken Sakurabayashi and W. Dynan for the pGCTD construct, and to Caroline Kane and Rodney Weilbaecher for the purified core Pol II preparations. T.P.C. was funded by a Fellowship from the University-wide Taskforce on AIDS.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL MATIJA@ITSA.UCSF.EDU; FAX (415) 502-5081.
References
- Akhtar A, Faye G, Bentley DL. Distinct activated and non-activated RNA polymerase II complexes in yeast. EMBO J. 1996;15:4654–4664. [PMC free article] [PubMed] [Google Scholar]
- Akoulitchev S, Makela TP, Weinberg RA, Reinberg D. Requirement for TFIIH kinase activity in transcription by RNA polymerase II. Nature. 1995;377:557–560. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]
- Alonso A, Derse D, Peterlin BM. Human chromosome 12 is required for optimal interactions between Tat and TAR of human immunodeficiency virus type 1 in rodent cells. J Virol. 1992;66:4617–4621. doi: 10.1128/jvi.66.7.4617-4621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau J, Xiao H, McCracken S, O’Hare P, Greenblatt J, Bentley D. Three functional classes of transcriptional activation domains. Mol Cell Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll R, Peterlin BM, Derse D. Inhibition of human immunodeficiency virus type 1 Tat activity by coexpression of heterologous trans-activators. J Virol. 1992;66:2000–2007. doi: 10.1128/jvi.66.4.2000-2007.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao DM, Gadbois EL, Murray PJ, Anderson SF, Sonu MS, Parvin JD, Young RA. A mammalian SRB protein associated with an RNA polymerase II holoenzyme. Nature. 1996;380:82–85. doi: 10.1038/380082a0. [DOI] [PubMed] [Google Scholar]
- Chiang C-M, Roeder RG. Cloning of an intrinsic human TFIID subunit that interacts with multiple transcriptional activators. Nature. 1995;267:531–536. doi: 10.1126/science.7824954. [DOI] [PubMed] [Google Scholar]
- Chun RF, Jeang KT. Requirements for RNA polymerase II carboxyl-terminal domain for activated transcription of human retroviruses human T-cell lymphotropic virus I and HIV-1. J Biol Chem. 1996;271:27888–27894. doi: 10.1074/jbc.271.44.27888. [DOI] [PubMed] [Google Scholar]
- Cismowski MJ, Laff GM, Solomon MJ, Reed SI. KIN28 encodes a C-terminal domain kinase that controls mRNA transcription in Saccharomyces cerevisiae but lacks cyclin-dependent kinase-activating kinase (CAK) activity. Mol Cell Biol. 1995;15:2983–2992. doi: 10.1128/mcb.15.6.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cujec TP, Cho H, Maldonado E, Meyer J, Reinberg D, Peterlin BM. The human immunodeficiency virus transactivator Tat interacts with the RNA polymerase II holoenzyme. Mol Cell Biol. 1997;17:1817–1823. doi: 10.1128/mcb.17.4.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Does HIV-1 Tat induce a change in viral initiation rights? Cell. 1993;73:417–420. doi: 10.1016/0092-8674(93)90126-b. [DOI] [PubMed] [Google Scholar]
- Dahmus ME. The role of multisite phosphorylation in the regulation of RNA polymerase II activity. Prog Nucleic Acid Res Mol Biol. 1994;48:143–179. doi: 10.1016/s0079-6603(08)60855-7. [DOI] [PubMed] [Google Scholar]
- ————— Phosphorylation of the C-terminal domain of RNA polymerase II. Biochim Biophys Acta. 1995;1261:171–182. doi: 10.1016/0167-4781(94)00233-s. [DOI] [PubMed] [Google Scholar]
- ————— Phosphorylation of mammalian RNA polymerase II. Methods Enzymol. 1996;273:185–193. doi: 10.1016/s0076-6879(96)73019-7. [DOI] [PubMed] [Google Scholar]
- Desai K, Loewenstein PM, Green M. Isolation of a cellular protein that binds to the human immunodeficiency virus Tat protein and can potentiate transactivation of the viral promoter. Proc Natl Acad Sci. 1991;88:8875–8879. doi: 10.1073/pnas.88.20.8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapkin R, Reinberg D. The multifunctional TFIIH complex and transcriptional control. Trends Biochem Sci. 1994;19:504–508. doi: 10.1016/0968-0004(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Drapkin R, LeRoy G, Cho H, Akoulitchev S, Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc Natl Acad Sci. 1996;93:6488–6493. doi: 10.1073/pnas.93.13.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaver WJ, Gileadi O, Li Y, Kornberg RD. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell. 1991;67:1223–1230. doi: 10.1016/0092-8674(91)90298-d. [DOI] [PubMed] [Google Scholar]
- Feaver WJ, Svejstrup JQ, Henry NL, Kornberg RD. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell. 1994;79:1103–1109. doi: 10.1016/0092-8674(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Jin P, Chamberlin HM, Morgan DO. Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell. 1995;83:47–57. doi: 10.1016/0092-8674(95)90233-3. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez LF, Mavankal G, Neveu JM, Lane WS, Ivanov D, Gaynor R B. Purification of a Tat-associated kinase reveals a TFIIH complex that modulates HIV-1 transcription. EMBO J. 1997;16:2836–2850. doi: 10.1093/emboj/16.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Hagmann M, Seipel K, Georgiev O, West MA, Litingtung Y, Schaffner W, Corden JL. RNA polymerase II C-terminal domain required for enhancer-driven transcription. Nature. 1995;374:660–662. doi: 10.1038/374660a0. [DOI] [PubMed] [Google Scholar]
- Hart CE, Ou CY, Galphin JC, Moore J, Bacheler LT, Wasmuth JJ, Petteway Jr S, Schochetman G. Human chromosome 12 is required for elevated HIV-1 expression in human-hamster hybrid cells. Science. 1989;246:488–491. doi: 10.1126/science.2683071. [DOI] [PubMed] [Google Scholar]
- Herrmann CH, Rice AP. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J Virol. 1995;69:1612–1620. doi: 10.1128/jvi.69.3.1612-1620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodo III HG, Blatti SP. Purification using polyethylenimine precipitation and low molecular weight subunit analyses of calf thymus and wheat germ DNA-dependent RNA polymerase II. Biochemistry. 1977;16:2334–2343. doi: 10.1021/bi00630a005. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JHJ, Egly J-M, Vermeulen W. TFIIH: A key component in multiple DNA transactions. Curr Opin Genet Dev. 1996;6:26–33. doi: 10.1016/s0959-437x(96)90006-4. [DOI] [PubMed] [Google Scholar]
- Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, Fan H. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol. 1993;67:6224–6233. doi: 10.1128/jvi.67.10.6224-6233.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol. 1996;134:963–970. doi: 10.1083/jcb.134.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Peterlin BM. Control of RNA initiation and elongation at the HIV-1 promoter. Annu Rev Biochem. 1994;63:717–743. doi: 10.1146/annurev.bi.63.070194.003441. [DOI] [PubMed] [Google Scholar]
- Kao S Y, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by Tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Kashanchi F, Piras G, Radonovich MF, Duvall JF, Fattaey A, Chiang CM, Roeder RG, Brady JN. Direct interaction of human TFIID with the HIV-1 transactivator Tat. Nature. 1994;367:295–299. doi: 10.1038/367295a0. [DOI] [PubMed] [Google Scholar]
- Kato H, Sumimoto H, Pognonec P, Chen CH, Rosen CA, Roeder RG. HIV-1 Tat acts as a processivity factor in vitro in conjunction with cellular elongation factors. Genes & Dev. 1992;6:655–666. doi: 10.1101/gad.6.4.655. [DOI] [PubMed] [Google Scholar]
- Keen NJ, Gait MJ, Karn J. Human immunodeficiency virus type-1 Tat is an integral component of the activated transcription-elongation complex. Proc Natl Acad Sci. 1996;93:2505–2510. doi: 10.1073/pnas.93.6.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK, Kane CM. Analysis of the signals for transcription termination by purified RNA polymerase II. Biochemistry. 1990;29:269–278. doi: 10.1021/bi00453a037. [DOI] [PubMed] [Google Scholar]
- Kim K-K, Chamberlin HM, Morgan DO, Kim S-H. Three-dimensional structure of human cyclin H, a positive regulator of the CDK-activating kinase. Nature Struct Biol. 1996;3:849–855. doi: 10.1038/nsb1096-849. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell. 1994;77:599–608. doi: 10.1016/0092-8674(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Kladis P, Sutton A, Solomon M. The CDK-activating kinase (CAK) from budding yeast. Cell. 1996;86:553–564. doi: 10.1016/s0092-8674(00)80129-4. [DOI] [PubMed] [Google Scholar]
- Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G. Multiple functional domains of Tat, the transactivator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 1989;17:3551–3561. doi: 10.1093/nar/17.9.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laspia MF, Rice AP, Mathews MB. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell. 1989;59:283–292. doi: 10.1016/0092-8674(89)90290-0. [DOI] [PubMed] [Google Scholar]
- Laybourn PJ, Dahmus ME. Transcription-dependent structural changes in the C-terminal domain of mammalian RNA polymerase subunit IIa/o. J Biol Chem. 1989;264:6693–6698. [PubMed] [Google Scholar]
- ————— Phosphorylation of RNA polymerase IIa occurs subsequent to interaction with the promoter and before the initiation of transcription. J Biol Chem. 1990;265:13165–13173. [PubMed] [Google Scholar]
- Liao SM, Zhang J, Jeffery DA, Koleske AJ, Thompson CM, Chao DM, Viljoen M, van Vuuren HJ, Young RA. A kinase-cyclin pair in the RNA polymerase II holoenzyme. Nature. 1995;374:193–196. doi: 10.1038/374193a0. [DOI] [PubMed] [Google Scholar]
- Lu H, Zawel L, Fischer L, Egly J-M, Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- Lu X, Welsh TM, Peterlin BM. The human immunodeficiency virus type 1 long terminal repeat specifies two different transcription complexes, only one of which is regulated by Tat. J Virol. 1993;67:1752–1760. doi: 10.1128/jvi.67.4.1752-1760.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madore SJ, Cullen BR. Functional similarities between HIV-1 Tat and DNA sequence-specific transcriptional activators. Virology. 1995;206:1150–1154. doi: 10.1006/viro.1995.1041. [DOI] [PubMed] [Google Scholar]
- Madore SJ, Cullen BR. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 Tat function. J Virol. 1993;67:3703–3711. doi: 10.1128/jvi.67.7.3703-3711.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela TP, Tassan JP, Nigg EA, Frutiger S, Hughes GJ, Weinberg RA. A cyclin associated with the CDK-activating kinase MO15. Nature. 1994;371:254–257. doi: 10.1038/371254a0. [DOI] [PubMed] [Google Scholar]
- Maldonado E, Reinberg D. News on initiation and elongation of transcription by RNA polymerase II. Curr Opin Cell Biol. 1995;7:352–361. doi: 10.1016/0955-0674(95)80090-5. [DOI] [PubMed] [Google Scholar]
- Maldonado E, Shiekhattar R, Sheldon M, Cho H, Drapkin R, Inostroza JA, Rickert P, Lees E, Anderson CW, Linn S, Reinberg D. A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature. 1996;381:86–89. doi: 10.1038/381086a0. [DOI] [PubMed] [Google Scholar]
- Marciniak RA, Sharp PA. HIV-1 Tat protein promotes formation of more processive elongation complexes. EMBO J. 1991;10:4189–4196. doi: 10.1002/j.1460-2075.1991.tb04997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavankal G, Ou SH, Oliver H, Sigman D, Gaynor RB. Human immunodeficiency virus type 1 and 2 proteins specifically interact with RNA polymerase II. Proc Natl Acad Sci. 1996;93:2089–2094. doi: 10.1073/pnas.93.5.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson S D, Wickens M, Bentley DL. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- Nelbock P, Dillon PJ, Perkins A, Rosen CA. A cDNA for a protein that interacts with the human immunodeficiency virus Tat transactivator. Science. 1990;248:1650–1653. doi: 10.1126/science.2194290. [DOI] [PubMed] [Google Scholar]
- O’Brien T, Hardin S, Greenleaf A, Lis JT. Phosphorylation of RNA polymerase II C-terminal domain and transcriptional elongation. Nature. 1994;370:75–77. doi: 10.1038/370075a0. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Sheline CT, Corden J, Jones KA, Peterlin BM. Trans-activation by human immunodeficiency virus Tat protein requires the C-terminal domain of RNA polymerase II. Proc Natl AcadSci. 1996;93:11575–11579. doi: 10.1073/pnas.93.21.11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipow V, Tassan JP, Nigg EA, Schibler U. A mammalian RNA polymerase II holoenzyme containing all components required for promoter-specific transcription initiation. Cell. 1995;83:137–146. doi: 10.1016/0092-8674(95)90242-2. [DOI] [PubMed] [Google Scholar]
- Parada CA, Roeder RG. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature. 1996;384:375–378. doi: 10.1038/384375a0. [DOI] [PubMed] [Google Scholar]
- Peterson SR, Dvir A, Anderson CW, Dynan WS. DNA binding provides a signal for phosphorylation of the RNA polymerase II heptapeptide repeats. Genes & Dev. 1992;6:426–438. doi: 10.1101/gad.6.3.426. [DOI] [PubMed] [Google Scholar]
- Poteet-Smith CE, Shabb JB, Francis SH, Corbin JD. Identification of critical determinants for autoinhibition in the pseudosubstrate region of type I alpha cAMP-dependent protein kinase. J Biol Chem. 1997;272:379–388. doi: 10.1074/jbc.272.1.379. [DOI] [PubMed] [Google Scholar]
- Ratnasabapathy R, Sheldon M, Johal L, Hernandez N. The HIV-1 long terminal repeat contains an unusual element that induces the synthesis of short RNAs from various mRNA and snRNA promoters. Genes & Dev. 1990;4:2061–2074. doi: 10.1101/gad.4.12a.2061. [DOI] [PubMed] [Google Scholar]
- Reardon JT, Ge H, Gibbs E, Sancar A, Hurwitz J, Pan ZQ. Isolation and characterization of two human transcription factor IIH (TFIIH)-related complexes: ERCC2/CAK and TFIIH. Proc Natl Acad Sci. 1996;93:6482–6487. doi: 10.1073/pnas.93.13.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol M, Kolb-Cheynel I, Egly J-M. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997;16:1628–1637. doi: 10.1093/emboj/16.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Adamczewski JP, Seroz T, Vermeulen W, Tassan JP, Schaeffer L, Nigg EA, Hoeijmakers JH, Egly JM. The MO15 cell cycle kinase is associated with the TFIIH transcription-DNA repair factor. Cell. 1994;79:1093–1101. doi: 10.1016/0092-8674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Selby MJ, Bain ES, Luciw PA, Peterlin BM. Structure, sequence, and position of the stem-loop in TAR determine transcriptional elongation by Tat through the HIV-1 long terminal repeat. Genes & Dev. 1989;3:547–558. doi: 10.1101/gad.3.4.547. [DOI] [PubMed] [Google Scholar]
- Serizawa H, Makela TP, Conaway JW, Conaway RC, Weinberg RA, Young RA. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature. 1995;374:280–282. doi: 10.1038/374280a0. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Ratnasabapathy R, Hernandez N. Characterization of the inducer of short transcripts, a human immunodeficiency virus type 1 transcriptional element that activates the synthesis of short RNAs. Mol Cell Biol. 1993;13:1251–1263. doi: 10.1128/mcb.13.2.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. 1995;374:283–287. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- Thompson NE, Steinberg TH, Aronson DB, Burgess RR. Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J Biol Chem. 1989;264:11511–11520. [PubMed] [Google Scholar]
- Thuret J-Y, Valay J-G, Faye G, Mann C. Civ 1(CAK in vivo), a novel cdk-activating kinase. Cell. 1996;86:565–576. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]
- Tong-Starksen SE, Luciw PA, Peterlin BM. Human immunodeficiency virus long terminal repeat responds to T cell activation signals. Proc Natl Acad Sci. 1987;84:6845–6849. doi: 10.1073/pnas.84.19.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veschambre P, Simard P, Jalinot P. Evidence for functional interaction between the HIV-1 Tat transactivator and the TATA box binding protein in vivo. J Mol Biol. 1995;250:169–180. doi: 10.1006/jmbi.1995.0368. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Chao DM, Imbalzano AN, Schnitzler GR, Kingston RE, Young RA. RNA polymerase II holoenzyme contains SWI/SNF regulators involved in chromatin remodeling. Cell. 1996;84:235–244. doi: 10.1016/s0092-8674(00)80978-2. [DOI] [PubMed] [Google Scholar]
- Wright S, Lu X, Peterlin BM. Human immunodeficiency virus type 1 Tat directs transcription through attenuation sites within the mouse c-myc gene. J Mol Biol. 1994;243:568–573. doi: 10.1016/0022-2836(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Wu-Baer F, Sigman D, Gaynor RB. Specific binding of RNA polymerase II to the human immunodeficiency virus trans-activating region RNA is regulated by cellular cofactors and Tat. Proc Natl Acad Sci. 1995;92:7153–7157. doi: 10.1073/pnas.92.16.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Herrmann CH, Rice AP. The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J Virol. 1996;70:4576–4584. doi: 10.1128/jvi.70.7.4576-4584.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankulov KY, Bentley DL. Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J. 1997;16:1638–1646. doi: 10.1093/emboj/16.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankulov K, Yamashita K, Roy R, Egly J-M, Bentley D. The transcriptional elongation inhibitor 5,6-dichlor-1-β-D-ribofuranosylbenzimidazole inhibits transcription factor IIH-associated protein kinase. J Biol Chem. 1995;270:23922–23925. doi: 10.1074/jbc.270.41.23922. [DOI] [PubMed] [Google Scholar]
- Yankulov KY, Pandes M, McCraken S, Bouchard D, Bentley D. TFIIH functions in regulating transcriptional elongation by RNA polymerase II in Xenopus oocytes. Mol Cell Biol. 1996;16:3291–3299. doi: 10.1128/mcb.16.7.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc Natl Acad Sci. 1996;93:6975–6980. doi: 10.1073/pnas.93.14.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Kumar KP, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes & Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Sharp PA. Tat-SF1: Cofactor for stimulation of transcriptional elongation by HIV-1 Tat. Science. 1996;274:605–610. doi: 10.1126/science.274.5287.605. [DOI] [PubMed] [Google Scholar]