

Abstract
ActRIB is a type I transmembrane serine/threonine kinase receptor that has been shown to form heteromeric complexes with the type II activin receptors to mediate activin signal. To investigate the function of ActRIB in mammalian development, we generated ActRIB-deficient ES cell lines and mice by gene targeting. Analysis of the ActRIB−/− embryos showed that the epiblast and the extraembryonic ectoderm were disorganized, resulting in disruption and developmental arrest of the egg cylinder before gastrulation. To assess the function of ActRIB in mesoderm formation and gastrulation, chimera analysis was conducted. We found that ActRIB−/− ES cells injected into wild-type blastocysts were able to contribute to the mesoderm in chimeric embryos, suggesting that ActRIB is not required for mesoderm formation. Primitive streak formation, however, was impaired in chimeras when ActRIB−/− cells contributed highly to the epiblast. Further, chimeras generated by injection of wild-type ES cells into ActRIB−/− blastocysts formed relatively normal extraembryonic tissues, but the embryo proper developed poorly probably resulting from severe gastrulation defect. These results provide genetic evidence that ActRIB functions in both epiblast and extraembryonic cells to mediate signals that are required for egg cylinder organization and gastrulation.
Keywords: Activin, serine/threonine kinase receptor, gene targeting, mesoderm induction, gastrulation
The early postimplantation development of the mouse embryo, which involves proliferation and differentiation of the three earliest cell types, including the primitive ectoderm, the primitive endoderm, and the trophectoderm, results in the formation of a cup-shaped embryo known as the egg cylinder. The bilaminar egg cylinder consists of the outer visceral endoderm layer derived from the primitive endoderm and the inner ectoderm layer with a morphological boundary between the distal embryonic ectoderm and the proximal extraembryonic ectoderm (Hogan et al. 1994). Cell signaling and cell–cell interactions between these primitive germ layers are likely to play important roles in mesoderm induction and axis formation during gastrulation.
Gastrulation of the mouse embryo begins with the formation of the primitive streak in the proximal posterior regions of the epiblast (embryonic ectoderm), which extends subsequently toward the distal tip of the egg cylinder. Mesoderm forms by ingression of the epiblast cells through the primitive streak. The anterior aspect of the primitive streak has been shown to be functionally equivalent to the dorsal blastopore lip of the Xenopus embryo, where the Spemann organizer is located (Beddington 1994; Tam et al. 1997). Formation and specification of mesodermal cells during gastrulation results in the establishment of the anteroposterior and dorsal-ventral body patterns. Whereas cell fate and gene expression before and during mouse gastrulation has been studied extensively, the signaling mechanisms that control mesoderm induction, primitive streak formation, and migration and specification of mesodermal cells remain largely unknown.
Studies of mesoderm induction in Xenopus laevis by use of the animal cap assay have identified several members of the transforming growth factor-β (TGFβ) family, including activins, Vg1, BMPs, and nodal-related proteins (Xnr) as potent mesoderm-inducing factors (Asashima et al. 1990; Smith et al. 1990; Thomsen et al. 1990; van den Eijnden-Van Raaij et al. 1990; Dale et al. 1992; Jones et al. 1992, 1995; Thomsen and Melton 1993). Although BMP4 has been shown to mediate induction of ventral types of mesoderm, activin, Vg1, and Xnr can induce various types of mesoderm, including dorsal cell types such as notochord and muscle. The implication for activin as an endogenous mesoderm-inducing factor came from the study which shows that expression of a truncated (kinase domain-deleted) type II activin receptor ActRIIB (previously called XAR1) in Xenopus embryos blocks all mesoderm formation (Hemmati-Brivanlou and Melton 1992). Later, it was shown that the truncated ActRIIB could also block mesoderm formation induced by Vg1 and BMP4 (Schulte-Merker et al. 1994; Chang et al. 1997). Recently, Dyson and Gurdon (1997) demonstrated that a secreted form of ActRIIB receptor, which specifically inhibits activin signaling but not Vg1 signaling, could block mesoderm formation in Xenopus.
Roles of activins and other TGFβ family factors in mammalian development have been analyzed by gene targeting in mice. A striking finding was that the zygotic activins A and B were shown to be dispensable for gastrulation as embryos lacking both activinβA and activinβB genes developed to term with no gross defects in mesoderm formation and patterning (Matzuk et al. 1995). Mutation analysis of two other TGFβ family ligands, BMP4 and nodal, however, showed that both factors are essential for early mouse development (Zhou et al. 1993; Winnier et al. 1995). The majority of BMP4-deficient embryos did not develop beyond the egg cylinder stage and showed little or no mesoderm, whereas some developed to the early somite stage exhibiting defects in the posterior regions of the embryo (Winnier et al. 1995). Disruption of the nodal gene resulted in defects in primitive streak formation (Conlon et al. 1994).
The action of TGFβ family factors is mediated by a family of transmembrane serine/threonine kinase receptors that fall into two classes (Kingsley 1994; Massagué 1996). Two type II receptors, ActRIIA and ActRIIB, are known as activin receptors on the basis of their high affinity for activin (Mathews and Vale 1991; Attisano et al. 1992). One of the type I receptors, ActRIB (also known as ALK4 or R2) (He et al. 1993; ten Dijke et al. 1993), can form heteromeric complexes with either type II activin receptor to specifically mediate activin signaling (ten Dijke et al. 1993, 1994a; Carcamo et al. 1994). The signal transduction mechanism for activin has been extensively studied and shown to be similar to that of TGFβ signaling (for review, see Derynck and Zhang 1996; Massagué 1996). On binding activin, the type II receptors form complexes with and phosphorylate type I receptors such as ActRIB, which in turn phosphorylates downstream Smad proteins such as Smad2. Activated Smad2 has been shown to enter nuclei and activate transcription of activin-responsive genes in cooperation with Smad4 and transcription factors such as Fast-1, a winged-helix transcription factor (Chen et al. 1996, 1997).ActRIB does not bind TGFβ or BMP4 in the presence of corresponding type II receptors (ten Dijke et al. 1994b; Liu et al. 1995), suggesting that ActRIB is specific for activin signaling. It has been shown that kinase-deficient ActRIB blocks activin-induced transcriptional activity (Tsuchida et al. 1995). Functional studies of ActRIB in Xenopus development have shown that overexpression of ActRIB induces dorsal mesoderm whereas truncated ActRIB (kinase-domain deleted) can inhibit mesoderm formation induced by activin (Armes and Smith 1997; Chang et al. 1997). Another type I receptor, ActRIA (also known as ALK2, R1, and Tsk 7L) (Attisano et al. 1993; Ebner et al. 1993; He et al. 1993; ten Dijke et al. 1993) can also bind activin in conjunction with the type II activin receptors (Attisano et al. 1993; ten Dijke et al. 1993, 1994a). Differing from ActRIB, it also binds BMPs in the presence of the BMP type II receptor (ten Dijke et al. 1994b; Liu et al. 1995) and the function of ActRIA in Xenopus development appears to be antagonistic to ActRIB (Armes and Smith 1997).
In this study, we analyzed ActRIB expression in early postimplantation mouse embryos and showed that the ActRIB receptor was predominantly expressed in the epiblast and the extraembryonic ectoderm before and during gastrulation. We generated ActRIB-deficient ES cell lines and mice by gene targeting and found that the function of ActRIB was required for the organization of the egg cylinder before gastrulation. Further, we showed that ActRIB functions in both embryonic ectoderm and extraembryonic cells to mediate signals required for early gastrulation. Thus, our results suggest that the function of ActRIB-mediated signaling pathway in gastrulation is conserved among vertebrate animals.
Results
Expression of ActRIB in early postimplantation mouse embryos
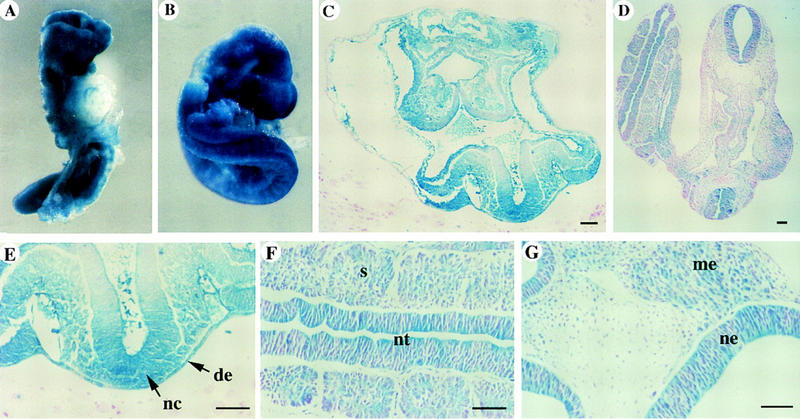
ActRIB expression in mid-gestation mouse embryos has been reported previously (Verschueren et al. 1995). Here, we analyzed the expression pattern of ActRIB earlier in development by in situ hybridization on sections of embryos isolated at different stages. In early postimplantation embryos (at E5.5–6.0), ActRIB was detected uniformly at low levels in the extraembryonic ectoderm and the epiblast, whereas the signal in the visceral endoderm was not above the background (Fig. 1A,B). During gastrulation (at E6.5–7.0), ActRIB expression continued to be detectable in the epiblast and extraembryonic ectoderm, whereas expression in the ectoplacental cone and proximal visceral endoderm was very weak (Fig. 1C–E). The distal endoderm cells, however, were found to be positive for ActRIB (Fig. 1C,E). At E7.5, all three embryonic germ layers, including the newly formed mesoderm, expressed ActRIB (Fig. 1F,G), and the signal in the embryonic ectoderm appeared to be stronger in the primitive streak region. ActRIB expression was not detected in the parietal endoderm throughout early development.
Figure 1.
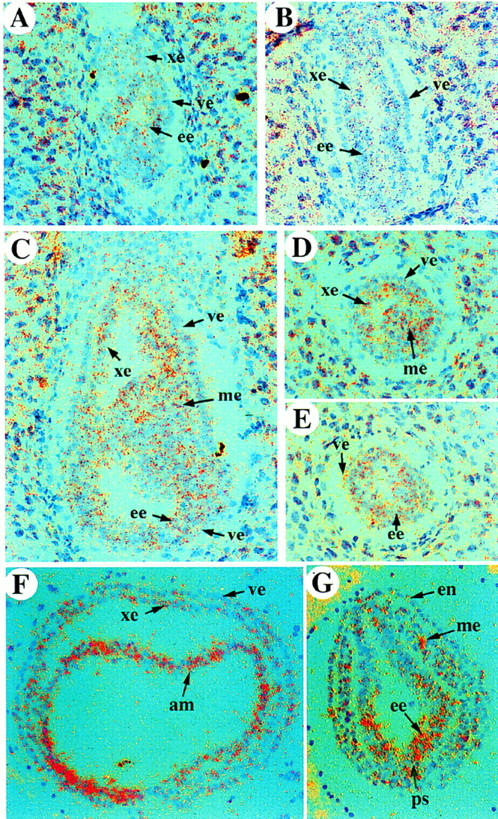
Expression of ActRIB in early postimplantation mouse embryos. In situ hybridization on sections of wild-type embryos from E5.5 to E7.5. Sagittal section of an E5.5 embryo (A), an E6.0 embryo (B), and sections of E6.5 embryos (C–E). (C) Sagittal section; (D) transverse section in the extraembryonic region, (E) or embryonic region. (F,G) Transverse sections in the extraembryonic (F) or embryonic (G) regions of an E7.5 embryo. (am) amniotic fold; (ee) embryonic ectoderm; (en) endoderm; (me) mesoderm; (ps) primitive streak; (ve) visceral endoderm; (xe) extraembryonic ectoderm.
Targeted disruption of the mouse ActRIB gene results in early embryonic lethality
To disrupt the ActRIB gene in mouse embryonic stem (ES) cells, a targeting vector was constructed in which a neomycin gene expression cassette was inserted into kinase subdomain VI, a highly conserved region among type I receptors (Fig. 2A). ES cells were transfected with the targeting vector and selected in G418-containing medium. Of 96 G418-resistant clones screened by Southern analysis with a 5′ external probe, seven were found to be positive for homologous recombination at the ActRIB locus. Two independent clones were injected into blastocysts and male chimeras were generated that transmitted the ActRIB mutation to their offspring. Phenotypes of mutants derived from both clones examined on the 129/Sv inbred or C57Bl/6 × 129/Sv hybrid genetic background were essentially identical. Mice heterozygous for the ActRIB mutation appeared grossly normal. Genotype analysis of progeny from heterozygote intercrosses revealed that 36% were wild type, 64% heterozygous, and none homozygous (Fig. 2B; Table 1), indicating that the ActRIB mutation was recessive embryonic lethal.
Figure 2.
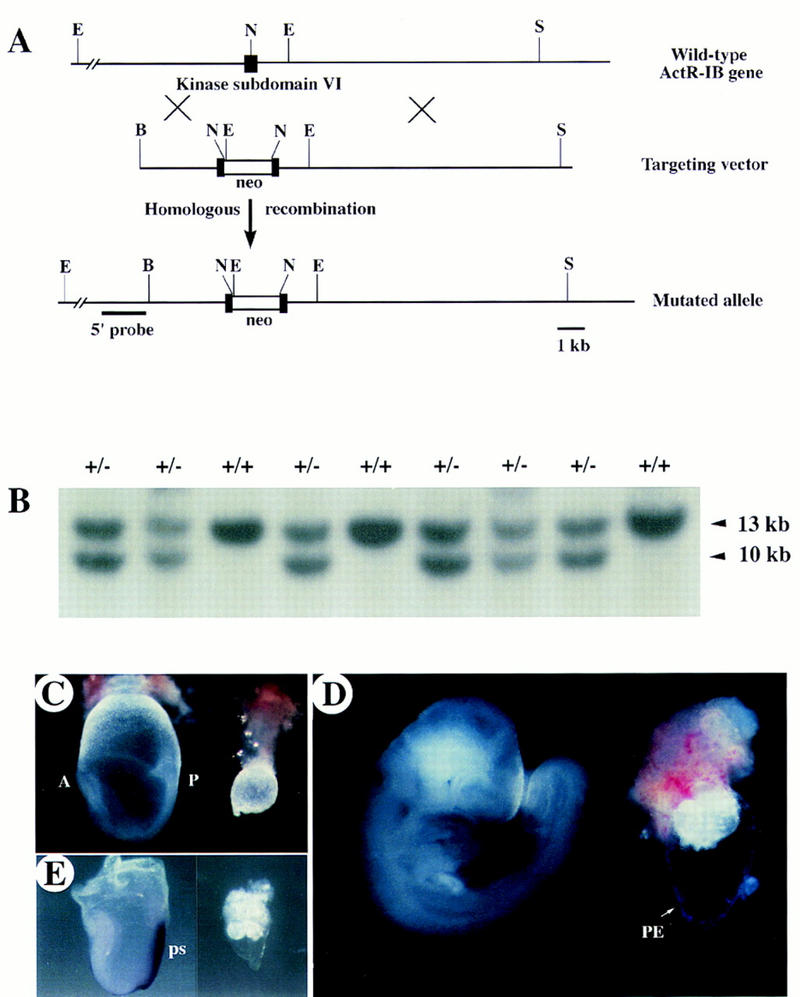
Disruption of ActRIB by homologous recombination results in early embryonic lethality. (A) (Top) Wild-type ActRIB locus shows the exon encoding the kinase subdomain VI (▪). (Middle) The targeting vector contains a 1.8-kb neo cassette (□ box), inserted at the NdeI site inside the kinase subdomain VI of the ActRIB gene. (Bottom) The mutated ActRIB locus. The 5′ external probe, a 1.6-kb genomic fragment, for Southern analysis is shown. (S) SalI; (R) EcoRI; (N) NdeI; (B) BstEII. (B) Genotype analysis of a litter of newborn mice from heterozygous mating. Tail genomic DNA was digested with EcoRI and blotted with the 5′ external probe. The genotypes are marked as +/+ for the wild-type and +/− for the heterozygote. No homozygous live-born mice were found. Arrowheads indicate the 13-kb wild-type and 10-kb mutant bands. (C,D) Gross morphology of normal embryos (left) and ActRIB−/− mutant littermates (right) at E7.5 (C) and late E8.5 (D). In the wild-type, anterior is marked as A and posterior as P. In the mutant embryos, no apparent anterior–posterior polarity was observed, whereas the parietal endoderm (PE) is present. (E). Analysis of T expression in E7.5 embryos by whole-mount in situ bybridization. Wild-type embryo (left); ActRIB−/− littermate (right). (ps) Primitive streak.
Table 1.
Genotype of offspring or embryos from heterozygote crosses
| Stage
|
Assay
|
Genotype
|
||
|---|---|---|---|---|
| +/+
|
+/−
|
−/−
|
||
| Newborn–adult | Southern | 131 | 235 | 0 |
| E9.5 | PCR/Southern | 9 | 28 | 0 |
| E8.5 | PCR | 16 | 28 | 11 |
| E7.5 | PCR | 9 | 17 | 12 |
To characterize the lethal phenotype, embryos from heterozygote intercrosses were examined at different days of gestation and genotyped (Table 1). Of 38 embryos examined at E7.5, 26 were morphologically normal, and 12 were abnormal. In the normal embryos the primitive streak was formed and the anteroposterior axis was well established, whereas the abnormal embryos were much smaller and lacked a discernible primitive streak (Fig. 2C). At E8.5, ∼25% of decidua contained abnormal conceptuses in which the embryos were either very small (Fig. 2D; Table 1) or completely resorbed. The abnormal embryos were confirmed to be ActRIB−/− by PCR analysis. No homozygous embryos were recovered at, or after, E9.5.
To determine whether mesoderm and the primitive streak were formed in ActRIB−/− embryos, expression of Brachyury (T), an early mesoderm marker (Wilkinson et al. 1990), was analyzed by in situ hybridization. We showed that T expression was not detected in E7.5 and E8.5 mutant embryos (Fig. 2E), suggesting that ActRIB−/− embryos were developmentally arrested prior to gastrulation.
ActRIB−/− embryos fail to form a normal embryonic egg cylinder
Histological sections of embryos from heterozygote intercrosses were analyzed at different developmental stages to determine the defects of the ActRIB mutants. At E5.0, no overt abnormal embryos were found (data not shown). The earliest abnormality was observed at E5.5 in ∼22% (6/27) of the embryos. In the wild-type embryos at this stage, the visceral endoderm cells were moderately vacuolated and in close contact with the neighboring ectodermal cells. In the mutants, however, the distal visceral endoderm cells appeared to be slightly more vacuolated and were detached from the epiblast, which appeared to contain fewer cells than that in a wild-type embryo (Fig. 3B). A small proamniotic cavity was present in the mutant embryos, indicating that initial cavity formation occurred (Fig. 3B).
Figure 3.
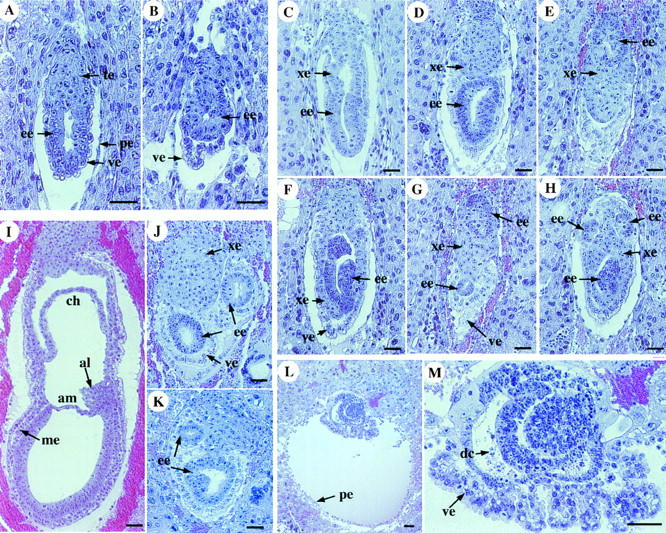
Histological analysis of ActRIB−/− embryos. Sagittal sections of the wild-type (A,C,I) and mutant embryos (B,D–H, J–M) at E5.5 (A,B), E6.5 (C–H), E7.5 (I–K) and E8.5 (L,M). In each panel, the ectoplacental cone (or the proximal region) is positioned towards the top. The distal visceral endoderm in the E5.5 mutant embryo is detached from the epiblast (B). The E6.5 mutant embryos contain only vacuolated columnar visceral endoderm cells (D–H). Germ layers or cell clusters that are stained darker are identified as epiblast through analysis of whole series of embryo sections. (M) An enlarged section of an E8.5 mutant embryo shown in L. Cell death of the epiblast in this mutant is apparent. (al) allantois; (am) amnion; (ch) chorion; (de) dead cells; (ee) embryonic ectoderm; (me) mesoderm; (pe) parietal endoderm; (ve) visceral endoderm; (xe) extraembryonic ectoderm. Bar, 50 μm.
At E6.5, the visceral endoderm of a normal embryo can be classified on the basis of their morphology into two subpopulations, the distal squamous cells surrounding the epiblast and the proximal columnar cells surrounding the extraembryonic ectoderm (Fig. 3C). In contrast, the mutant embryos (12/50) appeared to contain only vacuolated columnar visceral endoderm cells (Fig. 3D-H). The epiblast of some mutant embryos contained fewer cells than that of the wild-type embryos and often located ectopically, whereas the extraembryonic ectoderm cells, which originated from the polar trophectoderm, failed to form an epithelial layer (Fig. 3D–H) and often intruded into the distal region (Fig. 3E–H). In a three-dimensional view based on analyzing serial sections of the mutant embryos, the epiblast and the extraembryonic ectoderm were not fragmented, but intertwined and disorganized. The growth of the mutant embryos at E7.5 was further retarded. Unlike the wild-type embryos, in which the mesoderm layer, amnion, and chorion were formed (Fig. 3I), the mutant embryos showed defects similar to those observed at earlier stages and contained no mesoderm (Fig. 3J,K). In addition, the epiblast of some mutant embryos contained dying cells, indicative of tissue degeneration. At E8.5, the visceral endoderm layer in the mutant embryos was folded, likely resulting from extensive cell death and shrinkage of the ectoderm (Fig. 3L,M). No morphological defects were observed in the trophoblast or parietal endoderm of the mutant embryos throughout early development, consistent with the lack of ActRIB expression in those tissues.
To confirm the cell identities in the ActRIB mutants, we examined the expression pattern of molecular markers in the prestreak stage embryos. SSEA-1 is a monoclonal antibody recognizing cells of the inner cell mass (ICM) and its derivatives (Fox et al. 1981). In wild-type E6.5 embryos, SSEA-1 stained the embryonic ectoderm as well as the visceral endoderm, but not the extraembryonic ectoderm (Fig. 4B). In the mutants, SSEA-1 stained the visceral endoderm and clusters of cells separated by a mass of nonstained cells (Fig. 4F). These clusters of cells resembled morphologically the epiblast, whereas the nonstained cells were probably extraembryonic ectoderm on the basis of their morphology (Fig. 4E,F). We also examined the staining pattern of Troma-1, which is a monoclonal antibody recognizing the primitive endoderm and its derivatives including the visceral endoderm and parietal endoderm in normal postimplantation embryos (Kemler et al. 1981). In E6.5 wild-type embryos, the staining in the visceral endoderm surrounding the extraembryonic ectoderm was stronger than that surrounding the embryonic region (Fig. 4C), whereas in mutant embryos, the staining was strong in the distal part of the visceral endoderm (Fig. 4G). The staining pattern of Troma-1 in the mutant embryos indicated that the visceral endoderm was present but morphologically abnormal. Immunostaining with an antibody against laminin, a basement membrane protein produced in the parietal endoderm, revealed no significant difference between the wild-type and the mutant embryos (Fig. 4D,H). Taken together, these results confirmed histological analysis that the epiblast and the extraembryonic ectoderm were disorganized and the visceral endoderm failed to form the squamous cell type in the distal region of the egg cylinder.
Figure 4.
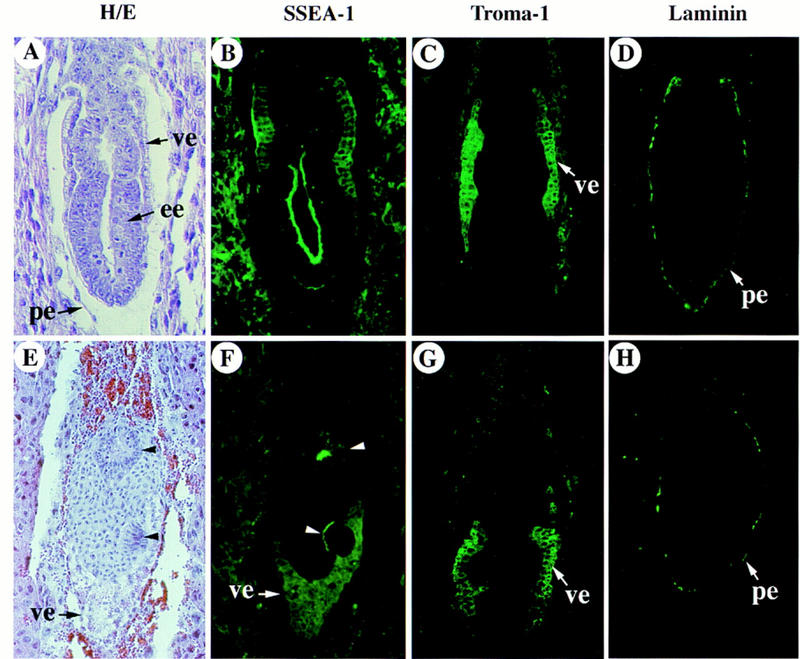
Analysis of expression of lineage-specific genes in the ActRIB−/− embryos. Hemotoxylin and eosin (H/E) stained sections (A,E) and immunostaining with SSEA-1 (B,F), Troma-1 (C,G) and anti-laminin (D,H) antibodies of the wild-type (A–D) and mutant (E–H) embryo sections at E6.5. The section in A is adjacent to that in B; the section in E is adjacent to that in F. Other sections are from different embryos. In each panel, the ectoplacental cone (or the proximal region) is positioned toward the top. The two clusters of stained epiblast cells in E are marked with arrowheads.
ActRIB-deficient ES cells were able to form mesoderm but not a primitive streak
Because development of ActRIB−/− embryos was arrested at the egg cylinder stage, the function of ActRIB in mesoderm formation and gastrulation could not be directly analyzed in mutant embryos. To circumvent this problem, we isolated ActRIB-deficient ES cells that were genetically marked with a lacZ transgene from Rosa 26 mice (Friedrich and Soriano 1991). Because the Rosa 26 lacZ transgene is expressed ubiquitously in the early embryo, it serves as a lineage marker for analyzing the contribution and tissue distribution of mutant cells in development. The lacZ+ ES cell lines were established by culturing delayed blastocysts isolated from female ActRIB+/− mice bred with male ActRIB+/− Rosa-26+/+. Of 12 lines isolated, one was confirmed to be Rosa+/− ActRIB−/− (expecting 1/4 to be Rosa+/− ActRIB−/−).ActRIB−/− ES cells were injected into wild-type blastocysts to generate chimeric embryos that were analyzed at various stages by X-gal staining followed by histological analysis of embryo sections.
When 5–10 cells were injected per blastocyst, the contribution of the mutant cells in most chimeras was 10%–50%. The majority of chimeric embryos were normal when examined at stages from E6.0 to E9.5 (Fig. 5A–E; Table 2). In late E7.5 chimeras, the lacZ-positive ActRIB−/− cells were found not only in the neural and surface ectoderm (Fig. 5C,D), but also in mesodermal tissues such as lateral plate mesoderm and allantois (Fig. 5C,D). ActRIB−/− cells, however, formed small patches and seemed to contribute preferentially to posterior and lateral mesoderm. In contrast, examination of several chimeras at E7.0 revealed that the ActRIB−/− cells did not contribute to the early mesoderm even when the epiblast consisted of ∼40% ActRIB−/− mutant cells (Fig. 5E). These results suggest that ActRIB−/− cells might not be competent to participate in mesoderm formation during early gastrulation, although they could contribute to mesoderm tissues at the late stage. When 15–20 cells were injected per blastocyst, the chimeric embryos with a high contribution (>80%) of mutant cells failed to undergo gastrulation (Table 2). Histological analysis of these embryos revealed a common feature that a relatively normal egg cylinder structure was formed, but a morphologically discernible primitive streak was absent (Fig. 5F), suggesting a functional requirment of ActRIB in primitive streak formation. To test whether ActRIB−/− cells were able to differentiate into mesodermal cells in a nonorganized structure, we generated teratomas from ActRIB−/− ES cells and found that the ActRIB−/− cells could form various types of mesoderm tissues such as muscle, fat, and cartilage (Fig. 6). Taken together, these results suggest that ActRIB-deficient cells retain capability to form mesoderm but appear to be impaired in their ability to form the primitive streak.
Figure 5.
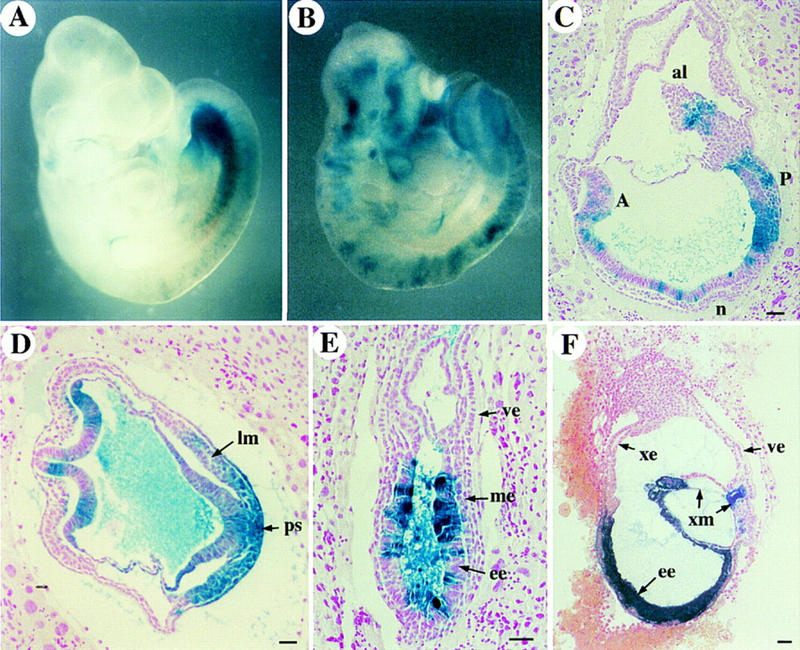
Chimeric embryos derived by injection of ActRIB−/− cells into wild-type blastocysts. The gross morphology of E9.5 embryos with low (A) or moderate (B) contribution of lacZ-labeled homozygous mutant cells appears normal. Sagittal (C) and transverse (D) sections of late E7.5 embryos show moderate contribution of mutant cells in allantois (al), mesoderm at posterior of the primitive streak (ps), and lateral mesoderm (lm). (E) An E7.0 chimeric embryo shows no blue staining of ActRIB−/− cells in mesoderm layer (me), even though the contribution in the adjacent embryonic ectoderm (ee) is relatively high. (F) A late E7.5 chimeric embryo section with very high contribution of mutant cells. No primitive streak or embryonic mesodermal cells were found, although some extraembryonic mesoderm (xm) cells were observed in an amniotic fold-like structure. Anterior and posterior of the embryo in C are marked as A and P, respectively. Bar, 50 μm.
Table 2.
Summary of chimera analysis
| Chimerasa
|
Dev. stage
|
No. of chimeras
|
Contributionb
|
|||
|---|---|---|---|---|---|---|
| <80% | >80% | |||||
| (normal) | (abnormal) | (normal) | (abnormal) | |||
| I | E6.0–6.5 | 4 | 4 | 0 | 0 | 0 |
| I | E7.0–7.5 | 34 | 17 | 4 | 2 | 11 |
| I | E8.5 | 12 | 7 | 0 | 0 | 5 |
| I | E9.5 | 8 | 6 | 0 | 0 | 2 |
| II | E8.5 | 6 | 2 | 0 | 4 | 0 |
| II | E9.5 | 11 | 7 | 0 | 4 | 0 |
Group I chimeric embryos were derived by injection of lacZ-marked ActRIB−/− cells into wild-type blastocysts (see Fig. 5). Group II chimeric embryos were derived by injection of ActRIB−/− (ALK-4+) cells into wild-type blastocysts (see Fig. 8).
Normal refers to embryos that formed normal body axes and differentiated tissues according to their developmental stages; abnormal refers to embryos that either did not form a primitive streak or mesoderm (E7.0–7.5) or displayed a severely distorted embryonic axis (E8.5–9.5).
Figure 6.
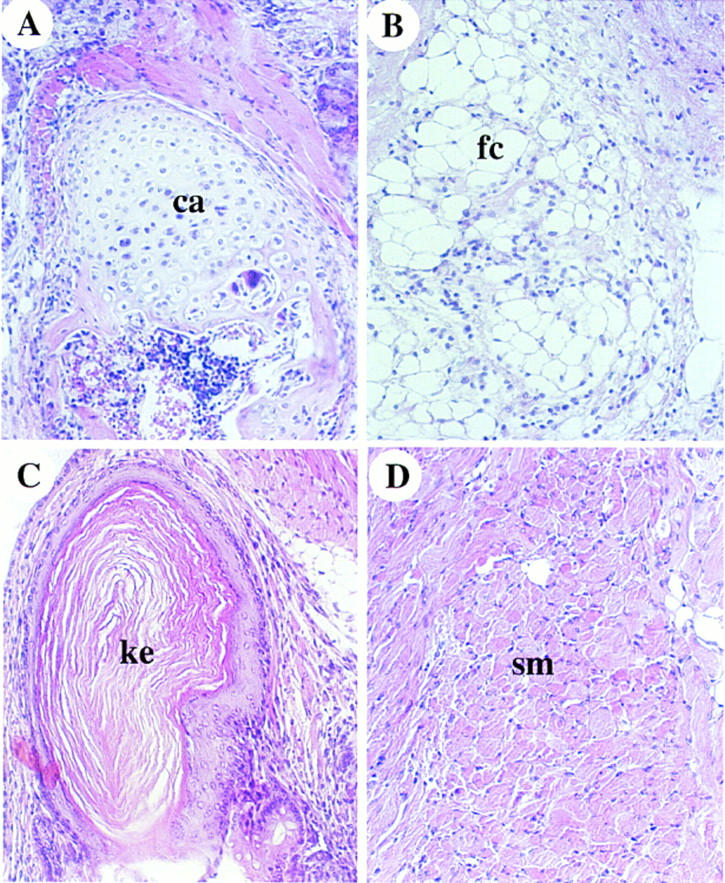
Teratomas derived from ActRIB−/− ES cells. Histology sections of teratomas derived from ActRIB−/− ES cells (A–D). Tissues such as cartilage (ca), fat cells (fc), keratinized epithelium (ke), and smooth muscle (sm) are shown.
The defects of ActRIB−/− cells can be rescued by a human ALK4 transgene
To confirm that the gastrulation defect of chimeric embryos resulted solely from the lack of ActRIB function, we tested whether expression of the ALK4 gene, the human homolog of the mouse ActRIB, could rescue the gastrulation defects. ALK4 tagged with an HA epitope at the amino terminus was expressed under the control of the promoter of the human elongation factor-1 α gene (EF-1α) (Fig. 7A). The EF-1α promoter was found to direct ubiquitous expression of a lacZ transgene in early mouse embryos (data not shown). Linearized DNA was transfected into the ActRIB−/− ES cells, and several cell lines resistant to hygromycin were isolated, among which four were further characterized. Western blot analysis of cell extracts by use of anti-HA antibody revealed that the HA-tagged ALK4 protein was detectable in all four lines (Fig. 7B), which were termed ActRIB−/− (ALK4+) lines. Southern blot analysis showed that each cell line contained one (lines 7, 21, and 42) or two (line 20) copies of the transgene, integrated at different genomic loci (Fig. 7C; data not shown). ES cells from lines 20, 21, and 42 were injected into wild-type blastocysts to generate chimeric embryos. Functional rescue of the gastrulation defect was achieved with lines 20 (see below) and 21 but not with line 42 (data not shown).
Figure 7.
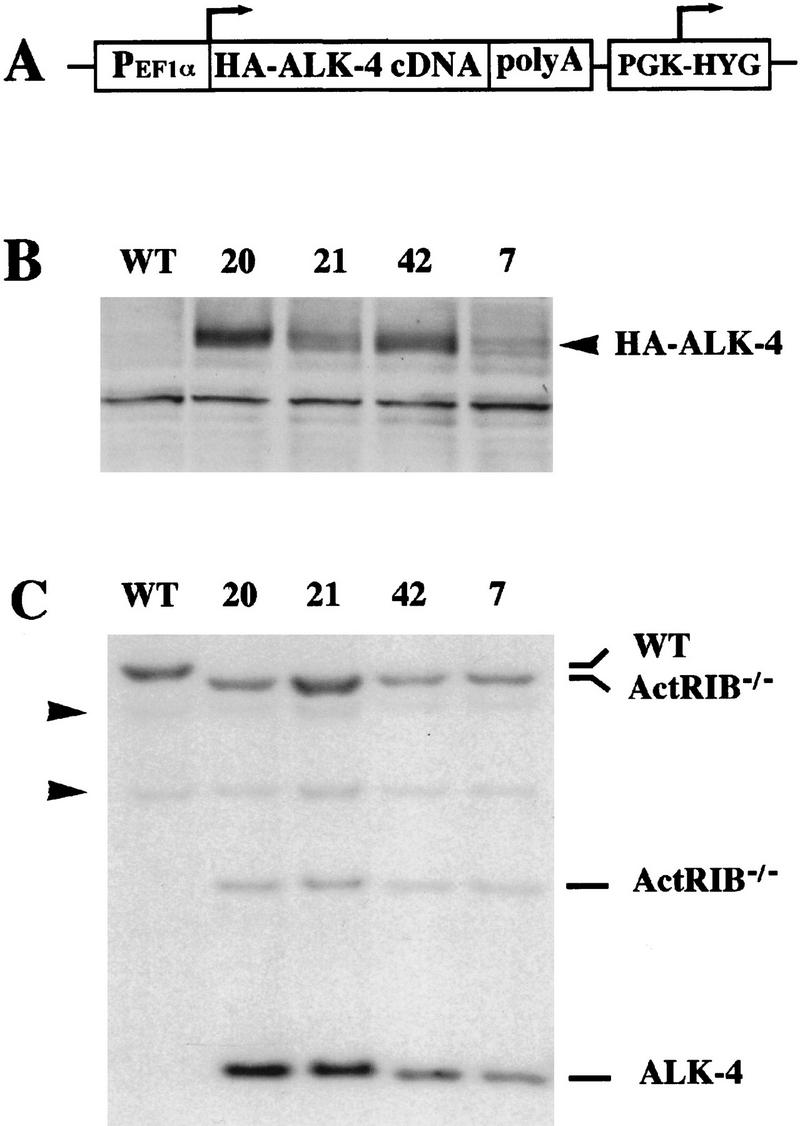
Expression of ALK4 transgene in ActRIB−/− ES cells. (A) A diagram of ALK4 transgene expression vector. The human ALK4 cDNA tagged with a triple HA epitope at the 3′ end of the coding sequence was inserted between the EF-1α promoter and SV40 polyadenylation signal followed by a hygromycin cassette. (B) Expression of HA–ALK4 protein in the transgenic ES cell lines was analyzed by immunoblotting the ES cell protein extracts with 12CA5, the monoclonal antibody against the HA epitope. The arrow head indicates the HA–ALK4 bands. The expression is higher in cell lines 20 and 42 than in lines 21 and 7. (C) Genotype of ALK4 transgenic ES cell lines and wild-type control cells. Genomic DNA from wild-type and the transgenic cell lines was digested with EcoRI and analyzed by Southern hybridization by use of a 1.6-kb HindIII–EcoRI ALK4 cDNA fragment as a probe. Cell lines 7, 21, and 42 each contain one copy and line 20 contains two copies of the transgene (data not shown). The bands representing the wild-type allele, the ActRIB mutant allele, and the ALK4 transgene are marked on the right. Arrowheads indicate two DNA fragments shared by both wild-type and ActRIB−/− mutant alleles.
Chimeras were generated by injection of ∼15 ES cells per blastocyst, and the contribution and distribution of the ActRIB−/− (ALK4+) cells was analyzed by X-gal staining followed by histological analysis of embryonic sections. The ES cell contribution in chimeras is summarized in Table 2. In contrast to the chimeric embryos derived from ActRIB−/− cells, many embryos with a high contribution of ActRIB−/− (ALK4+) cells showed normal gross morphology at E8.5 or E9.5 (Fig. 8A,B). Analysis of histological sections revealed that the ActRIB−/− (ALK4+) cells contributed extensively to all the embryonic tissues and the mesoderm components of the extraembryonic tissues (Fig. 8C–E). In contrast to the chimeras derived from ActRIB−/− cells, chimeras containing >80% of ActRIB−/− (ALK4+) cells were able to form normal body axes and differentiated tissues at E8.5 and E9.5. The notochord and somites in some embryos were colonized predominantly by the blue ActRIB−/− (ALK4+) cells (Fig. 8E,F). In tissues in which both wild-type and ActRIB−/− (ALK4+) cells were present, the ActRIB−/− (ALK4+) cells intermingled with the wild-type cells in a salt and pepper pattern, suggesting that the two types of cells were equally competent in forming the mesoderm tissues (Fig. 8F,G). Overall, these results indicated that the failure of ActRIB−/− cells to participate in primitive streak formation in chimeras could be rescued by the expression of human ALK-4.
Figure 8.
Rescuing ActRIB−/− ES cell defect with an ALK4 transgene in chimeras. The chimeric embryos were derived by injection of the ActRIB−/− (ALK4+) cells (line 20) into the wild-type blastocysts. X-gal-stained E8.5 (A) and E9.5 (B) embryos appear normal. (C,D) Histological sections of an E8.5 embryo (C) and an E9.5 (D) show high contribution of transgenic cells in almost all mesodermal cell types (C) or a salt and pepper pattern of intermingle of transgenic cells with the wild-type cells derived from the blastocyst (D). (E) An enlarged part of the embryo in C shows the notochord (nc) and definitive endoderm (de), completely derived from the transgenic cells. (F,G) Sections from an E9.5 embryo show the mesenchyme (me), neuroepithelium (ne), neural tube (nt), and somite (s). Bar, 67 μm.
ActRIB is also required in the extraembryonic cells for gastrulation
Because ActRIB expression was detected in the extraembryonic ectoderm, the following experiment was conducted to investigate whether its function was required in the extraembryonic tissues. We generated chimeric embryos in which the epiblast consisted predominantly of wild-type cells whereas the extraembryonic ectoderm and the visceral endoderm were derived from ActRIB−/− blastocysts on the basis of the observation described previously (Beddington and Robertson 1989). The prediction would be that if the ActRIB function was required in extraembryonic cells, the chimeric embryos would show defects during early development.
A lacZ-positive wild-type ES cell line R/S4 derived from Rosa 26 transgenic mice was used to generate chimeric embryos. R/S4 cells were competent to form chimeras with a high contribution in somatic tissues as well as in the germ line (Fig. 9C; data not shown). Blastocysts were harvested from ActRIB+/− crosses, in which one-fourth were expected to be ActRIB−/−. Approximately 15–20 cells were injected into each blastocyst and chimeras were dissected out at E9.5. We found that all 22 embryos dissected out contained well formed visceral yolk sac, among which, however, five showed little or no embryonic tissues (Fig. 9A,B). Southern analysis of DNA isolated from the parietal yolk sac that was derived entirely from the donor blastocyst, confirmed that these five abnormal chimeras were all derived from ActRIB−/− blastocysts (data not shown). Histological analysis showed that these abnormal chimeras contained a relatively normal yolk sac with extraembryonic mesoderm and blood cells and a severely retarded or degenerated embryo (Fig. 9B). Such deformed embryos were also found in chimeras dissected at E7.5 (Fig. 9D). Taken together, these results suggest that ActRIB also functions in the extraembryonic cells during gastrulation to regulate the development of the embryo proper.
Figure 9.
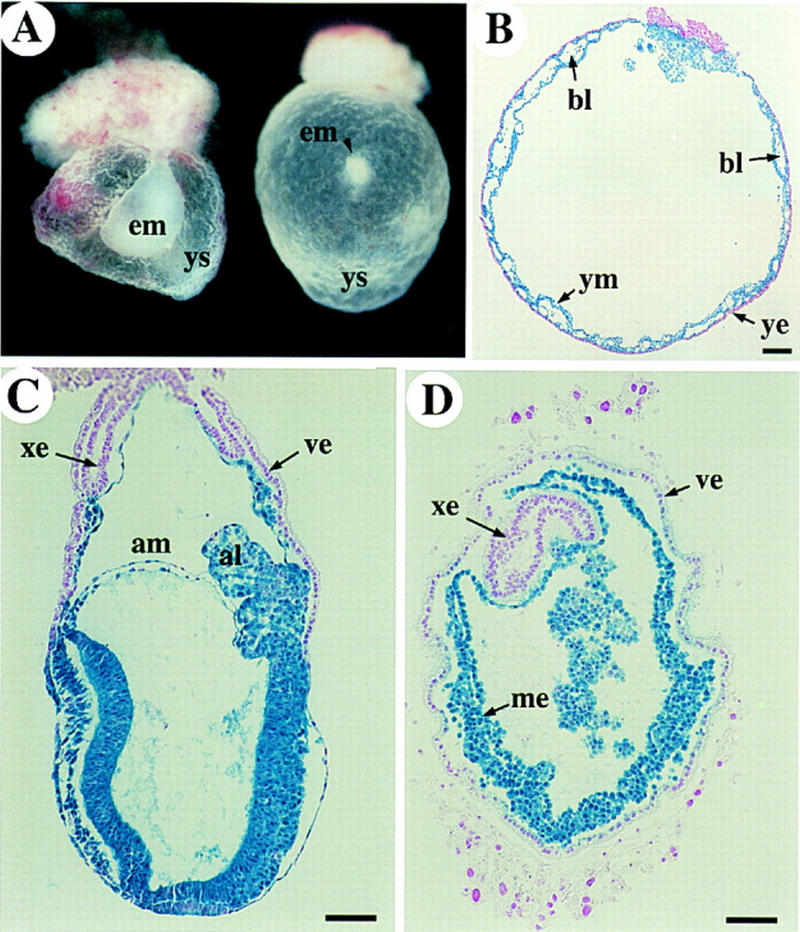
Chimeric embryos derived by injection of wild-type ES cells into the ActRIB−/−/ blastocysts. (A) Two E9.5 embryos were dissected and their parietal yolk sac DNA was genotyped as ActRIB−/−. The embryonic tissues (em) in the chimeras were very small. (B) E9.5 ActRIB−/− chimeric embryo was stained with X-gal and sectioned to show the visceral yolk sac mesoderm (ym) and blood cells (bl). The yolk sac mesoderm consists almost entirely of lacZ-marked wild-type cells; the yolk sac endoderm (ye) is derived from ActRIB−/− blastocyst. (C) Control E7.5 embryo generated by injecting the same wild-type ES cells into a wild-type or heterozygous blastocyst shows normal embryonic structures. (D) Transverse section of a late E7.5 abnormal chimera. Mesodermal cells were formed inside a yolk sac-like structure, but no obvious embryonic tissues were found. (bl) Blood cells; (ye) yolk sac endoderm; (ym) yolk sac mesoderm; (ys) yolk sac. Bar, 67 μm.
Discussion
ActRIB is required for egg cylinder organization
Early postimplantation development of the mouse embryo has been well characterized morphologically. Not until recent years, however, have the molecular mechanisms that control growth, differentiation, and organization of the early cell types begun to be uncovered. Targeted gene disruption has revealed essential functions for several genes in early mouse development. For instance, FGF4 has been found to be required for the proliferation of the inner cell mass cells (Feldman et al. 1995); E-cadherin and β-catenin are essential for cell adhesion in the primitive ectoderm (Haegel et al. 1995; Riethmacher et al. 1995). BMP4 and the type I BMP receptor, Bmpr (also called ALK-3), play a critical role in regulating proliferation of the epiblast (Mishina et al. 1995; Winnier et al. 1995). In this study, we showed that inactivation of ActRIB resulted in the disruption of the egg cylinder structure prior to gastrulation, indicating that ActRIB mediates a cellular function essential for early morphogenesis.
The morphological defects in ActRIB−/− embryos include the reduction in the number of epiblast cells in some embryos, the dissociation of the epiblast from the visceral endoderm layer, the ectopic localization of the epiblast and the extraembryonic ectoderm, the lack of a cavity in the extraembryonic ectoderm, and the lack of the squamous type of visceral endoderm. Some of the defects in ActRIB−/− are reminiscent of the phenotype of the nodal-deficient embryos (Conlon et al. 1991; Iannaccone et al. 1992). Interestingly, initial cavity formation occurred in the embryonic region, suggesting that signals from the visceral endoderm regulating the formation of proamniotic cavity was not disrupted (Coucouvanis and Martin 1995). Proliferation of the epiblast cells might be affected in the mutant embryos. It does not seem to be the primary defect that leads to other abnormalities, however, because a number of early mutants such as the BMP4, Bmpr, and Brca1 knockout mice, which display defects in epiblast proliferation, are able to form the normal egg cylinder structure (Mishina et al. 1995; Winnier et al. 1995; Hakem et al. 1996). Cell–cell or cell matrix adhesion plays a critical role in maintaining the epithelial structure. ActRIB has been shown to be involved in regulation of expression of extracellular matrix proteins that are essential components of the basement membrane (Cárcamo et al. 1994). It is possible that disruption of the ActRIB signaling pathway may affect expression of extracellular matrix proteins or cell adhesion molecules, resulting in disorganization of the epiblast and the extraembryonic ectoderm. So far, no knockout mutations of single extracellular matrix protein or cell adhesion molecule show similar defects as the ActRIB−/− embryos. Systematic analysis of expression of genes involved in cell–cell or cell-matrix adhesion in mutant embryos and identification of downstream target genes of the ActRIB signaling pathway in the future will help to elucidate the molecular mechanism for egg cylinder development.
The function of ActRIB in the epiblast is required for primitive streak formation
Gastrulation of the mouse embryo begins at E6.5 with the formation of the primitive streak and early mesoderm cells in the proximal posterior region of the epiblast. The primitive streak then elongates toward the distal tip of the egg cylinder allowing epiblast cells to ingress through and differentiate into different types of mesodermal cells (Lawson et al. 1991, 1992; Hogan et al. 1994). Which signaling molecules are involved in mesoderm induction and primitive streak formation and which tissue is the source of the inductive signals are questions that remain largely unsolved. In this study, we showed that the type I activin receptor ActRIB is not essential for mesoderm formation but appears to be required for primitive streak formation in the mouse.
Although T expression was not detected in E7.5 and E8.5 ActRIB−/− embryos (Fig. 2E), we found that ActRIB−/− cells were capable of forming mesoderm tissues in both chimeras and teratoma (Figs. 5, C and D, and 6), suggesting that ActRIB is not absolutely required for mesoderm formation. The lack of T expression in mutant embryos could be an indirect effect resulting from the disruption of the egg cylinder structure. Whereas ActRIB−/− cells could form mesoderm, they contributed poorly, if at all, to the early mesoderm in E7.0 chimeras (Fig. 5E,F). This result suggests that ActRIB−/− cells might not be as competent as the wild-type cells to form all types of mesoderm. One possibility is that ActRIB−/− cells might have lost the ability to participate in primitive streak formation. Consistent with this notion, we showed that most chimeric embryos, in which the contribution of ActRIB−/− ES cells to the epiblast exceeded 80%, failed to form a primitive streak and embryonic mesoderm cells (Fig. 5F; Table 2).
The defect of ActRIB−/− cells in primitive streak formation could be rescued by human ALK4 gene (Figs.7,8). We showed that ALK4 transgene was expressed in ActRIB−/− ES cells (Fig. 7) and presumably also in the epiblast during gastrulation of the chimeras, where ActRIB was normally expressed, thus rendering ActRIB−/− cells responsive to the signal required for primitive streak formation. This result confirmed that the defect of ActRIB−/− cells was indeed caused by disruption of the ActRIB gene and suggested a conserved function for the human ALK4 gene.
At present, it is not clear how ActRIB−/− cells fail to form the primitive streak and early mesoderm. Several possibilities are considered. Perhaps, ActRIB−/− epiblast cells were unable to delaminate; or the primitive streak was formed initially, but ActRIB−/− cells in the primitive streak failed to proliferate during elongation of the primitive streak; or ActRIB−/− cells were incapable of passing through the primitive streak to form early mesoderm cells. Further investigations are necessary to elucidate the molecular and cellular mechanisms by which the ActRIB-mediated signaling pathway regulates mouse gastrulation.
The function of ActRIB in the extraembryonic tissues is also required for gastrulation
An intriguing finding is that ActRIB also functions in the extraembryonic ectoderm or visceral endoderm to regulate embryonic development. We showed that, without exception, chimeric embryos derived by injection of the wild-type ES cells into the ActRIB−/− blastocysts failed to form any embryonic structures and were degenerated by E9.5, whereas the development of the extraembryonic tissues was relatively normal and the extraembryonic mesoderm was formed (Fig. 9). The arrest of embryonic growth suggests that gastrulation was severely impaired. If the ActRIB function was required solely in the epiblast, we would expect that a high contribution of wild-type ES cells would have rescued the defect of ActRIB−/− embryos. The failure of the wild-type cells to rescue ActRIB−/− embryos thus indicates that ActRIB also functions in the extraembryonic tissues during gastrulation, which is consistent with relatively high levels of ActRIB expression in the extraembryonic ectoderm during postimplantation development (Fig. 1).
It is quite peculiar that ActRIB functions in the extraembryonic cells, but is involved in regulation of embryonic development. Such a noncell autonomous action of ActRIB suggests that it may regulate expression or secretion of a signaling molecule that can act through a paracrine mechanism to regulate gastrulation of the embryo. The presence of the extraembryonic mesoderm in the chimeras (Fig. 8) suggests that the signal produced by the extraembryonic tissues is not required for mesoderm formation and migration to the extraembryonic region, but may play a critical role in maintaining the primitive streak or proliferation of the embryonic mesoderm,
The possible ligand(s) for ActRIB in early mouse development
Activin was the first TGFβ family molecule shown to have mesoderm inducing activity. Experiments with various truncated forms of type I (ALK4) or type II (ActRIIB) receptors to block mesoderm formation in Xenopus further support activin’s role as an endogenous mesoderm inducer (Hemmati-Brivanlou and Melton 1992; Armes and Smith 1997; Chang et al. 1997; Dyson and Gurdon 1997). Other TGFβ family signaling molecules such as Vg-1, BMP4, and Xnr also induce mesoderm formation in Xenopus animal cap assay. The unsolved problem is which one(s) play a key role in mesoderm induction and axis formation in normal development. In contrast to the role of activin in Xenopus early development, mice lacking activins A and B develop to term with no gross defects in mesoderm formation (Matzuk et al. 1995), indicating zygotic activins are not involved in mesoderm induction. Although transcripts of maternal activins are present abundantly in the decidual cells surrounding the egg cylinder (Albano et al. 1994), it is not clear whether they can participate in early mouse development. In this study, we provide genetic evidence that the type I activin receptor, ActRIB (ALK4), is required for egg cylinder organization and primitive streak formation, indicating that acitivin-like molecules are also essential for early development in mice.
Nodal and BMP4 have been shown to be essential for early mouse development. Could one of them be the ligand of ActRIB? We think that ActRIB is unlikely a type I receptor for BMP4 because BMP binds to two other type I receptors (ALK3 and ALK6) but not ActRIB (ALK4) in vitro (ten Dijke et al. 1994b), and a truncated ActRIB receptor does not inhibit BMP4 signaling in frog embryos (Chang et al. 1997). Moreover, the defects in BMP4-deficient embryos were different from those in ActRIB−/− embryos, but closely related to those of Bmpr (ALK3)-deficient mice (Mishina et al. 1995; Winnier et al. 1995).
In nodal-deficient embryos, T was ectopically expressed but primitive streak formation was disrupted, suggesting that nodal is probably not required for mesoderm induction but essential for primitive streak formation (Conlon et al. 1994). A recent study showed that chimeric embryos containing nodal-deficient extraembryonic cells were able to form the primitive streak, suggesting that nodal produced by the epiblast cells is required for primitive streak formation (Varlet et al. 1997). In this study, we provide strong evidence that ActRIB functions in the epiblast to regulate primitive streak formation. Whereas the nodal receptor has yet to be identified through ligand binding analysis, the similar defects in primitive streak formation in nodal−/− and ActRIB−/− embryos raises the possibility that ActRIB might function as the type I receptor for nodal. We have discovered recently that the type II activin receptors are also required for primitive streak formation and gastrulation in mice (S.P. Oh, H. Schrewe, M. Normura, J. Song, H. Lei, M. Okano, K.A. Goss, T. Gridley, and E. Li, in prep.). We speculate that nodal may signal via the complex of type II activin receptors and ActRIB to regulate primitive streak formation.
Materials and methods
Construction of the gene targeting vector
Mouse genomic DNA corresponding to the ActRIB gene was cloned by screening a 129/Sv mouse genomic library (Stratagene) with a 2.0-kb full-length rat cDNA probe (He et al. 1993). A 14-kb genomic DNA clone was isolated and subcloned into pBluescript KS vector. The targeting vector was constructed by inserting the PGK–neo–poly(A) cassette at the NdeI site in the exon encoding the conserved kinase subdomain VI. The final construct contained 3.0 kb of homologous genomic DNA on the 5′ side and 11.0-kb genomic DNA on the 3′ side of the neo gene that is in the same transcriptional direction as the ActRIB gene.
Construction of the ALK4 expression vector
The ALK4 transgene expression vector pEF-HGALK4 was constructed by inserting a 1.6-kb cDNA fragment, which contains the entire ALK4 coding sequences, between the HindIII and XbaI sites of plasmid pEF-BOS (Mizushima and Nagata 1990). The ALK4 gene was tagged with a triple HA epitope at the carboxyl terminus and its expression was controlled by the human EF-1α promoter and an SV40 polyadenylation signal. A 1.8-kb ClaI–XhoI DNA fragment containing a hygromycin cassette was inserted next to the SV40 polyadenylation signal for drug selection.
Generation of ActRIB mutant mice, ActRIB−/−, and ActRIB−/− (ALK4+) ES cell lines
To disrupt the ActRIB gene, the J1 ES cell line was electroporated with linearized targeting vector DNA and was selected in G418-containing medium as described (Li et al. 1992). The genotype of G418R clones was analyzed by Southern blot hybridization with a 1.6-kb NotI–BstEII genomic DNA fragment as a probe (See Fig. 1A). Recombinant ES clones were isolated and injected into C57BL/6 blastocysts to obtain chimeric mice as described previously (Li et al. 1992). Male chimeras with germ-line transmission were then bred to either 129/Sv females to establish an inbred strain or to C57BL/6 females to obtain hybrid F1 progeny.
To establish lacZ-marked ActRIB-deficient ES cell lines, Rosa-26+/+ mice (Friedrich and Soriano 1991; from Jackson Laboratory) were crossed to ActRIB+/− to obtain ActRIB+/− Rosa-26+/−, which were then backcrossed to Rosa-26+/+ to obtain ActRIB+/− Rosa-26+/+. Delayed blastocysts were isolated from ActRIB+/− females bred with ActRIB+/− Rosa-26+/+ males and ES cell lines were isolated from the inner cell mass grown in culture as described (Robertson 1987). The genotype of established ES cell lines with respect to ActRIB and Rosa 26 loci was determined by Southern analysis and X-gal staining (data not shown).
To produce ActRIB−/− (ALK4+) transgenic ES cells, ActRIB−/− ES cells were electroporated with a linearized transgene expression vector pEF-HGALK4 and selected in medium containing 100 μg/ml of hygromycin (GIBCO). The genotype of hygromycin-resistant clones was analyzed by Southern blot hybridization of EcoRI digested genomic DNA with a 1.6-kb HindII–EcoRI ALK-4 cDNA fragment. The expression of ALK4–HA fusion protein was analyzed by immunoblotting the ES cell protein extracts with 12CA5, the monoclonal antibody specific to the HA epitope (Boehringer Mannheim).
Histological analysis and immunohistochemistry
For histological analysis, embryos in uterus or dissected out of decidua were fixed in Bouin’s fixative, dehydrated, and embedded in paraffin as described by Kaufman (1992). Serial sections of 6 μm thickness were stained with hematoxylin and eosin.
For immunohistochemistry, decidua at E6.5 were fixed in 4% paraformaldehyde for 6 hr, dehydrated, embedded in paraffin, and cut at 6 μm thickness. Immunostaining was performed according to the protocol described by Hogan et al. (1994). The primary monoclonal or polyclonal antibodies used are Troma-1 (1:100); SSEA-1 (1:100) (from Developmental Studies Hybridoma Bank), and anti-laminin antibody (1: 200) (Sigma). Fluorescent-conjugated goat IgG to mouse or to rabbit (1: 500) (Cappel) was used as the secondary antibody.
In situ hybridization
Two ActRIB probes, one for the extracellular domain and the other for the 3′-untranslated region, were used in detecting ActRIB transcripts. The extracellular domain fragment spanning the region between nucleotides 119 and 389 was PCR amplified, inserted into pGEM-T, and transcribed as antisense RNA from the Sp6 promoter. The 3′-untranslated region between nucleotides 1600 and 1898 in a StuI–BstXI fragment was inserted into pBluescript II SK in antisense direction and transcribed from the T3 promoter (Verschueren et al. 1995). In situ hybridization with sectioned embryos was performed basically as described by Wilkinson (1992). Slides were exposed for 2 weeks and counterstained with hematoxylin.
For analysis of T expression, whole-mount in situ hybridization with a digoxigenin-labeled antisense RNA probe transcribed from BamHI-digested plasmid DNA pSK75 was performed as described (Wilkinson 1992).
Generation and analysis of chimeric embryos
ActRIB−/− mutant ES cells were injected into blastocysts from wild-type donors or ∼15 wild-type R/S4 cells were injected into blastocysts derived from ActRIB+/− crosses. Injected blastocysts were then implanted into uteri of pseudopregnant foster mothers. Generally, the development of in vitro manipulated blastocysts is delayed by ∼1 day. Embryos were dissected at different stages and stained with X-gal as described (Hogan et al. 1994). Embryos were fixed with Bouin’s solution or 4% paraformaldehyde after staining, and dehydrated. Most chimeric embryos were embedded in paraffin and sectioned as described earlier. Some were embedded in the acrylic resin JB4 (Polysciences) and sectioned at 5 μm. Nuclear fast red was used for counterstaining. Ratios of ES cell-derived tissues (blue) to host blastocyst-derived tissues (pink) were estimated from serial sections of each embryo.
Generation of ActRIB−/− ES cells derived teratocarcinomas
To produce teratocarcinomas, ∼1 × 106 ES cells were injected subcutaneously into the flanks of Balb/cBy-nu nude mice (Jackson Laboratories). Each mouse was injected with the wild-type cells on one side and the mutant cells on the other side. Tumors were dissected between 6 and 8 weeks when they were ∼0.5–1.5 cm in diameter. Teratomas were fixed in Bouin’s fixative. Dehydration, embedding, and sectioning were performed as described above.
Acknowledgments
We thank L. Mathews for HA-tagged human ALK-4 cDNA and S. Orkins for EF-1α promoter, A. McMahon for the Brachyury (T) probe, and C. Rouws, A. Loonstra, and L. Yu for excellent technical assistance. We are grateful to E. Robertson for insightful discussion, and E. Robertson, P. Tam, H. K. Defize, C. Mummery, and K. Lawson for critical comments on the manuscript. This work was supported by grants from Bristol-Myers Squibb (E.L.) and National Institutes of Health (P.K.D.). Z.G. and B.S.S. were recipients of the National Research Service Award and M.N. was a fellow of the Japanese Society for the Promotion of Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL en@cvrv.mgh.harvard.edu; FAX (617) 726-5806.
References
- Albano RM, Arkell R, Beddington RSP, Smith JC. Expression of inhibin subunits and follistatin during postimplantation mouse development: Decidual expression of activin and expression of follistatin in primitive streak, somites and hindbrain. Development. 1994;120:803–813. doi: 10.1242/dev.120.4.803. [DOI] [PubMed] [Google Scholar]
- Armes NA, Smith JC. The ALK-2 and ALK-4 activin receptors transduce distinct mesoderm-inducing signals during early Xenopus development but do not co-operate to establish thresholds. Development. 1997;124:3797–3804. doi: 10.1242/dev.124.19.3797. [DOI] [PubMed] [Google Scholar]
- Asashima M, Nakano H, Shimada K, Kinoshita K, Ishii K, Shibai H, Ueno N. Wilhelm Roux’s Arch Dev Biol. 1990;198:330–335. doi: 10.1007/BF00383771. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL, Cheifetz S, Massagué J. Novel activin receptors: Distinct genes and alternative mRNA splicing generate a repertoire of serine/threonine kinase receptors. Cell. 1992;68:97–108. doi: 10.1016/0092-8674(92)90209-u. [DOI] [PubMed] [Google Scholar]
- Attisano L, Carcamo J, Ventura F, Weis FMB, Massagué J, Wrana JL. Identification of human activin and TGF-beta type I receptor that form heteromeric kinase complexes with type II receptors. Cell. 1993;75:671–680. doi: 10.1016/0092-8674(93)90488-c. [DOI] [PubMed] [Google Scholar]
- Beddington RS. Induction of a second neural axis by the mouse node. Development. 1994;120:613–620. doi: 10.1242/dev.120.3.613. [DOI] [PubMed] [Google Scholar]
- Beddington RSP, Robertson EJ. An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Development. 1989;105:733–737. doi: 10.1242/dev.105.4.733. [DOI] [PubMed] [Google Scholar]
- Cárcamo J, Weis FM, Ventura F, Wieser R, Wrana JL, Attisano L, Massagué J. Type I receptors specify growth-inhibitory and transcriptional responses to transforming growth factor beta and activin. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Wilson PA, Mathews LS, Hemmati-Brivanlou A. A Xenopus type I activin receptor mediates mesodermal but not neural specification during embryogenesis. Development. 1997;124:827–837. doi: 10.1242/dev.124.4.827. [DOI] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-β signaling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Barth KS, Robertson EJ. A novel retrovirally induced embryonic lethal mutation in the mouse: Assessment of the developmental fate of embryonic stem cells homozygous for the 413.d proviral integration. Development. 1991;111:969–981. doi: 10.1242/dev.111.4.969. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development. 1994;120:1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR. Signals for death and survival: A two-step mechanism for cavitation in the vertebrate embryo. Cell. 1995;83:279–287. doi: 10.1016/0092-8674(95)90169-8. [DOI] [PubMed] [Google Scholar]
- Dale L, Howes G, Price BM, Smith JC. Bone morphogenetic protein 4: A ventralizing factor in early Xenopus development. Development. 1992;115:573–585. doi: 10.1242/dev.115.2.573. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y. Intracellular signaling: The mad way to do it. Curr Biol. 1996;6:1226–1229. doi: 10.1016/s0960-9822(96)00702-6. [DOI] [PubMed] [Google Scholar]
- Dyson S, Gurdon JB. Activin signaling has a necessary function in Xenopus early development. Curr Biol. 1997;7:81–84. doi: 10.1016/s0960-9822(06)00030-3. [DOI] [PubMed] [Google Scholar]
- Ebner R, Chen RH, Lawler S, Zioncheck T, Derynck R. Determination of type I receptor specificity by the type II receptors for TGF-β or activin. Science. 1993;262:900–902. doi: 10.1126/science.8235612. [DOI] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267:246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- Fox N, Damjanov I, Martinez-Hernandez A, Knowles BB, Solter D. Immunohistochemical localization of the early embryonic antigen (SSEA-1) in postimplantation mouse embryos and fetal and adult tissues. Dev Biol. 1981;83:391–398. doi: 10.1016/0012-1606(81)90487-5. [DOI] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mouse. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Federov L, Herrenknecht K, Kemler R. Lack of β-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. The tumor suppresser gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- He WW, Gustafson ML, Hirobe S, Donahoe PK. Developmental expression of four novel serine/threonine kinase receptors homologous to the activin/transforming growth factor-beta type II receptor family. Dev Dyn. 1993;196:133–142. doi: 10.1002/aja.1001960207. [DOI] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A, Melton DA. A truncated activin receptor inhibits mesoderm induction and formation of axial structure in Xenopus embryos. Nature. 1992;359:609–614. doi: 10.1038/359609a0. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. Summary of mouse development. [Google Scholar]
- Iannaccone PM, Zhou X, Khokha M, Boucher D, Kuehn MR. Insertional mutation of a gene involved in growth regulation of the early mouse embryo. Dev Dyn. 1992;194:198–208. doi: 10.1002/aja.1001940305. [DOI] [PubMed] [Google Scholar]
- Jones CM, Lyons KM, Lapan PM, Wright CV, Hogan BL. DVR-4 (bone morphogenetic protein-4) as a posterior-ventralizing factor in Xenopus mesoderm induction. Development. 1992;115:639–647. doi: 10.1242/dev.115.2.639. [DOI] [PubMed] [Google Scholar]
- Jones CM, Kuehn MR, Hogan BL, Smith JC, Wright CV. Nodal-related signals induce axial mesoderm and dorsalize mesoderm during gastrulation. Development. 1995;121:3651–3662. doi: 10.1242/dev.121.11.3651. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. The atlas of mouse development. San Diego, CA: Academic Press; 1992. [Google Scholar]
- Kemler R, Brulet P, Schnebelen M-T, Gaillard J, Jacob F. Reactivity of monoclonal antibodies against intermediate filament proteins during embryonic development. J Embryol Exp Morphol. 1981;64:45–60. [PubMed] [Google Scholar]
- Kingsley DM. The TGF-β superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes & Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Lawson KA, Pedersen RA. Clonal analysis of cell fate during gastrulation and early neurulation in the mouse. Postimplantation development in the mouse. CIBA Found Symp. 1992;165:3–26. doi: 10.1002/9780470514221.ch2. [DOI] [PubMed] [Google Scholar]
- Lawson KA, Meneses JJ, Pedersen RA. Clonal analysis of epiblast fate during germ layer formation in mouse embryo. Development. 1991;113:891–911. doi: 10.1242/dev.113.3.891. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Liu F, Ventura F, Doody J, Massagué J. Human type II receptor for bone morphogenic proteins (BMPs): Extension of the two-kinase receptor model to the BMPs. Mol Cell Biol. 1995;15:3479–3486. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGF-β signaling: Receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- Mathews LS, Vale WW. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell. 1991;65:973–982. doi: 10.1016/0092-8674(91)90549-e. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, Vassalli A, Bickenbach JR, Roop DR, Jaenisch R, Bradley A. Functional analysis of activins during mammalian development. Nature. 1995;374:354–356. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes & Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci. 1995;92:855–859. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson EJ. In: Teratocarcinomas and embryonic stem cells: A practical approach. Robertson EJ, editor. Oxford, UK: IRL Press; 1987. [Google Scholar]
- Schulte-Merker S, Smith JC, Dale L. Effects of truncated activin and FGF receptors and of follistatin on the inducing activities of BVg1 and activin: Does activin play a role in mesoderm induction? EMBO J. 1994;13:3533–3541. doi: 10.1002/j.1460-2075.1994.tb06660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Price BM, Van Nimmen K, Huylebroeck D. Identification of a potent Xenopus mesoderm-inducing factor as a homologue of activin A. Nature. 1990;345:729–731. doi: 10.1038/345729a0. [DOI] [PubMed] [Google Scholar]
- Tam, P.P., K.A. Steiner, S.X. Zhou, and G.A. Quinlan 1997. Lineage and functional analysis of the mouse organizer. Cold Spring Harbor Symp. Quant Biol. 62 (in press). [PubMed]
- ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, Heldin CH, Miyazono K. Activin receptor-like kinases: A novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene. 1993;8:2879–2887. [PubMed] [Google Scholar]
- ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, Heldin CH. Characterization of type I receptors for transforming growth factor-β and activin. Science. 1994a;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Yamashita H, Sampath TK, Reddi AH, Estevez M, Riddle DL, Ichijo H, Heldin CH, Miyazono K. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem. 1994b;269:16985–16988. [PubMed] [Google Scholar]
- Thomsen G, Woolf T, Whitman M, Sokol S, Vaughan J, Vale W, Melton DA. Activins are expressed early in Xenopus embryogenesis and can induce axial mesoderm and anterior structures. Cell. 1990;63:485–493. doi: 10.1016/0092-8674(90)90445-k. [DOI] [PubMed] [Google Scholar]
- Thomsen GH, Melton DA. Processed Vg1 protein is an axial mesoderm inducer in Xenopus. Cell. 1993;74:433–441. doi: 10.1016/0092-8674(93)80045-g. [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Vaughan JM, Wiater E, Gaddy-Kurten D, Vale WW. Inactivation of activin-dependent transcription by kinase-deficient activin receptors. Endocrinology. 1995;136:5493–5503. doi: 10.1210/endo.136.12.7588300. [DOI] [PubMed] [Google Scholar]
- van den Eijnden-Van Raaij AJ, van Zoelent EJ, van Nimmen K, Koster CH, Snoek GT, Durston AJ, Huylebroeck D. Activin-like factor from a Xenopus laevis cell line responsible for mesoderm induction. Nature. 1990;345:732–734. doi: 10.1038/345732a0. [DOI] [PubMed] [Google Scholar]
- Varlet I, Collignon J, Robertson EJ. Nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development. 1997;124:1033–1044. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- Verschueren K, Dewulf N, Goumans M, Lonnoy O, Feijen A, Grimsby S, Spiegle KV, Dijke PT, Moren A, Vanscheeuwijck P, et al. Expression of type I and type IB receptors for activin in midgestation mouse embryos suggests distinct functions in organogenesis. Mech Dev. 1995;52:109–123. doi: 10.1016/0925-4773(95)00395-h. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG. In situ hybridization: A practical approach. London, UK: Oxford University Press; 1992. [Google Scholar]
- Wilkinson DG, Bhatt S, Herrmann BG. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature. 1990;343:657–659. doi: 10.1038/343657a0. [DOI] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes & Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sasaki H, Lowe L, Hogan BL, Kuehn MR. Nodal is a novel TGF-β-like gene expressed in the mouse node during gastrulation. Nature. 1993;361:543–547. doi: 10.1038/361543a0. [DOI] [PubMed] [Google Scholar]