

SUMMARY
Pathogens adapt to the host environment by altering their patterns of gene expression. Microarray-based and genetic techniques used to characterize bacterial gene expression during infection are limited in their ability to comprehensively and simultaneously monitor genome-wide transcription. We used massively parallel cDNA sequencing (RNA-seq) techniques to quantitatively catalog the transcriptome of the cholera pathogen, Vibrio cholerae derived from two animal models of infection. Transcripts elevated in infected rabbits and mice relative to laboratory media derive from the major known V. cholerae virulence factors and also from genes and small RNAs not previously linked to virulence. The RNA-seq data was coupled with metabolite analysis of cecal fluid from infected rabbits to yield insights into the host environment encountered by the pathogen and the mechanisms controlling pathogen gene expression. RNA-seq-based transcriptome analysis of pathogens during infection produces a robust, sensitive, and accessible data set for evaluation of regulatory responses driving pathogenesis.
INTRODUCTION
Cholera is a severe and sometimes lethal diarrheal disease that has afflicted human populations for centuries and remains a significant threat to public health in many parts of the world. In addition to seasonal cholera epidemics on the Indian subcontinent, major cholera epidemics have occurred during the last two decades in several countries in Africa (Wkly Epidemiol Rec. WHO, 2010). The ongoing cholera epidemic in Haiti, which began in October 2010, has signaled the return of cholera to the western hemisphere (Chin et al., 2011).
Cholera is caused by Vibrio cholerae. This curved gram-negative rod has the unusual capacity to survive and multiply (colonize) in the human small intestine, where it produces cholera toxin (CT). This AB5 type toxin causes marked secretion of Cl− and water from intestinal epithelial cells into the bowel lumen and is the direct cause of cholera’s hallmark diarrhea (Sanchez and Holmgren, 2008). While many bacterial factors and processes contribute to V. cholerae’s capacity to colonize the small intestine (Ritchie and Waldor, 2009), a principal and essential factor is the type IV pilus TCP (Taylor et al., 1987). Two transcriptional regulators, ToxR and ToxT, are critical for coordinated expression of the genes encoding the biosynthesis of TCP and CT (ctxAB) (Krukonis and DiRita, 2003). Notably, both CT and TCP are encoded within mobile (or formerly mobile) genetic elements (Waldor and Mekalanos, 1996; Kovach et al., 1996).
Animal models have been valuable for exploring V. cholerae pathogenicity. Ligated rabbit ileal loops were used to demonstrate that cell-free supernatants from V. cholerae cultures contain an enterotoxic activity (now known to be CT) (De, 1959), however, this model circumvents the normal route of infection. Infant mice have been extremely useful for discovering genes that facilitate or are required for V. cholerae intestinal colonization (Ritchie and Waldor, 2009), such as those enabling production of TCP (Taylor et al., 1987; Herrington et al., 1988). However, one drawback of infant mice is that they do not develop overt diarrhea. In contrast, orogastric infection of cimetidene-treated infant rabbits with V. cholerae routinely leads to CT- and TCP-dependent cholera-like illness (Ritchie et al., 2010).
A central aim of studies of microbial pathogenesis is to understand how the host environment alters the global pattern of pathogen gene expression (Hsiao and Zhu, 2009). Both genetic and microarray-based high throughput approaches have been used to identify V. cholerae genes induced during infection (Merrell et al., 2002; Xu et al., 2003; Bina et al., 2003; Larocque et al., 2005). Genetic screens, which to date have relied on recombinase-based in vivo expression technology (RIVET), have been limited by bottle-necks in the host and a requirement that in vivo induced genes be transcriptionally silent in vitro, which hampered assessment of whether the TCP biosynthesis genes are induced in vivo (Lombardo et al., 2007). RIVET-based screens also do not allow for detection of genes that are transcriptionally silenced during infection. Fluorescent reporter based screens are useful for monitoring repression of gene expression in vivo but have been less useful in identifying in vivo induced genes (Hsiao et al., 2009). In contrast, microarray-based studies can detect both increases and decreases in gene expression during infection; however, microarrays usually do not contain complete representations of the genome (Merrell et al., 2002; Xu et al., 2003; Bina et al., 2003; Larocque et al., 2005). For example, all of the microarrays that have been used to analyze the V. cholerae transcriptome did not enable detection of non-coding RNAs. Furthermore, it is often difficult to compare microarray results that come from different laboratories as different approaches have been used to analyze data.
The development of massively parallel cDNA sequencing (RNA-seq) techniques is enabling deeper and more accurate assessment of transcriptomes from eukaryotes (Ozsolak and Milos, 2011) as well as bacteria (van Vliet, 2010; Sorek and Cossart, 2010). In contrast to hybridization-based methods such as microarrays, RNA-seq allows for unbiased annotation-independent detection of transcripts, increased sensitivity, and higher resolution (Croucher and Thompson, 2010). In bacterial pathogens, RNA-seq studies have been used to comprehensively map transcription start sites and operon structures (Cho et al., 2009; Sharma et al, 2010) and discover sRNAs (Sittka et al, 2008; Liu et al, 2009; Cho et al., 2009; Sharma et al, 2010; Irnov et al, 2010; Weissenmayer et al, 2011). However, to date this technology has not been applied to investigation of the global patterns of pathogen gene expression during infection of a mammalian host.
Here we used RNA-seq to generate comprehensive transcriptome profiles of V. cholerae during growth in the intestines of infant rabbits and infant mice as well as in laboratory cultures. Genes induced in vivo in both model hosts included all the known V. cholerae virulence factors including genes for CT and TCP biosynthesis as well as many genes encoding proteins and small RNAs not previously linked to infection. Contribution of the in vivo-induced genes identified in this study to intestinal colonization was also assessed. Furthermore, comparative analyses of metabolites present in culture supernatants and in cecal fluid from infected rabbits were used to infer explanations for some of the observed patterns of V. cholerae gene expression. Collectively, our findings indicate that RNA-seq is a powerful tool that can enable monitoring of pathogen gene expression during infection.
RESULTS AND DISCUSSION
V. cholerae, like other faculative pathogens, modulates its gene expression upon infection of its mammalian host. To gain understanding of V. cholerae’s adaptation to the host environment, we systematically cataloged the transcriptomes of bacteria grown in vivo and in vitro, using high throughput cDNA sequencing techniques. RNA was derived from organisms either grown in laboratory medium, isolated directly from the fluid that accumulates in the ceca of orally infected infant rabbits, or contained within small intestinal homogenates of orally infected infant mice. Strand-specific Illumina-based RNA-seq (Levin et al., 2010) was used for characterization of bacteria within cecal fluid, which reach densities of ~ 5 × 108 V. cholerae cfu/ml. Helicos-based sequencing, which required less manipulation of the relatively small amount of bacterial RNA obtained from the infant mouse intestinal homogenates, was used to assess V. cholerae transcript abundance in infected mice.
Between 8 and 12 million reads for each rabbit cecal sample (n=2) aligned to non-rRNA regions of the V. cholerae genome, and a similar number of reads were obtained for the in vitro grown control samples (Table 1). Fewer of the reads derived from murine samples (n=2, 284 and 61 thousand reads per intestinal sample) aligned to non-rRNA sequences in the V. cholerae genome, both because of the abundance of host and normal flora-derived transcripts and because rRNAs were not depleted prior to sequencing. Nonetheless, the reproducibility (R value) of the transcriptome data derived from the murine samples was equal to that obtained from the rabbit cecal fluid isolates (Table 1). Importantly, the correlation of technical replicates was very high (Table 1), suggesting that variations introduced during library construction and sequencing does not significantly contribute to differences in gene expression between samples. The correlation between replicate cultures grown in LB and sequenced by Helicos and Illumina, respectively, was somewhat lower (R=0.73), suggesting some platform-specific biases. The range of reads per ORF in the Illumina and Helicos datasets varied between 0 and greater than 100,000 and 8,000, respectively, reflecting a far more robust and sensitive dynamic measurement of expression than previously obtained in microarray-based approaches (Merrell et al., 2002; Xu et al., 2003; Bina et al., 2003; Larocque et al., 2005).
Table 1.
Summary of Illumina (top) and Helicos (bottom) RNAseq data.
| Growth condition | Sample | # aligned reads (millions) | # non-rRNA reads (millions) | R | |
|---|---|---|---|---|---|
| (tech reps) | (biol reps) | ||||
| LB | 1a* | 38.8 | 16.8 | > 0.99 | 0.93 |
| 1b* | 53.5 | 22.8 | |||
| 2 | 44.7 | 16.4 | |||
| M9 | 1a# | 54.8 | 18.5 | > 0.99 | 0.99 |
| 1b# | 54.3 | 19.0 | |||
| 2 | 47.9 | 20.0 | |||
| Rabbit | 1 | 39.6 | 12.0 | N/A | 0.84 |
| 2 | 27.8 | 8.5 | |||
| Growth condition | Sample | # aligned reads (millions) | # non-rRNA reads (thousands) | R (biol replicates) |
|---|---|---|---|---|
| LB | 1 | 5.2 | 449.1 | 0.99 |
| 2 | 4.0 | 362.8 | ||
| Mouse* | 1 | 4.5 | 283.7 | 0.83 |
| 2 | 1.0 | 61.2 |
Technical replicates
Each mouse sample had more than 100 million reads.
Regardless of sequencing depth and technology, comparative analyses revealed largely consistent global profiles for RNAs isolated from all test conditions (Fig. 1A–B and S1, Table S1). Heat maps of ranked coverage revealed numerous regions with transcript abundance that was uniformly high or uniformly low in vivo and in vitro. (Fig. 1A,B; inner 2 circles). Similarly, for both sequencing technologies, plots of RPKMO (reads per kilobasepair of gene per million reads aligning to annotated ORFs) were grossly similar for in vitro and in vivo-derived RNAs (Fig. 1A,B, 3rd and 4th circles). For example, chromosome I ori region transcripts were markedly more abundant than those from chromosome termini (Figs. 1A–B and S1), consistent with expected differences in copy number due to ongoing chromosome replication. Such differential expression was absent for chromosome II, which as previously observed yielded fewer transcripts overall than did chromosome I (Xu et al. 2003). Additionally, for all samples, very few transcripts corresponded to the 120 kb chrII superintegron (Heidelberg et al., 2000), suggesting this gene capture system is not routinely expressed in vitro or in vivo (Fig. 1A–B and S1).
Figure 1.

A–B) Profiles of V. cholerae gene expression in culture and during infection (A: rabbit (Illumina); B: mouse (Helicos)). Plots for chromosome I are on the left and for chromosome II are on the right and are based on data from two biological replicates for each condition. From inside to outside, the 6 circles in each plot correspond to the following: 1–2) heatmap of ranked coverage in 5 kb windows in vitro and in vivo, respectively 3–4) log2 of RPKMO (reads per kilobasepair of gene per million reads aligning to annotated ORFs) for each gene in vitro and in vivo, respectively. In circles 1–4 red, yellow, and blue correspond to windows/genes with high, middle, and low expression, respectively. 5) Regions encoding ribosomal proteins (black) or corresponding to indicated genomic islands. 6) Log2 of fold abundance in vivo vs. in vitro. Genes whose fold expression is statistically significant and > 4 fold higher or lower in vivo are highlighted in red and blue, respectively; the height of the bars corresponds to log2 of the differential abundance in vivo vs LB. Plots were created using Circos (Krzywinski et al., 2009). C) Strand-specific coverage per nucleotide across the genes within the TCP island. Blue and black lines represent read coverage sequenced and mapped from a representative rabbit transcript library and red and green lines represent those from a representative Illumina LB library. Read depth is plotted on a log2 scale to provide better definition of genomic regions with very high and low coverage. TCP genes are labeled and separated according to strand orientation. (see also Figure S1).
The variance analysis package DEseq (Anders and Huber, 2010) was used to systematically search the transcriptome data for the subset of genes with statistically significant (P < 1×10−5) and > 4-fold differential expression in vivo compared to in vitro (Fig. 2A–C and Table S2). Although expression of most of V. cholerae’s genes did not markedly differ when cells were obtained from an animal host rather than from in vitro cultures (Fig. 2A–C), 478 were found to be induced in vivo in at least one animal (Table S3), and 39 were induced in both rabbits and mice compared to M9 and/or LB media (Fig. 2E and Table 2). The lack of complete overlap between the sets of genes induced in the different animals likely reflects the different sites from which bacteria were isolated (small intestine vs cecal fluid) as well as host-specific differences.
Figure 2.

A–D) MA plots of V. cholerae RNA-seq data. In these plots, each point represents an annotated ORF. The log2 of the ratio of abundances of each ORF between the indicated conditions (M) is plotted against the average log2 of abundance of that ORF in all conditions (A). For each plot, M and A values were based on data from two biological replicates from each growth condition or animal model. Genes that are significantly differentially expressed (based on DESeq analyses) as well as several groups of genes that are mentioned in the text are highlighted with different symbols (see legend below plots). E) Venn diagram of genes over-expressed in mice and rabbits. Genes were considered over-expressed if their differential abundance between in vivo and in vitro samples was > 4-fold and had a P value < 1×10−5. The 39 genes in the overlap between rabbits and mice include 10 of the 13 genes in the ToxT regulon (Bina et al., 2003). (see also Figure S2 and Tables S2, S3, S4 and S5).
Table 2.
Relative in vivo fitness of V. cholerae mutants lacking genes induced in both rabbit and mouse *
| Locus No. # | Gene Name | Product | Fold Induction$ | CI^ | Reference | |
|---|---|---|---|---|---|---|
| Rabbit vs LB | Mouse vs LB | |||||
| vc0819 | aldA | Aldehyde dehydrogenase | 78 | 2784 | No defect | (Parsot and Mekalanos, 1991) |
| vc0820 | tagA | ToxR-activated gene A | 42 | 216 | No defect | (Parsot and Mekalanos, 1991) |
| vc0825 | tcpI | Toxin co-regulated pilus biosynthesis protein I | 63 | 302 | No defect | (Parsot and Mekalanos, 1991) |
| vc0828-0839 | tcpA,B, Q,C,R,D, S,T,E,F, toxT, tcpJ | Toxin co-regulated pilus biosynthesis proteins | 55–7200 | 42–1000 | <0.01 | (Peterson & Mekalanos, 1988; Kirn et al., 2003; Chiang and Mekalanos, 1998) |
| vc0840-0845 | acfB,C, orf4, tagE, acfAD | Accessory colonization factors, hypothetical protein and ToxR activated gene E | 11–109 | 7–58 | <0.08 except Vc0842-0843 not known | (Peterson & Mekalanos, 1988) |
| vc1456-1457 | ctxBA | Cholera enterotoxin subunits B and A | 340–400 | 50–380 | 0.4 (ctxA) | (Peterson & Mekalanos, 1988) |
| vc1773 | Hypothetical protein | 17 | 12 | 0.3 | ||
| vc1774 | Hypothetical protein | 11 | 14 | 1.5 | ||
| vc1776 | nanA | N-acetylneuraminate lyase | 21 | 14 | 0.06 | (Almagro-Moreno and Boyd, 2009) |
| vc2637 | Peroxiredoxin family protein | 12 | 9 | 1.2 | ||
| vca0183 | Nitric oxide dioxygenase | 10 | 8 | 1.4 | ||
| vca0241-0248 | Ascorbate specific PTS proteins | 9–150 | 7–26 | ~1.0 except Vca0242 – 0.3 | ||
| vca0556 | Hypothetical protein | 13 | 25 | 1.2 | ||
| vca0749 | glpC | Sn-glycerol-3-phosphate dehydrogenase subunit C | 11 | 5 | 0.7 | |
| vca1063 | speF | Ornithine decarboxylase | 8 | 8 | 1.6 | |
- This list corresponds to the 39 genes in the overlap region shown in Fig. 2b.
This represents the mean values derived from 2 samples from either animal model or from LB. The ranges correspond to the values for the genes listed.
CI, competitive indices were calculated as the output ratio of mutant vs WT cells divided by the input ratio of mutant vs WT cells in an infant mouse colonization assay. The values represent the means for at least 5 infant mice/group.
Notably, the set of genes most highly induced in both animals included all of the key known V. cholerae virulence factors and most of the genes controlled by the virulence linked transcriptional regulator ToxT (Krukonis and DiRita, 2003) (Fig. 2A–C). Expression of ctxAB and genes enabling production of TCP was induced 50- to more than 500-fold in rabbits and in mice (Fig. 1C, 2A–C and Table 2), and most of the remaining genes in the TCP pathogenicity island were overexpressed at least 20-fold in vivo (Fig. 1C, Table 2). Such induction has long been presumed to occur by cholera researchers, and was confirmed for a few genes using low-throughput approaches (Lee et al., 1999; Quinones et al., 2006). Quantitative RT-PCR based analyses of ctxA and tcpA expression in rabbit ligated loops yielded very similar magnitude of induction of these key virulence genes (vs LB) as we found (Nielsen et al., 2010). However, several earlier microarray-based studies failed to obtain evidence supporting a dramatic induction of ToxT-regulated genes (Merrell et al. 2002; Xu et al., 2003; Bina et al., 2003) although one study noted such induction in upper intestinal derived samples compared to stool samples (Larocque et al., 2005) and another study detected induction in non-luminal V. cholerae associated with the rabbit epithelium (Nielsen et al., 2010). Significantly, virulence gene induction was detectable in our RNA-seq studies even when infecting cells comprised only a small proportion of the isolated tissue, a condition likely to confound microarray-based analyses. Collectively, these data indicate that RNA-seq enables sensitive, comprehensive and quantitative characterization of bacterial gene expression during infection.
Given that many of the V. cholerae genes we observed to be induced in both animal models (e.g., the TCP island genes) are absolutely essential for colonization, we assessed whether other similarly induced genes are likewise required. The colonization capacities of 15 mutant strains, each lacking a single gene whose expression was induced in vivo in both models, were tested with in vivo competition assays in suckling mice. These assays revealed that vc1773, which encodes a hypothetical protein, promotes intestinal colonization (Table 2); interestingly, it is encoded within the Vibrio pathogenicity island 2 (VPI-2) along with nanA (vc1776), which was previously shown to have a role in vivo (Almagro-Morena and Boyd., 2009). Mutants lacking other genes that showed induction in vivo in both models were not markedly attenuated in their ability to colonize the infant mouse (Table 2).
Competition assays were also performed with mutants lacking a gene induced in just one of the two animal models, although only a subset of these genes were tested. Of 26 strains with insertion mutations that were tested, 8 displayed colonization defects, which ranged from 3 to 10 fold (Table S5); five of these were originally observed to be induced in rabbits and 3 in mice. Thus, similar to the genes that were induced in both animal models, only a subset of the genes induced in a single model promotes growth in the mouse intestine. Collectively, these observations indicate that other than the ToxT regulon, most genes with elevated transcript abundance in either or both rabbits and mice are apparently not critical for intestinal colonization. It is possible that expression of such genes facilitates V. cholerae’s survival upon shedding or its transmission to a new host (Schild et al., 2007); alternatively, induction may be a reflection of the host environment yet not serve as a critical adaptation to this environment. It should also be noted that many genes previously shown to contribute to V. cholerae colonization of the suckling mouse were not found to be induced in vivo in our analyses (Ritchie and Waldor, 2009). Many of these genes, including those encoding O-antigen biosynthesis, the RNA chaperone Hfq, and TolC, yielded abundant transcripts (coverage ranked in the top 20%) in vitro as well as in vivo (Table S1), suggesting that their functions are not specifically adapted for growth in the host.
To begin to understand the environment within the rabbit from which V. cholerae was isolated, we performed mass spectrometric analyses of cecal fluid from infected rabbits and of V. cholerae culture supernatants. The resulting data provided plausible explanations for the expression of some V. cholerae genes observed to be upregulated in the animal. For example, we detected long chain fatty acids in cecal fluid but not in culture supernatants (Fig. S3). These molecules may account for the elevated cecal expression of fadL (vc1042), a transporter of long chain fatty acids, and of the acyl CoA dehydrogenases vc1740 and fadE (vc2231) in rabbits compared to either LB or M9 (Fig. 2A&C, 3), as fatty acid degradation (Fad) gene expression may be induced to enable transport and metabolism of host derived long chain fatty acids. Similarly, in vivo induction of genes for glycerol transport (vca0137) and metabolism, including glycerol kinase (vca0744) and glycerol 3-phosphate dehydrogenase (vca0747-0749) may facilitate utilization of host-derived lipids.
Figure 3.
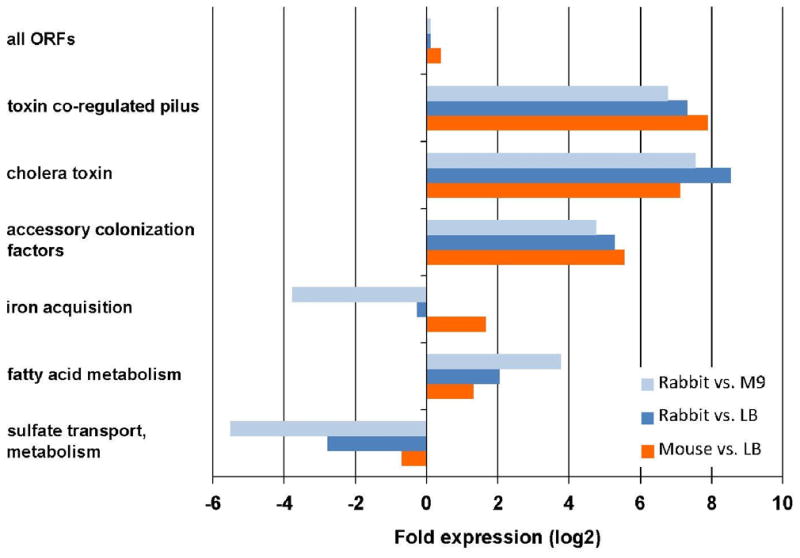
Differential expression of functionally related groups of genes in vivo compared to in culture. Fold expression was calculated based on the average M values calculated by DEseq for each group of genes. The following genes were included in each group: TCP: VC0825-0837; CTX: VC1456-1457; ACF: VC0840-0841, VC0844-0845; iron: VC0200, VCA0227-0230, VCA0911-0915, VC2209-2211, VC0771-VC0777; fatty acid: VC1042, VC1740, VC2231, VCA0137, VCA0744, VCA0747-0749; sulfate: VC2558, VC0538-0541, VC2559-2560, VC0384-0386. (see also Figure S3)
Environmental conditions are also likely to account for the lack of expression of iron uptake genes in bacteria isolated from infected rabbits. The majority of genes for both vibriobactin siderophore biosynthesis and transport (vc0474 (irgB), vc0475 (irgA), vc0771-0780) and iron III uptake (vc0200-0203, vca0229, vca0230) were found to be highly induced in the mouse small intestine relative to LB but were uninduced in the rabbit relative to LB (Figures 2A–B, 3 and Table S4). Transcripts for this set of genes were reduced in the rabbit and in LB media relative to in iron-poor M9 media (Fig. 2C,D). Collectively, these data suggest that iron is scarce in the mouse intestine but readily available within the rabbit cecum. Heme, which is detectable in the cecal fluid, may be a source of iron for the pathogen in the rabbit intestine (Fig S3).
Similarly, sulfate-containing compounds may be more available to bacteria within the rabbit than the mouse model. In the rabbit, there is reduced expression of the majority of genes (vc0538-0541, vc2558-2560, vc0384-0386) that are involved in the acquisition and utilization of sulfate for generation of reduced sulfur metabolites (such as thiols) (Fig. 2A,C; 3 and Table S4), perhaps because there is cysteine in the cecal fluid (Fig S3). In contrast, expression of these genes is equivalent in the mouse intestine to what was detected in vitro (Figures 2B and 3).
Transcription profiles derived from different growth conditions can also provide clues for deciphering transcriptional architecture and mechanisms that control transcription. For example, analysis of transcripts from the TCP island clearly demonstrates that tcpPH are not part of the operon that contains tcpA (Fig 1C). RNA-seq analysis also suggests that a predicted riboswitch upstream of a putative vitamin B12 receptor gene (vc0156) enables downregulation of this gene in vivo. Coverage plots of this region (Fig. 4) are consistent with transcription attenuation downstream of the putative riboswitch in vivo, but not in LB or M9 media, suggesting that B12 (cobalamin) may be available in the cecum (Nahvi et al., 2002). Searches for additional genes with such uneven sequence coverage may facilitate identification and characterization of riboswitches or other regulatory processes.
Figure 4.
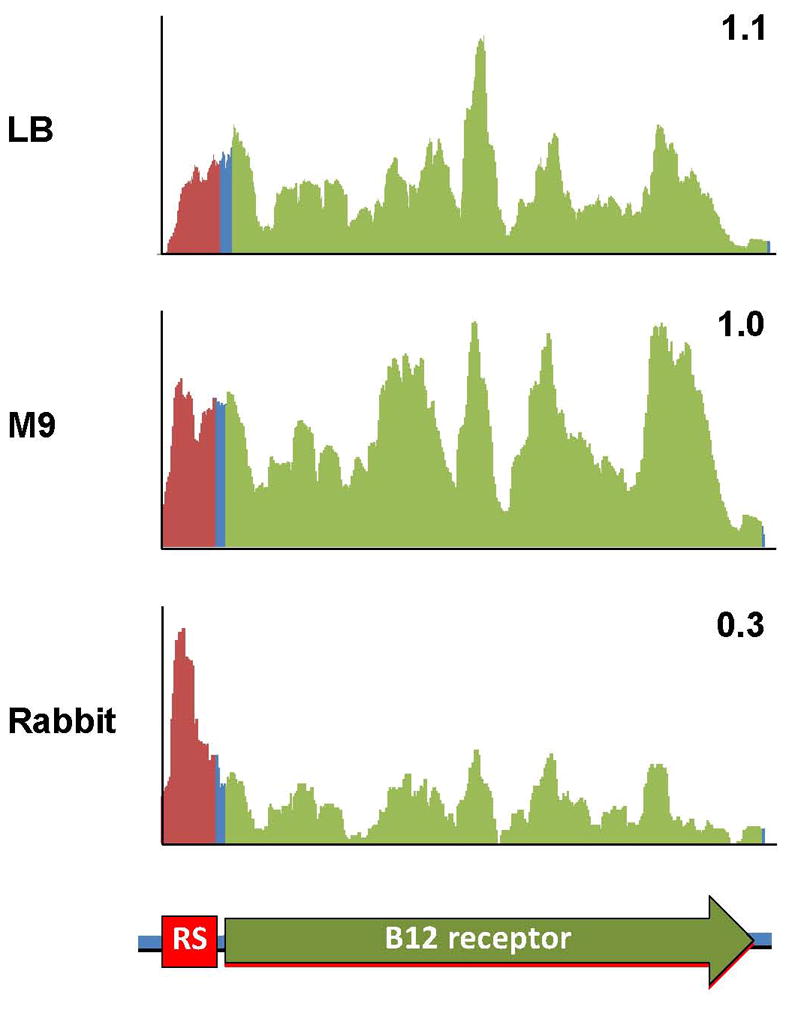
Coverage plots of RNA-seq reads aligning to a putative cobalamin-regulated riboswitch and its downstream gene. Y-axis is linear and values are arbitrary units corresponding to reads/position. The numbers in the corner of each plot correspond to the ratio of the abundance of ORF reads (green) vs. riboswitch region reads (red).
RNA-seq proved highly sensitive for detection of known ncRNAs, for discovery of new putative ncRNAs, and for identification of ncRNAs differentially regulated in vivo (Table S6, S7 and S8). Of the 45 V. cholerae regulatory RNAs previously characterized and/or predicted in the Rfam database, 42 were detected with greater than 50 reads in at least one sample (Table S1). These included the 4 rare Qrr sRNAs that govern V. cholerae quorum sensing (Lenz et al., 2004), which were detected in all samples, consistent with exponential phase growth and with expression of virulence genes in vivo. The iron-repressed sRNA RyhB was less abundant in the rabbit cecum and in LB than in M9 medium and the mouse intestine, providing further evidence that iron is not limiting in the cecum but is in the mouse intestine. We also identified seventy-seven putative intergenic sRNAs that had not been previously annotated (Table S6). Seven candidate or previously described sRNAs were overexpressed in rabbits (abundance increased 4-fold with P < .001) compared to LB and M9 (Table S7 and S8), including TarA, which was independently discovered and shown to promote intestinal colonization (Richard et al., 2010) and CsrC, another sRNA that contributes to quorum sensing (Lenz et al., 2005).
Taken together, our results suggest that growth of V. cholerae in vivo sets into action a complex transcriptional program that includes two main sets of genes. The first set of genes encodes many factors whose roles are central and/or specific to colonization or virulence. Most of these genes are induced in diverse hosts, often in response to the activator ToxT. However, a subset, such as those of the VSP-1 (Dziejman et al., 2002) and type VI secretion (T6S) genomic islands (Pukatzki et al., 2006) are induced in the mouse intestine but not in the rabbit cecum, and the signals that govern their expression remain to be identified. The second set of genes, which have not been linked to virulence, likely encode factors primarily involved in metabolic adaptations to environmental conditions that are host or niche specific. Both metabolomic and detailed transcriptomic analyses should facilitate further identification of the environmental cues that govern expression of these genes.
The detailed characterization of gene expression during infection that can be obtained using RNA-seq should have widespread utility in studies of pathogenesis. The approach allows for simultaneous genome-wide identification of transcription units, including rare transcripts and sRNAs, that are activated in vivo, and also those that are repressed. Furthermore, it can be used to analyze bacteria within infected tissues throughout infection, rather than requiring isolation of bacteria that are largely uncontaminated by host tissues. Consequently, the approach can simultaneously be used to monitor the physiology of a host in response to a pathogen as well as the transcriptome of commensal microbiota that co-exist with the pathogen at the site of infection (Rey et al., 2010). Transcript boundaries can also be mapped with nucleotide resolution, thereby facilitating identification of promoters, operons, and potential sites of transcription attenuation. Finally, the output of RNA-seq analyses can readily be standardized, which should facilitate comparisons among studies and laboratories, and promote a more comprehensive understanding of host-pathogen interactions.
EXPERIMENTAL PROCEDURES
Strains and growth conditions
A streptomycin-resistant derivative of the El Tor O1 V. cholerae clinical isolate C6706 was used for this study. For in vitro RNA preparations, the strain was grown to mid-exponential phase (O.D.600 ~0.4 – 0.6) in either LB or M9 media supplemented with 0.2% glucose and 0.1% casamino acids at 37°C. Where required the media was supplemented with 200μg/ml streptomycin and 40μg/ml of X-Gal.
Strand-specific dUTP library preparation for Illumina sequencing
10 μg of in vivo-derived bacterial RNA was sequentially treated with the MICROBEnrich and MICROBExpress kits (Ambion) to enrich for bacterial RNA and mRNA, respectively. 10μg of in vitro derived RNA was subjected only to the MICROBExpress kit. Strand-specific libraries were prepared using a dUTP second strand marking protocol (Parkhomchuk et al., 2009; Levin et al., 2010) with reagents from Invitrogen unless otherwise stated. Bacterial mRNA was fragmented using a RNA fragmentation kit (Ambion), which yielded fragments in the range of 60–200 nts. First strand cDNA was synthesized from 400ng of precipitated fragmented RNA, using 3μg of random primers, 4μg actinomycin D, and Superscript III. Following extraction and precipitation, second-strand cDNA was synthesized using dUTP (Applied Biosystems) rather than dTTP as described (Levin et al., 2010). Paired-end libraries for Illumina sequencing were prepared from purified cDNA (MinElute PCR purification kit; Qiagen) as recommended by Illumina, except that the size-selected adapter ligated cDNA was pre-incubated with 1μl Uracil-N-glycosylase (Applied Biosystems) at 37°C for 15 mins, followed by 95°C for 5 mins before the final PCR. PCR primers were removed using 1.8 volumes of AMPure XP beads (Beckman Coulter).
Library preparation for Helicos sequencing
cDNA was prepared and modified for Helicos sequencing according to the manufacturer’s protocol. A single stranded cDNA library was prepared using 1μg of purified RNA and 500ng of random hexamers (Invitrogen) and RNA was removed from the reaction using RNAseH and RNAseA. The cDNA library was sheared at 4°C using a Misonix 4000 with the amplitude set at 60% for 20 minutes (20 second on/off pulses), yielding primarily cDNA shorter than 200 nts according to Bioanalyzer (Agilent Technologies). Sheared cDNA was treated with terminal transferase (NEB) and dATP to generate the 100–200nt 3′ poly-A tail that is necessary for Helicos sequencing.
RNA-seq data analysis
Reads were aligned to chromosomes I and II of V. cholerae N16961 (RefSeq accession numbers NC_002505 and NC_002506) using MAQ 0.7.1-9 (Li et al., 2008) for Illumina reads and CLC_BIO (V4.5) for Helicos reads. The subsequent bioinformatics analysis included the following stages: 1) The number of reads aligning to each genomic position on each strand was calculated. 2) Each genomic position was annotated based on its location within the sense or antisense strand of ORFs, rRNA, tRNA, or regRNA or in an intergenic region. Positions where the antisense strand of one gene overlapped the sense strand of another were annotated as sense. Annotations of protein-encoding genes were based on RefSeq NC_002505.gff and NC_002506.gff; those of non-coding RNAs were derived from Rfam (v9.1) (Gardner et al., 2009) or from published data. 3) The total number of reads aligning to each category (e.g., ORFs, antisense to ORFs) and to each annotated gene was calculated. 4) Total reads/gene were normalized using RPKM ((reads/kb of gene)/(million reads aligning to genome)) or a variant we call RPKMO ((reads/kb of gene)/(million reads aligning to annotated ORFs)), enabling us to account not only for differences in the total number of reads obtained in each sample, but also for the often significant differences between samples in the proportions of reads corresponding to rRNAs (Table S1). 5) Putative transcription units (PTUs) were identified as stretches of 50–450 consecutive positions with read coverage > 20 that did not overlap annotated genes. Overlapping PTUs from biological replicates were resolved to give the final list of unique PTUs.
Identification of differentially expressed genes
Differentially expressed genes were identified using DEseq, a variance analysis package that was developed to infer statistically significant differences in gene expression data from high-throughput sequencing (Anders and Huber, 2010). Two biological replicates were included for each growth condition or animal model, and comparisons were conducted separately for Illumina and Helicos datasets. For each sample, the number of reads per gene was normalized by DEseq based on the total number of aligned reads for that sample. For calculation of A values, all genes with less than one read were assigned a value equal to ½ the normalized value for the gene with the lowest abundance in that sample and the mean of the log2 of normalized read/gene in each set of replicates was calculated. M, the log2 of the ratio of A between each condition, was then calculated based on these values.
Supplementary Material
HIGHLIGHTS.
Transcriptomes of V. cholerae from two in vivo infection models cataloged using RNA-seq
Transcripts elevated in vivo derive from the known major V. cholerae virulence factors
Elevated transcripts include genes and small RNAs not previously linked to virulence
RNA-seq coupled with host metabolite analysis explains pathogen gene expression patterns
Acknowledgments
We thank Brigid Davis for many helpful comments on the manuscript. We acknowledge Joshua Levin, Georgia Giannoukos, Brian Haas, Sara Chauvin, Sheila Fisher, and other members of the Broad Institute’s Genomic Sequencing Center for Infectious Diseases (GSCID) for their advice on library preparation and downstream sequence analysis. This work was funded by grants from NIAID (AI-018045, to JJM; R37 AI-42347, to MKW; AI-076608 to JL) and HHMI (to MKW). The Illumina sequencing has been funded in whole or in part with Federal funds from the NIAID, NIH, and Department of Health and Human Services, under Contract No. HHSN2722009000018C.
Footnotes
Additional methods are presented as supplemental information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almagro-Moreno S, Boyd EF. Sialic acid catabolism confers a competitive advantage to pathogenic Vibrio cholerae in the mouse intestine. Infect Immun. 2009;77:3807–3816. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc Natl Acad Sci U S A. 2003;100:2801–2806. doi: 10.1073/pnas.2628026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SL, Mekalanos JJ. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol. 1998;27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- Chin CS, Sorenson J, Harris JB, Robins WP, Charles RC, Jean-Charles RR, Bullard J, Webster DR, Kasarskis A, Peluso P, et al. The origin of the Haitian cholera outbreak strain. N Engl J Med. 364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BO. The transcription unit architecture of the Escherichia coli genome. Nat Biotechnol. 2009;27:1043–1049. doi: 10.1038/nbt.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholera vaccines: WHO position paper. Wkly Epidemiol Rec. 85:117–128. [PubMed] [Google Scholar]
- Croucher NJ, Thomson NR. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol. 2010;13:619–624. doi: 10.1016/j.mib.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De SN. Enterotoxicity of bacteria-free culture-filtrate of Vibrio cholerae. Nature. 1959;183:1533–1534. doi: 10.1038/1831533a0. [DOI] [PubMed] [Google Scholar]
- Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci U S A. 2002;99:1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–83. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao A, Zhu J. Genetic tools to study gene expression during bacterial pathogen infection. Adv Appl Microbiol. 2009;67:297–314. doi: 10.1016/S0065-2164(08)01009-5. [DOI] [PubMed] [Google Scholar]
- Irnov I, Sharma CM, Vogel J, Winkler WC. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010;38:6637–6651. doi: 10.1093/nar/gkq454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn TJ, Bose N, Taylor RK. Secretion of a soluble colonization factor by the TCP type 4 pilus biogenesis pathway in Vibrio cholerae. Mol Microbiol. 2003;49:81–92. doi: 10.1046/j.1365-2958.2003.03546.x. [DOI] [PubMed] [Google Scholar]
- Kovach ME, Shaffer MD, Peterson KM. A putative integrase gene defines the distal end of a large cluster of ToxR-regulated colonization genes in Vibrio cholerae. Microbiology. 1996;142(Pt 8):2165–2174. doi: 10.1099/13500872-142-8-2165. [DOI] [PubMed] [Google Scholar]
- Krukonis ES, DiRita VJ. From motility to virulence: Sensing and responding to environmental signals in Vibrio cholerae. Curr Opin Microbiol. 2003;6:186–190. doi: 10.1016/s1369-5274(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque RC, Harris JB, Dziejman M, Li X, Khan AI, Faruque AS, Faruque SM, Nair GB, Ryan ET, Qadri F, et al. Transcriptional profiling of Vibrio cholerae recovered directly from patient specimens during early and late stages of human infection. Infect Immun. 2005;73:4488–4493. doi: 10.1128/IAI.73.8.4488-4493.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–634. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol. 2005;58:1186–1202. doi: 10.1111/j.1365-2958.2005.04902.x. [DOI] [PubMed] [Google Scholar]
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JM, Livny J, Lawrence MS, Kimball MD, Waldor MK, Camilli A. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 2009;37:e46. doi: 10.1093/nar/gkp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MJ, Michalski J, Martinez-Wilson H, Morin C, Hilton T, Osorio CG, Nataro JP, Tacket CO, Camilli A, Kaper JB. An in vivo expression technology screen for Vibrio cholerae genes expressed in human volunteers. Proc Natl Acad Sci U S A. 2007;104:18229–18234. doi: 10.1073/pnas.0705636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. Host-induced epidemic spread of the cholera bacterium. Nature. 2002;417:642–645. doi: 10.1038/nature00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9:1043. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog. 2010:6. doi: 10.1371/journal.ppat.1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsot C, Mekalanos JJ. Expression of the Vibrio cholerae gene encoding aldehyde dehydrogenase is under control of ToxR, the cholera toxin transcriptional activator. J Bacteriol. 1991;173:2842–2851. doi: 10.1128/jb.173.9.2842-2851.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KM, Mekalanos JJ. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect Immun. 1988;56:2822–2829. doi: 10.1128/iai.56.11.2822-2829.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones M, Davis BM, Waldor MK. Activation of the Vibrio cholerae SOS response is not required for intestinal cholera toxin production or colonization. Infect Immun. 2006;74:927–930. doi: 10.1128/IAI.74.2.927-930.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, Gordon JI. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. 2010;285:22082–90. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AL, Withey JH, Beyhan S, Yildiz F, DiRita VJ. The Vibrio cholerae virulence regulatory cascade controls glucose uptake through activation of TarA, a small regulatory RNA. Mol Microbiol. 2010;78:1171–1181. doi: 10.1111/j.1365-2958.2010.07397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JM, Waldor MK. Vibrio cholerae interactions with the gastrointestinal tract: lessons from animal studies. Curr Top Microbiol Immunol. 2009;337:37–59. doi: 10.1007/978-3-642-01846-6_2. [DOI] [PubMed] [Google Scholar]
- Ritchie JM, Rui H, Bronson RT, Waldor MK. Back to the future: studying cholera pathogenesis using infant rabbits. MBio. 2010:1. doi: 10.1128/mBio.00047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci. 2008;65:1347–1360. doi: 10.1007/s00018-008-7496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe. 2007;2:264–277. doi: 10.1016/j.chom.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet. 11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet AH. Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol Lett. 2010;302:1–7. doi: 10.1111/j.1574-6968.2009.01767.x. [DOI] [PubMed] [Google Scholar]
- Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Weissenmayer BA, Prendergast JG, Lohan AJ, Loftus BJ. Sequencing illustrates the transcriptional response of Legionella pneumophila during infection and identifies seventy novel small non-coding RNAs. PLoS One. 2011;6:e17570. doi: 10.1371/journal.pone.0017570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Dziejman M, Mekalanos JJ. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc Natl Acad Sci U S A. 2003;100:1286–1291. doi: 10.1073/pnas.0337479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.