

Abstract
Colon cancer is the third most common cause of cancer and is the second leading cause of cancer deaths in the USA. Although inhibition of aldose reductase (AR) is known to prevent human colon cancer cell growth in nude mice xenografts, the role of AR in the regulation of cancer metastasis is not known. We now demonstrate the mechanisms by which AR regulates colon cancer metastasis in vitro and in vivo. Inhibition of AR prevented the epidermal growth factor (EGF) or fibroblast growth factor (FGF)-induced migration and invasion of human colon cancer (HT29; KM20) cells by >70% and also inhibited (>80%) the adhesion of the cancer cells to endothelial cells. Treatment of endothelial cells with AR inhibitors significantly (∼85%) downregulated the EGF or FGF-induced expression of Inter-Cellular Adhesion Molecule-1, Vascular cell adhesion molecule-1 and vascular endothelial-cadherin. Furthermore, liver metastasis of green fluorescent protein-labeled KM20 cells injected into the spleen of athymic nude mice was significantly (>65%) prevented by AR inhibitor, fidarestat or ARsiRNA delivered systemically into the mice. Similar results were observed with HT29 cells. AR inhibition or ablation also prevented (70–90%) the increase in the levels of matrix metalloproteinase-2, cyclin D1, CD31, CD34 and the activation of nuclear factor-kappa-binding protein in metastatic liver. Thus, our results indicate that AR regulates cancer cell adhesion, invasion and migration events which initiate metastasis and therefore, AR inhibition could be a novel therapeutic approach for the prevention of colon cancer metastasis.
Introduction
Colon cancer is one of the most common invasive malignancies and is the second leading cause of cancer-related deaths in the USA (1). Approximately 102 900 new cases of colon cancer and 51 370 colon cancer-related deaths are predicted for the year 2010 alone in the USA (1). Despite recent advancement in diagnostic, surgical and therapeutic techniques, the survival rate of colon cancer is poor due to recurrence of the disease (2). The lack of effective therapies for advanced colon cancer is related to poor understanding of the progression of the disease especially toward invasion, migration and metastasis (2–6). One of the major problems in the therapy of colon cancer is the prevention of metastasis, which is usually incurable and leads to speedy death of patients (2–6). Therefore, it is important to understand the mechanisms that cure/halt metastasis so that it could be prevented or significantly delayed.
The major sequential steps in the pathogenesis of tumor metastasis include: dissociation and intravasation of cells from a primary tumor into the circulation and survival of the cells in circulation, arrest in small vessels in cell-specific distant organs, adhesion to endothelial cells and extravagation into surrounding tissues, proliferation and vascularization of the metastatic tumor (5–7). The progression of metastasis occurs via blood or lymphatic system or by both. Altered expression of proteases, adhesion molecules, growth factors, cytokines, chemokines and angiogenic factors are known to play significant role in the tumor metastasis (7–13). In the tumor microenvironment, decreased expression of cell–cell adherent molecules such as E-cadherin, desmoplakin, cytokeratin and increased expression of N-cadherin, vimentin, fibronectin and matrix metalloproteinases (MMPs; family of human zinc-dependent endopeptidases) favors dissociation and motility of the tumor cell from the primary tumor site (7,12,13). Increased expression of MMPs correlates with tumor cell invasion and metastasis (12,13). Invasion of dissociated tumor cells occurs by several mechanisms: it may occur through thin-walled venules like lymphatic channels or hematogenous route. In the circulation, detached small tumor cell aggregates cause embolization of blood vessels (5). Tumor cells subsequently proliferate and extravagate into organ parenchyma by adhering to capillary endothelial cells or by adhering to subendothelial basement membrane by developing vascular network and evading the host immune system (2,11). Various factors such as MMPs, adhesion molecules, growth factors, cytokines, chemokines and angiogenic molecules involved in adhesion, invasion, migration and tumor metastasis are regulated by redox-sensitive transcription factor, nuclear factor-kappa-binding protein [NF-κB; (7–14)]. Reactive oxygen species (ROS)-mediated activation of NF-κB signaling is a key mechanism in tumorigenesis and mestastasis (14,15). Antioxidants such as N-acetyl cysteine, resveratrol, lycopene, green tea and epigallocatechin-3-gallate have been shown to attenuate the invasion and metastasis of various cancers including colon cancer via inhibition of NF-κB activation (16–20). Our recent studies with human colon cancer cells suggest that the polyol pathway enzyme—aldose reductase (AR), a member of aldo–keto reductase superfamily is a regulator of ROS signals induced by growth factors such as basic fibroblast growth factor (FGF) and platelet-derived growth factor and cytokines such as tumor necrosis factor-alpha (21–24).
We have recently demonstrated that AR besides reducing aldo-sugars, efficiently catalyzes the reduction of lipid aldehydes such as 4-hydroxy-trans-2-nonenal (HNE) and their glutathione (GSH) conjugates such as GS-HNE to 1, 4-dihydroxynonene (DHN) and GS-DHN, respectively with low (micromolar) Km compared with glucose with Km in millimolar range (23,25). We have shown earlier that inhibition of AR could prevent protein kinase C (PKC), NF-κB and activator protein-1 activation and the increase in cell growth caused by HNE and GS-HNE but not by GS-DHN (21,22,25). These studies suggested that the already reduced form of glutathione lipid aldehyde, GS-DHN is insensitive to AR inhibition and could be the main mediator of oxidative stress-induced NF-κB activation. Furthermore, we have demonstrated that AR inhibition as well as ablation by siRNA could prevent the growth factors such as FGF- and platelet-derived growth factor-induced activation of NF-κB, expression of cyclooxygenase-2 and production of prostaglandin-E2 in colon cancer cells (21,22). Most remarkably, in nude mice xenograft model, we have shown that inhibition of AR by ARsiRNA completely prevented growth of human colon adenocarcinoma cells (SW480) implanted subcutaneously (21). Recently, our results with azoxymethane model in male BALB/c mice showed that inhibition of AR by pharmacological inhibitor of AR as well as AR gene knockout in mice significantly prevented aberrant crypt foci formation and azoxymethane-induced expression of inflammatory markers, inducible nitric oxide synthase and cyclooxygenase-2 and preneoplastic marker proteins, cyclin D1 and beta-catenin and activation of transcription factor, NF-κB in mice colons (26). However, the involvement of AR in the metastatic progression of human colon cancer is unknown.
In the present study, we have demonstrated the mechanisms by which AR inhibition prevents colon cancer metastasis. Our in vitro results suggest that inhibition of AR could prevent cultured human colon cancer cells, HT29 adhesion, invasion and migration, which are important steps for metastasis initiation. Furthermore, our results show that inhibition of AR could prevent metastatic growth of human colon cancer cells, injected in the spleen of nude mice, in the liver of nude mice. These findings indicate that AR is an excellent novel therapeutic target for the prevention of metastatic spread of colon cancer.
Materials and methods
Materials
McCoy's 5A medium, minimum essential medium, Dulbecco's modified Eagle's medium-F12K, phosphate-buffered saline, penicillin/streptomycin solution, trypsin and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). Antibodies against AR, Cyclin D1, MMP2, CD34, phospho-p65 and Glyceraldehyde-3- phosphate dehydrogenase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Sorbinil and zopolrestat were gift from Pfizer (New York, NY). Fidarestat was obtained as a gift from Sanwa Kagaku Kenkyusho Co. Ltd (Tokyo, Japan). Cell invasion and migration assay kits were obtained from Chemicon International (Billerica, MA). Epidermal growth factor (EGF), FGF and other reagents used in western blot analysis were obtained from Sigma (St Louis, MO). AR-Sistable small interfering RNA was synthesized by Dharmacon Research (Chicago, IL). All other reagents used were of analytical grade.
Cell culture
Human colon cancer HT29 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA) and grown in McCoy’s 5A medium supplemented with 10% FBS and 1% penicillin/streptomycin. KM20 cells were obtained and grown in minimum Eagle medium supplemented with 10% FBS, 1% sodium pyruvate, 1% non-essential amino acids and 2% minimum essential medium essential vitamin as described (3,4). Human umbilical vascular endothelial cells (HUVEC) obtained from Cell Application Inc. (San Diego, CA) and grown in Dulbecco's modified Eagle's medium F12K medium containing 10% FBS and cultured at 37°C under an atmosphere containing 5% CO2. All the cells were examined for any mycoplasma and endotoxins contamination before the experiments.
Cell invasion assay
Invasive assay was performed using basement membrane extract (BME) as per the manufacturer instructions (Chemicon International). Briefly, 50 μl of basement membrane extract solution was added on top of 8 μ polyethylene terephthalate membrane to each well of 96-well plates and incubated at 37°C for 4 h to allow gel formation. Green fluorescent protein (GFP) transfected and untransfected HT29 or KM20 cells at 20 000 cells per well in basal medium with EGF (5 ng/ml) or FGF (10 ng/ml) with or without sorbinil or zopolrestat (20 μM) were plated on Matrigel. Controls cells received vehicle (dimethyl sulfoxide) solution only. After 24 h of incubation, invasion of cells toward bottom side of the well was measured using Calcein AM fluorescent dye at 480/520 nm or photographed invaded cells by fluoresce microscope (×100).
Cell migration (chemotaxis) assay
HT29 and KM20 cells were serum starved in McCoy’s medium with or without sorbinil or zopolrestat (20 μM) for 24 h and cell migration assay was performed as per the manufacturer instructions (Chemicon International). Briefly, 2 × 105 HT29 cells were plated per well of 24-well plate culture inserts containing 8.0 μm polycarbonate membrane. Subsequently, cells were treated with EGF (5 ng/ml) or FGF (10 ng/ml) with or without sorbinil or zopolrestat and the plate was transferred to 24-well plate which contained McCoy’s medium with growth factor ± sorbinil or zopolrestat and then the cells were incubated at 37°C under a 5% CO2 atmosphere. After 24 h, migrated cells were stained at bottom of the culture inserts and stain extracted into extraction buffer was measured calorimetrically at 560 nm. Migration of HUVEC in a scratch wound assay model was also performed as described elsewhere (27).
Cell adhesion assay
Cell adhesion assay was performed as described elsewhere (28). Briefly, HUVEC were plated in 96-well plate at 3 × 103 per well. Subconfluent cells were growth arrested in 0.1% FBS with or without AR inhibitor fidarestat (2 μM). After 24 h, EGF (5 ng/ml) or FGF (10 ng/ml) was added to the medium and the cells were incubated for another 24 h. All the control cells received vehicle solutions only. HT29 cells were harvested in serum-free medium and applied at 2500 cells per well to the HUVEC monolayer in 96-well plate for 6 h. The non-adherent cells were removed by rinsing the wells with serum-free medium, adherent cells were quantified by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. For fluorescent microscopic analysis of cell adhesion, HT29 cells were labeled with PKH67 green fluorescent dye (PHK67 cell linker kit; Sigma Co, St. Louis, MO) following manufacture instructions. Labeled HT29 cells and HT29-GFP cells were added to the growth factor-treated HUVEC, grown on chamber slides without and with presence and absence of AR inhibitors as described above and photographed using Nikon fluorescent microscope. The cell surface expression of adhesion molecules was measured by FACS analysis as described by Butts et al. (29).
In vivo metastasis
Four to six weeks-old male nude nu/nu mice were obtained from Harlan (Indianapolis, IN) and experiments were approved by the University of Texas Medical Branch Institutional Animal Care and Use Committee. The mice were fed standard chow (Formula Chow 5008; Purina Mills, St Louis, MO) and tap water ad libitum and allowed to acclimate for 1 week. Metastatic HT29-GFP or KM20-GFP cells were injected intrasplenically as described previously (3,4). Briefly, mice were anesthetized using isofluorane, a small left abdominal flank incision was created and the spleen was exteriorized. HT29-GFP or KM20-GFP cells (4 × 106 cells/100 μl) were injected into the spleen with a 27-gage needle. The spleen was returned to the abdomen and the wound was closed in one layer with wound clips. After 24 h, spleen was removed and animals were randomized into metastatic control, scrambled ARSiRNA and AR inhibitor (sorbinil, fidarestat and ARSiRNA) groups. Control group was fed with normal diet and AR inhibitor group fed with sorbinil (40 mg/kg body wt/day) in the diet, whereas fidarestat (50 mg/kg body wt/day) was given in drinking water. In siRNA treatment group, each mouse received (intravenous) Sistable control-SiRNA (AAAATCTCCCTAAAT CATACA) or ARSiRNA (AATCGGTGTCTCCAACTTCAA; 200 μg/mice) every 10 days for three times. Mice were killed after 35 days and metastasis in the liver was evaluated by using the Illumatool TLS (Lightools Research, Encinitas, CA).
Western blot analysis
To examine the expression of AR, cyclin D1, MMP2, CD34, phospho-p65, total-p65 and Glyceraldehyde-3- phosphate dehydrogenase proteins, western blot analysis was carried out. Equal amounts of protein from liver tissue extracts were subjected to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis followed by transfer of proteins to nitrocellulose filters and probing with the indicated antibodies. The antigen–antibody complex was detected by enhanced chemiluminescence (Pierce, Piscataway, NJ).
Reverse transcription—polymerase chain reaction analysis
Total RNA was isolated from liver samples by using RNeasy mini isolation kit (Qiagen, Valencia, CA). Total RNA (1.5 μg) sample was reverse transcribed with Omniscript and Sensiscript reverse transcriptase one-step reverse transcription–polymerase chain reaction(PCR) system with HotStarTaq DNA polymerase (Qiagen) at 55°C for 30 min followed by PCR amplification. The oligonucleotide primer sequences were as follows: 5′-ACCTGGATGCCGTCGTGGAC-3′ (sense) and 5′-TGTGGCAGCACCAGGGCAGC-3′ (antisense) for MMP2, 5′-TGTTTGCAAGCAGGACTTTG-3′ (sense) and 5′-ACGTCAGCCTCCACACTCTT-3′ (antisense) for cyclin D1, 5′-CCTGGAAGTCCCCTCCAGGGCAGG-3′ and 5′-GGTTGAAGTTGGAGATGCCAATAGC-3′ for AR and 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ (sense) and 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ (antisense) for Glyceraldehyde-3- phosphate dehydrogenase. PCR was carried out in a GeneAmp 2700 thermocycler (Applied Biosystems, Foster City, CA) under the following conditions: initial denaturation at 95°C for 15 min and 35 cycles of 94°C for 30 s, 62°C for 30 s and 72°C for 1 min and then 72°C for 5 min for final extension. PCR products were electrophoresed in 2% Agarose-1× TAE gels containing 0.5 μg/ml ethidium bromide. Bands were quantified using Kodak Image Station 2000R.
Immunohistochemical analysis
KM20-GFP liver metastatic samples were perfusion fixed with 4% paraformaldehyde and stored in 70% ethanol. Paraffin-embedded 5 μm tumor sections were stained with antibodies against AR, cyclin D1, MMP2, CD34, CD31 and phospho-p65 using DakoCytomation LSAB + System-HRP kit.
Statistical analysis
Data presented as mean ± SD and P values were determined by using an unpaired Student's t-test from Microsoft Office Excel 2007 software. P < 0.01 was considered as statistically significant.
Results
Inhibition of AR prevents invasion and migration of HT29 cells
To understand the role of AR in the metastasis of colon cancer, we first determined the effect of AR inhibition on cell growth, invasion and migration, which play a pivotal role in the establishment of tumor progression and metastasis of colon cancer cells. Cell growth was performed in regular 96-well plates and invasion assays were performed in chambered invasion kit containing matrigel. Stimulation of HT29 and KM20 cells with EGF or FGF for 16 h caused significant cell growth and inhibition of AR prevented the growth factor-induced cancer cell growth (Figure 1A). Similarly incubation of HT29 and KM20 cells with EGF and FGF caused significant invasion of cells through matrigel and inhibition of AR prevented the growth factor-induced invasive potential of metastatic HT29 and KM20 cells by >70% (Figure 1B). Furthermore, our results with in vitro migration of HT29 cells using transwell chemotaxis migration assay showed that inhibition of AR prevented EGF- as well as FGF-induced migration of HT29 cells through uncoated porous filters (Figure 1C and FigureS1 is available at Carcinogenesis Online). Similar results were observed when we normalized invasion and migration data with cell growth (Figure S2 is available at Carcinogenesis Online). These results indicate that AR inhibitors could prevent metastasis of colon cancer by inhibiting invasion and migration of colon cancer cells.
Fig. 1.
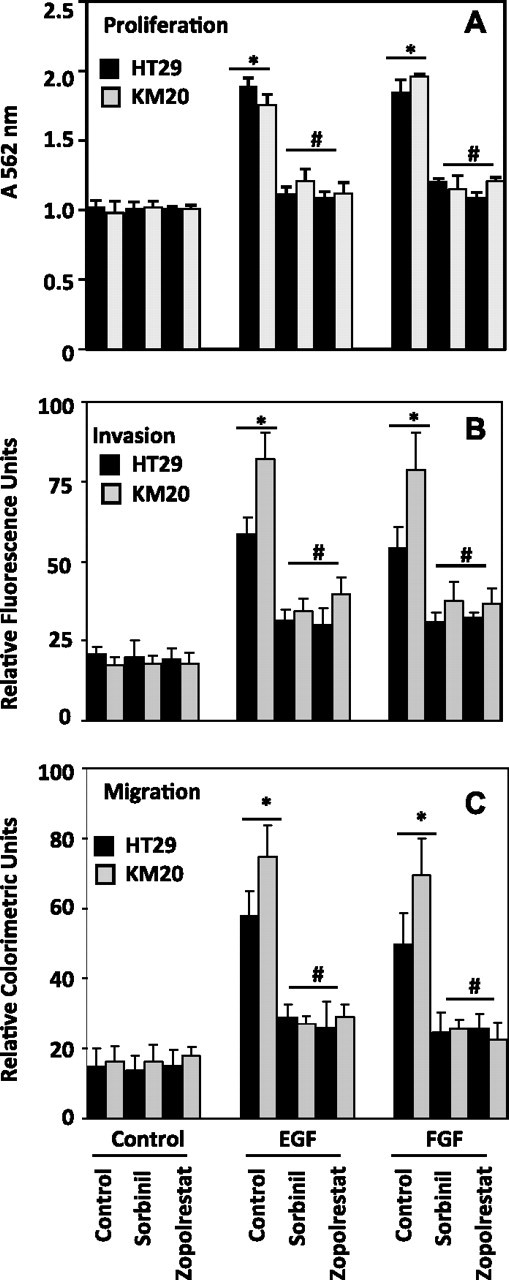
Inhibition of AR prevents growth factors-induced growth, invasion and migration of human colon cancer cells. (A) HT29-GFP and KM20-GFP cells (5000 cells per well in a 96-well plate) were kept in 0.1% serum for overnight followed by addition of EGF (5 ng/ml) or FGF (10 ng/ml) for additional 16 h in the absence and presence of AR inhibitors (20 μM). MTT absorbance was measured by recording absorbance at 560 nm. (B) GFP-transfected and untransfected HT29 or KM20 cells were plated at 20 000 cells per chambered matrigel well in a 96-well plate containing EGF (5 ng/ml) or FGF (10 ng/ml) with or without sorbinil or zopolrestat (20 μM). After 16 h of incubation, invasion of cells toward bottom side of the well was measured in situ using Calcein AM fluorescent dye at 480/520 nm. (C) Chemotaxis migration assay was performed in a plate containing 24-well culture inserts with 8.0 μm polycarbonate membrane. HT29 and KM20 cells which migrated to bottom of the culture inserts were stained and measured colorimetrically at 560 nm. Bars represent mean ± SEM (n = 4); *P < 0.001 versus control and #P < 0.01 versus growth factor-treated cells.
Inhibition of AR prevents growth factors-induced adhesion of human colon cancer HT29 cells to HUVEC
Since increased adhesion of cancerous cells to the endothelium is an important step to establish metastasis (7), we next determined the effect of AR inhibition on vascular adhesion of human colon cancer HT29 cells. As shown in the Figure 2A stimulation of HUVEC with growth factors such as EGF or FGF caused ∼60% increase in the adhesion of HT29 cells to HUVEC. However, pretreatment of HUVEC with AR inhibitor, fidarestat significantly attenuated the adhesion of HT29 cells to growth factors-stimulated HUVEC. These results were further confirmed by microscopic visualization of adhesion of fluorescent-labeled HT29 to HUVEC (Figure 2B). We next examined the effect of AR inhibition on adhesion of colon cancer cells stimulated with EGF or FGF followed by incubation with untreated HUVEC. Our results shown in Figure 2C indicate that stimulation of HT29 cells with growth factors caused increase in their adhesion to HUVEC and inhibition of AR prevented it (Figure 2C). Interestingly, the colon cancer cells adhesion to endothelial cells is more in the HUVEC cells prestimulated with growth factors as compared with HT29 cells prestimulated with growth factors. In both cases, AR inhibition demonstrates colon cancer cells adhesion to HUVEC.
Fig. 2.
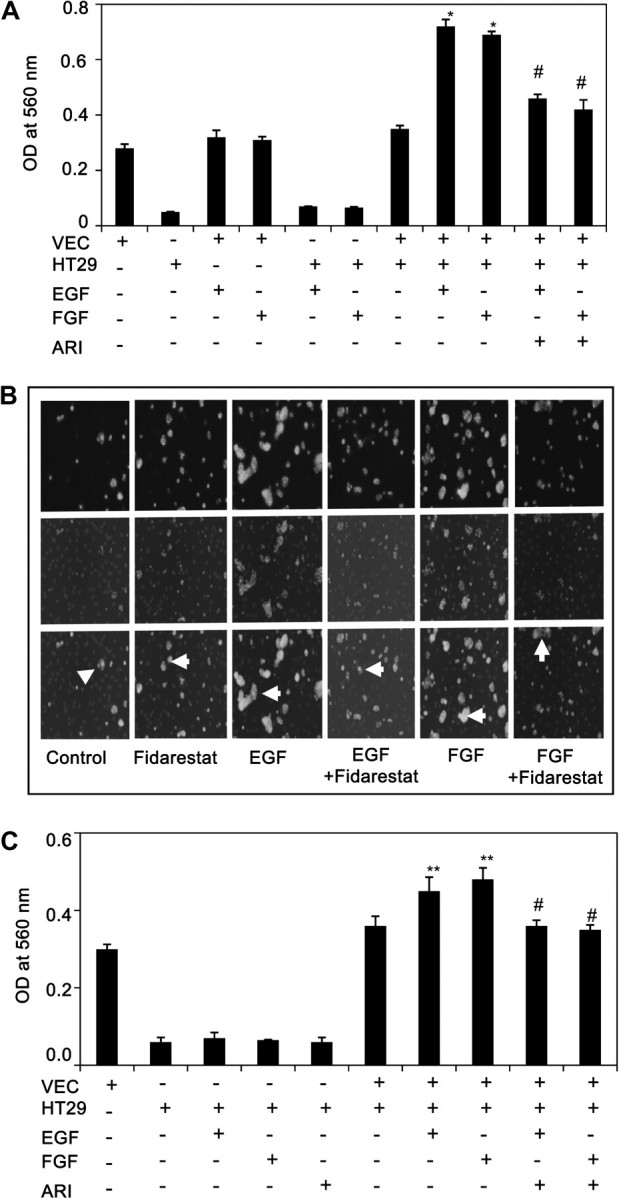
Inhibition of AR prevents growth factors-induced adhesion of HT29 cells to endothelial cells. (A) Adhesion of untreated HT29 cells to growth factor prestimulated HUVEC without or with AR inhibitors were quantified by MTT assay by measuring absorbance at 560 nm. (B) HT29 cells were labeled with PKH67 fluorescent (green) dye and incubated for 30 min with HUVEC, stimulated with growth factors for 16 h. Cells were washed to remove non-adhesive HT29 cells on HUVEC, fixed and mounted with 4′,6-diamidino-2-phenylindole. Adhesion of HT29 cells was visualized and photographed using Nikon fluorescent microscope. Arrows indicate the adhesion of HT29 cells (green) to HUVEC (only blue). (C) Adhesion of growth factor prestimulated HT29 cells without or with AR inhibitors to untreated HUVEC were quantified by MTT assay by measuring the absorbance at 560 nm. Bars represent mean ± SEM (n = 4); *P < 0.001; **P < 0.01 versus control and #P < 0.001 versus growth factor-treated cells.
Inhibition of AR prevents growth factors-induced expression of cell surface adhesion molecules in HUVEC
We next examined the endothelial cell surface expression of adhesion molecules by FACS analysis. As shown in the Figure 3A, stimulation of HUVEC with EGF or FGF for 24 h significantly increased the expression of Vascular cell adhesion molecule, Inter-Cellular Adhesion Molecule-1 and vascular endothelial-cadherin compared with untreated HUVEC. The increased expression of adhesion molecules was significantly attenuated by fidarestat (Figure 3A and B). However, AR inhibitor alone did not affect the basal levels of adhesion molecules. Since secretion of growth factors and cytokines by tumor cells is known to cause migration of endothelial cells which cause neovascularization (10), we determined the effect of AR inhibition on growth factors-induced migration of HUVEC. As shown in the Figure 3C treatment of HUVEC with EGF or FGF caused pronounced migration of endothelial cells in wound model assay.
Fig. 3.

Inhibition of AR prevents growth factors-induced expression of adhesion molecules in HT29 cells. (A) The cell surface expression of Vascular cell adhesion molecule, Inter-Cellular Adhesion Molecule and vascular endothelial-cadherin in HT29 cells and HUVEC were analyzed by FACS analysis. (B) The percentage of fluorescent-labeled cells was plotted in a bar diagram. (C) Inhibition of AR prevents growth factors-induced migration of HUVEC in a scratch wound assay. Wound scratches were made with a sterile pipette tip in the wells. After 72 h of incubation, photographs of the wells were taken using Nikon camera fixed to inverted microscope (×100). The line (-) in the figure indicates the level of HUVEC migration. The distance migrated as percentage of total distance was determined for quantitative assessments. Figures are shown from one of the three representative experiments. Bars represent mean ± SEM (n = 4); *P < 0.001 versus control and #P < 0.01 versus growth factor-treated cells.
Inhibition of AR prevents metastasis of human colon cancer cells in nude mice liver
Our in vitro studies clearly indicated that inhibition of AR could prevent invasion, migration and adhesion of colon cancer cells (HT29, KM20), which are critical steps that cause metastatic spread of tumor cells. Therefore, inhibition of AR could be useful to prevent metastasis in vivo. We have next determined the effect of AR inhibition on the liver metastasis of colon cancer using athymic nude mice model. In mice fed with control diet, markedly increased metastasis all over the liver was observed, whereas in mice given AR inhibitors or ARSiRNA fluorescence was significantly less (Figures 4A and 5A) indicating prevention of metastasis. The results were further quantified by measurement of fluorescence and values calculated as pixel numbers (Figures 4B and 5B). Results demonstrate a significant (>65%) decrease in tumor metastasis in the sorbinil and fidarestat fed mice as well as ARSiRNA-delivered systemically into the mice compared with control or AR-scrambled SiRNA which correlates with our qualitative assessment. Similarly, AR inhibitor, sorbinil also prevented liver metastasis of intrasplenical injection of GFP-transfected HT29 cells into the athymic mice (Figure S3 is available at Carcinogenesis Online). Furthermore, reverse transcription–PCR analysis of metastatic liver suggested that ablation of AR by ARSiRNA almost completely (>90%) inhibited the expression of AR (Figure 5C). Therefore, inhibition of AR could represent a unique strategy for the suppression of colorectal cancer metastasis.
Fig. 4.

Pharmacological inhibition of AR prevents metastatic tumor growth in nude mice. (A) KM20-GFP cells (4 × 106) were inoculated intraspleenically into athymic nude mice. After 24 h, spleenectomy was performed and the mice were given AR inhibitors, sorbinil (40 mg/kg body wt) in diet or fidarestat (50 mg/kg body wt) in drinking water. Mice were killed after 30 days. (B) Fluorescence pixel value of metastasized KM20-GFP cells in (A) was measured using Adobe Photoshop. (C) Illustration of macroscopic foci of KM20-GFP cells metastasized to the liver by hematoxylin and eosin staining. L, normal liver; M, metastasis. Bars represent mean ± SEM (n = 6); *P < 0.001 versus metastatic control. Color figure is available at Carcinogenesis online.
Fig. 5.

SiRNA ablation of AR prevents KM20 cells metastatic tumor growth in nude mice. (A) KM20-GFP cells (4 × 106) were inoculated intraspleenically into athymic nude mice. After 24 h, spleenectomy was performed and the mice were injected (intraperitoneally) with SiStable scrambled siRNA or ARSiRNA (200 μg/100 μl phosphate-buffered saline/mice) at day 1, day 10 and day 20. Mice were killed after 30 days. (B) Fluorescence, pixel value of metastasized KM20-GFP cells in (A) was measured using Adobe Photoshop. (C) Reverse transcription–PCR analysis of AR messenger RNA expression in nude mice liver. Bars represent mean ± SEM (n = 6); *P < 0.001 versus scrambled ARSiRNA. Color figure is available at Carcinogenesis online.
The extent of tumor metastatic growth and changes in morphological architecture in liver were investigated by studying the histopathology of hematoxylin and eosin stained liver sections. In control metastatic liver, multiple macroscopic foci of KM20-GFP cells were more compared with mice treated with AR inhibitor. Furthermore, the largest dimension of metastasis and the percentage of the metastatic cancer tissue over the total liver parenchyma were significantly decreased in mice treated with AR inhibitor compared with untreated metastatic group (Figure 4C). These results suggest that AR inhibition prevented metastatic growth in the liver.
Inhibition of AR prevents the expression of metastatic marker proteins, NF-κB activation and histopathological changes in nude mice liver
We next examined the expression of important proteins required for the metastatic growth of colon cancer. Our results showed a significant decrease in AR, MMP2, cyclin D1, CD34 expression and activation of p65 of NF-κB heterodimer in nude mice metastatic liver treated with fidarestat compared with untreated metastatic mice (Figure 6A). Similar results were observed when Immunohistochemistry analysis of liver sections was performed with AR, CD34, CD31, MMP2, cyclin D1 and phospho-p65 antibodies (Figure 6B and C). Consistent with the protein levels, messenger RNA levels of cyclin D1 and MMP2 were significantly increased in metastastic liver, whereas treatment of mice with fidarestat significantly prevented the expression of MMP2 and cyclin D1 (Figure 6A).
Fig. 6.
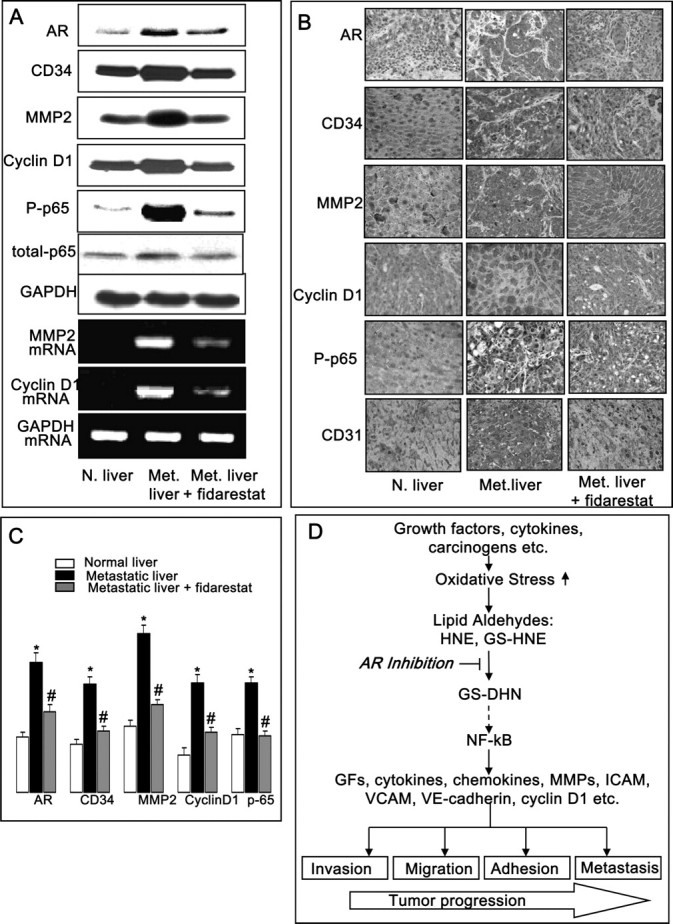
Inhibition of AR prevents expression of metastatic markers during colon cancer metastatic growth in nude mice liver. (A) Equal amounts of liver homogenates were subjected to western blot analysis using antibodies against AR, CD34, MMP2, cyclin D1, phospho-p65, total-p65 and Glyceraldehyde-3- phosphate dehydrogenase. Equal amounts of total RNA from liver were analyzed by reverse transcription–PCR for measuring messenger RNA levels of MMP2, cyclin D1 and Glyceraldehyde-3- phosphate dehydrogenase. (B) Cross sections of metastatic tumors were stained with anti-AR, anti-CD34, anti-CD31, anti-MMP2, anti-cyclin D1 and anti-phospho-p65. Immunoreactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Original magnification: ×400. (C) Percent staining was determined by measuring positive immunoreactivity per unit area. Arrows represent the area for positive staining for an antigen. The intensity of antigen staining was quantified by digital image analysis. Bars represent mean ± SEM (n = 4); *P < 0.001 versus normal liver control and #P < 0.001 versus metastatic liver. (D) Schematic representation of AR mediation in oxidative stress-induced tumor progression. Color figure is available at Carcinogenesis online.
Discussion
The mechanism of tumor metastasis involves a series of steps including escape of cells from primary tumor into blood or lymphatic system (intravasation), survival in the circulation and arrest in the secondary site (extravasation), vasucularization (angiogenesis) and growth of tumor [metastasis; (2,5–7)]. In this progression of cancer, any of the steps may become therapeutic target because inhibition of any one step would lead to disruption of entire metastatic cascade (2,9). The surgical resection and radiation therapy of primary colonic tumors are successful when tumor is locally confined. However, many of the patients are diagnosed when subclinical or clinical relevant liver metastasis has already occurred (2,9–11). During metastatic progression, dissociated cancer cells interact with various cellular components including blood cells and immune cells such as macrophages, lymphocytes and dendritic cells and induce embolization of blood vessels in the downstream organs (5–11). This mechanism is associated with generation of ROS and large quantities of proinflammatory cytokines, chemokines and growth factors (30,31). Indeed, involvement of ROS in cancer is well supported by epidemiological studies, which have demonstrated that several forms of cancer progression can be slowed down/prevented by using antioxidants as chemopreventive agents (32,33). However, the mechanisms by which ROS regulate cell functions or induce tissue injury remain unclear. Our recent studies demonstrate that AR plays a significant role in mediating ROS-induced inflammatory signals (21,23,34). Furthermore, using colon cancer cellular and animal models, we have shown that AR is an obligatory mediator of oxidative stress/inflammation induced by growth factors, cytokines and carcinogens (21,22,26). In the present report, we have demonstrated the involvement of AR in tumor cell adhesion, invasion, migration and metastasis using in vivo and in vitro models.
Tumor cell invasion and migration play a pivotal role in the progression of metastasis of cancer. Numerous studies with experimental metastasis models indicate that one of the most important components in the cell invasion is overexpression of proteases (12,13). Among the various proteases, MMPs are of particular importance because they degrade extracellular matrix and allow cells to overcome constrains of cell–cell and cell–matrix interaction. In addition, various reports show that expression of proteases is regulated by redox-sensitive transcription factors such as NF-κB during oxidative stress- or inflammation-induced pathogenesis (14,35,36). This is further supported by various studies, which have shown that antioxidants such as quercetin, resveratrol and lycopene prevent the invasion/migration of tumor cells via inhibiting expression of MMPs and activation of PKC/AKT/PI3K/NF-κB (13,16–20). Our results demonstrate that inhibition of AR significantly (70%) inhibits growth factor-induced invasion and migration of HT29 cells and increase in the expression of MMP2 in liver metastasis.
The successful progression of metastasis depends on the interaction of tumor cell with endothelial cell for the nutrients and oxygen and migration to other organs. Our in vitro results (Figures 1–3) demonstrate that inhibition of AR prevents the growth factors such as EGF- and FGF-induced migration of human colon cancer HT29 cells and adhesion of human colon cancer HT29 cells to endothelial cells by inhibiting the expression of adhesion molecules such as Inter-Cellular Adhesion Molecule, Vascular cell adhesion molecule and vascular endothelial-cadherin. Prevention of liver metastasis in nude mice by inhibition or ablation of AR which prevents the expression of the adhesion molecules suggest that AR is involved in adhesion of colon cancer cells to endothelial cells which would promote metastasis. Normally, human liver has extremely low levels of AR. However, AR is overexpressed in the liver under several oxidative stress and inflammation-related pathological conditions such as alcoholic liver cirrhosis, heart failure, myocardial ischemia, vascular inflammation, restenosis and cancer (23,37). Recently, Saraswat et al. (38) demonstrated increased expression of AR in various cancerous tissues such as lung, breast, prostate, cervix, ovarian, colon, etc. Furthermore, they showed that the specific activity of AR was higher in tumor areas than non-tumor regions of tissues. These results as well as our earlier studies demonstrating the prevention of tumor growth in nude mice by AR inhibition suggest that AR inhibitors could be excellent antitumor agents by preventing the processes required for metastasis (Figure 6D).
In the current study, we have shown that inhibition of metastasis by AR inhibitors was due to the prevention of cancer cell invasion, migration, adhesion and angiogenesis. Although in this study, we have shown that AR inhibition prevents invasion and adhesion in vitro, we have not clearly demonstrated the specific role of AR within the tumor-host cellular components because of the aggressiveness of the liver metastasis. Furthermore, the progression of metastatic cancer depends upon the establishment of an adequate blood supply to the tumor cells (39). Although we have demonstrated that AR inhibition prevents the expression of CD31 and CD34 potential markers for neovascularization in metastatic livers, the exact mechanism by which AR inhibition prevents angiogenesis in metastatic livers remains to be elucidated. However, in a separate study (40), we have recently shown that AR inhibition prevents Vascular endothelial growth factor- and FGF-induced capillary-like tube structure formation of HUVEC in vitro. Furthermore, AR inhibition also prevented the in vivo neovascularization in a matrigel plug model in rats. Our studies also indicate that AR inhibition prevents Vascular endothelial growth factor-induced increase of capillary-like structures formation, infiltration of endothelial cells and red blood cells in vivo. Thus, these studies indicate that AR inhibition also prevents angiogenesis, an important event in cancer metastasis.
Among the various experimental metastasis models, intrasplenic injection of colon cancer cells produces reproducible, rapid and efficient liver metastasis (39,41). Giavazzi et al. (42) demonstrated that metastatic potential of freshly isolated colon cancer cells is achieved after instrasplenic injection and not after intravenous, subcutaneous or intramuscular routes. Therefore, in present study, we injected the green fluorescent-labeled human colon cancer cells intraspleenically to athymic nude mice and followed the metastasis in the liver after spleenectomy (39,41). Our studies clearly demonstrate that inhibition of AR by two structurally different pharmacological inhibitors, sorbnil and fidarestat or by ablation of AR by SiRNA prevented the human colon cancer cells-induced metastatic growth in liver of nude mice (Figures 4 and 5). Thus, these results suggest that AR plays a pivotal role in the liver metastasis of the colon cancer. In the present study, we have though used two metastatic colon cancer cells HT29 and KM20 to examine the efficacy of AR inhibitors in prevention metastasis. However, given the heterogeneity of genetic and molecular alterations in colon cancer, and how they influence therapeutic responses, this study could be strengthened by using AR inhibitors to prevent cancer metastasis by using other human metastatic cancer cell lines. Furthermore, in this study, we have used AR inhibitors mainly used sorbinil and fidarestat which belong to the same class inhibitors and found that both the inhibitors are effective in preventing metastasis. As compared with sorbinil, fidarestat is a more potent and water soluble inhibitor and found to be safe for human use in phase-iii clinical studies for diabetic neuropathy. AR inhibitors have been shown to prevent NF-κB-dependent inflammatory and oxidative stress signals initiated by cytokines and growth factors (23).
In conclusion, our studies have demonstrated that AR plays a critical role of AR in colon cancer cells migration, invasion, adhesion, angiogenesis and the metastatic growth in liver. Since AR inhibitors such as fidarestat have already undergone phase-III clinical trials for the treatment of diabetic neuropathy and found to be safe without any major toxicity, our results provide basis for the emerging preclinical and clinical investigations of AR inhibitors for the chemotherapeutic interventions of colon cancer progression and metastasis.
Supplementary material
Supplementary Figures S1–S3 can be found at http://carcin.oxfordjournals.org/
Funding
Supported by in parts by NIH grants (CA129383 and DK36118 to S.K.S.) and GM71036 (to K.V.R.)
Supplementary Material
Acknowledgments
S.K.S. is a William Bowes Senior Fellow of the American Asthma Foundation.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AR
aldose reductase
- DHN
4-dihydroxynonene
- EGF
epidermal growth factor
- FGF
fibroblast growth factor
- GFP
green fluorescent protein
- HNE
4-hydroxy-trans-2-nonenal
- UVEC
human umbilical vascular endothelial cell
- MMP
matrix metalloproteinase
- NF-κB
nuclear factor-kappa-binding protein
- PCR
polymerase chain reaction
- ROS
reactive oxygen species
References
- 1.American Cancer Society. Cancer Facts and Figures 2010. Atlanta, GA: American Cancer Society; 2010. [Google Scholar]
- 2.Gupta GP, et al. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Rychahou PG, et al. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl Acad. Sci. USA. 2008;105:20315–20320. doi: 10.1073/pnas.0810715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rychahou PG, et al. Targeted molecular therapy of the PI3K pathway: therapeutic significance of PI3K subunit targeting in colorectal carcinoma. Ann. Surg. 2006;243:833–842. doi: 10.1097/01.sla.0000220040.66012.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langley RR, et al. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr. Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- 6.Chambers AF, et al. Steps in tumor metastasis: new concepts from intravital video microscopy. Cancer Metastasis Rev. 1995;14:279–301. doi: 10.1007/BF00690599. [DOI] [PubMed] [Google Scholar]
- 7.Zetter BR. Adhesion molecules in tumor metastasis. Semin. Cancer Biol. 1993;4:219–229. [PubMed] [Google Scholar]
- 8.Jung YD, et al. Role of the tumor microenvironment in mediating response to anti-angiogenic therapy. Cancer Metastasis Rev. 2000;19:147–157. doi: 10.1023/a:1026510130114. [DOI] [PubMed] [Google Scholar]
- 9.Fidler IJ, et al. Recognition and destruction of neoplastic cells by activated macrophages: discrimination of altered self. Biochim. Biophys. Acta. 1988;948:151–173. doi: 10.1016/0304-419x(88)90009-1. [DOI] [PubMed] [Google Scholar]
- 10.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002;29:15–28. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 12.Deryugina EI, et al. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 13.Mannello F, et al. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr. Cancer Drug Targets. 2005;5:285–298. doi: 10.2174/1568009054064615. [DOI] [PubMed] [Google Scholar]
- 14.Garg A, et al. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia. 2002;16:1053–1068. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- 15.Meyskens FL, Jr., et al. Activation of nuclear factor-kappa B in human metastatic melanomacells and the effect of oxidative stress. Clin. Cancer Res. 1999;5:1197–1202. [PubMed] [Google Scholar]
- 16.Sen T, et al. Multifunctional effect of epigallocatechin-3-gallate (EGCG) in downregulation of gelatinase-A (MMP-2) in human breast cancer cell line MCF-7. Life Sci. 2009;84:194–204. doi: 10.1016/j.lfs.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Yu H, et al. Resveratrol inhibits tumor necrosis factor-alpha-mediated matrix metalloproteinase-9 expression and invasion of human hepatocellular carcinoma cells. Biomed. Pharmacother. 2008;62:366–372. doi: 10.1016/j.biopha.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Huang CS, et al. Lycopene inhibits matrix metalloproteinase-9 expression and down-regulates the binding activity of nuclear factor-kappa B and stimulatory protein-1. J. Nutr. Biochem. 2007;18:449–456. doi: 10.1016/j.jnutbio.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Zafarullah M, et al. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang CS, et al. Inhibition of carcinogenesis by tea. Annu. Rev. Pharmacol. Toxicol. 2002;42:25–54. doi: 10.1146/annurev.pharmtox.42.082101.154309. [DOI] [PubMed] [Google Scholar]
- 21.Tammali R, et al. Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colon cancer cells. Cancer Res. 2006;66:9705–9713. doi: 10.1158/0008-5472.CAN-06-2105. [DOI] [PubMed] [Google Scholar]
- 22.Tammali R, et al. Aldose reductase regulates TNF-alpha-induced PGE2 production in human colon cancer cells. Cancer Lett. 2007;252:299–306. doi: 10.1016/j.canlet.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava SK, et al. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr. Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 24.Hers HG. The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochim. Biophys. Acta. 1956;22:202–203. doi: 10.1016/0006-3002(56)90247-5. [DOI] [PubMed] [Google Scholar]
- 25.Ramana KV, et al. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 26.Tammali R, et al. Aldose reductase deficiency in mice prevents azoxymethane-induced colonic preneoplastic aberrant crypt foci formation. Carcinogenesis. 2009;30:799–807. doi: 10.1093/carcin/bgn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang C, et al. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 28.Dianzani C, et al. Celecoxib modulates adhesion of HT29 colon cancer cells to vascular endothelial cells by inhibiting ICAM-1 and VCAM-1 expression. Br. J. Pharmacol. 2008;153:1153–1161. doi: 10.1038/sj.bjp.0707636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butts CL, et al. Evaluation of steroid hormone receptor protein expression in intact cells using flow cytometry. Nucl. Recept. Signal. 2007;5:1–6. doi: 10.1621/nrs.05007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feig DI, et al. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- 31.Ushio-Fukai M, et al. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikawa M, et al. Inhibition of metastatic tumor growth by targeted delivery of antioxidant enzymes. J. Control. Release. 2005;109:101–107. doi: 10.1016/j.jconrel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Liu Q, et al. Curcumin inhibits cell proliferation of MDA-MB-231 and BT-483 breast cancer cells mediated by down-regulation of NFkappaB, cyclinD and MMP-1 transcription. Phytomedicine. 2009;16:916–922. doi: 10.1016/j.phymed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Ramana KV, et al. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes. 2005;54:818–829. doi: 10.2337/diabetes.54.3.818. [DOI] [PubMed] [Google Scholar]
- 35.Naugler WE, et al. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tremblay PL, et al. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene. 2006;25:6563–6573. doi: 10.1038/sj.onc.1209664. [DOI] [PubMed] [Google Scholar]
- 37.Zeindl-Eberhart E, et al. Identification of tumor-associated protein variants during rat hepatocarcinogenesis, aldose reductase. J. Biol. Chem. 1994;269:14589–14594. [PubMed] [Google Scholar]
- 38.Saraswat M, et al. Overexpression of aldose reductase in human cancer tissues. Med. Sci. Monit. 2006;12:CR525–CR529. [PubMed] [Google Scholar]
- 39.Giatromanolaki A, et al. Angiogenesis in colorectal cancer: prognostic and therapeutic implications. Am. J. Clin. Oncol. 2006;29:408–417. doi: 10.1097/01.coc.0000221317.56731.4e. [DOI] [PubMed] [Google Scholar]
- 40.Tammali R, et al. Inhibition of aldose reductase prevents angiogenesis in vitro and in vivo. Angiogenesis. 2011 doi: 10.1007/s10456-011-9206-4. DOI 10.1007/s10456-011-9206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giavazzi R, et al. Experimental nude mouse model of human colorectal cancer liver metastases. J. Natl Cancer Inst. 1986;77:1303–1308. [PubMed] [Google Scholar]
- 42.Giavazzi R, et al. Metastatic behavior of tumor cells isolated from primary and metastatic human colorectal carcinomas implanted into different sites in nude mice. Cancer Res. 1986;46:1928–1933. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.