

Abstract
Telomeres protect against chromosomal breakage, fusion, and interchromosome bridges during cell division. Shortened telomeres have been observed in the lowest grade of pancreatic intraepithelial neoplasia. Genetically engineered mouse models of pancreatic neoplasia develop acinar-to-ductal metaplasia prior to the development of pancreatic intraepithelial neoplasia suggesting that acinar-to-ductal metaplasias can be an early precursor lesion to pancreatic cancer. Some human pancreatic intraepithelial neoplasias are associated with acinar-to-ductal metaplasias, and it has been suggested that these acinar-to-ductal metaplasias arise as a consequence of growth of adjacent pancreatic intraepithelial neoplasias. Since the earliest known genetic lesions of pancreatic intraepithelial neoplasias is shortened telomeres we compared the telomere lengths of acinar-to-ductal metaplasia lesions, pancreatic intraepithelial neoplasias and adjacent normal cells of human pancreata to determine if acinar-to-ductal metaplasias could be precursors to pancreatic intraepithelial neoplasia. We used quantitative fluorescent in situ hybridization to measure the telomere length of cells from pancreatic lesions and adjacent normal pancreata from 22 patients, including 20 isolated acinar-to-ductal metaplasias, 13 pancreatic intraepithelial neoplasias associated with acinar-to-ductal metaplasias, and 12 pancreatic intraepithelial neoplasias. Normalized mean telomere fluorescence was significantly different among the cell types analyzed; 12.6±10.2 units in normal acinar cells, 10.2±6.4 in ductal cells, 8.4±5.9 in fibroblasts, 9.4±7.3 in isolated acinar-to-ductal metaplasias, 4.1±2.9 in pancreatic intraepithelial neoplasia-associated acinar-to-ductal metaplasias, and 1.6±1.9 in pancreatic intraepithelial neoplasias, respectively (p<0.001, ANOVA with randomized block design). Telomeres were significantly shorter in pancreatic intraepithelial neoplasia-associated acinar-to-ductal metaplasias (p<0.05, post-hoc Duncan test) and in pancreatic intraepithelial neoplasias (p<0.05), than in normal cells, or isolated acinar-to-ductal metaplasias. Thus, shortened telomeres are found in pancreatic intraepithelial neoplasia-associated acinar-to-ductal metaplasias, but not in isolated acinar-to-ductal metaplasia lesions. These results indicate that isolated acinar-to-ductal metaplasias are not a precursor to pancreatic intraepithelial neoplasia, and support the hypothesis that pancreatic intraepithelial neoplasia-associated acinar-to-ductal metaplasias arise secondary to pancreatic intraepithelial neoplasia lesions.
INTRODUCTION
Pancreatic ductal adenocarcinoma is the 4th leading cause of cancer death in the United States.1 The detection and treatment of early-stage pancreatic ductal adenocarcinomas and its precursor lesions is likely to be the best way to reduce the mortality of the patients from this deadly disease.2
There is considerable evidence supporting a multistep progression model of pancreatic carcinogenesis, whereby progressive genetic and epigenetic changes accumulate as pancreatic intraepithelial neoplasias (PanINs) undergo increasing grades of histological changes culminating in infiltrating pancreatic ductal adenocarcinoma.3, 4 The most common genetic alterations of pancreatic ductal adenocarcinomas arise in PanINs including mutational activation of the KRAS oncogene,5 and inactivation of tumor suppressor genes p16/CDKN2A,5 SMAD4/DPC4,6 TP53,7, BRCA2,8 and shortened telomeres.9 Of these changes, telomere shortening is found in ~90% of PanIN-1 lesions and is considered to be one of the earliest genetic events in pancreatic carcinogenesis, while inactivation of TP53, SMAD4/DPC4, and BRCA2 generally only become evident in PanIN-3 lesions.10 DNA hypermethylation events that lead to gene silencing in pancreatic cancers are also evident in precursor PanIN and intraductal papillary mucinous neoplasm (IPMN) lesions, including p16, SPARC, TSLC1 and others, 11-14 but in general these methylation alterations are more prevalent in higher grade PanINs and do not occur earlier than telomere shortening.
Acinar-to-ductal metaplasias are composed of a mixture of cells with acinar cells and ductal morphology, often containing cells with a morphologic appearance suggestive of acinar cells transforming into cells with ductal differentiation. The isolated acinar-to-ductal metaplasia lesion, also known as acinar ectasia was first described as uremic exocrine pancreatopathy, because it was observed in patients with advanced uremia,15 although it has also been observed in other disease settings.16 Acinar-to-ductal metaplasias have been receiving more attention recently because of reports indicating they are found in association with PanIN lesions. 17, 18 Brune et al. observed prominent acinar-to-ductal metaplasia lesions in areas of lobulocentric parenchymal atrophy. 18 Shi et al. reported acinar-to-ductal metaplasia lesions in association with parenchymal changes in patients with multiple PanINs and a strong family history of pancreatic cancer. In this setting the degree of pancreatic acinar atrophy was variable from focal acinar-to-ductal metaplasia and partial acinar atrophy to total replacement of lobular units by acinar-to-ductal metaplasia, fibrotic stroma, and aggregates of islet cells and complete loss of acinar cells.19 Zhu et al. described histologic features of acinar-to-ductal metaplasia occurring adjacent to PanIN and found that acinar-to-ductal metaplasia lesions contained both acinar-like cells with zymogen granules and duct-like cells with mucinous cytoplasm and showed its association with PanINs.17
Telomeres are structures of hexameric repeated DNA sequences, TTTAGG in humans complexed with numerous telomere binding proteins at the termini of linear chromosomes.20 Telomeres protect against chromosomal breakage, fusion, and interchromosome bridges during cell division. Dysfunction of telomeres gives rise to chromosomal instability.21-27 Once telomeres become critically short, telomeric fusions may occur between chromosomal arms, and these fusions can create ring or dicentric chromosomes leading to anaphase bridges during mitosis.23 Breakage of anaphase bridges generates highly recombinogenic free DNA ends, fusion of broken ends and chromosomal rearrangements that can be self-perpetuating 22, 23 and are typical of many cancers. Telomere shortening will cause senescence unless senescence pathways such as p53 activation are overcome.28 Later in neoplastic evolution, neoplastic clones up-regulate the enzyme telomerase which facilitates cellular immortalization.29-31 Pancreatic adenocarcinomas have very short telomeres,23 complex karyotypes, numerous chromosomal abnormalities and high fractional allelic losses, supporting an important role for telomeric dysfunction as a driving force in pancreatic carcinogenesis.23, 32-39
Several genetically engineered mouse models of pancreatic adenocarcinomas reproduce the histologic progression of human pancreatic ductal adenocarcinomas.40 In these models, acinar-to-ductal metaplasias can be observed even before mPanIN lesions and have been implicated as a possible precursor to mPanIN lesions 17, 40-43. On the other hand, we have previously microdissected human PanIN-associated acinar-to-ductal metaplasia lesions and analyzed them for KRAS gene mutations to help identify the origin of these PanIN-associated acinar-to-ductal metaplasia lesions.44 KRAS gene mutations were only detected in acinar-to-ductal metaplasia lesions associated with PanINs. Mutations were not identified in isolated acinar-to-ductal metaplasias suggesting that the KRAS gene mutations occurring in human PanIN-associated acinar-to-ductal metaplasia lesions probably represent retrograde extension of PanINs, rather than evidence that isolated acinar-to-ductal metaplasias are neoplastic and that the PanINs arose from their adjacent acinar-to-ductal metaplasias.
In-situ evaluation of acinar-to-ductal metaplasias can provide more accurate analysis of the cellular origin of molecular alterations. We therefore evaluated telomere length in isolated acinar-to-ductal metaplasia lesions, in PanIN-associated acinar-to-ductal metaplasia lesions and in normal pancreatic cells using fluorescence in situ hybridization in order to determine if acinar-to-ductal metaplasia lesions are precursor lesions to PanINs.
METHODS
Case Selection and Histologic Features of Isolated Acinar-to-ductal Metaplasias and PanIN-Associated Acinar-to-ductal Metaplasias
Pancreatoduodenectomy (Whipple operation) specimens from 2004 to 2006 were reviewed for the presence of either PanIN-associated acinar-to-ductal metaplasias or isolated acinar-to-ductal metaplasia lesions. Acinar-to-ductal metaplasia lesions were classified as isolated acinar-to-ductal metaplasia (also previously known as acinar ectasia) and PanIN-associated acinar-to-ductal metaplasias based on histologic features.18, 44 Histologically, isolated acinar-to-ductal metaplasias are characterized by dilatation of acini such that they resemble ductules and flattening of acinar cells with loss of zymogen granules.16 PanIN-associated acinar-to-ductal metaplasias, as their name implies, are acinar-to-ductal metaplasias that are in direct cellular contact with, or are in close proximity to PanINs. Some metaplastic cells contain mucin with flattened nuclei and some acinar-to-ductal metaplasias contain cytoplasmic eosinophilic zymogen granules.44 Representative images of isolated acinar-to-ductal metaplasias and PanIN-associated acinar-to-ductal metaplasias are depicted in Figure 1.
Figure 1.
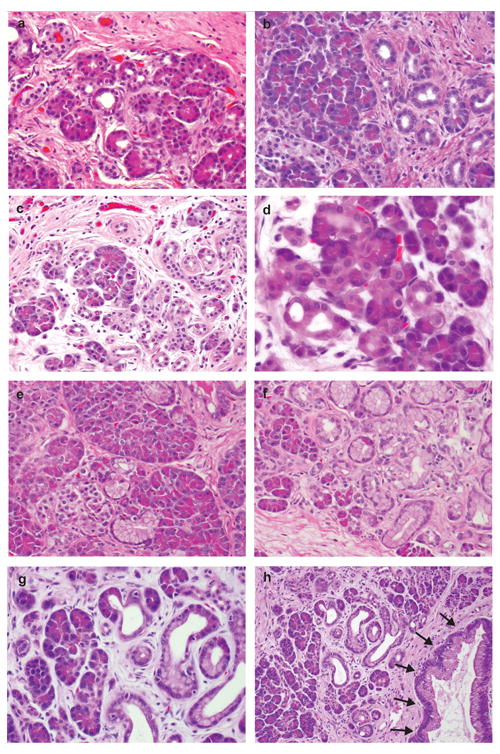
Representative images of acinar-to-ductal metaplasia. a-d) Isolated acinar-to-ductal metaplasia. Metaplastic acinar-to-ductal metaplasia cells make luminal structures. Some metaplastic cells contain eosinophilic cytoplasmic granules which are characteristic of acinar cells. E-h) PanIN-associated acinar-to-ductal metaplasias. Metaplastic cells contain supranuclear mucin, others have eosinophilic cytoplasmic granules. g) Higher magnification of h. h) Shows spectrum from normal acini, isolated acinar-to-ductal metaplasias, PanIN-associated acinar-to-ductal metaplasias, and PanIN (arrows).
PanINs are microscopic papillary or flat noninvasive epithelial neoplasms arising in a pancreatic ducts, composed of cuboidal to columnar cells with varying amounts of mucin and various degrees of cytologic or structural atypia.4 PanIN lesions can be further classified as PanIN-1, PanIN-2, and PanIN-3 according to the degree of cytologic and architectural atypia.4 A similar classification system has been developed for precursor lesions in genetically engineered mouse models.45 We were interested in studying the earliest events in human pancreatic adenocarcinomas, therefore the PanIN lesions selected for analysis in this study were low-grade PanIN lesions, predominantly PanIN-1 lesions, which are low-grade, flat, or papillary epithelial lesions composed of tall columnar cells with basally located nuclei and abundant supranuclear mucin. A small number of PanIN-2 lesions were also included. PanIN-2 lesions are structurally slightly more complex than PanIN-1 lesions, and they have more nuclear changes including loss of nuclear polarity, nuclear hyperchromasia, variation in nuclear size, nuclear crowding, and nuclear pseudostratification. All samples were collected with approval from the Johns Hopkins Committee for Clinical Investigation.
Tissue Procurement and Telomere Fluorescence In Situ Hybridization (FISH)
From selected cases, 2 serial sections (5 μm) were cut from formalin-fixed, paraffin-embedded tissue blocks and placed on poly L-lysine charged slides. One section was stained with hematoxylin and eosin and used as a guide slide. Telomere FISH was performed as described previously.9,46 In brief, deparaffinized, 5-μm tissue sections on charged slides underwent rehydration in a series of graded ethanol solutions, immersion in 2% Tween-20 for 1min, and heat-induced antigen retrieval for 20 minutes in citrate buffer. Slides were then cooled down to room temperature for 10 min, and dehydrated in a series of graded ethanol solutions. FISH was performed on sections by co-hybridization of a custom-made N-terminal Cy3-labeled, telomere specific peptide nucleic acid (PNA) probe (N-CCCTAACCCTAACCCTAA-C) and two N-terminal FITC-labeled, centromere specific, PNA probes (N-ATTCGTTGGAAACGGGA-C, N-CACAAAGAAGTTTCTGAG-C; Applied Biosystems, Framingham, MA). PNA probes were applied together, each at a concentration of 300 ng/ml in diluent (70% formamide, 10mM Tris pH 7.5, 0.5% B/M Blocking Reagent; Boehringer-Manheim) and slides denatured at 83°C for 4 min, followed by 2 hour hybridization at room temperature. Sections were washed twice at room temperature for 15 min with PNA wash buffer (70% formamide, 10mM Tris pH 7.5, and 0.1% BSA), followed by 5 min washes 3 times in PBS with 0.1% Tween. Nuclei were counterstained for 1 min at room temperature with a 1:10,000 dilution in water of a 5 mg/ml stock solution of DAPI (4’-6-diamidino-2-phenlindole; a DNA-binding dye; Sigma). Sections were mounted using Prolong Anti-Fade Mounting Media solution (Invitrogen).
Microscopy and Image Analysis
Telomere FISH produced a speckled pattern of nuclear signals in all cases examined, in keeping with results previously described 47. Digital fluorescent telomere signals were visualized using a Nikon 50i epifluorescence microscope equipped with X-Cite series 120 illuminator (EXFO Photonics Solutions Inc., Ontario, CA) and a 40x/1.4 NA non-oil immersion Neofluar lens. Fluorescence excitation/emission filters were as follows: Cy3 excitation, 546 nm/10 nm BP; emission, 578 nm LP (Carl Zeiss Inc.); DAPI excitation, 330 nm; emission, 400 nm via an XF02 fluorescence set (Omega Optical, Brattleboro, VT); Alexa Fluor 488 excitation, 475 nm; emission, 535 nm via a combination of 475RDF40 and 535RDF45 filters (Omega Optical). Grayscale images were captured for presentation using Nikon NIS-Elements software and an attached Photometrics CoolsnapEZ digital camera, pseudo-colored and merged. Integration times typically ranged from 500 ms to 800 ms for Cy3 (telomere) and FITC (centromere) signal capture, 50 ms to 100 ms for the DAPI counterstain. The accuracy of the scoring was evaluated by having two of the authors (SMH, CMH) score a random number of ~100 cells from multiple slides –no significant differences were noted between each observer. Quantification of the digitized fluorescent telomere signals was accomplished by use of a semi-automated algorithm, written for the open source image analysis software package, ImageJ (http://rsb.info.nih.gov/ij/) and telomere counting software, Telometer (http://bui2.win.ad.jhu.edu/telometer/).
For a given 12-bit grayscale image, cells that contained detectable telomere signals that were in sharp focus were chosen for analysis. Image processing was performed as follows. Cy3 telomere signals were extracted by using a rolling ball filter to create a binary mask that was then re-applied to the original image following background subtraction. Telomeric signals identified by the segment mask, having sizes above the threshold for background noise, and that were contained within the area inscribed by the nuclear DAPI signal, were then measured; data for each telomeric spot within that particular nucleus was tabulated. Cells were manually circled either individually or in small groups.
The final data consisted of telomere pixel areas and intensities as well as DAPI nuclear total pixel area and total intensity. These data were then exported to Microsoft Excel (Microsoft Corp., Redmond, WA, USA) for further analysis. The measurement used for comparative purposes was the summed intensities of all telomeric pixels for a given cell divided by the total DAPI intensity of that cell. This ratio of total telomere signal/total DAPI signal was considered as normalized mean telomere fluorescence for each cell. This in situ approach to quantify relative telomere lengths in samples of preneoplastic and cancerous lesions has been well established and validated previously.47
Telomere lengths measured as more than 3 standard deviations above the normal range were excluded from analysis because these measurements were considered as likely to be inaccurate. Using this criterion the telomere lengths of 4.9% cells (344/7066), representing a similar number of cells of each cell type were excluded.
Statistical methods
Statistical analyses were performed using SAS (version 9.13) and R 2.10.1 (http://www.r-project.org).48 The disease type, number of cells analyzed and summary statistics of telomere fluorescence intensity (mean and range) were tabulated for each individual. Plots of the telomere lengths were provided to graphically depict the intra- and inter-patient variability. Replicate measurements were made for each individual to more accurately assess the individual distributional characteristics. The summary statistics, including mean, range, 25th–75th quartiles, and inter-quartile range (IQR) of telomere length of acinar cells were calculated for the individual medians and individual IQRs for each disease subtype. Normalized mean telomere fluorescence between and among normal acinar cells, ductal cells, fibroblasts, isolated acinar-to-ductal metaplasias, PanIN-associated acinar-to-ductal metaplasias, and PanINs was compared by ANOVA with randomized block design and Duncan’s tests. Normalized mean telomere fluorescence between PanIN-associated acinar-to-ductal metaplasias and PanINs according to the disease of the patients and grade of PanINs were compared by Student T-test. Linear regression analysis was performed to examine the relationship between patient age and the relative telomere length in normal acinar cells. A p-value of less than 0.05 was considered statistically significant.
RESULTS
Characteristics of Cases
Telomere lengths were analyzed in normal pancreatic cells, cells characterized by acinar-to-ductal metaplasia and PanIN lesions from 22 patients (mean ±SD age, 63.5±6.5, range, 44 to 72 years), including 13 men (59%) and 9 women (41%). The diagnosis of these 22 patients included 7 with pancreatic ductal adenocarcinoma, 3 with adenocarcinoma of the ampulla of Vater, 2 with cholangiocarcinoma, 3 with an intraductal papillary mucinous neoplasm, 3 with PanINs, 2 with chronic pancreatitis, 1 with a pancreatic endocrine neoplasm, and 1 with a serous cystadenoma. The analyzed included 62 isolated acinar-to-ductal metaplastic lesions from 20 patients, 34 PanIN-associated acinar-to-ductal metaplastic lesions from 13 patients, 15 PanIN lesions from 12 patients and normal pancreatic cells, including 72 areas of acinar cells from 22 patients, 13 areas of normal pancreatic ductal cells from 13 patients, and 22 areas with normal fibroblasts from 99 different lesions of 22 patients. A total of 7066 nuclei were selected for computer assisted image analysis, including nuclei from 1968 acinar cells, 383 ductal cells, 1315 fibroblasts, 1951 cells from isolated acinar-to-ductal metaplasias, 986 cells from PanIN-associated acinar-to-ductal metaplasias, and 463 cells from PanINs.
Comparison of Telomere Lengths
Representative images of telomere FISH are depicted in Figure 2. Normalized mean telomere fluorescence was significantly different among the cell types analyzed; 12.6±10.2 (mean±S.D) units in normal acinar cells, 10.2±6.4 units in ductal cells, 8.4±5.9 units in fibroblasts, 9.4±7.3 units in isolated acinar-to-ductal metaplasias, 4.1±2.9 units in PanIN-associated acinar-to-ductal metaplasias, and 1.6±1.9 units in PanINs, respectively (p<0.001, ANOVA with randomized block design). Telomeres were significantly shorter in PanIN-associated acinar-to-ductal metaplasias (p<0.05, post hoc Duncan test) and in PanINs (p<0.05), than in acinar cells, ductal cells, fibroblasts, or isolated acinar-to-ductal metaplasia. However, there was no significant difference in the relative telomere lengths among the other cell types, including between PanIN-associated acinar-to-ductal metaplasias and PanINs (Figure 3).
Figure 2.
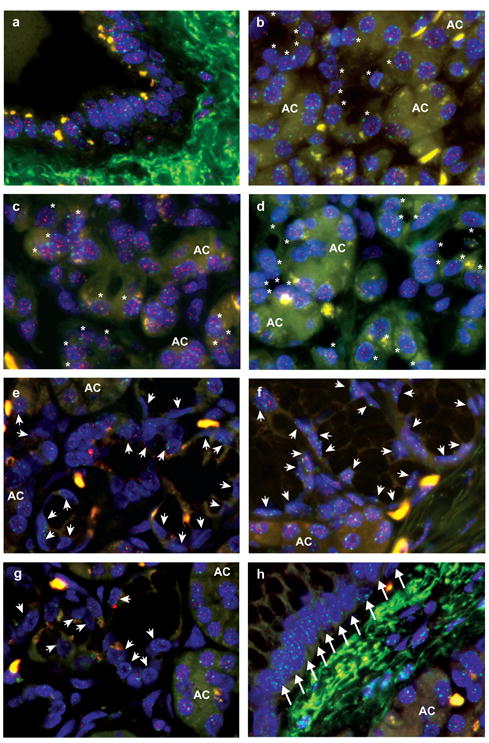
Representative images of telomere FISH. Nuclei are stained with DAPI (blue). The telomeres are stained with a Cy3-labeled telomere specific FISH probe (small red dots in nuclei) and the centromeres are stained with FITC-labeled centromere specific FISH probes (small green dots in nuclei). Telomeric signals in isolated acinar-to-ductal metaplasias (asterisks) are retained and are similar to those in acinar (AC) and ductal cells. However, telomeric signals in PanIN-associated acinar-to-ductal metaplasias (arrowheads) and in PanIN (arrows) are weaker than those in acinar (AC) and ductal cells. a) Ductal cells. B-d) Isolated acinar-to-ductal metaplasias (asterisks) and acinar cells (AC). E-g) PanIN-associated acinar-to-ductal metaplasias (arrowheads) and acinar cells (AC). h) PanIN (arrows) and acinar cells (AC).
Figure 3.
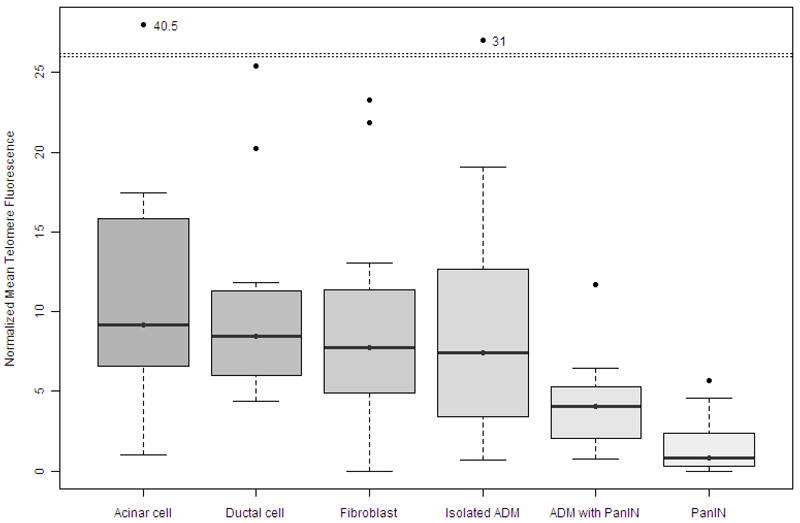
The normalized mean telomere fluorescence of normal, metaplastic and neoplastic lesions of the pancreas; Telomeres are significantly shorter in PanIN-associated acinar-to-ductal metaplasias (p<0.05, post hoc Duncan test) and in PanINs (p<0.05), than they are in acinar cells, ductal cells, fibroblasts, or isolated acinar-to-ductal metaplasia. However there was no significant difference of the telomere lengths among other cell types.
We also compared the normalized mean telomere fluorescence of isolated acinar-to-ductal metaplasias, PanIN-associated acinar-to-ductal metaplasias, and PanIN lesions according to each patient’s underlying disease. We dichotomized patients into two groups: those with cancers, and those without cancers, including patients with intraductal papillary mucinous neoplasm, PanINs, chronic pancreatitis, and serous cystadenomas. As shown in Figure 4, there was no significant difference in the normalized mean telomere fluorescence of these lesions based on the patients underlying disease state (cancer vs. non-cancer). Normalized mean acinar-to-ductal metaplasia telomere fluorescence was 10.8±6.0 units in patients with cancer and 9.2±6.7 units in those without cancer (p=0.29, Student T-test). Normalized mean telomere fluorescence of PanIN-associated acinar-to-ductal metaplasias was 5.1±4.9 units in patients with cancer and 3.8±2.2 units in those without cancer (p=0.36). The normalized mean telomere length of the PanIN lesions was 1.9±2.1 units in patients with cancer and 0.6±0.9 units in those without cancer (p=0.13).
Figure 4.
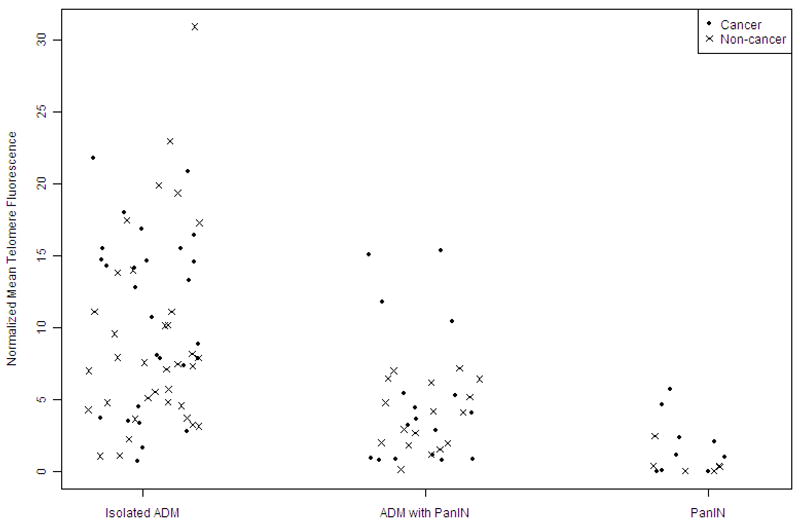
The normalized mean telomere fluorescence of metaplastic and precursor lesions of the pancreas based on underlying patient diagnosis. Each dot represents the normalized mean telomere fluorescence of cells from patients with cancers. Each x-mark represents the normalized mean acinar-to-ductal metaplasia telomere fluorescence of cells from patients without cancer. Telomere lengths were not significantly different in isolated acinar-to-ductal metaplasia lesions, PanIN-associated acinar-to-ductal metaplasias, and PanINs by underlying patient diagnosis.
When telomere length of PanINs was compared by grade of PanINs, there was no significant difference of telomere length based on PanIN grades (PanIN-1, normalized mean telomere fluorescence, 1.0±1.5 units; PanIN-2, 2.1±2.2 units, p=0.27, Student T-test).
Because telomere length gradually shortens with increasing age, we also correlated telomere length in normal pancreatic acinar cells by patient age but we did not find any relationship between telomere length and patient age (R2=0.0079, p=0.69), although our study was not powered for this type of analysis.
DISCUSSION
Our understanding of pancreatic carcinogenesis is fundamentally based on our understanding of the histologic progression of pancreatic precursor lesions and their associated genetic alterations.49 Genetically engineered mouse models have helped to confirm the human pancreatic cancer progression model and these models have also provided support for the ductal origin of pancreatic adenocarcinomas.40, 42, 43, 50, 51 However, there are several observations arising from genetically engineered mouse models of pancreatic neoplasia that have been interpreted to indicate pancreatic acinar cells are a source of pancreatic ductal adenocarcinomas. For example, the LSL-KRASG12D/+p48Cre/+ mice (LSL= LoxP-Stop-LoxP) which have mutant KRAS (G12D) developmentally expressed in pancreatic ductal and acinar cells develop acinar-to-ductal metaplasia lesions and early mouse PanIN (mPanIN) causing some investigators to conclude that acinar-to-ductal metaplasia lesions and PanIN lesions arise from the same molecular pathways.17 Another group similarly demonstrated that the conditional expression of an endogenous KRASG12V oncogene in cells of acinar and centroacinar origin results in the development of the full spectrum of mPanINs and infiltrating carcinoma.50 In addition, with Ela-CreERT2Tg/+; LSL-KrasG12D mice Habbe et al. targeted the Elastase- and Mist1-expressing acinar cells and found that these mice developed acinar-to-ductal metaplasia with mPanIN lesions of all histological grades.42 However, targeting oncogenic KRAS to mature ductal cells using the cytokeratin19 promoter failed to induce mPanINs or infiltrating adenocarcinomas.52
Until genetically engineered mouse model studies implicated acinar-to-ductal metaplasias as a possible precursor lesion to mPanINs, human acinar-to-ductal metaplasias were not a major focus of surgical pathologists or cancer researchers. Now it is recognized that acinar-to-ductal metaplasias can occur in close association with PanIN lesions. In our recent report of KRAS mutations in acinar-to-ductal metaplasia lesions KRAS mutations were identified in 63% (12 of 19) of PanIN-associated acinar-to-ductal metaplasias and in 74% (14 of 19) of PanINs. All of the PanIN-associated acinar-to-ductal metaplasias harbored the identical mutation to their corresponding PanIN lesion. No KRAS gene mutations were identified in isolated acinar-to-ductal metaplasia lesions or in normal acinar cells.44 Because KRAS gene mutations were not detected in isolated acinar-to-ductal metaplasias, it was concluded that acinar-to-ductal metaplasias associated with PanINs are likely to represent retrograde extension of PanIN lesions, although the possibility that acinar-to-ductal metaplasias could be precursor lesions to PanINs could not be ruled out.
In the present study, we find evidence that PanIN-associated acinar-to-ductal metaplasias have short telomeres typical of PanINs and other neoplastic lesions, although the mean telomere length of PanIN-associated acinar-to-ductal metaplasias was intermediate between normal length and the length in PanINs. We also provide further evidence that isolated acinar-to-ductal metaplasias do not have short telomeres supporting previous evidence that these lesions do not genetic alterations to indicate that they are neoplastic clones. The simplest explanation for our results is that isolated acinar-to-ductal metaplasias are not a precursor to PanIN lesions. However, we considered an alternative possibility that while many acinar-to-ductal metaplasias are not genetically unstable, a subset of acinar-to-ductal metaplasias could become genetically unstable and rapidly acquire shortened telomeres and then rapidly evolve into PanIN. If this were so it would be hard to identify an isolated acinar-to-ductal metaplastic lesion that was genetically unstable. This explanation is hard to reconcile the frequent coexistence of mutant KRAS and telomere shortening in PanIN-associated acinar-to-ductal metaplasias. Since telomere shortening is more prevalent in early PanINs than KRAS mutations, telomere shortening is thought to evolve prior to KRAS mutations, so if the PanIN-associated acinar-to-ductal metaplasias were a precursor to their adjacent PanINs one would expect that these lesions would harbor much fewer KRAS mutations than their adjacent PanIN-1 lesions, but our previous report found these PanIN-associated acinar-to-ductal metaplasias had a similar prevalence of KRAS gene mutations as their adjacent PanINs. Thus, the lack of identification of isolated acinar-to-ductal metaplasias with short telomeres and the similar prevalence of mutant KRAS in PanIN-associated acinar-to-ductal metaplasias and their adjacent PanIN-1 lesions supports the hypothesis that the genetic alterations found in PanIN-associated acinar-to-ductal metaplasias represent retrograde extension of the nearby PanINs into the acinar-to-ductal metaplasia area.
Further studies using acinar and ductal markers applied to these lesions may help to differentiate between the possibilities of retrograde extension of PanIN into acinar cells versus acinar-to-ductal metaplasias as true precursors to PanINs.
In summary, PanIN-associated acinar-to-ductal metaplasias have short telomeres, but isolated acinar-to-ductal metaplasia lesions do not. These results support the hypothesis that isolated acinar-to-ductal metaplasias are not a precursor to PanIN.
Acknowledgments
This work was supported by NIH grants (P50-CA62924, R01-CA120432, RO1-CA97075, and), and the Michael Rolfe Foundation.
Footnotes
This work was presented in part at the 99th annual meeting of the United States & Canadian Academy of Pathology, Washington, D. C. in March 2010.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Canto MI, Goggins M, Hruban RH, Petersen GM, Giardiello FM, Yeo C, et al. Screening for early pancreatic neoplasia in high-risk individuals: a prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:766–81. doi: 10.1016/j.cgh.2006.02.005. quiz 665. [DOI] [PubMed] [Google Scholar]
- 3.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–86. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, et al. An illustrated consensus on the classification of PanINand intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–87. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–3. [PubMed] [Google Scholar]
- 6.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]
- 7.Luttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Kloppel G, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001;158:1677–83. doi: 10.1016/S0002-9440(10)64123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–71. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541–7. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hruban RH, Maitra A, Goggins M. Update on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol. 2008;1:306–16. [PMC free article] [PubMed] [Google Scholar]
- 11.Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Yeo CJ, et al. Preproenkephalin and p16 gene CpG island hypermethylation in PanIN(PanIN) and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160:1573–81. doi: 10.1016/S0002-9440(10)61104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato N, Fukushima N, Hruban RH, Goggins M. CpG island methylation profile of PanIN. Mod Pathol. 2008;21:238–44. doi: 10.1038/modpathol.3800991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansen M, Fukushima N, Rosty C, Walter K, Altink R, van Heek T, Hruban R, Offerhaus JG, Goggins M. Aberrant Methylation of the 5’ CpG Island of TSLC1 is Common in Pancreatic Ductal Adenocarcinoma and is First Manifest in High-Grade PanINs. Cancer Biology and Therapy. 2002;1:293–6. doi: 10.4161/cbt.84. [DOI] [PubMed] [Google Scholar]
- 14.Hong SM, Kelly D, Griffith M, Omura N, Li A, Li CP, et al. Multiple genes are hypermethylated in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2008;21:1499–507. doi: 10.1038/modpathol.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abu-Alfa A, Ivanovich P, Mujais SK. Uremic exocrine pancreopathy. Nephron. 1988;48:94–100. doi: 10.1159/000184885. [DOI] [PubMed] [Google Scholar]
- 16.Hruban RH, Pitman MS, Klimstra DS. Tumors of the Pancreas, AFIP Atlas of Tumor Pathology Fourth Series Fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007. [Google Scholar]
- 17.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–73. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brune K, Abe T, Canto M, O’Malley L, Klein AP, Maitra A, et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am J Surg Pathol. 2006;30:1067–76. [PMC free article] [PubMed] [Google Scholar]
- 19.Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med. 2009;133:365–74. doi: 10.5858/133.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harley CB, Villeponteau B. Telomeres and telomerase in aging and cancer. Curr Opin Genet Dev. 1995;5:249–55. doi: 10.1016/0959-437x(95)80016-6. [DOI] [PubMed] [Google Scholar]
- 21.Blackburn EH. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005;579:859–62. doi: 10.1016/j.febslet.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 22.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–5. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 23.Gisselsson D, Jonson T, Petersen A, Strombeck B, Dal Cin P, Hoglund M, et al. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci U S A. 2001;98:12683–8. doi: 10.1073/pnas.211357798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hackett JA, Greider CW. Balancing instability: dual roles for telomerase and telomere dysfunction in tumorigenesis. Oncogene. 2002;21:619–26. doi: 10.1038/sj.onc.1205061. [DOI] [PubMed] [Google Scholar]
- 25.Klapper W, Parwaresch R, Krupp G. Telomere biology in human aging and aging syndromes. Mech Ageing Dev. 2001;122:695–712. doi: 10.1016/s0047-6374(01)00223-8. [DOI] [PubMed] [Google Scholar]
- 26.O’Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell. 2002;2:149–55. doi: 10.1016/s1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]
- 27.Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–9. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 28.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 31:9–18. doi: 10.1093/carcin/bgp268. Epub 2009 Nov 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 30.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 31.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–5. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 32.Griffin CA, Hruban RH, Morsberger LA, Ellingham T, Long PP, Jaffee EM, et al. Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res. 1995;55:2394–9. [PubMed] [Google Scholar]
- 33.Brat DJ, Hahn SA, Griffin CA, Yeo CJ, Kern SE, Hruban RH. The structural basis of molecular genetic deletions. An integration of classical cytogenetic and molecular analyses in pancreatic adenocarcinoma. Am J Pathol. 1997;150:383–91. [PMC free article] [PubMed] [Google Scholar]
- 34.Abe T, Fukushima N, Brune K, Boehm CSN, Matsubayashi H, Canto M, Petersen GM, Hruban RH, Goggins M. Genome wide allelotypes of familial pancreatic adenocarcinomas and familial and sporadic intraductal papillary mucinous neoplasms. Clin Cancer Res. 2007;13:6019–25. doi: 10.1158/1078-0432.CCR-07-0471. [DOI] [PubMed] [Google Scholar]
- 35.Calhoun ES, Hucl T, Gallmeier E, West KM, Arking DE, Maitra A, et al. Identifying Allelic Loss and Homozygous Deletions in Pancreatic Cancer without Matched Normals Using High-Density Single-Nucleotide Polymorphism Arrays. Cancer Res. 2006;66:7920–8. doi: 10.1158/0008-5472.CAN-06-0721. [DOI] [PubMed] [Google Scholar]
- 36.Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, Troup WJ, Romm JM, Doheny K, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004;64:871–5. doi: 10.1158/0008-5472.can-03-2756. [DOI] [PubMed] [Google Scholar]
- 37.Nowak N, Gailea D, Conroya J, McQuaida D, Cowella J, Cartera R, et al. Genome Wide aberrations in pancreatic ductal adenocarcinoma. Cancer Genet Cyto. 2005;161:36–50. doi: 10.1016/j.cancergencyto.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Brat DJ, Hahn SA, Griffin CA, Yeo CJ, Kern SE, Hruban RH. The structural basis of molecular genetic deletions. An integration of classical cytogenetic and molecular analyses in pancreatic adenocarcinoma. Am J Pathol. 1997;150:383–91. [PMC free article] [PubMed] [Google Scholar]
- 39.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 41.Morris JPt, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–20. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, et al. Spontaneous induction of murine PanIN(mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–8. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Shi C, Hong SM, Lim P, Kamiyama H, Khan M, Anders RA, et al. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Mol Cancer Res. 2009;7:230–6. doi: 10.1158/1541-7786.MCR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 46.Vander Griend DJ, Konishi Y, De Marzo AM, Isaacs JT, Meeker AK. Dual-label centromere and telomere FISH identifies human, rat, and mouse cell contribution to Multispecies recombinant urogenital sinus xenografts. Prostate. 2009;69:1557–64. doi: 10.1002/pros.21001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meeker AK, Hicks JL, Platz EA, March GE, Bennett CJ, Delannoy MJ, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002;62:6405–9. [PubMed] [Google Scholar]
- 48.Team RDC. R:A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- 49.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 51.Murtaugh LC, Leach SD. A case of mistaken identity? Nonductal origins of pancreatic “ductal” cancers. Cancer Cell. 2007;11:211–3. doi: 10.1016/j.ccr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 52.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–9. [PubMed] [Google Scholar]