

Abstract
Sestrins (Sesns) are a family of highly conserved stress-responsive proteins, transcriptionally regulated by p53 and forkhead transcription factor that exhibit oxidoreductase activity in vitro and can protect cells from oxidative stress. However, their major biochemical and physiological function does not appear to depend on their redox (reduction and oxidation) activity. Sesns promote activation of adenosine-5′-monophosphate (AMP)-dependent protein kinase in both mammals and flies. Stress-induced Sesn expression results in inhibition of the target of rapamycin complex 1 (TORC1) and the physiological and pathological implications of disrupting the Sesns-TORC1 crosstalk are now being unravelled. Detailing their mechanism of action and exploring their roles in human physiology point to exciting new insights to topics as diverse as stress, cancer, metabolism and aging.
Keywords: aging, p53, redox, Sestrin, target of rapamycin
Introduction
Every organism exists in a constantly changing environment and faces frequent challenges that threaten its survival and can compromise its well-being. In spite of huge differences in lifespan amongst different species and even between individuals within one species, the existence of all individuals depends on so-called stress responses, whose main goal is to provide protection against life-threatening challenges and restore physiological homeostasis. Well-studied stress responses include the heat-shock response (Morimoto, 1998), the unfolded protein response (Ron & Walter, 2007) or the induction of metallothioneins in response to toxic heavy metals (Karin, 1985). In addition to providing protection against acute insults, many of the products of these stress responses have normal physiological functions and their expression may provide both stress adaptation as well as increased overall fitness.
This review deals with a small group of stress-inducible proteins called Sestrins (Sesns), whose physiological functions are just starting to be appreciated and understood.
The Discovery of Sestrins
The tumour suppressor protein p53 has attracted much attention since its identification (Lane & Crawford, 1979) and molecular cloning (Chumakov et al, 1982; Oren & Levine, 1983), mainly because the gene is mutated and/or inactivated in most human cancers (Levine, 1997). p53 protein accumulation and activity are induced by genotoxic stress (Levine, 1997) as well as oxidative (Sablina et al, 2005) and oncogenic stresses (Lowe et al, 2004). p53 is a well-established transcription factor with tumour suppressive properties as its absence in mice results in spontaneous tumours (Donehower et al, 1992). Many p53 target genes have been thoroughly characterized and implicated in its tumour suppressive functions (Vousden & Lane, 2007; Vousden & Prives, 2009), but so far not a single target gene whose ablation results in tumorogenesis has been identified. Excessive p53 activity in response to severe DNA damage can lead to cell death through induction of pro-apoptotic target genes such as Puma and Noxa (Vousden & Prives, 2009), whereas more modest p53 activation inhibits cell cycle progression mostly through induction of p21 (Vousden & Prives, 2009). More recently, p53 was found to control glycolysis through induction of TP53-induced glycolysis and apoptosis regulator (TIGAR) (Bensaad et al, 2006). In 1994, Kley's group isolated a new p53 target gene that was named p53-activated gene 26 (PA26) (Buckbinder et al, 1994). PA26 did not show any similarity to previously known genes but a close homologue of the PA26 gene named hypoxia-inducible gene 95 (Hi95) was identified several years later (Budanov et al, 2002) (Fig 1). PA26, Hi95 and a third member of the family, found as an open reading frame by in silico analysis (Budanov et al, 2002; Peeters et al, 2003), comprise a small gene family whose members were named Sesns after Sestri Levante, a small town on the Ligurian coast of Italy where, during a human genetics course, researchers discovered the amino acid sequence homology between the three proteins (Peeters et al, 2003) (Fig 2). In this review, we refer to the Sesns as Sesn1, Sesn2 and Sesn3. Sesn3 was also found as a gene regulated by serum and growth factors (Nogueira et al, 2008). Whereas Sesn1 and Sesn2 are mainly responsive to p53 (Budanov et al, 2002; Velasco-Miguel et al, 1999), Sesn3 is activated by FoxO transcriptional factors (Nogueira et al, 2008), which can also contribute to Sesn1 induction (Nogueira et al, 2008; Tran et al, 2002). The Sesns are highly conserved and Caenorhabditis elegans and Drosophila melanogaster orthologues were found by analysis of the corresponding genomes (see Box 1 and Fig 2) (Budanov et al, 2002). Whereas vertebrates contain three Sesn genes, invertebrates in general have a single Sesn with no Sesn genes found in yeast (Lee et al, 2010) (Fig 2).
Figure 1. Historical timeline of Sestrin research.

Since the discovery of Sesns as stress-inducible proteins, many studies shed light on understanding the physiological function of the molecule.
Figure 2. Conservation of Sestrins.
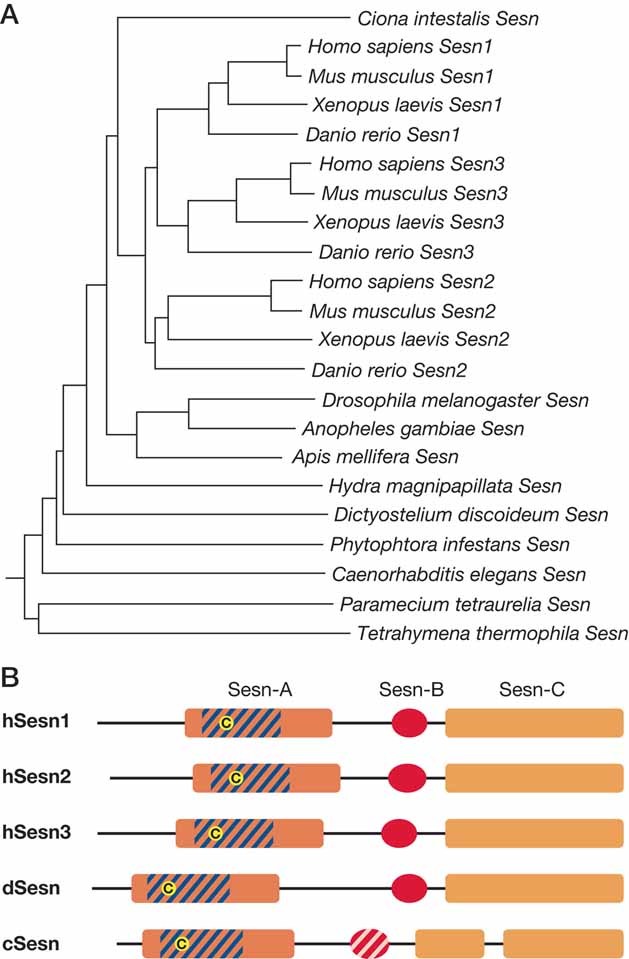
- Multiple alignment was performed using CLUSTALW algorithm. Phylogenic tree was constructed using Neighbour-Joining methods. Sesn paralogue diversification is only observed within the vertebrate branches.
- Putative domain structure of Sesn-family proteins. Sesn-A domain corresponds to 81–221 a.a., Sesn-B domain to 273–311 a.a. and Sesn-C domain to 325–492 a.a. of human Sesn1 short isoform protein, which were estimated from multiple sequence alignments. Human Sesn1 long isoform is N-terminally extended from Sesn1 short isoform. Part of Sesn-A domain shows similarity to bacterial AhpD family proteins (dashed with blue) and contains a conserved cystein residue (yellow circle), which may be critical for antioxidant function of the protein. Caenorhabditis elegans Sesn (cSesn) has a less conserved Sesn-B domain (dashed with pink).
BOX 1: Isoforms, structure and expression of Sestrins.
Sesn1 is considered the primordial member of the family and most similar to invertebrate Sesns (Lee et al, 2010). The SESN1 gene is transcribed into three mRNAs from three different promoters, such that each transcript contains a unique first exon and nine common exons. The transcripts encode three different proteins: Sesn1-T1 (551 a.a., 68 kDa), Sesn1-T2 (491 a.a., 55 kDa) and Sesn1-T3 (426 a.a., 48 kDa), which vary in their amino (N)-terminal portions. Only the T2 and T3 isoforms are p53-inducible (Velasco-Miguel et al, 1999). Two major transcripts of the Xenopus Sesn1 gene were also identified within Xenopus (Hikasa & Taira, 2001), but no alternative transcripts were found in zebrafish (Peeters et al, 2003). The SESN2 gene by contrast is transcribed into a single mRNA species. Sesn2 mRNA encodes a 480 a.a. protein which runs as a 60-kDa band on SDS–polyacrylamide gel electrophoresis (Budanov et al, 2002). According to the Uniprot database (http://www.uniprot.org/uniprot/P58005#P58005-2), there are two alternatively spliced forms of SESN3, encoding two different protein products: 492 a.a. (57.3 kDa) and 317 a.a. (36 kDa). Sesns are located on different chromosomes: SESN1 on 6q21, SESN2 on 1p35.3, and SESN3 on 11q21 in the human genome (Budanov et al, 2002).
Structural predictions in silico using the GLOBE program (http://cubic.bioc.columbia.edu/predictprotein) suggest that Sesns are globular proteins composed mostly of α-helical regions. Predicted highly conserved α-helices are connected by the less conserved hinge regions (Budanov et al, 2002). However, no obvious known structural motifs or folds appear to be present. The Sesns contain several putative Ser/Thr and Tyr phosphorylation sites, including casein kinase 2 (CK2), protein kinase C (PKC) and protein kinase A (PKA) sites (Budanov et al, 2002). Recent phosphoproteomic analysis revealed that Drosophila Sesn (dSesn) is phosphorylated at multiple sites in vivo (Zhai et al, 2008).
Sesns are ubiquitously expressed in all adult tissues, although at different levels (Budanov et al, 2002; Peeters et al, 2003; Velasco-Miguel et al, 1999). Interestingly, Sesn1 and Sesn2 are most highly expressed in skeletal muscle (Velasco-Miguel et al, 1999) and dSesn is also highly expressed in skeletal muscle (Lee et al, 2010). The amount of dSesn expression increases upon maturation to imago stage and aging (Lee et al, 2010).
Glossary
Alzheimer's disease
Age-related neurodegenerative disease, characterized pathologically by protein aggregates known as plaques and tangles consisting mainly of the proteins amyloid beta and phosphorylated tau. The main symptoms are memory loss and reduced cognitive functioning.
Apoptosis
Programmed cell death characterized by shrinkage of the cell, condensation of chromatin and fragmentation of the cell into membrane-covered bodies that are eliminated by phagocytosis.
Autophagy
Tightly regulated process for degradation of intracellular constituents in lysosome helping to maintain cellular integrity. Divided into three groups: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy is the most studied form of autophagy characterized by encapsulation of part of cytoplasm or organelles into two-membrane vesicles followed fusion with lysosome.
Cancer
Disease featuring abnormal and uncontrolled proliferation of a group of cells resulting in invasive growths or tumour followed by spread throughout the body.
Chronic obstructive pulmonary disease
Progressive lung disease encompassing chronic bronchitis and emphysema (destruction of lung tissue) and characterized by narrowing airways and limitation of air flow through the lungs.
Fat droplets
Microaggregates of lipids (mainly triglycerides) visible within cells.
Genotoxic stress
Reaction of cells or organism on DNA-damage induced by genotoxins, irradiation or intrinsic inability to support DNA integrity.
Glycolysis
Metabolic pathway that converts glucose into pyruvate through a sequence of reactions involving 10 intermediates, releasing energy in form of ATP and NADH.
Hepatosteatosis
Fat deposition in the liver that exceeds 5% of the total weight of liver or at visualization with more that 5% of hepatocytes containing fatty droplets at light microscopic examination.
Inflammation
A poorly specific immune response of body tissues to infection, irritation or other injuries, characterized by pain, swelling, redness and heat.
Ischaemic injury
Damage and/or dysfunction of tissue induced by restriction of blood supply resulting in shortage of oxygen, glucose and some other nutrients.
Lipogenesis
Metabolic process by which acetyl-CoA is converted to fat. Lipogenesis encompasses the process of fatty acid synthesis and subsequent triglyceride synthesis. It plays an important role in conservation of energy in form of fat.
Mitophagy
Autophagy selective for degradation of mitochondria.
Oncogenic stress
Reaction of organism to hyperactivation of oncogenes to prevent uncontrolled proliferation of potentially detrimental procarcinogenic cells.
Oxidative burst
Rapid but temporal release of ROS by cells to stimulate cell signalling or combat infections.
Oxidative stress
An imbalance between production and decomposition of ROS results in accumulation of ROS followed by oxidative damage of cellular constituents.
Parkinson's disease
Age-related neurodegenerative disease, characterized pathologically by protein aggregates in the brain, mainly composed of alpha-synuclein, known as Lewy bodies. The main symptoms are reduced motor skills and movement defects.
Pulmonary emphysema
Progressive disease of lung causing shortness of breath. The disease is characterized by destruction of lung tissue around alveoli, making them unable to hold their functional shape upon exhalation.
Reactive oxygen species
Inorganic or organic chemically reactive molecules containing oxygen in form of oxygen ion, radical or peroxide. Work as second messengers in cell signalling under physiological concentration, but cause oxidative stress when accumulated.
Sarcopenia
Degenerative loss of skeletal muscle tissue and skeletal muscle strength as a consequence of aging.
Second messengers
Small molecules that convey and amplify signals from receptors on cell surface to effector molecules.
Translation
Process of protein biosynthesis on a ribosome during which mRNA directs incorporation of amino acids into polypeptide chain.
Type II diabetes
A metabolic medical condition characterized by insulin resistance and high blood glucose levels.
Sestrins and Redox Control
The only structural similarity identified between Sesns and other proteins is limited to a conserved N-terminal region that contains a motif found in prokaryotic proteins, including the Mycobacterium tuberculosis AhpD protein (Budanov et al, 2004) (Fig 2B). AhpD is a component of alkyl-hydroperoxidereductase which provides protection against reactive oxygen species (ROS) (Bryk et al, 2002). More specifically, AhpD is responsible for regeneration of a thiol-specific peroxidase AhpC that is oxidized during reduction of peroxides and reactive nitrogen species (Bryk et al, 2002). The mammalian peroxiredoxin (Prx) family consists of six members, whose catalytic cycle include oxidation of a catalytic cysteine (Cys)-SH to Cys-SOH, followed by formation of a S—S bridge that is reduced back to Cys-SH by the thioredoxin system (Rhee et al, 2005). During an oxidative burst, the Prxs can be over-oxidized to cysteine sulfinic acid (Cys-SO2H), resulting in their inactivation. In their oxidized form (Cys-SO2H), Prxs can be rescued by a special sulfinyl-reductase system comprised of sulphiredoxins (Biteau et al, 2003; Chang et al, 2004). Given their similarity to AhpD, the Sesns were examined for effects on ROS accumulation. Depletion of Sesn1 or Sesn2 by gene silencing in cultured cells resulted in ROS accumulation, whereas Sesn1/2 overexpression reduced ROS levels (Budanov et al, 2004) (Fig 3A). Sesns are now known to modulate Prx regeneration in cancer cell lines (Budanov et al, 2004), macrophages (Essler et al, 2009) and neurons, where they prevent oxidative stress caused by N-methyl-d-aspartate (NMDA) receptor activation (Papadia et al, 2008) (Fig 3A). Sesns (in particular Sesn2) can restore the activity of over-oxidized Prxs (Cys-SO2) (Budanov et al 2004), although it was reported that Sesn is not an enzyme that is directly involved in reduction of over-oxidized Prxs, but rather play an auxiliary role in the process (Rhee et al, 2008). Thus Sesns likely inhibit ROS accumulation through maintenance of Prxs activity, although other antioxidant mechanisms of action performed by Sesns cannot be ruled out at this stage.
Figure 3. Regulators of Sestrin expression.
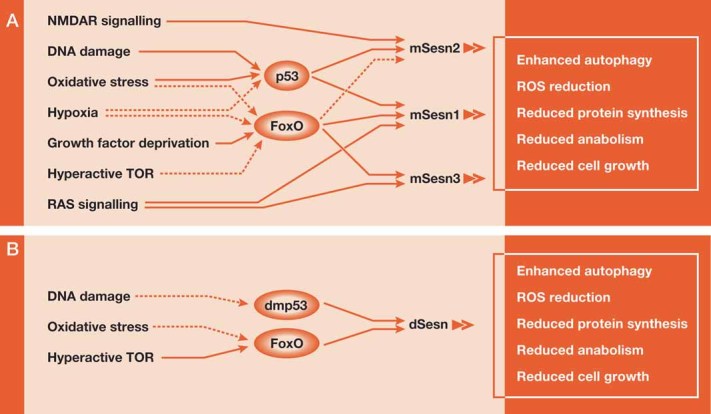
- Regulation of Sesn family gene expression in mammals. Stress-regulated transcription factors, p53 and FoxO were shown to be involved in the regulation of Sesn proteins.
- Regulation of Sesn in Drosophila. Solid lines depict confirmed mechanism of inducing Sesns. Dotted lines depict potential transcriptional mechanisms for Sesn regulation.
Although ROS can cause severe damage to many cellular constituents (Finkel & Holbrook, 2000), they may also operate as second messengers involved in the regulation of cell proliferation (Finkel, 2003; Martindale & Holbrook, 2002). For instance, oncogenic Ras proteins induce ROS accumulation and ROS are thought to be important effectors of Ras GTPase (Alexandrova et al, 2006; Irani et al, 1997). Ras activation results in down-regulation of Sesn1 and Sesn3 expression and this may contribute to Ras-induced ROS accumulation (Kopnin et al, 2007). The ability of Ras to cause down-regulation of Sesn1 and Sesn3 may be related to their positive regulation by FoxO transcription factors (Fig 3) (Chen et al, 2010; Nogueira et al, 2008; Tran et al, 2002). FoxO activity is subject to negative regulation by Ras through the AKT/protein kinase B (PKB) and ERK protein kinases, which retain FoxO proteins in the cytoplasm (Yang & Hung, 2009). Furthermore, Sesn3 was found to be responsible for some of the antioxidant activity of FoxO3A, and ectopic expression of activated AKT increases ROS accumulation through down-regulation of Sesn3 expression (Nogueira et al, 2008). Sesns are not the only antioxidant proteins activated by FoxO. The list also includes superoxide dismutase 2 (SOD2) and catalase, so Sesns may cooperate with other antioxidant proteins to provide a robust antioxidant defense system triggered by FoxO in response to different environmental stimuli (Salih & Brunet, 2008).
Sesn1 and Sesn2 are regulated by p53 (Fig 3A), and p53 is also known to activate antioxidant responses (Sablina et al, 2005). In addition to Sesn1 and Sesn2, p53 can induce expression of glutathione peroxidase 1 (Hussain et al, 2004), SOD2 (Hussain et al, 2004), aldehyde dehydrogenase 4A1 (ALDH4A1) (Yoon et al, 2004), p53-inducible protein 1 (INP1) (Cano et al, 2009), TIGAR (Bensaad et al, 2006), catalase (O'Connor et al, 2008) and glutaminase 2 (Hu et al, 2010), congruently with its ability to elevate antioxidant defenses. Interestingly, under severe stress conditions p53 induces ROS production and activates expression of pro-oxidant proteins involved in induction of apoptosis, such as PIG3, PUMA and Bax (Polyak et al, 1997; Vousden & Lane, 2007). Nevertheless, ROS accumulation is enhanced in p53-deficient cells (Ding et al, 2007; Sablina et al, 2005) and as a result, p53-deficient cells exhibit elevated DNA oxidation and chromosomal instability (Kopnin et al, 2007; Sablina et al, 2005). The antioxidant activity of p53 is also important for protecting retinal cells against oxidative stress and cell death (O'Connor et al, 2008). The importance of p53 in antioxidant defense is supported by in vivo analysis of transgenic mice with extra copies of p53 and the p53 regulator Arf1 that show reduced oxidative damage and ROS accumulation (Matheu et al, 2007). Expression of Sesn1 and Sesn2 is increased in these mice which also display an increased longevity phenotype. Despite the lack of direct evidence for a role of Sesns in the delay of aging, the plethora of data on the protective role of Sesns against oxidative stress in cell culture and their role in delaying aging-associated pathologies in Drosophila (see below) make them good candidates for the anti-aging activity in this mouse model.
Many recent studies suggest that age-associated neurodegenerative diseases, such as Alzheimer's disease (Querfurth & LaFerla, 2010) and Parkinson's disease (Schapira & Tolosa, 2010) are associated with the accumulation of oxidative stress. Hence, the antioxidant activity of Sesns may exert a neuroprotective role during aging as well as upon neuronal injury. Lending further credit to this hypothesis, Sesn2 is highly induced in mouse brains that suffered ischaemic injury (Budanov et al, 2002). In addition, Sesn2 is also induced by neuronal synaptic activity through NMDA receptor signalling thereby mediating intrinsic antioxidant defenses triggered by synaptic activity (Papadia et al, 2008) (Fig 3). dSesn-null Drosophila (Lee et al, 2010) show an obvious muscle degeneration phenotype, strikingly similar to the ones observed in Parkin-null and PTEN induced putative kinase 1 (PINK)-null mutant flies (Clark et al, 2006; Greene et al, 2003; Park et al, 2006). Parkin and PINK are homologues of human genes for familial parkinsonism and the pathologies caused by loss of either dSesn, Parkin or PINK are suppressed by administration of antioxidants such as vitamin E (Bier, 2006; Lee et al, 2010; Wang et al, 2006). Altogether, we believe that Sesn's antioxidant roles, as well as other protective roles that will be described below, are functioning to attenuate age- or brain injury-induced neurodegenerative diseases.
Sestrins and TOR Signalling
During a search for signalling pathways that may be affected by the redox (reduction and oxidation) activity of Sesn, we found that a major target for Sesn is the pathway that controls the activity of target of rapamycin (TOR) kinase (Budanov & Karin, 2008) (see Box 2, Fig 4). The regulation of TOR activity by Sesn, however, does not depend on Sesn redox activity (Budanov & Karin, 2008; Lee et al, 2010). TOR is a critical regulator of cell growth, proliferation, translation, metabolism and autophagy, first identified as a protein kinase whose activity is inhibited by the macrolide rapamycin (Alexander et al, 2010; Blagosklonny, 2008; Chen et al 2009; Wullschleger et al, 2006). In mammals, mTOR is present in two different complexes, TORC1 and TORC2. p53 is a known inhibitor of TORC1 and Sesn1 and Sesn2 are essential for this activity (Budanov & Karin, 2008) (Fig 5). Ectopic expression of Sesn1 or Sesn2 in different experimental settings results in strong inhibition of TORC1 activity, monitored by phosphorylation of p70S6K, 4E-binding protein (4EBP)1 and S6 proteins (Budanov & Karin, 2008). Two different splice forms of Sesn3 and dSesn can also inhibit the activity of TORC1 (Budanov & Karin, 2008; Chen et al, 2010; Lee et al, 2010) and, importantly, inhibition of Sesn1 or Sesn2 expression prevents the suppression of TORC1 activity otherwise imposed by genotoxic stress and p53 (Budanov & Karin, 2008; Wempe et al, 2010).
BOX 2 The TOR signalling pathway.
TOR (or mTOR in mammals) forms two distinct complexes called TORC1 and TORC2, which differ in the presence of the adaptor proteins Raptor and PRAS40 in TORC1 and the adaptor proteins Rictor, Sin1 and Protor-1 in TORC2 (Laplante & Sabatini, 2009b) (Fig 4). Curiously, rapamycin inhibits TORC1 activity, whereas TORC2 activity is considered to be rapamycin-independent (Wullschleger et al, 2006). TORC1 phosphorylates the protein kinase p70S6K1 and the 4EBP proteins, which are involved in regulation of translation, whereas TORC2 phosphorylates AKT, a critical regulator of metabolism and viability (Laplante & Sabatini, 2009b; Wullschleger et al, 2006).
TORC1 activity is regulated by a small GTPase, Ras homologues enriched in brain (Rheb), which binds and activates TORC1 upon guanosine-5′-triphosphate (GTP) loading (Wullschleger et al, 2006). Rheb itself is subject to negative control by the tuberous sclerosis 1 and 2 proteins (TSC1 and TSC2), which form a stable heterodimer that functions as a GTPase activating protein (Laplante & Sabatini, 2009b; Wullschleger et al, 2006). The TSC1:TSC2 complex is a nodal point for TORC1 regulation, because it integrates numerous inputs including the protein kinases AKT, ERK and RSK that function as negative regulators of TSC1:TSC2, thereby leading to TORC1 activation (Laplante & Sabatini, 2009b). Importantly, TSC1:TSC2 activity is positively regulated by AMP-activated protein kinase (AMPK), which is a major nutrient sensor (Shaw, 2009). Nutrient starvation, caloric restriction (CR) and exercise, all of which lead to ATP depletion and an increase in the AMP to ATP ratio, can activate AMPK (Shaw, 2009). AMPK activation results in inhibition of TORC1 activity through phosphorylation of TSC2 (Inoki et al, 2003) and Raptor (Gwinn et al, 2008). AMPK itself is a protein complex composed of a catalytic α subunit, and regulatory β and γ subunits (Shaw, 2009) and its activation can be brought about by different mechanisms including phosphorylation by upstream kinases such as LKB1, Ca2+/calmodulin-dependent kinase and transforming growth factor-β-activated kinase 1 (TAK1), and through interaction with regulator proteins such as KSR2 (Costanzo-Garvey et al, 2009; Wang & Guan, 2009). Curiously, p53 activation was found to inhibit TORC1 through activation of AMPK (Feng et al, 2005).
Figure 4. Overview of TOR signalling.
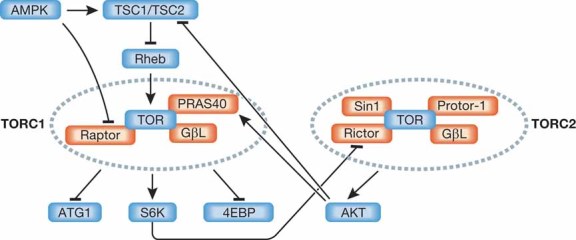
TOR kinase can be found in two distinct complexes in cells. TORC1 is rapamycin-sensitive but TORC2 is rapamycin-insensitive, and they are regulated differently. While TORC2 can induce TORC1 activation, hyperactivation of TORC1 can lead to suppression of TORC2. Signalling molecules are shaded in blue and TOR cofactors are shaded in red.
Figure 5. Role of Sestrins in cell signalling.
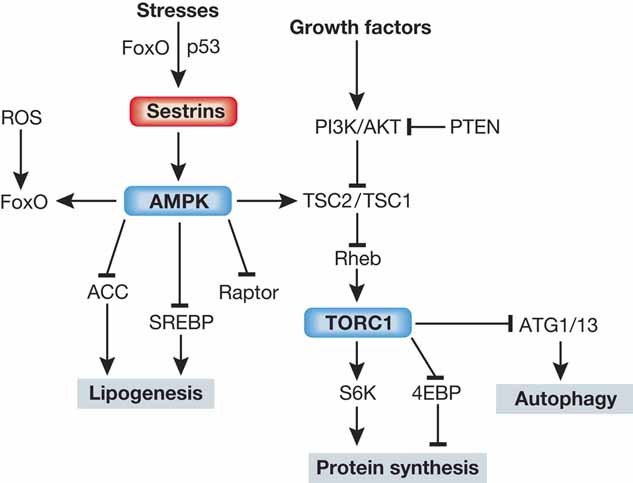
Sesn regulates AMPK-TORC1 signalling and controls cellular metabolism. While Sesn-dependent activation of AMPK and inhibition of TOR suppresses protein and lipid anabolism, Sesn can activate autophagic catabolism, which can provide additional energy sources, remove excessive lipid droplets or protein aggregates, and eliminate damaged mitochondria.
Inhibition of TOR is, however, independent of the anti-oxidant effects of the Sesns. Expression of a Sesn2 redox-defective mutant (C125S) (Budanov et al, 2004) led to as much inhibition of TORC1 as overexpression of wild type Sesn2 in human cell lines and similar findings were made in Drosophila (Budanov & Karin, 2008; Lee et al, 2010). To understand how Sesn inhibits TORC1, we examined the role of the small GTPase Rheb and the TSC1:TSC2 complex (see Box 2) in this process. Ectopically expressed Sesn2 decreased GTP loading of endogenous Rheb and overexpression of Rheb abrogated the inhibition of TORC1 by Sesn1/2 (Budanov & Karin, 2008). We also found that Sesn1/2 inhibits TORC1 in a TSC1:TSC2-dependent manner (Budanov et al, 2004). Importantly, Sesn1/2 induces AMPK phosphorylation at T172 and enhances AMPK-mediated phosphorylation of the TSC1:TSC2 complex (Fig 5) (Budanov & Karin, 2008). The mechanism by which Sesns activate AMPK is yet to be elucidated and it is not even clear whether it is liver kinase B (LKB1)-dependent or not. In some cells, Sesn overexpression can lead to AMPK phosphorylation in the absence of LKB1, whereas in others it is LKB1 dependent (Wei Wang and Michael Karin, unpublished data). Most likely through interaction with AMPK, Sesns facilitate activation by upstream stimuli or autophosphorylation.
TORC1 regulates many intracellular processes, including translation, cell growth, proliferation and autophagy (Wullschleger et al, 2006). Ectopic expression of Sesn2 inhibits phosphorylation of 4EBP1 and stimulates its binding to the translation initiation factor eIF-4E, thereby inhibiting the initiation of mRNA translation (Budanov & Karin, 2008). Accordingly, Sesn2 prevents translation of c-Myc and Cyclin D1 mRNAs, blocking cell proliferation (Budanov & Karin, 2008). Sesn1 and Sesn2 may also play a critical role in regulating cap-dependent protein synthesis in response to ionizing irradiation (Braunstein et al, 2009). Another important function of TORC1 is inhibition of macroautophagy (hereafter called autophagy) and Sesns may play a role here too. Autophagy is a process of sequestration and lysosomal degradation of intracellular organelles (including mitochondria) and cytoplasmic content (Klionsky et al, 2008). Autophagy plays an important role in the control of cell viability by providing necessary nutrients during starvation and in monitoring the health and integrity of organelles, especially the mitochondrion. Defective autophagy results in accumulation of damaged mitochondria which produce ROS and thereby can lead to oxidative stress and cellular damage (Gottlieb & Carreira, 2010). TORC1 inhibits autophagy through phosphorylation-mediated inhibition of the ATG1 (ULK1) protein kinase, and ATG13 proteins (Hosokawa et al, 2009; Jung et al, 2009; Wullschleger et al, 2006). shRNA-mediated knockdown of Sesn2 inhibits p53-dependent autophagy in response to nutrient depletion and rapamycin (Maiuri et al, 2009). Moreover, lithium, an inhibitor of inositol monophosphatase causing decreased phospho-inositol-1,4,5-triphosphate levels, and thapsigargin, an inhibitor of an endoplasmatic reticulum (ER) Ca2+-ATPase responsible for release of ER-stored Ca2+ to the cytoplasm, induce autophagy in a Sesn2-dependent manner (Maiuri et al, 2009). Overexpression of Sesn2 induces autophagy as indicated by conversion of the autophagic protein LC3-I (18K) into LC3-II (16K) form (Andrei V. Budanov, Jun Hee Lee and Michael Karin, unpublished data).
Sesn-dependent suppression of TORC1 signalling was also demonstrated in Drosophila. When overexpressed in mammalian 293 cells, dSesn functions like its mammalian counterpart; it increases the activity of AMPK towards TSC2 and suppresses p70S6K phosphorylation and activity (Lee et al, 2010). dSesn overexpression in flies decreases cell size (Lee et al, 2010), which is a general phenomenon associated with inhibition of TORC1 (Wullschleger et al, 2006). Genetic epistasis results fit well into a model where dSesn-mediated cell size suppression occurs between AKT and p70S6K, where the AMPK-TSC2 axis acts; as reducing expression of TSC1, TSC2 or AMPK hampered dSesn-dependent cell growth inhibition (Lee et al, 2010). Interestingly, expression of mammalian Sesn1 and Sesn2 in Drosophila also led to reduced cell size (Lee et al, 2010), implying that the Sesn-dependent regulation of TORC1 signalling is conserved throughout the metazoan system.
More importantly, Sesn also appears to be induced by hyperactivation of TORC1 signalling (Fig 5). dSesn expression is elevated upon TORC1 activation (mediated by overexpression of a constitutively active insulin receptor or Rheb, or by genetic ablation of TSC1 and PTEN) (Lee et al, 2010). Moreover, activation of TORC1 induces ROS accumulation and subsequent activation of a JNK-FoxO signalling module, which is responsible for dSesn induction (Budanov et al, 2004; Lee et al, 2010). TORC1-induced Sesn induction was also observed in mammalian cell lines (Andrei V. Budanov, Jun Hee Lee and Michael Karin, unpublished data). As it stands, Sesns are not only stress-dependent inhibitors of TORC1 activity, but also negative feedback regulators of TORC1 signalling. Interestingly, Sesns induced by ROS are inhibitors of ROS accumulation, thus they can control ROS levels trough their intrinsic antioxidant activity in parallel with the inhibition of TORC1 and stimulation of autophagy (mitophagy). This system might provide a dual control for regulation of Sesns expression and Sesns-mediated control of ROS accumulation, allowing cells to tightly adjust Sesns expression to the amounts of ROS (Fig 5). In the future it would be interesting to address the role of each individual pathway in regulation of Sesn expression. Below we will discuss the importance of this TORC1-Sesn-AMPK-TORC1 feedback loop.
Implications of the Sestrin-TORC1 Crosstalk
Aging
All multicellular organisms suffer from progressive decline in bodily function and physiology through a process called aging. Human aging is associated with many diseases such as type II diabetes, cancer, chronic inflammation, cardiovascular diseases and various degenerative diseases, such as sarcopenia, Alzheimer's disease and other forms of neurodegeneration and dementia. A general feature of aged cells and tissues is oxidative stress and accumulation of oxidized and modified proteins and protein aggregates, which can be the driving forces behind age-associated diseases. Thus, if we refer to aging as a slow but progressive increase in oxidative stress, it is conceivable that a stress response is activated during aging to impact this process.
There are two well established ways to control or attenuate age-associated disorders: these are the inhibition of the insulin/IGF1 signalling pathway and CR, both of which potentiate AMPK activation and inhibit TORC1 (Narasimhan et al, 2009). CR prevents the onset of various age-associated diseases including cancer, diabetes, cardiovascular diseases and neuronal degeneration in rhesus monkeys, a model species closely related to humans (Colman et al, 2009). Both CR and inhibition of the insulin/IGF pathway inhibit TORC1 activity and this inhibition increases lifespan in yeast, worms, flies and mice (Harrison et al, 2009; Kaeberlein et al, 2005; Kapahi et al, 2004). For example, mutation and inactivation of S6 kinase (S6K), a downstream target of TORC1, increased lifespan and decreased the incidence of diabetic phenotypes in mouse model (Selman et al, 2009). TORC1 overactivity has been associated with cancer, type II diabetes, obesity, cardiac hypertrophy and neurodegenerative diseases (Rosner et al, 2008; Stanfel et al, 2009). Amongst the many TORC1-controlled outputs, the ones most critical for affecting life- and healthspan are autophagy and mRNA translation (Hashimoto et al, 2009; Vellai, 2009). The role of mRNA translation is not well defined but an overall increase in translation could cause a decrease in fidelity of protein synthesis, increasing the overall burden of misfolded proteins hence contributing to oxidative stress (Kapahi et al, 2010). Inhibition of TORC1 could also decrease the translation of mRNAs encoding particular proteins involved in cell growth and metabolism and thereby affect the rate of metabolism, ROS production and intracellular damage. Another mechanism that affects aging is autophagy, which helps prevent the accumulation of age-associated sub-cellular abnormalities. Strikingly, the activity of p53, a positive regulator of autophagy and a suppressor of ROS, is decreased with age (Feng et al, 2007; Feng et al, 2005; Sablina et al, 2005). Conversely, increased p53 activity in mice through extra transgenic copies of the ARF1 and p53 genes is accompanied by increased expression of Sesn1/2 and can delay aging and age-associated disorders and reduce ROS accumulation (Matheu et al, 2007).
Given all of the above, as well as Sesns’ crosstalk with TORC1, it is expected that Sesns will be able to modulate aging and age-related disease processes (Fig 6) (Topisirovic & Sonenberg, 2010). Studying the physiological role of mammalian Sesns is complicated by the biochemical and regulatory redundancy of their function and expression, but having only a single Sesn gene (dSesn) Drosophila provide a simpler model to understand Sesn function (Lee et al, 2010). dSesn-null flies do not exhibit any developmental abnormalities, suggesting that dSesn is dispensable for normal growth. We cannot exclude, however, developmental roles for mammalian Sesns. For instance, Sesn1 appears to be involved in notochord development (Hikasa & Taira, 2001) and development of left–right asymmetry (Peeters et al, 2003). Inactivation of dSesn led to accelerated tissue aging and reduced health span, characterized by accumulation of triglycerides, muscle degeneration, cardiac malfunction and oxidative stress (Lee et al, 2010). These phenotypes are similar to those associated with inhibition of AMPK activity and activation of TORC1 (Stanfel et al, 2009). In the fly, dSesn was induced upon chronic TORC1 activation, which leads to ROS accumulation via activation of JNK and FoxO signalling (Lee et al, 2010). Thus, any condition that leads to ROS accumulation may be able to induce dSesn expression and possibly Sesn1 and Sesn3 through this pathway. Interestingly, JNK activation is known to extend lifespan through FoxO (Wang et al, 2005) and it is likely that Sesns mediate the effect of JNK and FoxO on longevity as well as ARF/p53.
Figure 6. Sestrins as suppressors of age-associated diseases.
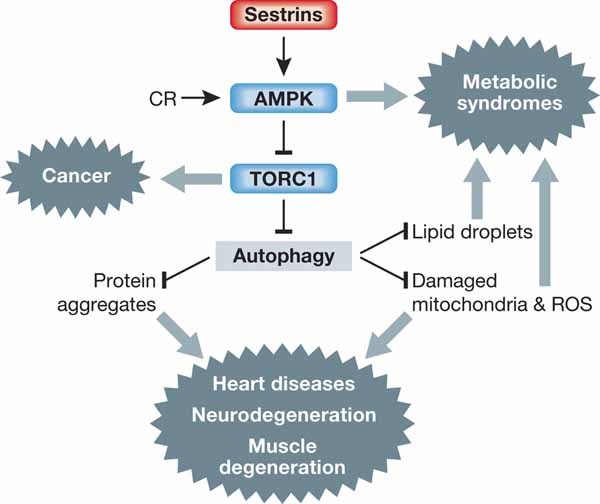
Both Sesns and CR can activate AMPK, inhibit TORC1 and induce autophagy, which are generally considered to be beneficial for preventing aging and age-associated pathologies.
Metabolism
Recent literature indicates that TORC1 hyperactivity is involved in various metabolic syndromes, such as obesity and type II diabetes (Wullschleger et al, 2006). TORC1 can enhance lipid synthesis by augmenting the activity of the lipogenic transcription factor sterol response element binding protein (SREBP), and by increasing expression of its downstream targets: fatty acid synthase, fatty acid carboxylase, acetyl-CoA synthase and acetyl-CoA carboxylase (ACC) (Laplante & Sabatini, 2009a). In addition to this, inhibition of autophagy through TORC1 activation can stimulate lipid accumulation because autophagy is required for the removal of fat droplets in liver (Singh et al, 2009a). Correspondingly, TORC1 inhibition with rapamycin reduced high fat diet-induced obesity and hepatosteatosis (Chang et al, 2009) as well as carbon tetrachloride-induced hepatic fibrosis (Bridle et al, 2009). Interestingly, autophagy plays an opposite role in lipid accumulation in adipose tissue and mice with adipose-specific knockout of autophagic ATG7 gene are lean and have decreased white adipose mass and enhanced insulin sensitivity (Singh et al, 2009b; Zhang et al, 2009). Autophagy is involved in differentiation of white adipose tissue (WAT) and adipose-specific ATG7 KO mice demonstrate conversion of WAT to brown adipose tissue (BAT). BAT uses fatty acids for autonomous energy expenditure and heat generation, supporting a lean body mass phenotype (Singh et al, 2009b).
dSesn-null flies have increased lipid accumulation in the fat body, the Drosophila homologue of the mammalian liver, which is associated with decreased AMPK activity and increased TOR activity (Lee et al, 2010). Inhibition of TOR by rapamycin or activation of AMPK with AICAR or metformin prevented fat accumulation (Lee et al, 2010). Increased lipogenesis in dSesn-null flies was associated with transcriptional up-regulation of SREBP and its downstream targets (Lee et al, 2010). Since TORC1 signalling pathway is known to be involved in obesity-induced hepatosteatosis, accumulation of WAT and diabetic progression in mammals (Dann et al, 2007; Laplante & Sabatini, 2009a; Laplante & Sabatini, 2009b), it will be interesting to determine the role of Sesns in regulation of lipid metabolism in different metabolically active organs such as liver, adipose tissue and muscle. It is worth knowing whether mammalian Sesns have a role in pathogenesis of age- and obesity-related metabolic disorders.
Muscle and neurodegeneration
dSesn-null flies exhibit cardiac arrhythmia, accompanied by increased heart period and heart diameter, that are very similar to the phenotypes of TORC1 hyperactivation (Lee et al, 2010; Wessells et al, 2004, 2009). Analysis of actin fibres demonstrated deterioration of the heart structure in dSesn-null flies. Also skeletal muscle is affected in dSesn-null flies as they suffered from age-dependent structural degeneration with accumulation of dysfunctional mitochondria and degenerated sarcomeres (Lee et al, 2010), a phenotype observed in extremely old flies too, albeit with reduced penetrance (Takahashi et al, 1970). Importantly, feeding of dSesn-null larvae with rapamycin or the AMPK activator AICAR prevents or attenuates most of these degenerative phenotypes (Lee et al, 2010).
Mammalian Sesns are expressed in the heart and muscle (Velasco-Miguel et al, 1999) and, in particular, Sesn1 is highly expressed in muscle. Rapamycin protects the heart from failure in hypoxia-reperfusion injury mouse model of heart attack (Khan et al, 2006) and AMPK activity is also dysregulated in human cardiomyopathy (Dyck & Lopaschuk, 2006). It is possible that Sesns exhibit a protective role in the mammalian muscle and heart too (through AMPK signalling) and could be responsible for example for preventing cardiac dysfunction during aging and obesity.
Autophagy can also prevent diverse neurodegenerative diseases associated with accumulation of protein aggregates, such as Alzheimer's, Parkinson's and Huntington's diseases (Querfurth & LaFerla, 2010; Sarkar & Rubinsztein, 2008; Schapira & Tolosa, 2010). Therefore, autophagy defects may be a general cause of neurodegenerative diseases and induction of autophagy may have therapeutic value for treating neurodegenerative disorders caused by accumulation of aggregated proteins, damaged mitochondria and ROS. Rapamycin, as a potent TORC1 inhibitor, can strongly induce autophagy and administration of rapamycin to Drosophila and mouse models of Huntington's (Ravikumar et al, 2004) and Parkinson's disease (Malagelada et al, 2010; Tain et al, 2009) ameliorates the degenerative phenotypes of these animal models. Since Sesns are intrinsic feedback inhibitors of TORC1 signalling and inducers of autophagy, and can be induced by a variety of stresses including oxidative and proteotoxic stresses, it is very likely that Sesns are functioning to attenuate the onset and progression of the neurodegenerative diseases by stimulating autophagy. In support of this hypothesis, Sesn2 was found to be up-regulated in response to exposure of neuroblastoma CHP134 cells to amyloid β(1–42) peptides, responsible for Alzheimer's disease (Hara et al, 2006; Kim et al, 2003). In addition, Sesns were also suggested to mediate the neuroprotective effects against oxidative stress conferred by p53, NMDA receptor and rosiglitazone, an antidiabetic drug and insulin sensitizer (Doonan et al, 2009; O'Connor et al, 2008; Papadia et al, 2008). Collectively, the autophagy-inducing role of Sesns, as well as their antioxidant function could contribute to the attenuation of the age-associated neurodegenerative diseases.
Cancer
Cancer cells require acceleration of anabolic processes such as protein and lipid biosynthesis, and increased energy production (Jones & Thompson, 2009). The TORC1 pathway is often activated in human cancers and rapamycin exhibits anti-tumorigenic activity (Guertin & Sabatini, 2007). mTOR is considered a potential drug target in cancer, stimulating the development of rapamycin analogues (Faivre et al, 2006; Guertin & Sabatini, 2007). TORC1 can support the neoplastic phenotype by increasing the rate of protein and lipid synthesis as well as glycolysis and mitochondrial respiration (Wullschleger et al, 2006). In addition, TORC1 inhibits autophagy, a process that protects mitochondria and peroxisomes from damage. Thus ROS accumulation in cells experiencing chronic TORC1 activation can accelerate tumour development by increasing DNA damage.
Because Sesns can suppress TORC1 activation, inhibit ROS accumulation, reduce genomic instability and stimulate autophagy, they may have tumour suppressor activity and could be part of the tumour suppressive gene network activated by p53 and FoxO. Accordingly, TORC1-stimulated hyperplasic cell growth was enhanced in the absence of dSesn (Lee et al, 2010), indicating that Sesns are negative regulators of cell growth. Overexpression of Sesn1 or Sesn2 suppressed hyperactive TORC1-induced cancerous cell growth (Budanov & Karin, 2008; Budanov et al, 2002). Sesn2-deficient mouse embryonic fibroblasts were significantly more susceptible to E1A + H-Ras-induced oncogenic transformation than wild type counterparts, suggesting that Sesn2 may have tumour suppressive activity (Budanov & Karin, 2008). Knockdown of Sesn2, as well as p53, accelerated A549 cancer cell growth in a tumour xenograft model (Sablina et al, 2005). Sesn2 was also shown to suppress angiogenesis and vascular endothelial growth factor (VEGF) production in the same model system (Andrei V. Budanov, AA Sablina and PM Chumakov, unpublished). Indeed, the human SESN1 (6q21) and SESN2 (1p35) loci are frequently deleted in cancer (Ragnarsson et al, 1999; Velasco-Miguel et al, 1999), and Sesn1 and Sesn2 expression is down-regulated in lung adenocarcinomas (Garber et al, 2001). Moreover, the Sesn3 promoter was found to be methylated in 20% of endometrial cancers (Zighelboim et al, 2007). Moreover, it was also shown that Sesn2 can counteract TGFβ signalling pathway (Wempe et al, 2010), which may have both tumour-suppressive and metastasis promoting activity depending on context (Ikushima & Miyazono, 2010). All of these findings indicate that the putative tumour suppressive function of Sesns merits further investigation.
Sestrins and TGFβ Signalling in the Lung
The TGFβ signalling is associated with chronic obstructive pulmonary disease (COPD), a leading cause of human mortality upon chronic exposure to cigarette smoke and other air pollutants (Morty et al, 2009). Inactivation of a short splice variant of LTBP4S, a member of the latent TGFβ-binding protein family, causes pulmonary emphysema associated with defective TGFβ signalling in mice (Sterner-Kock et al, 2002). Interestingly, Wempe et al recently demonstrated that Sesn2 deficiency partially suppresses pulmonary emphysema through stimulation of TGFβ signalling (Wempe et al, 2010). Sesn2 suppresses the TGFβ pathway in lung fibroblasts although we do not know the exact molecular mechanism. Whether other Sesns regulate this pathway in other contexts and tissues is unclear thus far but their potential pathological function should be assessed in other fibrotic diseases, especially liver fibrosis that can be enhanced by obesity.
Concluding Remarks and Future Perspectives
The link between stress and aging has just started to be explored. Sesns are evolutionarily conserved stress-inducible genes that encode antioxidant proteins involved in the regulation of the AMPK-TORC1 axis. It is clear that the AMPK-TORC1 axis is regulated by Sesns and it provides a major conduit for their function. However, despite its physiological relevance, the exact mechanism(s) by which Sesns promote AMPK activation is so far unclear. It will also be important to better understand, using genetic approaches, the role of autophagy, especially mitophagy, in Sesn action and the pathological consequence of Sesn depletion in mammals. dSesn protects the fly from early aging and age-associated disorders through regulation of lipid metabolism, cardiac and muscle function. The role of Sesns in regulation of physiology, aging and lifespan in vertebrates is yet to be examined, but the ongoing generation of Sesn1 and Sesn3 knockout mice, as well as combined Sesn1, Sesn2 and Sesn3 double and triple knockout mice will provide much needed resources for such studies. The essential contribution of the TORC1 pathway to development, carcinogenesis, immunity, metabolism and neurodegenerative diseases make the Sesns potential regulators of all these processes. The impact of Sesns on stress responses and age-associated diseases needs now to be tested in different mouse models of cancer, progeroid syndromes, metabolic derangements and neurodegenerative diseases, and some of these experiments have already been initiated. The role(s) of Sesns in lifespan extension by CR and in the maintenance of muscle and cardiac function by exercise are other pressing questions.
Pending issues
What is the 3D molecular structure of Sesns?
What is the precise molecular mechanism underlying the Sesn-induced activation of AMPK?
How are regulation of redox balance and energy sensing by Sesns linked?
What is the mechanism of regulation of autophagy by Sesns?
What are the physiological roles of Sesns in suppressing age-associated pathologies conserved in mammals? Can it be demonstrated in mouse models of cancer, progeroid syndromes, metabolic derangements and neurodegenerative diseases?
What is the isoform-specific role of mammalian Sesns?
Can we find some Sesn mutations or SNP in human patients suffering from early onset of the age-associated diseases?
How is the expression of Sesns changed in different types of cancer? Are there any potential mechanisms of Sesn inactivation? Does Sesn inactivation play any role during carcinogenesis?
What is the mechanism of TGFβ regulation and what is the role of Sesns in TGFβ-dependent processes including fibrosis, inflammation and carcinogenesis?
The modulation of Sesn activity or expression with small molecule or peptide Sesn analogues might represent a handle to further understand and eventually prevent and treat some of the most common diseases now considered to be an inevitable part of aging.
Acknowledgments
This work was supported by grants and fellowships from the NIH and the Superfund Research Program (CA118165, ES006376 and P42-ES010337 to M.K.; DK082080 to A.V.B.), Korea Research Foundation (KRF-2007-357-C00096 to J.H.L.), Human Frontier Science Program Organization (LT00653/2008-L to J.H.L). M.K. is an American Cancer Society professor. We thank Ryan Holzer for help in manuscript preparation.
The authors declare that they have no conflict of interest.
Abbreviations
- 4E-BP
4E-binding protein
- ACC
acetyl-CoA carboxylase
- AICAR
aminoimidazole carboxamide ribonucleotide
- ALDH4
aldehyde dehydrogenase 4 gene
- AMPK
AMP-activated protein kinase
- BAT
brown adipose tissue
- COPD
chronic obstructive pulmonary disease
- CR
caloric restriction
- Cys
Cystein
- dmp53
Drosophila melanogaster p53
- GPX1
gluthatione peroxidase 1
- FoxO
forkhead box transcription factor belonging to O-subclass
- JNK
c-Jun N-terminal kinase
- Hi95
hypoxia-inducible gene 95
- IGF1
insulin-like growth factor 1
- IGF-BP3
IGF1-binding protein 3
- INP1
p53-inducible protein 1
- mTOR
mammalian TOR
- NAD(P)H
nicotineamide adenine dinucleotide phosphate
- NMDA
N-methyl-d-aspartate
- PA26
p53-activated protein 26
- PI3K
phosphatidylinositol-3-kinase
- PINK
PTEN induced putative kinase 1
- PTEN
phosphatase and tensin homologue
- Prx
peroxiredoxin
- Redox
reduction and oxidation
- Rheb
Ras homologue enriched in brain
- ROS
reactive oxygen species
- S6K
S6 kinase
- Sesn
Sestrin
- SOD
superoxide dismutase
- TAK1
TGFβ-activated kinase 1
- TIGAR
TP53-induced glycolysis and apoptosis regulator
- TOR
target of rapamycin
- TORC1
TOR complex 1
- TORC2
TOR complex 2
- TSC
tuberous sclerosis complex protein
- WAT
white adipose tissue.
References
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova AY, Kopnin PB, Vasiliev JM, Kopnin BP. ROS up-regulation mediates Ras-induced changes of cell morphology and motility. Exp Cell Res. 2006;312:2066–2073. doi: 10.1016/j.yexcr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bier E. Antioxidants put Parkinson flies back in the PINK. Proc Natl Acad Sci USA. 2006;103:13269–13270. doi: 10.1073/pnas.0606288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7:3344–3354. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol. 2009;29:5645–5656. doi: 10.1128/MCB.00711-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle KR, Popa C, Morgan ML, Sobbe AL, Clouston AD, Fletcher LM, Crawford DH. Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl. 2009;15:1315–1324. doi: 10.1002/lt.21804. [DOI] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Seizinger BR, Kley N. Gene regulation by temperature-sensitive p53 mutants: identification of p53 response genes. Proc Natl Acad Sci USA. 1994;91:10640–10644. doi: 10.1073/pnas.91.22.10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated Sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ, et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res. 2009;69:219–226. doi: 10.1158/0008-5472.CAN-08-2320. [DOI] [PubMed] [Google Scholar]
- Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109:496–503. doi: 10.1254/jphs.08215fp. [DOI] [PubMed] [Google Scholar]
- Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- Chen C, Jeon S, Bhaskar P, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N. FoxOs inhibit mTORC1 and activate akt by inducing the expression of Sestrin3 and rictor. Cell. 2010;18:592–604. doi: 10.1016/j.devcel.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest. 2010;90:762–773. doi: 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumakov PM, Iotsova VS, Georgiev GP. [Isolation of a plasmid clone containing the mRNA sequence for mouse nonviral T-antigen] Dokl Akad Nauk SSSR. 1982;267:1272–1275. [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann SG, Selvaraj A, Thomas G. mTOR complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Ding B, Chi SG, Kim SH, Kang S, Cho JH, Kim DS, Cho NH. Role of p53 in antioxidant defense of HPV-positive cervical carcinoma cells following H2O2 exposure. J Cell Sci. 2007;120:2284–2294. doi: 10.1242/jcs.002345. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Doonan F, Wallace DM, O'Driscoll C, Cotter TG. Rosiglitazone acts as a neuroprotectant in retinal cells via up-regulation of Sestrin-1 and SOD-2. J Neurochem. 2009;109:631–643. doi: 10.1111/j.1471-4159.2009.05995.x. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally. J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essler S, Dehne N, Brune B. Role of Sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009;583:3531–3535. doi: 10.1016/j.febslet.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA. 2007;104:16633–16638. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Carreira RS. Autophagy in health and disease: V. Mitophagy as a way of life. Am J Physiol Cell Physiol. 2010;299:203–210. doi: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ookuma S, Nishida E. Lifespan extension by suppression of autophagy genes in Caenorhabditis elegans. Genes Cells. 2009;14:717–726. doi: 10.1111/j.1365-2443.2009.01306.x. [DOI] [PubMed] [Google Scholar]
- Hikasa H, Taira M. A Xenopus homolog of a human p53-activated gene, PA26, is specifically expressed in the notochord. Mech Dev. 2001;100:309–312. doi: 10.1016/s0925-4773(00)00519-0. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. Cellular context-dependent “colors” of transforming growth factor-beta signaling. Cancer Sci. 2010;101:306–312. doi: 10.1111/j.1349-7006.2009.01441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, III, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Metallothioneins: proteins in search of function. Cell. 1985;41:9–10. doi: 10.1016/0092-8674(85)90051-0. [DOI] [PubMed] [Google Scholar]
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Kim JR, Lee SR, Chung HJ, Kim S, Baek SH, Kim JH, Kim YS. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: involvement of RTP801 in amyloid beta-peptide toxicity. Exp Mol Med. 2003;35:403–411. doi: 10.1038/emm.2003.53. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopnin PB, Agapova LS, Kopnin BP, Chumakov PM. Repression of Sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability. Cancer Res. 2007;67:4671–4678. doi: 10.1158/0008-5472.CAN-06-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009a;19:R1046–1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009b;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8:1571–1576. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morty RE, Konigshoff M, Eickelberg O. Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:607–613. doi: 10.1513/pats.200908-087RM. [DOI] [PubMed] [Google Scholar]
- Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19:R657–666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, Wallace DM, O'Brien CJ, Cotter TG. A novel antioxidant function for the tumor-suppressor gene p53 in the retinal ganglion cell. Invest Ophthalmol Vis Sci. 2008;49:4237–4244. doi: 10.1167/iovs.08-1963. [DOI] [PubMed] [Google Scholar]
- Oren M, Levine AJ. Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen. Proc Natl Acad Sci USA. 1983;80:56–59. doi: 10.1073/pnas.80.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Peeters H, Debeer P, Bairoch A, Wilquet V, Huysmans C, Parthoens E, Fryns JP, Gewillig M, Nakamura Y, Niikawa N, et al. PA26 is a candidate gene for heterotaxia in humans: identification of a novel PA26-related gene family in human and mouse. Hum Genet. 2003;112:573–580. doi: 10.1007/s00439-003-0917-5. [DOI] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Ragnarsson G, Eiriksdottir G, Johannsdottir JT, Jonasson JG, Egilsson V, Ingvarsson S. Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival. Br J Cancer. 1999;79:1468–1474. doi: 10.1038/sj.bjc.6690234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Woo HA, Bae SH, Park S. Sestrin 2 is not a reductase for cysteine sulfinic acid of peroxiredoxins. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2360. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008;659:284–292. doi: 10.1016/j.mrrev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC. Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol. 2010;6:309–317. doi: 10.1038/nrneurol.2010.52. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–1074. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, et al. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002;16:2264–2273. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Philpott DE, Miquel J. Electron microscope studies on aging Drosophila melanogaster. 3. Flight muscle. J Gerontol. 1970;25:222–228. doi: 10.1093/geronj/25.3.222. [DOI] [PubMed] [Google Scholar]
- Topisirovic I, Sonenberg N. Cell biology. Burn out or fade away. Science. 2010;327:1210–1211. doi: 10.1126/science.1187497. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene. 1999;18:127–137. doi: 10.1038/sj.onc.1202274. [DOI] [PubMed] [Google Scholar]
- Vellai T. Autophagy genes and ageing. Cell Death Differ. 2009;16:94–102. doi: 10.1038/cdd.2008.126. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, Zhang Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Wang W, Guan KL. AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 2009;196:55–63. doi: 10.1111/j.1748-1716.2009.01980.x. [DOI] [PubMed] [Google Scholar]
- Wempe F, De-Zolt S, Koli K, Bangsow T, Parajuli N, Dumitrascu R, Sterner-Kock A, Weissmann N, Keski-Oja J, von Melchner H. Inactivation of Sestrin 2 induces TGF-{beta} signaling and partially rescues pulmonary emphysema in a mouse model of COPD. Dis Model Mech. 2010 doi: 10.1242/dmm.004234. [DOI] [PubMed] [Google Scholar]
- Wessells R, Fitzgerald E, Piazza N, Ocorr K, Morley S, Davies C, Lim HY, Elmen L, Hayes M, Oldham S, et al. d4eBP acts downstream of both dTOR and dFoxo to modulate cardiac functional aging in Drosophila. Aging Cell. 2009;8:542–552. doi: 10.1111/j.1474-9726.2009.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004;36:1275–1281. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004;49:134–140. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- Zhai B, Villen J, Beausoleil SA, Mintseris J, Gygi SP. Phosphoproteome analysis of Drosophila melanogaster embryos. J Proteome Res. 2008;7:1675–1682. doi: 10.1021/pr700696a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zighelboim I, Goodfellow PJ, Schmidt AP, Walls KC, Mallon MA, Mutch DG, Yan PS, Huang TH, Powell MA. Differential methylation hybridization array of endometrial cancers reveals two novel cancer-specific methylation markers. Clin Cancer Res. 2007;13:2882–2889. doi: 10.1158/1078-0432.CCR-06-2367. [DOI] [PubMed] [Google Scholar]