

Abstract
DNA damage is an important mechanism in carcinogenesis, so genes related to maintaining genomic integrity may influence papillary thyroid cancer (PTC) risk. Candidate gene studies targeting some of these genes have identified only a few polymorphisms associated with risk of PTC. Here, we expanded the scope of previous candidate studies by increasing the number and coverage of genes related to maintenance of genomic integrity. We evaluated 5077 tag single-nucleotide polymorphisms (SNPs) from 340 candidate gene regions hypothesized to be involved in DNA repair, epigenetics, tumor suppression, apoptosis, telomere function and cell cycle control and signaling pathways in a case–control study of 344 PTC cases and 452 matched controls. We estimated odds ratios for associations of single SNPs with PTC risk and combined P values for SNPs in the same gene region or pathway to obtain gene region-specific or pathway-specific P values using adaptive rank-truncated product methods. Nine SNPs had P values <0.0005, three of which were in HDAC4 and were inversely related to PTC risk. After multiple comparisons adjustment, no SNPs remained associated with PTC risk. Seven gene regions were associated with PTC risk at P < 0.01, including HUS1, ALKBH3, HDAC4, BAK1, FAF1_CDKN2C, DACT3 and FZD6. Our results suggest a possible role of genes involved in maintenance of genomic integrity in relation to risk of PTC.
Introduction
Thyroid cancer incidence has been steadily increasing worldwide and is now the fifth most common cancer diagnosed in women in the USA (1). Papillary thyroid cancer (PTC), the most common histological type, accounts for the majority of this increased incidence (2). Exposure to ionizing radiation at a young age is the strongest known risk factor for papillary thyroid cancer (3). In addition, family history studies have suggested that thyroid cancer has a greater familial risk than other common cancers with the reported relative risk estimates of 3- to 4-fold or higher (4–8). Because radiation is known to induce DNA damage, an important mechanism in carcinogenesis, heritable polymorphic variation in genes related to maintenance of genomic integrity has been hypothesized to affect risk of thyroid cancer. While two genome-wide association studies have identified only thyroid transcription factor gene polymorphisms both in persons exposed (9) and unexposed (10) to ionizing radiation, candidate gene studies have identified promising polymorphisms in other pathways including DNA repair and other genomic integrity-related pathways (11–20). For example, several studies reported associations with thyroid cancer risk for single-nucleotide polymorphism (SNPs) in the DNA repair genes XRCC1 (13–15,18) and XRCC3 (11,16,18) as well as other loci (RET, PTEN) involved in the mitogen-activated protein (MAP) kinase and AKT-signaling pathways (12,20). While providing initial clues to determinants of genetic susceptibility to PTC, these early studies were relatively small or had a limited selection of genes, suggesting that additional genetic determinants of PTC risk remain to be discovered.
Here, we expand the scope of previous candidate gene studies of thyroid cancer by increasing the number and coverage of genes belonging to several pathways related to maintenance of genomic integrity. Specifically, we examined risk of PTC in relation to 5077 common tag SNPs in a selected set of 340 genes involved in DNA repair, cell cycle control, apoptosis, tumor suppression, telomere function, epigenetics, Wnt/beta-catenin signaling, MAP kinase signaling and PI3K/AKT signaling using data from a common genotyping platform that was developed to evaluate genetic susceptibility to a variety of rare cancers (21,22).
Materials and methods
Study population
Our cases included thyroid cancers diagnosed within the US Radiologic Technologists (USRT) cohort as described previously (23) and cancers diagnosed and treated at the University of Texas M. D. Anderson Cancer Center (UTMDACC) (13). Controls for both types of cases were selected from the USRT study (24). Briefly, the USRT study was initiated in 1984 and included 146 022 radiologic technologists nationwide who were certified for at least 2 years by the American Registry of Radiologic Technologists between 1926 and 1982. Three questionnaires were administered (1984–1989, 1994–1998 and 2003–2005) to collect information on health outcomes (including self-reports of thyroid cancer), demographic characteristics, medical history, work practices and other environmental risk factors. Participant response to questionnaires has consistently been ∼70%. Thyroid cancer cases reported on any of the three questionnaires were recruited for blood collection. Of 415 living thyroid cancer cases eligible for recruitment in 2005, 66 would not agree to release their medical records, and the pathology records of an additional 25 could not be obtained. In addition, there were 54 refusals and 19 non-responders to an invitation to provide blood samples and 19 who sent their samples after recruitment had closed, leaving 232 confirmed thyroid cancer cases who donated blood samples for the study (55.9% collected from all self-reported cases or 71.6% of the confirmed cases). The 491 controls used in the current study are the same as the controls used for our previous thyroid cancer study (18). Included in the case group were 243 serially recruited histologically confirmed incident PTC cases from the UTMDACC who presented for care between 1999 and 2005 and subsequently underwent surgery (13). Each UTMDACC PTC case provided demographic and exposure information, including radiation exposure (defined as ever received medical radiation exposure to the whole body, head or neck), through a self-administered questionnaire (12). Study controls were selected from the USRT cohort originally to match the USRT cases (90.6% female). The proportion of females among the UTMDACC cases was lower (64.1%) than in the USRT cohort. To ensure that the cases from the two studies were comparable, we compared their average age at diagnosis, smoking status and tumor size and found that the two case series were similar. Both studies were reviewed and approved by their respective Institutional Review Boards and all subjects provided informed consent.
Variable collection and data harmonization
Data concerning demographics, health history, family history of cancer and other risk factors were collected by self-administered mailed questionnaires or telephone interview in the USRT study or a self-administered questionnaire at the time of blood collection in the UTMDACC study. Variables defined similarly in both studies were race and histological type of thyroid cancer. However, the majority of analysis variables had to be harmonized, so that they were comparable across the two studies (cigarette smoking, alcohol consumption, prior exposure to therapeutic radiation and family history of any cancer or specifically of thyroid cancer). Because controls were matched to cases on year of birth (+/− 2 years) and to enable time-appropriate partitioning of exposures (such as cigarette smoking), we assigned a referent age to controls to correspond to the time of a case’s diagnosis. Specifically, a control for a specific case was randomly selected from case-defined strata of sex and year of birth. If no control could be found for a specific case, then matching on sex and year of birth was relaxed (in that order). The algorithm was repeated until referent age was assigned to every control. All exposures in controls were accounted for up to the referent age and ignored thereafter.
Laboratory methods
DNA extraction.
In the USRT study, venipuncture whole blood samples were shipped with a temperature-stabilizing pack overnight to the processing laboratory in Frederick, MD. At UTMDACC, venipuncture whole blood samples were collected and processed in the clinic. Both studies extracted DNA from peripheral blood leucocytes using Qiagen Mini Kits (Qiagen, Valencia, CA) (2,3) according to manufacturer’s instructions.
Genotyping.
Genotyping of 27 904 SNPs, including tag SNPs in 1316 candidate genes and their surrounding regions (within 20 kb 5′ of the start of transcription at the first exon and 10 kb 3′ of the last exon) and intergenic SNPs included on the platform because of associations with several cancers, was performed at the NCI Core Genotyping Facility (Advanced Technology Center, Gaithersburg, MD; http://cgf.nci.nih.gov/) using the custom-designed iSelect Infinium assay (Illumina, San Diego, CA,www.illumina.com), which was designed before the results of the two thyroid cancer genome-wide association studies (9,10) were available. Tag SNPs were selected for target gene regions from the common SNPs (minor allele frequency > 5%) genotyped by the HapMap Project (Data Release 20/Phase II, NCBI Build 36.1, assembly dbSNPb126) in the Caucasian population (CEU) using TagZilla, which is a part of the GLU, software package (http://code.google.com/p/glu-genetics/) based on the pairwise binning method of Carlson et al. (25) with a binning threshold of r2 > 0.8.
Quality control.
Of 27 904 SNPs included in the genotyping platform, 722 failed genotyping (no amplification or clustering) and 208 had monoallelic calls and were excluded from the analysis. In addition, SNPs with <95% concordance (n = 656) or <90% completion (n = 740) among the 48 randomly inserted quality control replicates that ranged in size from 6 to 12 samples per individual were also excluded. We further excluded 740 SNPs that failed Hardy–Weinberg Equilibrium test (P < 0.00001). Of the 947 study participants with DNA specimens (n = 232 USRT cases, n = 223 UTMDACC cases, n = 492 USRT controls), we excluded subjects if their samples failed genotyping (n = 18), were not assayed (n = 4), or had <90% completion rate (n = 15).
Final set of tag SNPs, subpathways and analytic population
Following quality control-related exclusions, we excluded in the analysis 1607 intergenic SNPs that were included on the platform but had been previously implicated in etiology of non-thyroid cancers and 5706 tag SNPs with the minor allele frequency <10% or the lowest achievable significance level computed from the marginal totals >1−30 (26). After all exclusions, there were 17 525 tag SNPs in 1129 gene regions available for analysis. Of these, there were 5077 SNPs in the 340 gene regions related to pathways involved with genomic integrity maintenance and the subject of current analysis. The candidate gene regions were subdivided into nine a priori-defined pathways (DNA repair, cell cycle control, apoptosis, tumor suppression, telomere function, epigenetic, Wnt/beta-catenin signaling, MAP kinase signaling and PI3K/AKT signaling) based on evidence of biological and/or functional relatedness of the genes obtained from various publically available databases, including NCBI Entrez Gene (www.ncbi.nlm.nih.gov/sites/entrez) and GeneCards (www.genecards.org). The DNA repair pathway was further divided into narrower subpathway categories including direct reversal of damage, base excision repair, homologous recombination, mismatch repair, non-homologous end joining, other conserved damage response genes, etc. When assigning gene regions with a broad range of known functions, we allowed for allocation to multiple pathways. A complete list of all candidate gene regions evaluated in current analyses and their pathway allocation is available in supplementary Table A, Carcinogenesis Online.
To minimize potential for population stratification and phenotypic heterogeneity of thyroid cancer cases, we also excluded in the analysis individuals with non-European ancestry (n = 97) and cases with follicular thyroid cancer (n = 17) leaving 344 papillary thyroid cancer cases (n = 202 USRT and n = 142 UTMDACC) and 452 controls of European ancestry with validated genotyping results. Allele frequencies for papillary thyroid cancer cases of European ancestry were largely similar between the USRT and the UTMDACC study sites and between males and females, so these groups were combined for genetic analyses.
Statistical analysis
We organized the analytic approach to proceed from examining the relationship of individual SNPs with PTC risk to examining relationships at the gene region level, pathway (e.g. apoptosis) and subpathway (e.g. direct reversal of damage in the DNA repair pathways) level and, finally, overall. We chose this approach to maximize our ability to detect small SNP effects that could only be appreciated in the aggregate. For examples, several SNPs in the same gene region might not individually contribute to PTC risk, but all of the SNPs with small effects taken together at the gene region level could be significant.
SNP-based associations.
Logistic regression models were used to calculate odds ratios and 95% confidence intervals of the association of PTC risk with each SNP genotype, coded as 0, 1, 2, with 0 denoting the homozygous genotype with common allele as the referent category. We calculated the linear Ptrend for SNP genotype in crude models and models adjusted for sex, attained age in four categories (<35, 35–44, 45–54, 55+ years) and year of birth (<1940, 1940–1949, 1950+) as an ordinal variable. We also evaluated the effect of additional adjustment for cigarette smoking (ever/never), alcohol consumption (ever/never), and body mass index as a continuous variable. The results from models with additional adjustments were essentially similar and we chose to present the results from basic models adjusted for sex, attained age and year of birth throughout the manuscript. We adjusted for multiple comparisons using the false discovery rate control method (27).
Gene region- and pathway-based analyses.
We combined SNP-specific P values of linear trend within the same gene region using an adaptive rank-truncated product method (28). This method accounts for the linkage disequilibrium (LD) structure within the gene region and allows for flexibility in assumptions about the number of SNPs to include in the P value calculation. For the subset of genes with the combined P value for gene region (Pregion) <0.01, we evaluated pairwise indices of LD (D′ and r2) in controls using the Haploview package (29) and used a haplotype-sliding window approach (with windows composed of three SNPs) to evaluate potential loci in small genetic regions that may have been overlooked with a single locus analysis (30). Gene region level P values were combined into the P values associated with the nine pathways as well as the DNA repair specific categories or subpathways (Ppathway). Lastly, we evaluated the significance of the overall group of genomic integrity pathways combining all gene region level based P values (Poverall). Subpathway/pathway-based analyses were repeated including and excluding gene regions with multiple allocations.
Statistical analyses were conducted in SAS version 9.1 (SAS Institute, Cary, NC) and in R, except where otherwise noted.
Results
The characteristics of 344 cases of PTC and 452 controls are summarized in Table I. There was a lower proportion of females among cases compared with controls (79.7 versus 93.6%). The distribution of referent age in cases and controls was comparable. On average, cases were less likely to smoke or drink alcohol or to have a family history of cancer but more likely to have a family history of thyroid cancer or a higher body mass index.
Table I.
Characteristics of the study population by case and control status, USRT Study and University of Texas M. D. Anderson Cancer Center Study
| Characteristic | Cases (N = 344) n (%) | Controls (N = 452) n (%) | P-value |
| Study | na | ||
| Radiologic Technologists | 202 (58.7) | 452 (100.0) | |
| M. D. Anderson Cancer Center | 142 (41.3) | 0 | |
| Gender | <0.001 | ||
| Female | 274 (79.7) | 423 (93.6) | |
| Male | 70 (20.3) | 29 (6.4) | |
| Attained/referent age, year | 0.360 | ||
| 19–25 | 26 (7.6) | 30 (6.6) | |
| 26–35 | 73 (21.2) | 103 (22.8) | |
| 36–45 | 113 (32.8) | 159 (35.2) | |
| 46–55 | 77 (22.4) | 105 (23.2) | |
| 56–65 | 43 (12.5) | 49 (10.8) | |
| 66–79 | 12 (3.5) | 6 (1.3) | |
| Smoking statusa | 0.001 | ||
| Never | 208 (60.6) | 234 (52.2) | |
| Former | 87 (25.4) | 101 (22.5) | |
| Current | 48 (14.0) | 113 (25.2) | |
| Alcohol consumptiona | 0.159 | ||
| Never | 179 (52.0) | 213 (47.1) | |
| Ever | 158 (45.9) | 221 (48.9) | |
| Unknown | 7 (2.0) | 18 (4.0) | |
| Number of relatives with cancera | 0.002 | ||
| None | 170 (49.4) | 171 (37.8) | |
| One | 107 (31.1) | 180 (39.8) | |
| Two | 39 (11.3) | 64 (14.2) | |
| Three or more | 8 (5.2) | 33 (7.3) | |
| Unknown | 10 (2.9) | 4 (0.9) | |
| Number of relatives with thyroid cancer | <0.001 | ||
| None | 319 (92.7) | 442 (97.8) | |
| At least one | 15 (4.4) | 6 (1.3) | |
| Unknown | 10 (2.9) | 0 | |
| Adopted | 0 | 4 (0.9) | |
| Mean (SD) | Mean (SD) | ||
| BMI (kg/m2)a | 26.1 (6.0) | 24.4 (4.7) | <0.001 |
BMI, body mass index; na, not applicable.
As of referent age. See text for description of referent age assignment.
SNP-based associations
While the observed distribution of P values of linear trend for all 5077 SNPs was not statistically different from the expected uniform (null) distribution, there was some suggestion of departure from the null in the area of lowest P values (Figure 1). Nine SNPs in six gene regions were associated with risk of PTC at Ptrend <0.0005 (Table II; a complete list of all SNP-based Ptrend values is available in supplementary Table C, Carcinogenesis Online). Three of the nine SNPs (rs6749348, rs507159 and rs7584828) associated with reduced risk of PTC were in the HDAC4 gene and did not appear to be in LD with one another (D′ range 29–47, r2 range 0.01–0.06) (Figure 2). The remaining six SNPs were in the genetic regions HUS1 (rs2708906), BAK1 (rs493871), ALKBH3 (rs10838192), DACT3 (rs314659), MGMT (rs4751109) and FAF1_CDKN2C (rs11587909). After multiple comparisons adjustment, none of the SNP-based Ptrend values remained significant.
Fig. 1.

Observed and expected distributions for the results of test of linear trend with 5077 tag SNPs in 340 gene regions in genomic integrity-related genes in a case–control study of PTC risk.
Table II.
Odds ratio and 95% confidence interval, adjusted for sex, attained age and year of birth, for the tag SNPs associated with papillary thyroid cancer risk at Ptrend <0.0005
| Target gene | SNP | Genotype | Odds ratio (95% CI) | Ptrenda/Ptrend FDRb |
| HDAC4 | rs6749348 | GG | Ref. | 7.83E-05/0.2359 |
| GA | 0.41 (0.26–0.65) | |||
| AA | 0.28 (0.03–2.46) | |||
| HDAC4 | rs507159 | GG | Ref. | 0.0002/0.2359 |
| GC | 0.68 (0.49–0.93) | |||
| CC | 0.39 (0.23–0.67) | |||
| HDAC4 | rs7584828 | GG | Ref. | 0.00046/0.2775 |
| GA | 0.55 (0.38–0.79) | |||
| AA | 0.33 (0.08–1.28) | |||
| HUS1 | rs2708906 | AA | Ref. | 0.0001/0.2359 |
| AG | 1.55 (1.11–2.18) | |||
| GG | 2.40 (1.51–3.82) | |||
| BAK1 | rs493871 | GG | Ref. | 0.0001/0.2359 |
| GA | 1.41 (0.98–2.04) | |||
| AA | 2.29 (1.49–3.50) | |||
| ALKBH3 | rs10838192 | TT | Ref. | 0.0003/0.2775 |
| TC | 1.74 (1.25–2.41) | |||
| CC | 2.33 (1.05–5.17) | |||
| DACT3 | rs314659 | GG | Ref. | 0.0004/0.2775 |
| GA | 1.59 (1.16–2.18) | |||
| AA | 2.18 (1.26–3.77) | |||
| MGMT | rs4751109 | CC | Ref. | 0.0004/0.2775 |
| CA | 1.66 (1.15–2.39) | |||
| AA | 2.50 (1.24–5.03) | |||
| FAF1_CDKN2C | rs11587909 | CC | Ref. | 0.00049/0.2775 |
| CT | 1.36 (0.99–1.88) | |||
| TT | 2.44 (1.46–4.08) |
CI, confidence interval.
SNP-based linear Ptrend calculated based on the three-level genotype (0, 1 and 2) in logistic regression models adjusted for sex, attained age and year of birth.
False discovery rate (FDR) corrected linear Ptrend.
Fig. 2.
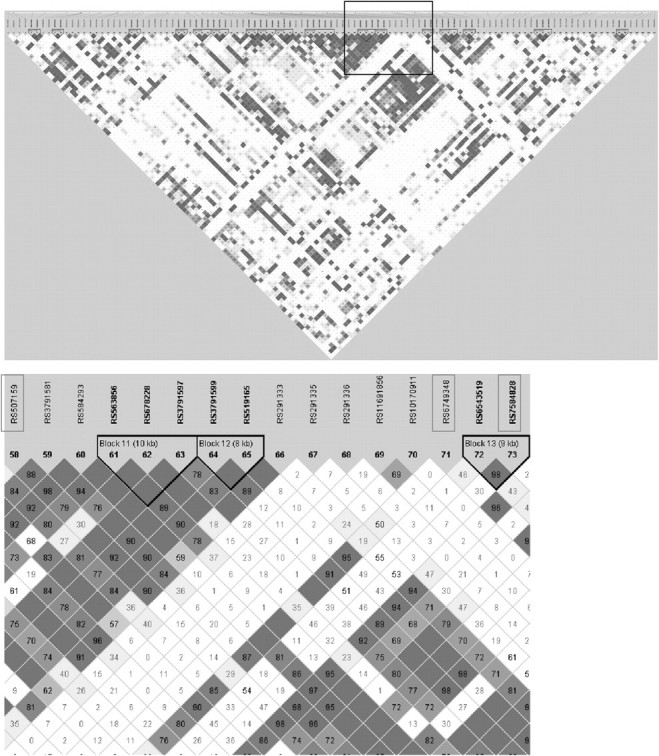
HDAC4 sequential scan and variable-sized sliding window analysis (with detailed view of the top three SNPs blocked, below) in the cases and controls used for this study.
Gene region-based associations
Consistent with individual SNP analyses, six gene regions (HUS1, HDAC4, ALKBH3, BAK1, FAF1_CDKN2C, DACT3) containing the most significant SNPs were associated with PTC risk at Pregion <0.01 (Table III). In addition, one gene (FZD6) containing several moderately significant SNPs was associated with PTC risk at Pregion <0.01. Two of the seven promising gene regions are involved in DNA repair (HUS1, ALKBH3), two in apoptosis (BAK1, FAF1_CDKN2C), two in Wnt/beta-catenin-signaling (FZD6, DACT3) and one in the epigenetic (HDAC4) pathway. After multiple comparisons adjustment, none of the gene regions were significant at Pregion <0.10. The results of all gene region-based analyses are available in supplementary Table D, Carcinogenesis Online. For a more detailed description of the top-ranked genes, see supplementary Table B, Carcinogenesis Online.
Table III.
Significance levels (P values) among those gene regions with uncorrected Pregion <0.01 for an association with papillary thyroid cancer
| Gene/region | Chromosome location | Number of SNPs | Pregiona/Pregion FDRb |
| HUS1 | 7p13–p12 | 8 | 0.0009/0.3060 |
| HDAC4 | 2q37.3 | 111 | 0.0025/0.3128 |
| ALKBH3 | 11p11.2 | 25 | 0.0041/0.3128 |
| FZD6 | 8q22.3–q23.1 | 13 | 0.0045/0.3128 |
| BAK1 | 6p21.3 | 25 | 0.0046/0.3128 |
| FAF1_CDKN2C | 1p32–p33 | 22 | 0.0060/0.3254 |
| DACT3 | 19q13.32 | 9 | 0.0067/0.3254 |
Gene based P values (Pregion) calculated using the adaptive rank-truncated product method.
False discovery rate (FDR) corrected linear Pregion.
Pathway-based associations
The apoptosis pathway was the only pathway significantly associated with risk of PTC (Ppathway = 0.039) (Table IV). However, after excluding four gene regions (including two of the top gene region-based hits BAK1 and FAF1_CDKN2C) overlapping with other pathways, the apoptosis pathway-based P value was no longer significant, suggesting that the excluded gene regions were primarily responsible for the association. While the DNA repair pathway as a whole was not statistically significantly associated with PTC risk, the DNA repair subpathways direct reversal of DNA damage and other conserved damage response genes, were associated with risk (Ppathway = 0.002 and Ppathway = 0.017, respectively). Finally, the genomic integrity pathway as a whole was of borderline statistical significance (Poverall = 0.074 and Poverall = 0.061 including and excluding overlap, respectively).
Table IV.
Pathway-based significance levels (P values) for each genomic integrity pathway analyzed, with and without accounting for overlapping genes between pathways
| With overlapa |
Without overlap |
|||
| Ppathwayb | Number of genes/SNPs | Ppathwayb | Number of genes/SNPs | |
| Overall | 0.074 | 338/5042 | 0.061 | 324/4530 |
| Pathways | ||||
| Apoptosis | 0.039 | 25/322 | 0.372 | 21/264 |
| Epigenetic | 0.126 | 28/679 | 0.119 | 26/667 |
| Wnt/beta-catenin signaling | 0.141 | 39/725 | 0.120 | 34/602 |
| DNA repair | 0.233 | 151/1979 | 0.345 | 145/1857 |
| Direct reversal of damage | 0.002 | 2/91 | ||
| Other conserved damage response genes | 0.017 | 9/74 | ||
| Base excision repair | 0.164 | 19/295 | ||
| Non-homologous end joining | 0.281 | 9/143 | ||
| Rad6 pathway | 0.285 | 5/45 | ||
| Other suspected DNA repair genes | 0.325 | 9/105 | ||
| Homologous recombination | 0.642 | 20/242 | ||
| Chromatin structure | 0.657 | 2/21 | ||
| DNA polymerase | 0.664 | 15/151 | ||
| Nucleotide excision repair | 0.677 | 30/404 | ||
| Editing processing nucleases | 0.731 | 6/61 | ||
| Fanconi anemia | 0.737 | 9/100 | ||
| Genes defective in diseases associated with sensitivity to DNA damaging agents | 0.805 | 4/61 | ||
| Mismatch repair | 0.817 | 9/147 | ||
| Tumor suppressor | 0.368 | 28/379 | 0.312 | 21/289 |
| Cell cycle control | 0.486 | 27/435 | 0.717 | 20/359 |
| MAPK signaling | 0.668 | 17/296 | 0.707 | 12/157 |
| PI3K/AKT signaling | 0.671 | 6/61 | 0.671 | 6/61 |
| Telomere | 0.733 | 17/166 | 0.733 | 17/166 |
Some genes were allocated to more than one pathway. To calculate P values without multiple pathway allocations, these specific genes were dropped from their respective pathways.
Pathway- or subpathway-based P values (Ppathway) calculated using the adaptive rank-truncated product method.
Discussion
We evaluated the associations of tag SNPs in candidate genes from several interrelated pathways involved in maintenance of genomic integrity, including DNA repair, epigenetic mechanisms, telomere function, apoptosis, cell cycle control, tumor suppression and the MAPK, PI3K/AKT and Wnt/beta-catenin cell-signaling pathways with risk of PTC. The group of genes as a whole was suggestively associated with risk of PTC (Poverall = 0.074/Poverall = 0.061). We found nine tag SNPs in seven gene regions that were associated with PTC at Ptrend <0.0005. The strongest associations were seen for SNPs in the histone deacetylase 4 gene HDAC4, in the DNA repair checkpoint gene HUS1 and in the apoptosis gene BAK1 (Ptrend < 0.0001). Gene region-based analyses showed that all three of these gene regions (HDAC4, HUS1 and BAK1) were significant at Pregion <0.005. While after formal correction for multiple comparisons neither individual SNP- nor gene region-based results remained statistically significant, several of our findings with suggestive P values are of potential biologic or clinical interest.
Specifically, the observed thyroid cancer associations with multiple polymorphisms in HDAC4 were intriguing. HDAC4 is a histone deacetylation gene that has the capacity to alter chromosome structure and silence gene transcription by limiting access of transcription factors to DNA, particularly tumor suppressor genes, thereby deactivating tumor suppression activity (31,32). Histone deacetylase inhibitors are demonstrated anticancer agents whose main mechanism of action is the transcriptional reactivation of tumor suppressor genes that have been turned off through histone deacetylation (33). Several studies of thyroid tumor cells have demonstrated the ability of HDAC inhibitors to facilitate radioactive iodine uptake (34–36) and suppress growth and proliferation (37–40). However, no studies have been published on associations between HDAC polymorphisms and thyroid cancer risk.
HUS1 has been linked with other cancers, namely breast and ovarian (41,42), and is part of the Rad9-Rad1-Hus1 (911) cell cycle checkpoint complex that plays a key role in all checkpoint responses to DNA damage (43). In vitro studies have shown that human cells exposed to ionizing and ultraviolet radiation have higher levels of the 911 protein complex compared with unexposed cells (44,45) and that this relationship is dose dependent. No studies previously have reported a relationship between HUS1 polymorphisms and thyroid cancer.
We also found variants in two genes postulated to play a role in apoptosis, BAK1 and FAF1, had suggestive P values for an association with an increased risk of PTC. Previous research suggests that these genes may play a role in carcinogenesis of certain cancers, including testicular cancer and chronic lymphocytic leukemia (46,47), myeloma (48) and mantle cell lymphoma (49). Moreover, BAK1 expression appears to be upregulated in thyroid tumor cells (50–2).
Our strongest pathway-based findings for the DNA repair direct reversal of damage pathway (53) was driven by two of our top SNP-based findings—tag SNPs rs10838192 in ALKBH3 and rs4751109 in MGMT. While little is known about ALKBH3 in relation to cancer, several candidate gene studies have linked MGMT polymorphisms to risk of head and neck cancer (54,55), glioma (56–58) and esophageal cancer (59,60). Moreover, two studies have found an association between MGMT hypermethylation and PTC (61,62).
In interpreting the results of our study, several strengths and limitations need to be considered. Our study had high participation rates minimizing potential for selection bias. Because survival rates for PTC are exceptionally high, survival bias is unlikely. To minimize concerns about population stratification, all analyses were limited to individuals of European ancestry. Moreover, cases from the two studies were similar with regard to age at diagnosis, smoking status, tumor size and allele frequencies. Although radiation exposure is an established risk factor for thyroid cancer, our cases are unlikely to be radiation related because doses are low, most of the exposure occurred in adulthood, and no dose–response has been observed in the USRT population (63,64), and only five of the UTMDACC cases were exposed to radiation from self-report of radiotherapy (12). Thus, the results of our study should be internally valid. However, extrapolation of our findings to the general population requires caution. Thyroid cancer incidence in the USRT cohort was higher than in the general population, with a standardized incidence rate ratio of 1.7. This difference is probably due to increased screening in the USRT cohort as the proportion of small thyroid tumors was higher among the cohort (30%) compared with the general population (15%) based on the SEER registries database (23) but comparable with that in the UTMDACC cases.
Given that PTC cases were less likely to be cigarette smokers or alcohol drinkers and had higher average body mass index that controls, we explored whether these factors may have influenced the associations of interest. When added to the models, these variables did not meaningfully change the risk estimates, and therefore, are unlikely to confound our main findings. Other strengths of our study include thorough selection of genes related to a variety of genomic integrity pathways (53,65–67) and nearly complete representation of DNA repair genes. Relative to genome-wide association studies, the coverage of selected gene regions was higher, although we could have missed important associations with SNPs not included within the genotyping platform. While among the larger studies with respect to the number of thyroid cancer cases and controls, our study had limited power to detect weak associations, especially for less common genetic variants. Another limitation is the use of tag SNPs that are themselves unlikely to be the disease-related SNPs but are assumed to be in LD with the causal variant. To address these limitations, we excluded SNPs with minor allele frequency <10% and relied on robust gene/pathway adaptive rank-truncated product methods combining SNP-specific P values of trend to confirm associations with risk of PTC.
In summary, our results suggest that genetic alterations in the pathway involved in maintenance of genomic integrity may contribute to thyroid cancer susceptibility.
Supplementary material
Supplementary Tables A–D can be found at http://carcin.oxfordjournals.org/
Funding
This research was supported in part by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health and by a grant from the American Thyroid Association (Principal Investigator: E.M.S.). This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E.
Supplementary Material
Acknowledgments
We are grateful to the radiologic technologists who participated in the USRT Study; Jerry Reid of the American Registry of Radiologic Technologists for continued support of this study; Diane Kampa and Allison Iwan of the University of Minnesota for data collection and study coordination; Liliana Mugartegui for patient recruitment, data collection and study coordination at the University of Texas M. D. Anderson Cancer Center; Laura Bowen of Information Management Systems for biomedical computing statistical support. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- LD
linkage disequilibrium
- SNP
single-nucleotide polymorphism
- USRT
US Radiologic Technologist
- UTMDACC
University of Texas M. D. Anderson Cancer Center
References
- 1.Jemal A, et al. Cancer Statistics, 2010. CA Cancer J. Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Davies L, et al. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA. 2006;295:2164–2167. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 3.Ron E, et al. Thyroid cancer. In: Schottenfeld D, JF Fraumeni J, editors. Cancer Epidemiology and Prevention. New York, NY: Oxford University Press; 2006. [Google Scholar]
- 4.Hemminki K, et al. Familial risk of cancer by site and histopathology. Int. J. Cancer. 2003;103:105–109. doi: 10.1002/ijc.10764. [DOI] [PubMed] [Google Scholar]
- 5.Dong C, et al. Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families. Int. J. Cancer. 2001;92:144–150. [PubMed] [Google Scholar]
- 6.Frich L, et al. Familial occurrence of nonmedullary thyroid cancer: a population-based study of 5673 first-degree relatives of thyroid cancer patients from Norway. Cancer Epidemiol. Biomarkers Prev. 2001;10:113–117. [PubMed] [Google Scholar]
- 7.Goldgar DE, et al. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J. Natl Cancer Inst. 1994;86:1600–1608. doi: 10.1093/jnci/86.21.1600. [DOI] [PubMed] [Google Scholar]
- 8.Hrafnkelsson J, et al. Familial non-medullary thyroid cancer in Iceland. J. Med. Genet. 2001;38:189–191. doi: 10.1136/jmg.38.3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi M, et al. The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum. Mol. Genet. 2010;19:2516–2523. doi: 10.1093/hmg/ddq123. [DOI] [PubMed] [Google Scholar]
- 10.Gudmundsson J, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 2009;41:460–464. doi: 10.1038/ng.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturgis EM, et al. Radiation response genotype and risk of differentiated thyroid cancer: a case-control analysis. Laryngoscope. 2005;115:938–945. doi: 10.1097/01.MLG.0000163765.88158.86. [DOI] [PubMed] [Google Scholar]
- 12.Ho T, et al. RET polymorphisms and haplotypes and risk of differentiated thyroid cancer. Laryngoscope. 2005;115:1035–1041. doi: 10.1097/01.MLG.0000162653.22384.10. [DOI] [PubMed] [Google Scholar]
- 13.Ho T, et al. Association of XRCC1 polymorphisms and risk of differentiated thyroid carcinoma: a case-control analysis. Thyroid. 2009;19:129–135. doi: 10.1089/thy.2008.0153. [DOI] [PubMed] [Google Scholar]
- 14.Akulevich NM, et al. Polymorphisms of DNA damage response genes in radiation-related and sporadic papillary thyroid carcinoma. Endocr. Relat. Cancer. 2009;16:491–503. doi: 10.1677/ERC-08-0336. [DOI] [PubMed] [Google Scholar]
- 15.Chiang FY, et al. Association between polymorphisms in DNA base excision repair genes XRCC1, APE1, and ADPRT and differentiated thyroid carcinoma. Clin. Cancer Res. 2008;14:5919–5924. doi: 10.1158/1078-0432.CCR-08-0906. [DOI] [PubMed] [Google Scholar]
- 16.Bastos HN, et al. Association of polymorphisms in genes of the homologous recombination DNA repair pathway and thyroid cancer risk. Thyroid. 2009;19:1067–1075. doi: 10.1089/thy.2009.0099. [DOI] [PubMed] [Google Scholar]
- 17.Silva SN, et al. Association of polymorphisms in ERCC2 gene with non-familial thyroid cancer risk. Cancer Epidemiol. Biomarkers Prev. 2005;14:2407–2412. doi: 10.1158/1055-9965.EPI-05-0230. [DOI] [PubMed] [Google Scholar]
- 18.Siraj AK, et al. RAD52 polymorphisms contribute to the development of papillary thyroid cancer susceptibility in Middle Eastern population. J. Endocrinol. Invest. 2008;31:893–899. doi: 10.1007/BF03346438. [DOI] [PubMed] [Google Scholar]
- 19.Adjadj E, et al. Germ-line DNA polymorphisms and susceptibility to differentiated thyroid cancer. Lancet Oncol. 2009;10:181–190. doi: 10.1016/S1470-2045(09)70020-8. [DOI] [PubMed] [Google Scholar]
- 20.Santarpia L, et al. Genetic alterations in the RAS/RAF/mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways in the follicular variant of papillary thyroid carcinoma. Cancer. 2010;116:2974–2983. doi: 10.1002/cncr.25061. [DOI] [PubMed] [Google Scholar]
- 21.Wang SS, et al. Common genetic variants and risk for HPV persistence and progression to cervical cancer. PLoS One. 2010;5:e8667. doi: 10.1371/journal.pone.0008667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang HP, et al. Common genetic variation in the sex hormone metabolic pathway and endometrial cancer risk: pathway-based evaluation of candidate genes. Carcinogenesis. 2010;31:827–833. doi: 10.1093/carcin/bgp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sigurdson AJ, et al. Cancer incidence in the US radiologic technologists health study, 1983–1998. Cancer. 2003;97:3080–3089. doi: 10.1002/cncr.11444. [DOI] [PubMed] [Google Scholar]
- 24.Lönn S, et al. Papillary thyroid cancer and polymorphic variants in TSHR- and RET-related genes: a nested case-control study within a cohort of US radiologic technologists. Cancer Epidemiol. Biomarkers Prev. 2007;16:174–177. doi: 10.1158/1055-9965.EPI-06-0665. [DOI] [PubMed] [Google Scholar]
- 25.Carlson CS, et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46:515–522. [PubMed] [Google Scholar]
- 27.Benjamini Y, et al. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat Soc. Ser. B Stat. Methodol. 1995;57:289–300. [Google Scholar]
- 28.Yu K, et al. Pathway analysis by adaptive combination of P-values. Genet. Epidemiol. 2009;33:700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett JC, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 30.Mathias RA, et al. A graphical assessment of p-values from sliding window haplotype tests of association to identify asthma susceptibility loci on chromosome 11q. BMC Genet. 2006;7:38. doi: 10.1186/1471-2156-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan L, et al. HDAC4 inhibits the transcriptional activation of mda-7/IL-24 induced by Sp1. Cell. Mol. Immunol. 2010;7:221–226. doi: 10.1038/cmi.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozdag H, et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 2006;7:90. doi: 10.1186/1471-2164-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villar-Garea A, et al. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int. J. Cancer. 2004;112:171–178. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]
- 34.Furuya F, et al. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology. 2004;145:2865–2875. doi: 10.1210/en.2003-1258. [DOI] [PubMed] [Google Scholar]
- 35.Kitazono M, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(-) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 2001;86:3430–3435. doi: 10.1210/jcem.86.7.7621. [DOI] [PubMed] [Google Scholar]
- 36.Zarnegar R, et al. Increasing the effectiveness of radioactive iodine therapy in the treatment of thyroid cancer using Trichostatin A, a histone deacetylase inhibitor. Surgery. 2002;132:984–990. doi: 10.1067/msy.2002.128690. discussion 990. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg VL, et al. Histone deacetylase inhibitors promote apoptosis and differential cell cycle arrest in anaplastic thyroid cancer cells. Thyroid. 2001;11:315–325. doi: 10.1089/10507250152039046. [DOI] [PubMed] [Google Scholar]
- 38.Greenberg VL, et al. Butyrate alters the expression and activity of cell cycle components in anaplastic thyroid carcinoma cells. Thyroid. 2001;11:21–29. doi: 10.1089/10507250150500621. [DOI] [PubMed] [Google Scholar]
- 39.Mitsiades CS, et al. Novel histone deacetylase inhibitors in the treatment of thyroid cancer. Clin. Cancer Res. 2005;11:3958–3965. doi: 10.1158/1078-0432.CCR-03-0776. [DOI] [PubMed] [Google Scholar]
- 40.Xiao X, et al. Notch1 mediates growth suppression of papillary and follicular thyroid cancer cells by histone deacetylase inhibitors. Mol. Cancer Ther. 2009;8:350–356. doi: 10.1158/1535-7163.MCT-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de la Torre J, et al. Expression of DNA damage checkpoint protein Hus1 in epithelial ovarian tumors correlates with prognostic markers. Int. J. Gynecol. Pathol. 2008;27:24–32. doi: 10.1097/pgp.0b013e31812dfaef. [DOI] [PubMed] [Google Scholar]
- 42.Vega A, et al. Evaluating new candidate SNPs as low penetrance risk factors in sporadic breast cancer: a two-stage Spanish case-control study. Gynecol. Oncol. 2009;112:210–214. doi: 10.1016/j.ygyno.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 43.Sancar A, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 44.Zou L, et al. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, et al. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005;24:4728–4735. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enjuanes A, et al. Genetic variants in apoptosis and immunoregulation-related genes are associated with risk of chronic lymphocytic leukemia. Cancer Res. 2008;68:10178–10186. doi: 10.1158/0008-5472.CAN-08-2221. [DOI] [PubMed] [Google Scholar]
- 47.Rapley EA, et al. A genome-wide association study of testicular germ cell tumor. Nat. Genet. 2009;41:807–810. doi: 10.1038/ng.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker BA, et al. A compendium of myeloma associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116:e56–e65. doi: 10.1182/blood-2010-04-279596. [DOI] [PubMed] [Google Scholar]
- 49.Bea S, et al. Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood. 2009;113:3059–3069. doi: 10.1182/blood-2008-07-170183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bröcker M, et al. Expression of apoptosis-related proteins in thyroid tumors and thyroid carcinoma cell lines. Exp. Clin. Endocrinol. Diabetes. 1996;104(suppl. 4):20–23. doi: 10.1055/s-0029-1211694. [DOI] [PubMed] [Google Scholar]
- 51.Todaro M, et al. High levels of exogenous C2-ceramide promote morphological and biochemical evidences of necrotic features in thyroid follicular cells. J. Cell. Biochem. 2002;86:162–173. doi: 10.1002/jcb.10203. [DOI] [PubMed] [Google Scholar]
- 52.Wang SH, et al. IFN[gamma] sensitization to TRAIL-induced apoptosis in human thyroid carcinoma cells by upregulating Bak expression. Oncogene. 2003;23:928–935. doi: 10.1038/sj.onc.1207213. [DOI] [PubMed] [Google Scholar]
- 53.Wood RD, et al. Human DNA repair genes, 2005. Mutat. Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z, et al. Polymorphisms of the DNA repair gene MGMT and risk and progression of head and neck cancer. DNA Repair (Amst) 2010;9:558–566. doi: 10.1016/j.dnarep.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang W-Y, et al. Selected genetic polymorphisms in MGMT, XRCC1, XPD, and XRCC3 and risk of head and neck cancer: a pooled analysis. Cancer Epidemiol. Biomarkers Prev. 2005;14:1747–1753. doi: 10.1158/1055-9965.EPI-05-0162. [DOI] [PubMed] [Google Scholar]
- 56.Wiencke JK, et al. Molecular features of adult glioma associated with patient race/ethnicity, age, and a polymorphism in O6-methylguanine-DNA-methyltransferase. Cancer Epidemiol. Biomarkers Prev. 2005;14:1774–1783. doi: 10.1158/1055-9965.EPI-05-0089. [DOI] [PubMed] [Google Scholar]
- 57.Felini MJ, et al. DNA repair polymorphisms XRCC1 and MGMT and risk of adult gliomas. Neuroepidemiology. 2007;29:55–58. doi: 10.1159/000108919. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, et al. Association and interactions between DNA repair gene polymorphisms and adult glioma. Cancer Epidemiol. Biomarkers Prev. 2009;18:204–214. doi: 10.1158/1055-9965.EPI-08-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma WJ, et al. DNA polymorphism and risk of esophageal squamous cell carcinoma in a population of North Xinjiang, China. World J. Gastroenterol. 2010;16:641–647. doi: 10.3748/wjg.v16.i5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akbari MR, et al. Candidate gene association study of esophageal squamous cell carcinoma in a high-risk region in Iran. Cancer Res. 2009;69:7994–8000. doi: 10.1158/0008-5472.CAN-09-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishida E, et al. DNA hypermethylation status of multiple genes in papillary thyroid carcinomas. Pathobiology. 2007;74:344–352. doi: 10.1159/000110028. [DOI] [PubMed] [Google Scholar]
- 62.Schagdarsurengin U, et al. CpG island methylation of tumor-related promoters occurs preferentially in undifferentiated carcinoma. Thyroid. 2006;16:633–642. doi: 10.1089/thy.2006.16.633. [DOI] [PubMed] [Google Scholar]
- 63.Simon SL, et al. Estimating historical radiation doses to a cohort of U.S. radiologic technologists. Radiat. Res. 2006;166 doi: 10.1667/RR3433.1. (1 Pt 2): 174–192. [DOI] [PubMed] [Google Scholar]
- 64.Zabel EW, et al. Thyroid cancer and employment as a radiologic technologist. Int. J. Cancer. 2006;119:1940–1945. doi: 10.1002/ijc.22065. [DOI] [PubMed] [Google Scholar]
- 65.Charames GS, et al. Genomic instability and cancer. Curr. Mol. Med. 2003;3:589–596. doi: 10.2174/1566524033479456. [DOI] [PubMed] [Google Scholar]
- 66.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 67.Paulsen RD, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.