

Abstract
Traditionally, the single most unique feature of the immune system has been attributed to its capability to discriminate between self (e.g., host proteins) and nonself (e.g., pathogens). More recently, an emerging immunologic concept involves the notion that the immune system responds via a complex system for sensing signals of danger, such as pathogens or host-derived signals of cellular distress (e.g., ischemia), while remaining unresponsive to nondangerous motifs. Experimental studies have provided strong evidence that the production and signaling effects of extracellular adenosine are dramatically enhanced during conditions of limited oxygen availability as occurs during ischemia. As such, adenosine would fit the bill of signaling molecules that are enhanced during situations of cellular distress. In contrast to a danger signal, we propose here that extracellular adenosine operates as a countermeasure, in fact as a safety signal, to both restrain potentially harmful immune responses and to maintain and promote general tissue integrity during conditions of limited oxygen availability. Antioxid. Redox Signal. 15, 2221–2234.
Introduction
Ischemia is a pathologic condition that occurs in the setting of a wide variety of diseases, including myocardial infarction (26), acute kidney injury (AKI) (43), and intestinal ischemia-reperfusion injury (50). Similarly, ischemia reperfusion injury of the graft is an important problem during solid organ transplantation surgery, including the kidneys, the lungs, the heart, the pancreas, and the liver (28). During an ischemic event, disruption of the arterial blood supply for a vascular bed or a specific organ causes an imbalance between oxygen and nutrition supply and demand, resulting in profound tissue hypoxia (30). Particularly, in organs with a blood supply system that is supported by end arteries as opposed to a rich collateral system of blood supply, an ischemic event will take a particularly severe course (30). If blood flow can be restored, the ischemic event is followed by a reperfusion phase that in many instances will further increase tissue injury and aggravate the immunologic response (122). Global organ ischemia can also occur in the setting of solid organ transplantation, where a donor organ is explanted and stored under cold conditions (cold ischemia time), rewarmed during the operation (warm ischemia time), and reperfused. Ischemia and reperfusion injury will result in an immunologic response that drives very robust elevations of inflammatory mediators (43–45), and the recruitment of inflammatory cells of the innate (37) or adaptive immune system (71) into the ischemic organ. For example, a recent study in patients undergoing kidney transplantation demonstrated that graft inflammation correlates with ischemia time, and that a Toll-like receptor (TLR)4 loss-of-function mutation confers improved kidney function after transplantation (69). This study gives an example that controlling pathologic inflammatory response occurring in an a sterile environment—as is the case during solid organ transplantation—represents an important therapeutic target. However, it is important to point out that while ischemia and ischemia-reperfusion injury will trigger an acute inflammatory response (66, 70), tissue hypoxia will also elicit adaptive and anti-inflammatory responses (12, 32, 39). In fact, the characterization of hypoxia-elicited anti-inflammatory signaling pathways is currently an area of intense investigation, as such pathways could represent ideal therapeutic approaches to treat pathologic inflammation during ischemia (19, 24, 25, 29, 33, 35).
At the most fundamental level, any insults to the physiological integrity of tissues will precipitate a coordinated cascade of processes that converge toward the limitation of further damage, preservation of residual functionality, and promotion of tissue repair. In as much as these processes engage the immune system, immune reactivity to suitable stimuli is a precondition for any further modulation of destructive, protective, and restorative mechanisms. The determinants of immune reactivity, as opposed to an absence of reactivity described by terms such as ignorance or tolerance, have long posed a conceptual challenge to immunologists, and here the proposed distinction between self and nonself constitutes one of the most fundamental and influential concepts in the science of immunology. However, among the more consequential challenges to the self/nonself discrimination as a driving force for immune reactivity (or absence thereof) is Polly Matzinger's danger hypothesis that stipulates an induction and regulation of immune responses as contingent upon the particular nature of the consequences arising from interactions between organisms and specific stimuli (77). Thus, immune reactivity is not limited to nonself but rather focused on the breakdown of constituents that pose potential harm to the host, and involves in addition to adaptive and innate immune responses, the specific context of affected tissues and organ systems (78, 102). The danger hypothesis nominally emphasizes the importance of potentially harmful stimuli and the subsequent generation of appropriate immune responses, yet unrestrained immunity can cause considerable damage itself and thus requires effective regulation of immune responses. As such, damage associated molecular pattern molecules are derived molecules that can initiate and perpetuate immune response in the noninfectious inflammatory response. They serve as the Signal 0 similar to Pathogen-associated molecular pattern molecules that drive initiation and perpetuation of an inflammatory response. For example, extracellular adenosine triphosphate (ATP) released from apoptotic cells can function as a find-me signal to attract inflammatory cells (particularly phagocytes), and the balance between extracellular ATP release and its conversion to adenosine is an important aspect of purinergic danger signaling (27). While the nature of specific danger signals is subject to a host of current investigations, we propose here that extracellular adenosine operates as a countermeasure, in fact as a safety signal, to both restrain potentially harmful immune responses and to maintain and promote general tissue integrity.
Extracellular adenosine is a signaling molecule that has an intimate relationship to tissue hypoxia. Hypoxia is a pathological condition in which the body as a whole (generalized hypoxia) or a region of the body (tissue hypoxia) is deprived of adequate oxygen supply. As specifically discussed in the present review, tissue hypoxia can occur in the context of ischemia, where a restriction in blood supply, generally due to factors in arterial blood vessels (e.g., a blood clot) causes tissue deprivation of oxygen and other metabolic supplies. As such, hypoxia or ischemia will enhance extracellular pathways that coordinate phosphohydrolysis-dependent adenosine production from its precursor molecules (22, 33, 48, 65, 94, 110, 112). Moreover, hypoxia will further elevate extracellular adenosine signaling by delaying adenosine transport and intracellular metabolism (17, 29, 73, 81, 82). In addition of the four known adenosine receptors (Ars; A1AR, A2AAR, A2BAR, and A3AR), both A2 receptors are transcriptionally induced by hypoxia (2, 67). In addition, extracellular adenosine signaling events have been implicated in modulating immune responses, particularly in the context of acute tissue hypoxia as occurs during ischemia (25, 26, 34, 36, 43–45, 47–52, 65, 100). In support of this hypothesis, several studies implicate extracellular adenosine signaling in dampening excessive inflammation and attenuation of collateral tissue damage during ischemia (Fig. 1) (41, 85, 104, 105, 107, 111). In this review, we discuss the transcriptional control and the specific pathways that drive extracellular adenosine accumulation and signaling during ischemia, and develop the hypothesis that extracellular adenosine may serve as an endogenous danger signal for the presence of hypoxia-elicited inflammation. Moreover, we discuss the possibility of targeting these pathways to treat specific diseases that involve ischemia-reperfusion injury.
FIG. 1.
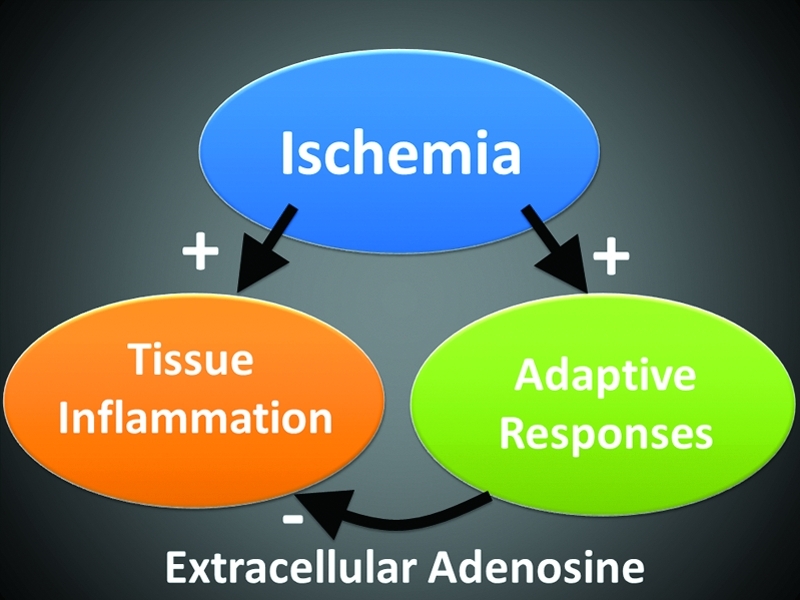
Tissue inflammation and protection during ischemia-reperfusion injury. Ischemia-reperfusion injury is associated with tissue inflammation and organ dysfunction. At the same time, tissue hypoxia within ischemic organs can elicit endogenous protective pathways that are critical in dampening inflammation and help to adapt ischemic organs to hypoxia. In the present review, we propose that extracellular adenosine signaling represents a hypoxia-elicited signaling mechanism that dampens inflammation and injury. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Adenosine: A Signaling Molecule
Adenosine belongs to the chemical group of nucleosides. Nucleosides are composed of a nucleobase—adenine in the case of adenosine—covalently linked to a ribose sugar (Fig. 2). Adenine is a purine—heterocyclic aromatic organic compound consisting of a pyrimidine ring fused to an imidazole ring. Purines are the most widely distributed kind of nitrogen-containing heterocycle in nature.
FIG. 2.
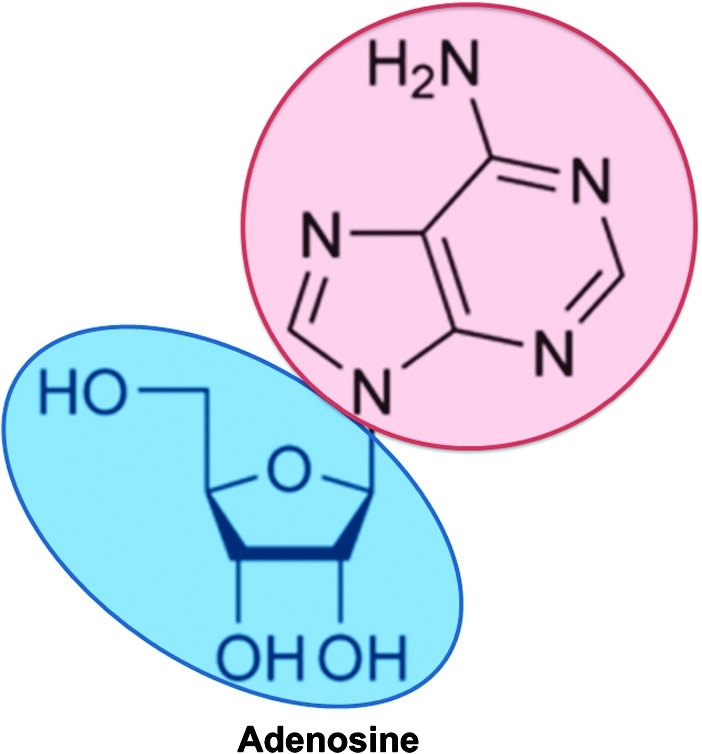
Adenosine. Adenosine belongs to the chemical group of nucleosides composed of the purine base adenosine (red) attached to a ribose sugar. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
In the intracellular compartment, adenosine is a central building block for molecules central to the storage of genetic information or energy metabolism. For example, adenosine is the molecular core of adenosine phosphates, such as 5′-adenosine monophosphate (AMP) or 5′-ATP (Fig. 3), both of them central to cellular energy metabolism and energy transfer. Adenosine derivatives like ATP are also implicated in the regulation of multiple cell functions, for example, by participating in the protein phosphorylation. Similarly, adenosine is part of the molecular backbone of RNA, and—in its deoxyribose form—of DNA. Extracellular adenosine functions mainly as a signaling molecule. Interestingly, this observation was made long before the discovery of RNA or DNA. In 1927, Drury and Szent-Gyorgyi from the University of Cambridge, United Kingdom, performed an experiment where they injected extracts from cardiac tissues intravenously into a whole animal. They were surprised to notice a transient disturbance of the cardiac rhythm and slowing of the heart rate (20). Following several purification steps, the authors were able to identify the biologically active compound of the extract as an adenine compound (20, 28). The term adenine was coined in 1885 by Nobel laureate Albrecht Kossel to describe a component originally derived from the pancreas (aden: gland, -ine commonly used in 19th century for derived substances). At present, four different ARs have been described that represent G-protein coupled membrane-spanning receptors. Signaling events through these receptors have been associated with a wide range of biological functions, such as the regulation of the heart-rate (64, 76, 92, 120), vascular tone (6), the sensation of pain, and the regulation of immune functions of the innate or adaptive immune system (28, 85, 104–107). For example, the signaling effects of extracellular adenosine signaling are utilized in a clinical setting when treating patients with supraventricular tachycardia. This cardiac arrhythmia is characterized by an elevated heart rate that originates from a center higher than the ventricles (supraventricular). To treat patients with this form of arrhythmia, adenosine is injected as a rapid intravenous bolus and results in a complete heart block that lasts ∼5–10 s. When the heartbeat recovers, the hope is that the arrhythmia is terminated, and a normal sinus rhythm prevails (18). Here, we will discuss how endogenous adenosine signaling is altered by conditions of ischemia or hypoxia, and how these pathways contribute to attenuating hypoxia-induced inflammation of ischemic organs.
FIG. 3.

Adenosine triposphate. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Extracellular Adenosine Stems from Extracellular Nucleotide Phosphohydrolysis
It is now well established that during conditions of limited oxygen availability or inflammation, such as occurs during ischemia and reperfusion injury, extracellular nucleotide levels are elevated. In fact, intracellular ATP levels are tightly controlled and relatively high (4–8 mM). During adverse conditions, ATP (or adenosine diphosphate [ADP]) can be released from essentially every type of cell in different manners. This can include leakage from cells that are undergoing apoptosis or necrosis during ischemia. This can also involve vesicular release as has been described as the key mechanism of ADP release from platelets (28). Moreover, there are many examples for specific pathways responsible for the controlled or regulated ATP release. For example, and given the association of polymorphonuclear leukocytes (PMNs) with adenine-nucleotide/nucleoside signaling in the inflammatory milieu, a study from our laboratory investigated the hypothesis that PMNs could function as a source of extracellular ATP. Initial studies using high-performance liquid chromatography and luminometric ATP detection assays revealed that PMNs release ATP through activation-dependent pathways. After excluding lytic ATP release, we used pharmacological strategies to reveal a potential mechanism involved in PMN-dependent ATP release (e.g., verapamil, dipyridamole, brefeldin A, 18-alpha-glycyrrhetinic acid, and connexin-mimetic peptides). These studies showed that PMN ATP release occurs through connexin 43 (Cx43) hemichannels in a protein/phosphatase-A-dependent manner. Findings in human PMNs were confirmed in PMNs derived from induced cx43−/− mice, whereby activated PMNs release <15% of ATP relative to littermate controls, whereas Cx43 heterozygote PMNs were intermediate in their capacity for ATP release. Taken together, these studies identify a role for Cx43 in activated PMN ATP release, therein contributing to the innate metabolic control of the inflammatory milieu (31). We made similar findings in studies of ATP release from hypoxic vascular endothelial cells (39). Other examples of extracellular ATP release include stretch-dependent ATP release from bladder umbrella cells (114), or glia cell-dependent ATP release via P2X7 receptors (108). Together and in conjunction with studies that measured ATP or ADP levels in ischemic organs, there is strong evidence that conditions of inflammation or hypoxia are associated with robust elevations of extracellular ATP levels (5–7, 41, 45, 48, 65, 104, 107).
Once released into the extracellular compartment, ATP/ADP is rapidly converted to adenosine (Fig. 2). Extracellular ATP/ADP-phosphohydrolysis is mainly achieved enzymatically by ecto-nucleoside triphosphate diphosphohydrolases (E-NTPDases), a recently described family of ubiquitously expressed membrane-bound enzymes (95, 124). The catalytic sites of plasma membrane expressed E-NTPDases 1–3 and 8 are exposed to the extracellular milieu, the others are intracellular (95). The presumptive biological role of plasma membrane-bound E-NTPDases (E-NTPDase 1–3 and 8) is to fine-tune extracellular nucleotide levels. For example, E-NTPDase1 (CD39) plays an important role in vascular endothelial function by blocking platelet aggregation via the phosphohydrolysis of ATP and ADP from the blood to maintain vascular integrity (75, 89). At the same time, E-NTPDase1 is also important in the maintenance of platelet functionality by preventing platelet P2Y1-receptor desensitization. As such, mice gene targeted for E-NTPDase1 (cd39−/− mice) show prolonged bleeding time with minimally perturbed coagulation parameters (38). Of significant physiological relevance is the fact that the E-NTPDase-end product, AMP, serves as the major metabolic substrate for 5′-ecto-nucleotidase (CD73)-dependent generation of extracellular adenosine (33). Thus, E-NTPDase expression and function are key-regulators of extracellular adenosine signaling.
The final step of extracellular adenosine generation is achieved via the CD73. The enzyme consists of a dimer of two identical 70-kDa subunits bound by a glycosyl phosphatidyl inositol linkage to the external face of the plasma membrane. In addition to its role in extracellular adenosine generation, CD73 has been used as a marker of lymphocyte differentiation. Due to its GPI-anchor, there are situations when CD73 can come off the membrane and recent studies have implicated soluble CD73 as a biomarker of human disease (84). Studies of gene-targeted mice for CD73 have found many phenotypic manifestations that are at mainly attributed to the enzymatic function of CD73 (8, 11, 16, 22, 26, 44, 49, 52, 68, 112). While CD73 is expressed very widely, highest expressional levels can be found in the heart, the liver, the kidney, the lungs, the brain, and particularly the intestine (112). Interestingly, the intestine is also a site that is highest in adenosine deaminase (ADA) activity (79). As such, the intestine appears to be the tissue with the highest capacity for adenosine production adenosine deamination. Taken together, these studies point toward a pathway for extracellular adenosine generation that involves extracellular release of nucleotides, particularly ATP and ADP, followed by a two-step enzymatic phosphohydrolysis via CD39 and CD73 resulting in the extracellular liberation of adenosine (Fig. 4).
FIG. 4.
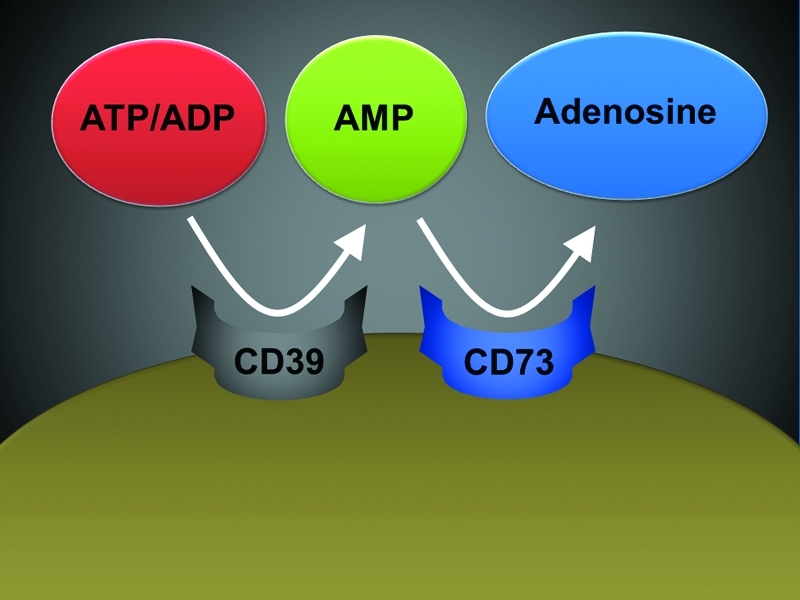
Extracellular adenosine generation. In the extracellular compartment, adenosine mainly stems from phosphohydrolysis of precursor molecules. Specifically, this involves conversion of ATP/ADP via the ecto-apyrase CD39 to AMP, followed by a second conversion step catalyzed by the 5′-ecto-nucleotidase CD73. ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CD39, ecto-apyrase, ENTPDase 1; CD73, 5′-ecto-nucleotidase. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Alternative Pathways for Extracellular Adenosine Generation
While studies in gene-targeted mice with defects in the nucleotide phosphohydrolysis pathway (cd39−/− or cd73−/− mice) demonstrate defects in extracellular adenosine protection and signaling, there are also alternative enzymatic pathways that have been implicated in the extracellular production of adenosine. For example, a recent study discovered that renal injury drives the release of 2′,3′-cyclic AMP (cAMP) (positional isomer of 3′,5′-cAMP) into the interstitium. This finding motivated the hypothesis that renal injury leads to activation of an extracellular 2′,3′-cAMP-adenosine pathway (i.e., metabolism of extracellular 2′,3′-cAMP to 3′-AMP and 2′-AMP, which are metabolized to adenosine, a retaliatory metabolite) (59, 60). These recent studies point toward the possibility that there may be alternative mechanisms for extracellular adenosine generation other than the CD39/CD73 pathway. While there is very strong genetic and pharmacologic evidence for a critical role of CD39/CD73 in extracellular adenosine generation and signaling, the physiologic relevance for extracellular cyclic adenosine nucleotides and their relationship to the CD39/CD73 pathway have yet to be determined. Moreover, there is evidence that alkaline phosphatase may play an important role in some tissues for extracellular adenosine generation (88).
Adenosine Receptors
Once generated in the extracellular space, adenosine can signal through four ARs: A1AR, A2AAR, A2BAR, and A3AR (28). ARs are G-protein-coupled receptors that can function through alterations in adenylate cyclase activity, thereby resulting in attenuation (A1AR and A3AR) or elevation (A2AAR and A2BAR) of cAMP levels (54, 55). Due to the fact that viable gene-targeted mice were successfully generated during the past decade, considerable progress could be made toward better defining the physiological roles of these receptors. For example, the A1AR has been implicated in the adenosine-induced heartblock. As such, intravascular adenosine treatment that causes heart block in wild-type mice does not alter the heart rates of A1AR−/− mice (64, 76, 93). An initial report of A2AAR−/− mice implicated this receptor in psychoactivity (72). This study found that A2AAR−/− mice were viable and bred normally. Their exploratory activity was reduced, whereas caffeine, which normally stimulates exploratory behavior, became a depressant of exploratory activity. Knockout animals scored higher in anxiety tests, and male mice were much more aggressive toward intruders (72). Later studies of A2AAR−/− mice focused on the role of this receptor in mediating inflammatory responses (85, 111). Studies in A2BAR−/− mice have implicated this receptor in attenuating inflammation and vascular injury (118, 119), and in hypoxia-elicited tissue adaptation (21, 26, 42, 47, 50, 100). Other reports indicate a potentially detrimental role of A2BAR signaling in chronic injury models, particularly during chronic obstructive lung disease, pulmonary fibrosis, or alcohol induced fatty liver disease (4, 87, 109, 123). Genetic studies in mice with targeted disruption of the A3AR have implicated this receptor in pro- and anti-inflammatory responses (80, 99). At present, studies in mice with tissue-specific deletion of individual ARs will allow to move field forward toward better understanding the tissue-specific contributions, and the timing of adenosine signaling events in models of disease (Fig. 5).
FIG. 5.

Examples for extracellular adenosine signaling. Extracellular adenosine can signal through four distinct adenosine receptors: A1AR, A2AAR, A2BAR, and A3AR. Examples for signaling events are displayed. AR, adenosine receptor. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Termination of Extracellular Adenosine Signaling
After activation of ARs, extracellular adenosine signaling is terminated in a two-step process. As first step, adenosine is transported from the extracellular toward the intracellular compartment. This is mainly achieved via nucleoside transporters (73), the equilibrative nucleoside transporters (ENTs, diffusion-limited channels) and concentrative nucleoside transporters (sodium-dependent transporters). Particularly, ENTs have been implicated in terminating extracellular adenosine signaling. For example, the ENT inhibitor dipyridamole is clinically used to for stress echocardiography, where inhibition of coronary ENTs results in elevated extracellular adenosine levels, and adenosine-dependent coronary vasodilatation. Similarly, enhanced extracellular adenosine concentrations may also account for the antithrombotic effects of oral dipyridamole treatment that is used in combination with aspirin to prevent recurrent stroke (98). Once within the cytosole, adenosine is rapidly metabolized to inosine via the ADA, or to AMP via the adenosine kinase. Together with adenosine transporters, its rapid intracellular metabolism accounts for adenosine's relatively short half-life. It is important to point out that both adenosine transport and metabolism represent important mechanisms to fine-tune extracellular adenosine signaling events. For example, attenuation of adenosine transport or metabolism can account for significant increases in extracellular adenosine concentrations and signaling effects (29, 73, 81, 82).
An additional mechanism of extracellular adenosine breakdown involves the ADA. While acute increases in adenosine are important to counterbalance excessive inflammation or vascular leakage, chronically elevated adenosine levels may be toxic. Thus, we reasoned that clearance mechanisms might exist to offset deleterious influences of chronically elevated adenosine. Guided by microarray results revealing induction of endothelial ADA mRNA in hypoxia, we used in vitro and in vivo models of adenosine signaling, confirming induction of ADA protein and activity—however, at later time points than hypoxia induction of CD73 or CD39 occurs. Further studies in human endothelia revealed that ADA-complexing protein CD26 is coordinately induced by hypoxia, effectively localizing ADA activity at the endothelial cell surface. Moreover, ADA surface binding was effectively blocked with glycoprotein 120 (gp120) treatment, a protein known to specifically compete for ADA-CD26 binding. Functional studies of murine hypoxia revealed that inhibition of ADA with deoxycoformycin (dCF) enhances protective responses mediated by adenosine (vascular leak and neutrophil accumulation). Analysis of plasma ADA activity in pediatric patients with chronic hypoxia undergoing cardiac surgery demonstrated a 4.1 ± 0.6-fold increase in plasma ADA activity compared with controls. Taken together, these results reveal induction of ADA as innate metabolic adaptation to chronically elevated adenosine levels during hypoxia. In contrast, during acute hypoxia associated with vascular leakage and excessive inflammation, ADA inhibition may serve as therapeutic strategy (32).
Effect of Hypoxia on Extracellular Adenosine Accumulation and Signaling
In the present review, we are developing the concept that adenosine functions as a safety signal in the context of disrupted tissue integrity, but discussed here with an emphasis on ischemia and reperfusion injury. To function as a safety signal during ischemia, adenosine would have to be produced or released during conditions that are characteristic for ischemia (such as limited oxygen availability). In contrast, in nondangerous situations, extracellular adenosine production should be attenuated. Consistent with this concept, we will present experimental evidence in the following paragraph that ischemia drives a molecular switch that ultimately results in ischemia-elicited enhancement of extracellular adenosine levels and signaling events.
Critical coordinators in this pathway are hypoxia-inducible factors (HIFs) (32, 39, 46, 53, 67, 70, 107, 116). HIFs are α/β heterodimers that bind hypoxia response elements (HREs) at target gene loci under hypoxic conditions (Fig. 6). In the presence of oxygen, HIFs are inactivated by posttranslational hydroxylation of specific amino acid residues within their α subunits. Prolyl hydroxylation promotes interaction with the von Hippel–Lindau protein (pVHL) E3 ubiquitin ligase complex and proteolytic inactivation by proteasomal degradation, whereas asparaginyl hydroxylation blocks coactivator recruitment. These hydroxylation steps are catalyzed by a set of nonheme Fe(II)-dependent and 2-oxoglutarate-dependent dioxygenases (prolyl hydroxylases, PHDs) whose absolute requirement for molecular oxygen confers sensitivity to hypoxia (101). HIF-1α was the original HIF isoform identified by affinity purification using oligonucleotides from the erythropoietin locus (115), whereas HIF-2α and HIF-3α were identified by homology searches or screens for interaction partners with HIF-1β. HIF-3α is the more distantly related isoform and, in certain splicing arrangements, encodes a polypeptide that antagonizes HRE-dependent gene expression. However, HIF-1α and HIF-2α are closely related, and both activate HRE-dependent gene transcription (117). Nevertheless, knockout studies in mice demonstrate that HIF-1α and HIF-2α play nonredundant roles, and inactivation of each one results in a distinctly different phenotype. This may result, in part, from differences in tissue-specific and temporal patterns of induction of each isoform (57, 101), but, not uncommonly, both isoforms are expressed within a given cell type, and the results of several studies suggest that HIF-1α and HIF-2α may have distinct transcriptional targets (90, 91). For example, the transcription of genes encoding enzymes that operate in a coordinated way in the glycolytic pathway appears to be driven by HIF-1α and not HIF-2α (58).
FIG. 6.

HIF-dependent alteration of gene expression during hypoxia. Under normoxic conditions, hydroxylation of proline residues within the α-subunit promotes HIF-α association with the von Hippel Lindau gene product, leading to HIF-α destruction via the ubiquitin/proteasome pathway. This is achieved by oxygen-sensing PHDs. In addition, hydroxylation of an asparagine residue by FIH blocks association with coactivators (e.g., p300). In hypoxia, substrate availability for PHDs and FIH is decreased, thereby preventing hydroxylation-dependent destruction of HIF-α, and allowing coactivator binding. Thus, HIF-α subunits (both HIF-1α and HIF-2α) escape proteolysis, dimerize with HIF-1β, recruit coactivators, and activate transcription via binding to the promoter region of hypoxia-responsive genes (hypoxia-responsive elements). This leads to the transcriptional activation of genetically controlled survival pathways, including angiogenesis, erythropoiesis, metabolism, mitochondrial functions, or adenosine generation and signaling. FIH, factor inhibiting HIF; HREs, hypoxia-responsive elements; HIF, hypoxia-inducible factor; PHDs, prolyl-hydroxylases; pVHL, von Hippel Lindau gene product. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
HIFs play a key role in coordinating enhanced adenosine signaling events during conditions of ischemia. This interaction occurs on multiple levels. In 2002, a series of studies from the research laboratory of Sean Colgan identified a previously unrecognized HIF-binding site within the promoter region of the CD73 gene (110). The authors performed studies with promoter constructs, site-directed mutagenesis, and HIF loss- and gain-of-function approaches to reveal a functional role of HIF-1α in the transcriptional induction of CD73. While the notion that hypoxia can enhance the effects of extracellular adenosine signaling has been long suspected, these studies provided the first mechanistic connection between hypoxia signaling and adenosine generation (110). Moreover and consistent with the concept of adenosine as a danger signal that dampens inflammation during ischemia, CD73-deficient mice that were subsequently generated by the laboratory of Linda Thompson show increased vascular leakage and inflammatory cell accumulations in multiple organs when exposed to conditions of limited oxygen availability (37, 112). In fact, these studies were among the first to provide genetic in vivo evidence that extracellular adenosine generation and signaling can counterbalance hypoxia-induced inflammation (Fig. 7) (33, 112).
FIG. 7.

Nucleotide metabolism and nucleoside signaling during hypoxia. Under conditions of limited oxygen availability—such as occurs during ischemia—diminished oxygen supply promotes the transcriptional induction of CD39, CD73, and ARs (particularly, the A2BAR). Under these conditions, extracellular nucleotides in the form of ATP or ADP can be released by different cell types, for example, activated neutrophils. Via a two-step enzymatic reaction involving CD39 and CD73, this process results in the liberation of extracellular adenosine. Adenosine generated in this fashion is available for activation of surface ARs, particularly the A2BAR. A2BAR signaling can elicit increases in intracellular cyclic AMP, resulting, for example, in enhanced barrier function. PMN, polymorphonuclear leukocyte (neutrophil). (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Serendipitously, a series of additional studies confirmed that HIFs regulate not only the pace-maker enzyme for extracellular adenosine generation, but also a whole molecular pathway responsible for ischemia-elicited enhancement of adenosine signaling. This occurs on multiple levels. In addition to CD73-dependent adenosine generation, HIFs drive AR expression during conditions of limited oxygen availability. As such, it is important to point out that several studies indicate that transcriptional induction of ARs represents an important adaptation to enhance extracellular adenosine signaling, for instance, during acute lung injury (23). As such, studies demonstrate that the A2BAR is selectively induced during hypoxia exposure of human endothelial or epithelial cells (33, 67), or in murine studies of ischemia-reperfusion injury (25, 26, 50). Again, mechanistic studies to define the transcriptional pathway of A2BAR induction determined a critical contribution of HIF-1α as transcriptional regulator (67). Interestingly, a more recent study also demonstrated that the A2AAR is induced by conditions of limited oxygen availability. Again, transcriptional studies determined a role of HIFs in this response. But in contrast to the A2BAR that is induced by HIF-1α, these studies determined a critical role of HIF-2α in A2AAR induction during conditions of limited oxygen availability (2).
In addition to alterations of AR expression, HIFs also coordinate a transcriptionally regulated responses that delays adenosine uptake. This occurs on the level of adenosine transporters, and also on the level of enzymatic intracellular adenosine metabolism. Studies measuring the capacity of vascular endothelia or intestinal epithelia to transport adenosine that is added to their supernatants revealed delayed adenosine uptake into cells that were previously exposed to hypoxia (29, 82). More mechanistic studies utilizing transport of radio-labeled adenosine in combination with siRNA repression of individual adenosine transporters found that ENT1 and ENT2 are the predominant adenosine transporters in vascular endothelia, or intestinal epithelia, respectively (29, 82). While in most instances HIFs transcriptionally induce target genes, binding of HIF to the promoter region of ENT1 or ENT2 dampens gene transcription, ultimately resulting in adenosine transporter repression. From a functional point of view, this endogenous pathway provides an additional mechanism to increase extracellular adenosine signaling by prolonging the extracellular half-life of adenosine that accumulates on the extracellular surface.
In addition to HIF-dependent changes of adenosine transporters, previous studies have also determined an influence of hypoxia on intracellular adenosine signaling. A very elegant study from the laboratory of Juergen Schrader determined that hypoxia-elicited inhibition of the adenosine kinase drives extracellular adenosine levels of the heart. In addition, other studies with promoter constructs, and a combination of in vitro and in vivo HIF loss- and gain-of-function revealed a hypoxia-responsive element within the AK promoter, thereby showing yet another HIF-dependent pathway that drives extracellular adenosine levels. Finally, previous studies found that the neuronal guidance molecule netrin-1 can enhance extracellular adenosine signaling events—particularly through the A2BAR (13, 97). Netrins belong to a group of guidance cue molecules that play an important role in brain development (103). These molecules have more recently been implied in the regulation of inflammatory events (10, 74). Consistent with the role of HIFs in enhancing extracellular adenosine signaling, we identified a HIF-binding site within the promoter region of netrin-1, and functional studies demonstrated HIF-1α in inducing mucosal netrin-1 expression and attenuation of hypoxia-induced inflammation (97). These studies identified a role of a neuronal guidance molecule outside the brain. Under the control of HIFs, and by enhancing extracellular A2BAR signaling, netrin-1 dampens hypoxia-induced inflammation of mucosal organs (Fig. 8) (97).
FIG. 8.

Ischemia-induced inflammation enhances mucosal netrin-1 production. During ischemia-elicited inflammation (e.g., during intestinal ischemia), HIF is stabilized and coordinates the transcriptional induction of netrin-1. As such, epithelial-released netrin-1 can attenuate neutrophil accumulation into the ischemic intestine. This process involves enhancement of adenosine signals through the A2BAR. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Taken together, the above studies demonstrate that hypoxia coordinates transcriptional mechanism under the control of HIFs that result in enhanced extracellular adenosine production and signaling, whereas adenosine transport and metabolism after AR activation are delayed. As such, the stage is set for adenosine to function as a safety signal during conditions of limited oxygen availability—such as typically occurs during ischemia (Fig. 9).
FIG. 9.

Effect of ischemia on extracellular adenosine. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Inflammatory Cell Adenosine Signaling
For adenosine to function as safety signal, we will need to discuss the tissue-specific roles of adenosine signaling in the context of potentially dangerous situations that will require a careful tuning of pathophysiological responses that preserve residual functional integrity, promote tissue repair, and limit pathology. Here, adenosine signaling may exert cytoprotective effects either directly on parenchymal cells or indirectly through the modulation of immune cell activities. To answer this question, studies in bone marrow chimeric mice for individual ARs with deletion of a specific AR on either on the inflammatory cells or on the radio-resistant tissues have defined contribution of adenosine tissue versus immune cell signaling events. These studies have provided evidence for both scenarios: in some cases, biological adenosine responses involve signaling events on cardiac myocytes, epithelial cells, or endothelial cells (21, 23, 24, 43); in other cases, AR activation dampens inflammation via activation of inflammatory cell ARs (16, 21, 23, 43, 71, 97, 121). Here, we will discuss two examples of how adenosine affects immune cell functions.
A first example for an important role of adenosine signaling in modulating immune cell functions occurs via AR activation expressed on PMNs. A lot of original research work in this area comes from the laboratory of Bruce Crohnstein. His research work identified adenosine as a physiologic modulator of superoxide anion generation by human neutrophils via activation of the A2AR expressed on human neutrophils. His group also provided the first in vivo evidence for a functional role of the A2A receptor in dampening inflammatory responses (14), which was subsequently confirmed in very elegant genetic studies from the laboratory of Misha Sitkovsky (85). Consistent with the theme of the present review, other studies identified PMN-elicited adenosine signaling events in PMN transmigration during conditions of limited oxygen availability (37). Hypoxia and ischemia are well-documented inflammatory stimuli and result in tissue PMN accumulation. Likewise, increased tissue adenosine levels are commonly associated with hypoxia, and given the anti-inflammatory properties of adenosine, the authors of this study hypothesized that adenosine production via adenine nucleotide metabolism at the vascular surface triggers an endogenous anti-inflammatory response during hypoxia. Initial in vitro studies indicated that endogenously generated adenosine, through activation of PMN adenosine A2A and A2B receptors, functions as an antiadhesive signal for PMN binding to microvascular endothelia. Intravascular nucleotides released by inflammatory cells undergo phosphohydrolysis via hypoxia-induced CD39 (CD39 converts ATP/ADP to AMP) and CD73 (CD73 converts AMP to adenosine). Extensions of these in vitro findings using cd39−/− or cd73−/− mice revealed that extracellular adenosine produced through adenine nucleotide metabolism during hypoxia is a potent anti-inflammatory signal for PMNs in vivo. These findings identify CD39 and CD73 as critical control points for endogenous adenosine generation and implicate A2AAR and A2BAR signaling in an endogenous feedback mechanism to attenuate excessive tissue PMN accumulation (37). Subsequent studies to identify the contribution of individual ARs exposed gene-targeted mice for each AR to ambient hypoxia (8% oxygen over 4 h) and studied hypoxia-elicited inflammation (21). These studies observed a relatively selective role for the A2BAR in dampening hypoxia-induced inflammation. Additional experiments in bone marrow chimeric A2BAR mice suggested a predominant role of vascular A2BARs in dampening hypoxia-elicited vascular leakage, whereas hypoxia-associated increases in tissue neutrophils were, at least in part, mediated by A2BAR-expressing hematopoietic cells. Taken together, these studies provide pharmacologic and genetic evidence for a role of PMN adenosine signaling as central control point of hypoxia-associated inflammation. Similarly, a recent study identified the neuronal guidance molecule netrin-1 as an alternative pathway of enhancing adenosine signaling events via A2BARs expressed on PMN. In fact, exogenous treatment with netrin-1 dampened hypoxia-associated inflammation in mucosal organs of wild-type but not of A2BAR−/− mice. Moreover, studies in bone marrow chimeric mice for the A2BAR suggested that netrin-1-elicited attenuation of PMN trafficking into hypoxic organs requires an interaction of the myeloid A2BAR with netrin-1 (97). Taken together, there is now considerable evidence that in situations of ischemia or hypoxia, enhancement of extracellular adenosine levels and PMN-dependent AR signaling dampens acute immune responses.
As a second example, recent studies from the laboratory of Simon Robson have implicated adenosine generation and signaling in a subpopulation of T lymphocytes—T regulatory cells (T reg cells). Studies of T reg cells have been limited by the dearth of specific surface markers to identify this group of cells in an in vivo setting. In this landmark study from the laboratory of Simon Robson, his team shows that the expression of CD39 in concert with CD73 distinguishes CD4(+)/CD25(+)/Foxp3(+) T reg cells from other T cells. These ectoenzymes generate pericellular adenosine from extracellular nucleotides. The coordinated expression of CD39/CD73 on T reg cells and the adenosine A2A receptor on activated T effector cells generates immunosuppressive loops, indicating roles in the inhibitory function of T reg cells. Consequently, T reg cells from cd39−/− null mice show impaired suppressive properties in vitro and fail to block allograft rejection in vivo. The authors conclude that CD39 and CD73 are surface markers of T reg cells that impart a specific biochemical signature characterized by adenosine generation that has functional relevance for cellular immunoregulation (16).
Adenosine in Ischemia-Reperfusion Injury
In the above paragraphs we have discussed research studies that provide strong evidence for the fact that extracellular adenosine levels and signaling events are enhanced during conditions of limited oxygen availability such occurs during ischemia-reperfusion injury. We have also provided evidence that adenosine signaling events can dampen immune responses, via activation of ARs expressed either on the tissues or on immune cells. Thus, the stage is set to take a look at adenosine generation and signaling during ischemia and reperfusion injury in different organ systems. In fact, these studies are consistent with the concept that adenosine can function as an endogenous safety signal to dampen hypoxia-induced inflammation and tissue injury during ischemia. While adenosine signaling has been implicated in many models of ischemia-reperfusion injury, we will focus in the present review on extracellular adenosine in myocardial, hepatic, and renal ischemia reperfusion injury.
Myocardial ischemia
Myocardial ischemia represents a major health problem of Western countries. Current therapeutic interventions focus mainly on early and persistent coronary reperfusion, and additional pharmacological strategies to increase resistance to myocardial ischemia are currently the areas of intense investigation (40, 56, 113). Here, several studies from the adenosine field provide evidence for a cardio-protective role of extracellular adenosine during myocardial ischemia-reperfusion injury. Many of these studies combine two different experimental approaches: first, they utilize pharmacologic or genetic approaches to study the role of adenosine production and signaling during myocardial ischemia and reperfusion injury; second, they utilize an experimental approach to elicit ischemia-driven cardio-adaptive responses to identify novel therapeutic targets. In fact, a powerful strategy for cardio-protection would be to recapitulate the consequences of ischemic preconditioning (IP) (83), where short and repeated episodes of ischemia and reperfusion before myocardial infarction result in attenuation of infarct size. Despite multiple attempts to identify the underlying molecular mechanisms, pharmacological strategies utilizing such pathways have yet to be further defined and introduced into clinical practice (63).
Several genetic and pharmacological studies provide strong evidence that attenuation of extracellular adenosine generation via the CD39 and CD73 pathway results in attenuated cardio-protection by IP treatment, and pharmacologic strategies that will enhance this pathway are associated with attenuated myocardial infarct sizes (28). For example, a study in dogs demonstrated that inhibition of CD73 is associated with abolished cardio-protection by ischemic preconditioning (62). Similarly, studies in gene-targeted mice for CD39 or CD73 revealed larger infarct sizes after 60 min of in situ ligation of the left coronary artery and 2 h of reperfusion, in conjunction with abolished cardio-protection by IP treatment (26, 65). Moreover, intravascular treatment with compounds that enhance extracellular generation of adenosine from ATP/ADP (soluble apyrase from potatos) or phosphohydrolysis of AMP to adenosine (soluble nucleotidase from snake venom) attenuated myocardial infarct sizes. Consistent with studies on ambient hypoxia exposure (33, 110), cardiac studies of CD39/CD73 transcript and protein levels indicated transcriptional induction of this pathway with myocardial ischemia (26, 65). In contrast to the role of HIF in transcriptional induction of CD73/A2ARs, some of these studies identified a central role of the transcription factor SP1 for the induction of CD39 during hypoxia or myocardial ischemia (34).
Consistent with the concept of adenosine signaling as a safety signal to dampen tissue inflammation and collateral damage, while at the same time promoting adaptation and repair, genetic studies have implicated all four ARs in cardio-protection from ischemia-reperfusion injury (28). Several very elegant studies from the laboratory of John Headrick in collaboration with Michael Blackburn demonstrate a functional role of the A1AR in cardio-protection from ischemia. While mice with overexpression of the A1AR are protected from ischemia, A1AR knockout mice have attenuated ischemia tolerance (76, 92, 93). Functional studies indicate that intrinsically activated A1ARs appear to enhance postischemic contractility and limit cardiac cell death (93). Studies from the research laboratory of Joel Linden suggest an important role of the A2AAR in cardio-protection from ischemia (121). In the course of these studies, Dr. Linden's research team created A2AAR bone marrow chimeric mice that they treated with a highly specific agonist of the A2AAR. Their results indicate that A2AAR activation on bone marrow-derived cells, specifically T or B lymphocytes, is responsible for the infarct-sparing and anti-inflammatory effects of ATL146e administered at the time of reperfusion after coronary occlusion. These studies were among the first to suggest that AR activation on cells of the adaptive immune system dampens myocardial ischemia-reperfusion injury. These findings are in strong agreement with a role of adenosine signaling as a safety signal.
In addition to the A1AR and A2AAR, other studies provided pharmacological and genetic evidence for the A2BAR in cardio-protection by ischemia (25, 26). While the A2BAR is the most adenosine-insensitive AR, there are two factors that may allow the A2BAR to become functionally relevant during myocardial ischemia. First, adenosine levels are dramatically elevated during myocardial ischemia; therefore, the concentration of extracellular adenosine can reach levels sufficient to activate the A2BAR. Second, the A2BAR is transcriptionally induced during myocardial ischemia, or ischemic preconditioning via a transcriptional pathway involving cardiac HIFs (25). Studies of in situ myocardial ischemia or ischemic preconditioning provide strong evidence for cardio-protection via A2BAR signaling. In fact, treatment of wild-type mice with an A2BAR selective AR agonist (BAY 60-6583) is associated with an ∼50% reduction of myocardial infarct size, but no change in infarct size in A2BAR−/− mice (26).
Taken together, several studies provide strong evidence that during myocardial ischemia or ischemic preconditioning of the heart, extracellular adenosine levels are elevated, and AR activation dampens myocardial inflammation and tissue injury. Moreover, other studies provide evidence for cardio-protection from ischemia by attenuating adenosine uptake or metabolism (17, 96). Together, these studies are consistent with the concept of adenosine signaling serving as a danger signal to dampen hypoxia-driven inflammation and tissue injury of the heart after myocardial ischemia and reperfusion injury.
Hepatic ischemia reperfusion injury
Hepatic ischemia reperfusion injury plays an important clinical role during liver transplantation and major liver resection. For instance, hepatic ischemia injury increases the risk for early organ dysfunction and acute rejection after liver transplantation, and novel therapeutic approaches to dampen hepatic ischemia reperfusion injury are currently an area of intense research. Similarly to studies in myocardial ischemia reperfusion injury, there is now strong pharmacologic and genetic evidence for a protective role of extracellular adenosine production during hepatic ischemia reperfusion injury (48, 52). Many of these findings are also consistent with a role of extracellular adenosine generation and signaling in intestinal ischemia-reperfusion injury (49, 50). A very elegant study from the laboratory of Joel Linden investigated the role of AR signaling in intestinal ischemia-reperfusion injury (71). Here, the team of Dr. Linden observed that liver reperfusion injury is reduced by lymphocyte depletion or activation of A2AARs with the selective agonist ATL146e (71). Specifically, this study found a surprising role for adenosine-dependent inhibition of natural killer T (NKT) cells, a sub-population of lymphocytes representing about 0.2% of peripheral blood T-cells. NKT cells recognize the nonpolymorphic CD1d molecule, an antigen-presenting molecule that binds self- and foreign lipids and glycolipids. This study provides strong evidence that the activation of NKT cells by a CD1d-dependent mechanism plays a central role in initiating the inflammatory cascade responsible for reperfusion injury in the liver and that these cells are key targets of A2AAR agonist protection in hepatic ischemia-reperfusion injury (28, 71).
Renal ischemia-reperfusion injury
Renal ischemia-reperfusion injury represents a major source for morbidity and mortality in hospitalized patients. In fact, renal ischemia-reperfusion injury is among the leading causes of acute kidney injury (AKI). For example, this can frequently occur in patients who are having major surgeries, and particularly patients with pre-existing renal disease will suffer greatly from an additional ischemic injury. Moreover, pharmacologic approaches to protect the kidneys from ischemia-reperfusion injury are very limited. Similar to studies in myocardial or hepatic ischemia-reperfusion injury, extracellular adenosine generation via CD39 and CD73 has been implicated in kidney protection from ischemia (44, 45). Moreover, different ARs have been investigated in kidney protection from ischemia. Studies from the laboratory of H. Thomas Lee have provided strong evidence for kidney protection from ischemia via activation of renal A1ARs (61, 86). Genetic deletion of the A1AR increased renal injury after ischemia-reperfusion injury, suggesting that receptor activation is protective in vivo. Recently, the team of Dr. Lee tested the hypothesis that by expressing the human-A1AR in A1AR knockout mice, ischemia reperfusion injury could be attenuated. Renal ischemia-reperfusion was induced in knockout mice 2 days after intrarenal injection of saline or a lentivirus encoding enhanced green fluorescent protein human-A1AR. The authors found that the latter procedure induced a robust expression of the reporter protein in the kidneys of knockout mice. Mice with kidney-specific human-A1AR reconstitution had significantly lower plasma creatinine, tubular necrosis, apoptosis, and tubular inflammation as evidenced by decreased leukocyte infiltration, pro-inflammatory cytokine, and intercellular adhesion molecule-1 expression in the kidney after injury compared to mice injected with saline or the control lentivirus (61). Other studies found kidney protection via the A2AAR expressed in inflammatory cells (15), whereas a recent study indicated that vascular A2BARs protect the kidneys from ischemia (43). In the later study, a comparison of all four AR knockout mice during renal ischemic preconditioning pointed toward a selective role of the A2BAR. Utilizing a previously described mouse model that was generated by the laboratory of Katya Ravid, allowing analysis of A2BAR expression in vivo (118, 119), the authors found that the A2BAR is expressed mainly within the vasculature of the renal architecture. Moreover, studies in bone marrow chimeric mice pointed toward a contribution of radio-resistant A2BAR signaling to kidney protection from ischemia (43).
Summary and Future Challenges
This review highlights the role of extracellular adenosine production and signaling as a transcriptionally controlled metabolic pathway in organ protection from ischemia. Based on the fact that extracellular adenosine generation is dramatically enhanced under ischemic conditions, we propose that adenosine serves as a safety signal during ischemia reperfusion injury, as well as other clinically relevant scenarios involving acute tissue damage, by promoting tissue protection and dampening of inflammation. In the specialized literature, adenosine is occasionally referred to as a danger signal, making it sometimes difficult to understand whether its generation after tissue injury or ischemia will have a cytotoxic or cytoprotective effect. However, in the context of acute tissue injury (such as ischemia, hypoxia, or acute inflammation), most evidence points toward a tissue protective role of adenosine generation and signaling (1, 9, 24, 28, 34, 36, 42, 48, 50, 64, 82, 94, 100). Therefore, we feel that the term of a safety rather than a danger signal seems more appropriate in this context.
It is important to point out that the role of adenosine signaling in more chronic models of injury could be quite different. For example, a series of very elegant studies from the laboratory of Michael Blackburn point toward a pro-fibrotic and detrimental role of adenosine generation and signaling in chronic lung injury (3, 4, 109, 123). Moreover, other potentially unwanted side effects of enhancing extracellular adenosine signaling could include alterations in blood pressure, heart-rate or sleep-awake cycle (120), fatty liver disease (87), or alterations in platelet function, thromboregulation, or bleeding (38, 51, 89). As such, future studies will have to define the exact time frame, dosing, and potential side effects of adenosine therapeutics. As such, we believe an important challenge for the future of the adenosine field will be to translate some of the discussed experimental studies into a clinical scenario, for example, with the goal to treat patients suffering from ischemia-reperfusion injury, or to improve outcomes of solid organ transplantation.
Abbreviations Used
- A1/A2A/A2B/A3
adenosine receptor subtype 1/2A/2B/3
- ADA
adenosine deaminase
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- AR
adenosine receptor (A1AR, A2AAR, A2BAR, A3AR)
- ATP
adenosine triphosphate
- cAMP
cyclic AMP
- CD39
ecto-apyrase, ENTPDase 1
- CD73
5′-ecto-nucleotidase
- ENT
equilibrative nucleoside transporter
- HIF
hypoxia-inducible factor
- PMN
polymorphonuclear leukocyte (neutrophil)
Acknowledgments
The authors want to acknowledge Shelley A. Eltzschig for the artwork during article preparation. The present review is supported by U.S. National Institutes of Health Grants R01-HL092188, R01-DK083385, and R01HL098294, and by Foundation for Anesthesia Education and Research Grants to H.K.E., and a research fellowship by the Deutsche Forschungsgemeinschaft (GR2121/1-1) to A.G.
References
- 1.Aherne CM. Kewley EM. Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.05.016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad A. Ahmad S. Glover L. Miller SM. Shannon JM. Guo X. Franklin WA. Bridges JP. Schaack JB. Colgan SP. White CW. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci U S A. 2009;106:10684–10689. doi: 10.1073/pnas.0901326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn MR. Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol Sci. 2003;24:66–70. doi: 10.1016/S0165-6147(02)00045-7. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn MR. Vance CO. Morschl E. Wilson CN. Adenosine receptors and inflammation. Handb Exp Pharmacol. 2009;193:215–269. doi: 10.1007/978-3-540-89615-9_8. [DOI] [PubMed] [Google Scholar]
- 5.Burnstock G. Purinergic signaling and vascular cell proliferation and death. Arterioscler Thromb Vasc Biol. 2002;22:364–373. doi: 10.1161/hq0302.105360. [DOI] [PubMed] [Google Scholar]
- 6.Burnstock G. Vessel tone and remodeling. Nat Med. 2006;12:16–17. doi: 10.1038/nm0106-16. [DOI] [PubMed] [Google Scholar]
- 7.Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008;7:575–590. doi: 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- 8.Castrop H. Huang Y. Hashimoto S. Mizel D. Hansen P. Theilig F. Bachmann S. Deng C. Briggs J. Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H. Yang D. Carroll SH. Eltzschig HK. Ravid K. Activation of the macrophage A2b adenosine receptor regulates tumor necrosis factor-alpha levels following vascular injury. Exp Hematol. 2009;37:533–538. doi: 10.1016/j.exphem.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cirulli V. Yebra M. Netrins: beyond the brain. Nat Rev Mol Cell Biol. 2007;8:296–306. doi: 10.1038/nrm2142. [DOI] [PubMed] [Google Scholar]
- 11.Colgan SP. Eltzschig HK. Eckle T. Thompson LF. Physiological Roles of 5′-Ectonucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colgan SP. Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corset V. Nguyen-Ba-Charvet KT. Forcet C. Moyse E. Chedotal A. Mehlen P. Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature. 2000;407:747–750. doi: 10.1038/35037600. [DOI] [PubMed] [Google Scholar]
- 14.Cronstein BN. Naime D. Ostad E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest. 1993;92:2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day YJ. Huang L. McDuffie MJ. Rosin DL. Ye H. Chen JF. Schwarzschild MA. Fink JS. Linden J. Okusa MD. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deaglio S. Dwyer KM. Gao W. Friedman D. Usheva A. Erat A. Chen JF. Enjyoji K. Linden J. Oukka M. Kuchroo VK. Strom TB. Robson SC. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decking UKM. Schlieper G. Kroll K. Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- 18.Delacretaz E. Clinical practice. Supraventricular tachycardia. N Engl J Med. 2006;354:1039–1051. doi: 10.1056/NEJMcp051145. [DOI] [PubMed] [Google Scholar]
- 19.Dieterich HJ. Weissmuller T. Rosenberger P. Eltzschig HK. Effect of hydroxyethyl starch on vascular leak syndrome and neutrophil accumulation during hypoxia. Crit Care Med. 2006;34:1775–1782. doi: 10.1097/01.CCM.0000218814.77568.BC. [DOI] [PubMed] [Google Scholar]
- 20.Drury AN. Szent-Gyorgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68:213–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckle T. Faigle M. Grenz A. Laucher S. Thompson LF. Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckle T. Fullbier L. Wehrmann M. Khoury J. Mittelbronn M. Ibla J. Rosenberger P. Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. doi: 10.4049/jimmunol.178.12.8127. [DOI] [PubMed] [Google Scholar]
- 23.Eckle T. Grenz A. Laucher S. Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckle T. Koeppen M. Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 2009;24:298–306. doi: 10.1152/physiol.00022.2009. [DOI] [PubMed] [Google Scholar]
- 25.Eckle T. Kohler D. Lehmann R. El Kasmi KC. Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 26.Eckle T. Krahn T. Grenz A. Kohler D. Mittelbronn M. Ledent C. Jacobson MA. Osswald H. Thompson LF. Unertl K. Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 27.Elliott MR. Chekeni FB. Trampont PC. Lazarowski ER. Kadl A. Walk SF. Park D. Woodson RI. Ostankovich M. Sharma P. Lysiak JJ. Harden TK. Leitinger N. Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eltzschig HK. Abdulla P. Hoffman E. Hamilton KE. Daniels D. Schonfeld C. Loffler M. Reyes G. Duszenko M. Karhausen J. Robinson A. Westerman KA. Coe IR. Colgan SP. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med. 2005;202:1493–1505. doi: 10.1084/jem.20050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eltzschig HK. Collard CD. Vascular ischaemia and reperfusion injury. Br Med Bull. 2004;70:71–86. doi: 10.1093/bmb/ldh025. [DOI] [PubMed] [Google Scholar]
- 31.Eltzschig HK. Eckle T. Mager A. Kuper N. Karcher C. Weissmuller T. Boengler K. Schulz R. Robson SC. Colgan SP. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 32.Eltzschig HK. Faigle M. Knapp S. Karhausen J. Ibla J. Rosenberger P. Odegard KC. Laussen PC. Thompson LF. Colgan SP. Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood. 2006;108:1602–1610. doi: 10.1182/blood-2006-02-001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eltzschig HK. Ibla JC. Furuta GT. Leonard MO. Jacobson KA. Enjyoji K. Robson SC. Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eltzschig HK. Kohler D. Eckle T. Kong T. Robson SC. Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113:224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eltzschig HK. Macmanus CF. Colgan SP. Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc Med. 2008;18:103–107. doi: 10.1016/j.tcm.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eltzschig HK. Rivera-Nieves J. Colgan SP. Targeting the A2B adenosine receptor during gastrointestinal ischemia and inflammation. Expert Opin Ther Targets. 2009;13:1267–1277. doi: 10.1517/14728220903241666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eltzschig HK. Thompson LF. Karhausen J. Cotta RJ. Ibla JC. Robson SC. Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 38.Enjyoji K. Sevigny J. Lin Y. Frenette PS. Christie PD. Esch JS., 2nd Imai M. Edelberg JM. Rayburn H. Lech M. Beeler DL. Csizmadia E. Wagner DD. Robson SC. Rosenberg RD. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med. 1999;5:1010–1017. doi: 10.1038/12447. [DOI] [PubMed] [Google Scholar]
- 39.Faigle M. Seessle J. Zug S. El Kasmi KC. Eltzschig HK. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE. 2008;3:e2801. doi: 10.1371/journal.pone.0002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frangogiannis NG. Ren G. Dewald O. Zymek P. Haudek S. Koerting A. Winkelmann K. Michael LH. Lawler J. Entman ML. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 41.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 42.Frick JS. MacManus CF. Scully M. Glover LE. Eltzschig HK. Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182:4957–4964. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grenz A. Osswald H. Eckle T. Yang D. Zhang H. Tran ZV. Klingel K. Ravid K. Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Medicine. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grenz A. Zhang H. Eckle T. Mittelbronn M. Wehrmann M. Kohle C. Kloor D. Thompson LF. Osswald H. Eltzschig HK. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol. 2007;18:833–845. doi: 10.1681/ASN.2006101141. [DOI] [PubMed] [Google Scholar]
- 45.Grenz A. Zhang H. Hermes M. Eckle T. Klingel K. Huang DY. Muller CE. Robson SC. Osswald H. Eltzschig HK. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J. 2007;21:2863–2873. doi: 10.1096/fj.06-7947com. [DOI] [PubMed] [Google Scholar]
- 46.Haeberle HA. Durrstein C. Rosenberger P. Hosakote YM. Kuhlicke J. Kempf VA. Garofalo RP. Eltzschig HK. Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS ONE. 2008;3:e3352. doi: 10.1371/journal.pone.0003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hart M. Jacobi B. Schittenhelm J. Henn M. Eltzschig HK. A2B adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol. 2009;182:3965–3968. doi: 10.4049/jimmunol.0802193. [DOI] [PubMed] [Google Scholar]
- 48.Hart ML. Gorzolla IC. Schittenhelm J. Robson SC. Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol. 2010;184:4017–4024. doi: 10.4049/jimmunol.0901851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hart ML. Henn M. Kohler D. Kloor D. Mittelbronn M. Gorzolla IC. Stahl GL. Eltzschig HK. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008;22:2784–2797. doi: 10.1096/fj.07-103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hart ML. Jacobi B. Schittenhelm J. Henn M. Eltzschig HK. Cutting edge: A2B adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol. 2009;182:3965–3968. doi: 10.4049/jimmunol.0802193. [DOI] [PubMed] [Google Scholar]
- 51.Hart ML. Kohler D. Eckle T. Kloor D. Stahl GL. Eltzschig HK. Direct treatment of mouse or human blood with soluble 5′-nucleotidase inhibits platelet aggregation. Arterioscler Thromb Vasc Biol. 2008;28:1477–1483. doi: 10.1161/ATVBAHA.108.169219. [DOI] [PubMed] [Google Scholar]
- 52.Hart ML. Much C. Gorzolla IC. Schittenhelm J. Kloor D. Stahl GL. Eltzschig HK. Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology. 2008;135:1739–1750.e3. doi: 10.1053/j.gastro.2008.07.064. [DOI] [PubMed] [Google Scholar]
- 53.Hartmann H. Eltzschig HK. Wurz H. Hantke K. Rakin A. Yazdi AS. Matteoli G. Bohn E. Autenrieth IB. Karhausen J. Neumann D. Colgan SP. Kempf VA. Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology. 2008;134:756–767. doi: 10.1053/j.gastro.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Hasko G. Csoka B. Nemeth ZH. Vizi ES. Pacher P. A(2B) adenosine receptors in immunity and inflammation. Trends Immunol. 2009;30:263–270. doi: 10.1016/j.it.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hasko G. Linden J. Cronstein B. Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann HC. Update and rationale for ongoing acute myocardial infarction trials: combination therapy, facilitation, and myocardial preservation. Am Heart J. 2006;151:S30–S39. doi: 10.1016/j.ahj.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 57.Holmquist-Mengelbier L. Fredlund E. Lofstedt T. Noguera R. Navarro S. Nilsson H. Pietras A. Vallon-Christersson J. Borg A. Gradin K. Poellinger L. Pahlman S. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 58.Hu CJ. Wang LY. Chodosh LA. Keith B. Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson EK. Ren J. Gillespie DG. Dubey RK. Extracellular 2′,3′-cyclic adenosine 5′-monophosphate is a potent inhibitor of preglomerular vascular smooth muscle cell and mesangial cell growth. Hypertension. 2010;56:151–158. doi: 10.1161/HYPERTENSIONAHA.110.152454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jackson EK. Ren J. Mi Z. Extracellular 2′,3′-cAMP is a source of adenosine. J Biol Chem. 2009;284:33097–33106. doi: 10.1074/jbc.M109.053876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim M. Chen SW. Park SW. D'Agati VD. Yang J. Lee HT. Kidney-specific reconstitution of the A1 adenosine receptor in A1 adenosine receptor knockout mice reduces renal ischemia-reperfusion injury. Kidney Int. 2009;75:809–823. doi: 10.1038/ki.2008.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitakaze M. Hori M. Morioka T. Minamino T. Takashima S. Sato H. Shinozaki Y. Chujo M. Mori H. Inoue M, et al. Infarct size-limiting effect of ischemic preconditioning is blunted by inhibition of 5′-nucleotidase activity and attenuation of adenosine release. Circulation. 1994;89:1237–1246. doi: 10.1161/01.cir.89.3.1237. [DOI] [PubMed] [Google Scholar]
- 63.Kloner RA. Rezkalla SH. Preconditioning, postconditioning and their application to clinical cardiology. Cardiovasc Res. 2006;70:297–307. doi: 10.1016/j.cardiores.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 64.Koeppen M. Eckle T. Eltzschig HK. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS ONE. 2009;4:e6784. doi: 10.1371/journal.pone.0006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohler D. Eckle T. Faigle M. Grenz A. Mittelbronn M. Laucher S. Hart ML. Robson SC. Muller CE. Eltzschig HK. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–1794. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 66.Kong T. Eltzschig HK. Karhausen J. Colgan SP. Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of {beta}2 integrin gene expression. PNAS. 2004;101:10440–10445. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong T. Westerman KA. Faigle M. Eltzschig HK. Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006;20:2242–2250. doi: 10.1096/fj.06-6419com. [DOI] [PubMed] [Google Scholar]
- 68.Koszalka P. Ozuyaman B. Huo Y. Zernecke A. Flogel U. Braun N. Buchheiser A. Decking UK. Smith ML. Sevigny J. Gear A. Weber AA. Molojavyi A. Ding Z. Weber C. Ley K. Zimmermann H. Godecke A. Schrader J. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- 69.Kruger B. Krick S. Dhillon N. Lerner SM. Ames S. Bromberg JS. Lin M. Walsh L. Vella J. Fischereder M. Kramer BK. Colvin RB. Heeger PS. Murphy BT. Schroppel B. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A. 2009;106:3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhlicke J. Frick JS. Morote-Garcia JC. Rosenberger P. Eltzschig HK. Hypoxia inducible factor (HIF)-1 Coordinates induction of Toll-like receptors TLR2 and TLR6 during Hypoxia. PLoS ONE. 2007;2:e1364. doi: 10.1371/journal.pone.0001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lappas CM. Day YJ. Marshall MA. Engelhard VH. Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203:2639–2648. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ledent C. Vaugeois JM. Schiffmann SN. Pedrazzini T. El Yacoubi M. Vanderhaeghen JJ. Costentin J. Heath JK. Vassart G. Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 73.Loffler M. Morote-Garcia JC. Eltzschig SA. Coe IR. Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arterioscler Thromb Vasc Biol. 2007;27:1004–1013. doi: 10.1161/ATVBAHA.106.126714. [DOI] [PubMed] [Google Scholar]
- 74.Ly NP. Komatsuzaki K. Fraser IP. Tseng AA. Prodhan P. Moore KJ. Kinane TB. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14729–14734. doi: 10.1073/pnas.0506233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marcus AJ. Broekman MJ. Drosopoulos JHF. Islam N. Alyonycheva TN. Safier LB. Hajjar KA. Posnett DN. Schoenborn MA. Schooley KA. Gayle RB. Maliszewski CR. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–1360. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matherne GP. Linden J. Byford AM. Gauthier NS. Headrick JP. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischemia. Proc Natl Acad Sci U S A. 1997;94:6541–6546. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 78.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 79.Mohamedali KA. Guicherit OM. Kellems RE. Rudolph FB. The highest levels of purine catabolic enzymes in mice are present in the proximal small intestine. J Biol Chem. 1993;268:23728–23733. [PubMed] [Google Scholar]
- 80.Montesinos MC. Desai A. Delano D. Chen JF. Fink JS. Jacobson MA. Cronstein BN. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003;48:240–247. doi: 10.1002/art.10712. [DOI] [PubMed] [Google Scholar]
- 81.Morote-Garcia JC. Rosenberger P. Kuhlicke J. Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 82.Morote-Garcia JC. Rosenberger P. Nivillac NM. Coe IR. Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 83.Murry CE. Jennings RB. Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 84.Niemela J. Ifergan I. Yegutkin GG. Jalkanen S. Prat A. Airas L. IFN-beta regulates CD73 and adenosine expression at the blood-brain barrier. Eur J Immunol. 2008;38:2718–2726. doi: 10.1002/eji.200838437. [DOI] [PubMed] [Google Scholar]
- 85.Ohta A. Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 86.Park SW. Chen SW. Kim M. Brown KM. D'Agati VD. Lee HT. Protection against acute kidney injury via A1 adenosine receptor-mediated Akt activation reduces liver injury after liver ischemia and reperfusion in mice. J Pharmacol Exp Ther. 2010;333:736–747. doi: 10.1124/jpet.110.166884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng Z. Borea PA. Wilder T. Yee H. Chiriboga L. Blackburn MR. Azzena G. Resta G. Cronstein BN. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest. 2009;119:582–594. doi: 10.1172/JCI37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Picher M. Burch LH. Hirsh AJ. Spychala J. Boucher RC. Ecto 5′-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem. 2003;278:13468–13479. doi: 10.1074/jbc.M300569200. [DOI] [PubMed] [Google Scholar]
- 89.Pinsky DJ. Broekman MJ. Peschon JJ. Stocking KL. Fujita T. Ramasamy R. Connolly ES., Jr. Huang J. Kiss S. Zhang Y. Choudhri TF. McTaggart RA. Liao H. Drosopoulos JH. Price VL. Marcus AJ. Maliszewski CR. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rankin EB. Biju MP. Liu Q. Unger TL. Rha J. Johnson RS. Simon MC. Keith B. Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reichelt ME. Shanu A. Willems L. Witting PK. Ellis NA. Blackburn MR. Headrick JP. Endogenous adenosine selectively modulates oxidant stress via the A1 receptor in ischemic hearts. Antioxid Redox Signal. 2009;11:2641–2650. doi: 10.1089/ars.2009.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reichelt ME. Willems L. Molina JG. Sun CX. Noble JC. Ashton KJ. Schnermann J. Blackburn MR. Headrick JP. Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circ Res. 2005;96:363–367. doi: 10.1161/01.RES.0000156075.00127.C3. [DOI] [PubMed] [Google Scholar]
- 94.Reutershan J. Vollmer I. Stark S. Wagner R. Ngamsri KC. Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009;23:473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- 95.Robson SC. Wu Y. Sun X. Knosalla C. Dwyer K. Enjyoji K. Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin Thromb Hemost. 2005;31:217–233. doi: 10.1055/s-2005-869527. [DOI] [PubMed] [Google Scholar]
- 96.Rose JB. Naydenova Z. Bang A. Eguchi M. Sweeney G. Choi DS. Hammond JR. Coe IR. Equilibrative Nucleoside Transporter 1 (ENT1) plays an essential role in cardioprotection. Am J Physiol Heart Circ Physiol. 2010;298:H771–H777. doi: 10.1152/ajpheart.00711.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rosenberger P. Schwab JM. Mirakaj V. Masekowsky E. Mager A. Morote-Garcia JC. Unertl K. Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 98.Sacco RL. Diener HC. Yusuf S. Cotton D. Ounpuu S. Lawton WA. Palesch Y. Martin RH. Albers GW. Bath P. Bornstein N. Chan BP. Chen ST. Cunha L. Dahlof B. De Keyser J. Donnan GA. Estol C. Gorelick P. Gu V. Hermansson K. Hilbrich L. Kaste M. Lu C. Machnig T. Pais P. Roberts R. Skvortsova V. Teal P. Toni D. Vandermaelen C. Voigt T. Weber M. Yoon BW. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008;359:1238–1251. doi: 10.1056/NEJMoa0805002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Salvatore CA. Tilley SL. Latour AM. Fletcher DS. Koller BH. Jacobson MA. Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 100.Schingnitz U. Hartmann K. Macmanus CF. Eckle T. Zug S. Colgan SP. Eltzschig HK. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010;184:5271–5279. doi: 10.4049/jimmunol.0903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schofield CJ. Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 102.Schroder K. Tschopp J. The inflammasomes. Cell. 140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 103.Serafini T. Colamarino SA. Leonardo ED. Wang H. Beddington R. Skarnes WC. Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- 104.Sitkovsky M. Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 105.Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009;30:102–108. doi: 10.1016/j.it.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 106.Sitkovsky MV. Kjaergaard J. Lukashev D. Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008;14:5947–5952. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]
- 107.Sitkovsky MV. Lukashev D. Apasov S. Kojima H. Koshiba M. Caldwell C. Ohta A. Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 108.Suadicani SO. Brosnan CF. Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. 2006;26:1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sun CX. Zhong H. Mohsenin A. Morschl E. Chunn JL. Molina JG. Belardinelli L. Zeng D. Blackburn MR. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Synnestvedt K. Furuta GT. Comerford KM. Louis N. Karhausen J. Eltzschig HK. Hansen KR. Thompson LF. Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thiel M. Chouker A. Ohta A. Jackson E. Caldwell C. Smith P. Lukashev D. Bittmann I. Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thompson LF. Eltzschig HK. Ibla JC. Van De Wiele CJ. Resta R. Morote-Garcia JC. Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Walsh MC. Bourcier T. Takahashi K. Shi L. Busche MN. Rother RP. Solomon SD. Ezekowitz RA. Stahl GL. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol. 2005;175:541–546. doi: 10.4049/jimmunol.175.1.541. [DOI] [PubMed] [Google Scholar]
- 114.Wang ECY. Lee J-M. Ruiz WG. Balestreire EM. von Bodungen M. Barrick S. Cockayne DA. Birder LA. Apodaca G. ATP and purinergic receptor-dependent membrane traffic in bladder umbrella cells. J Clin Invest. 2005;115:2412–2422. doi: 10.1172/JCI24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang G. Jiang B. Rue E. Semenza G. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wellmann S. Bettkober M. Zelmer A. Seeger K. Faigle M. Eltzschig HK. Buhrer C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun. 2008;372:892–897. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 117.Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 118.Yang D. Koupenova M. McCrann DJ. Kopeikina KJ. Kagan HM. Schreiber BM. Ravid K. The A2b adenosine receptor protects against vascular injury. Proc Natl Acad Sci U S A. 2008;105:792–796. doi: 10.1073/pnas.0705563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang D. Zhang Y. Nguyen HG. Koupenova M. Chauhan AK. Makitalo M. St. Jones MR. Hilaire C. Seldin DC. Toselli P. Lamperti E. Schreiber BM. Gavras H. Wagner DD. Ravid K. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang JN. Chen JF. Fredholm BB. Physiological roles of A1 and A2A adenosine receptors in regulating heart rate, body temperature, and locomotion as revealed using knockout mice and caffeine. Am J Physiol Heart Circ Physiol. 2009;296:H1141–H1149. doi: 10.1152/ajpheart.00754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang Z. Day YJ. Toufektsian MC. Ramos SI. Marshall M. Wang XQ. French BA. Linden J. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111:2190–2197. doi: 10.1161/01.CIR.0000163586.62253.A5. [DOI] [PubMed] [Google Scholar]
- 122.Yellon DM. Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 123.Zhou Y. Mohsenin A. Morschl E. Young HW. Molina JG. Ma W. Sun CX. Martinez-Valdez H. Blackburn MR. Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol. 2009;182:8037–8046. doi: 10.4049/jimmunol.0900515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]