

Abstract
Caspases are fundamental components of the mammalian apoptotic machinery, but the precise contribution of individual caspases is controversial. CPP32 (caspase 3) is a prototypical caspase that becomes activated during apoptosis. In this study, we took a comprehensive approach to examining the role of CPP32 in apoptosis using mice, embryonic stem (ES) cells, and mouse embryonic fibroblasts (MEFs) deficient for CPP32. CPP32ex3−/− mice have reduced viability and, consistent with an earlier report, display defective neuronal apoptosis and neurological defects. Inactivation of CPP32 dramatically reduces apoptosis in diverse settings, including activation-induced cell death (AICD) of peripheral T cells, as well as chemotherapy-induced apoptosis of oncogenically transformed CPP32−/− MEFs. As well, the requirement for CPP32 can be remarkably stimulus-dependent: In ES cells, CPP32 is necessary for efficient apoptosis following UV- but not γ-irradiation. Conversely, the same stimulus can show a tissue-specific dependence on CPP32: Hence, TNFα treatment induces normal levels of apoptosis in CPP32 deficient thymocytes, but defective apoptosis in oncogenically transformed MEFs. Finally, in some settings, CPP32 is required for certain apoptotic events but not others: Select CPP32ex3−/− cell types undergoing cell death are incapable of chromatin condensation and DNA degradation, but display other hallmarks of apoptosis. Together, these results indicate that CPP32 is an essential component in apoptotic events that is remarkably system- and stimulus-dependent. Consequently, drugs that inhibit CPP32 may preferentially disrupt specific forms of cell death.
Keywords: Apoptosis, caspase, CPP32, cell death, MEFs
Programmed cell death (PCD), or apoptosis, is a basic feature of all nucleated animal cells and is essential for normal development and tissue homeostasis (Nagata 1997). Because apoptosis is under genetic control, mutations that disrupt apoptotic programs can produce diseases. For example, inappropriate or excessive cell death can lead to neurodegenerative disorders such as Alzheimer’s or Huntington’s disease, whereas reductions in cell death contribute to lymphoproliferative disorders or neoplasia (Green and Martin 1995; Steller 1995; Goldberg et al. 1996). Many toxic agents, including most compounds used to treat cancer, induce apoptosis (Mesner et al. 1997). Hence, factors that influence apoptotic cell death may contribute to the outcome of cancer therapy. Because dysregulation of apoptosis contributes to a variety of human disorders (Thompson 1995), compounds that promote or inhibit specific forms of apoptosis may be of considerable therapeutic value.
Important insights into the molecular control of apoptosis have come from studies of the nematode Caenorhabditis elegans (Hengartner and Horvitz 1994a; Hengartner 1996). For example, loss-of-function mutations in ced9 lead to inappropriate cell death, whereas gain-of-function mutations block virtually all cell death. In contrast, loss-of-function mutations of either ced3 or ced4 result in a global suppression of cell death, implying that these genes encode key components of the cellular machinery required for PCD. Counterparts to each of these C. elegans genes have been identified in mammalian systems. bcl2 is homologous to ced9, ced3 is related to interleukin-1β converting enzyme (ICE), and recently, Apaf1 has been shown to be homologous to ced4 (Yuan et al. 1993; Hengartner and Horvitz 1994b; Vaux et al. 1996; Zhou et al. 1997). Despite this conservation, the complexity of apoptotic control in mammalian systems appears to be far greater than in C. elegans, even at the level of the central cell death machinery. bcl2 is a member of a much larger gene family that includes proteins that both promote and inhibit cell death (Reed 1997). Furthermore, at least 10 members of the ICE gene family, now called caspases, have been identified (Alnemri et al. 1996).
Considerable evidence suggests that, as in C. elegans, caspases are essential components of the mammalian cell death machinery (Steller 1995; Nicholson et al. 1995). Because the proteolytic cleavage of proteins is largely irreversible, activation of these enzymes may represent a rate-limiting step in apoptosis. Thus, elucidating the role and regulation of caspases is essential for a complete understanding of apoptosis. Nevertheless, the existence of multiple caspases, often coexpressed in the same cell type and coactivated during apoptosis, raises several fundamental questions. Do the different caspases function in a tissue-specific manner? Can they participate in some forms of cell death but not others? Do individual caspases have specific tasks in the destruction of the cell, or are they functionally redundant? Finally, from a therapeutic perspective: Would the inhibition of key caspases have global effects on apoptosis or disrupt only certain forms of cell death?
Of the caspases identified to date, CPP32 (caspase 3) is closest in sequence homology and substrate specificity to the ced3 (Xue et al. 1996). Furthermore, CPP32 has been shown to be one of the major activated caspases present in apoptotic cells, suggesting that it plays a prominent role in the cell death process (Faleiro et al. 1997). Despite this prediction, mice deficient for CPP32 were reported to have neuronal defects, but no other obvious developmental abnormalities were described (Kuida et al. 1996).
To better establish the role of CPP32 in various forms of apoptosis, we took a comprehensive approach toward evaluating the consequences of CPP32 deficiency in a variety of settings. To this end, we deleted exon 3 of the CPP32 gene and generated CPP32ex3−/− mice as well as CPP32ex3−/− embryonic stem (ES) cells and CPP32ex3−/− primary mouse embryonic fibroblasts (MEFs). We conclude that the requirement for CPP32 in apoptosis is tissue-specific and can even be stimulus-specific within the same cell type. Moreover, in select cell systems, CPP32 is strictly required for chromatin condensation and DNA degradation, but not for other features of apoptotic cell death. Consequently, these studies document the fundamental importance of CPP32 in many forms of apoptosis, and underscore the complexity of apoptotic control in mammalian systems. The remarkable variability with which inactivation of CPP32 affects apoptosis suggests that inhibitors that target CPP32—and perhaps other caspases as well—may have potential utility in blocking specific forms of cell death.
Results
Generation of CPP32ex3 mutant cells and mice
The CPP32 gene was disrupted in ES cells by use of a targeting vector that deleted exon 3 of CPP32 gene, leading to the introduction of termination codons in all three reading frames. The neomycin resistance gene (neo) was inserted in the targeting vector such that neo was flanked by 0.6 kb and 2.1 kb of genomic DNA 5′ and 3′ of CPP32 exon 3, respectively (Fig. 1A). The targeting construct was electroporated to ES cells, and three of 800 G418-resistant colonies analyzed by PCR and Southern blotting were found to be heterozygous at the CPP32 locus. Two of these ES cell clones were used to generate chimeric mice, and both successfully contributed to the germ line.
Figure 1.



Targeted disruption of the CPP32 locus. (A) A 9.4-kb HindIII DNA fragment of the mouse CPP32 wild-type locus (top). Exon (Ex) 2 and 3 (open boxes) and HindIII (H) sites are shown. The targeting construct (middle) and the position of (neo), which replaces CPP32 exon 3. neo is flanked by 0.6-kb and 2.1-kb genomic DNA at the 5′ and 3′ ends of the CPP32 locus, respectively. The recombinant CPP32ex3 allele (bottom) shows the 2.3-kb HindIII fragment present in the mutated allele. Positions of the primers and probe used to genotype the mutant are shown. (B) Southern blot analysis of representative genomic tail DNA from one litter of CPP32ex3 heterozygous intercrosses (left) and Southern blot analysis of DNA from heterozygous and homozygous CPP32ex3 ES cells. DNA was digested with HindIII and hybridized with 5′ external probe. The 9.4-kb band representative of the CPP32 wild-type (wt) and the 2.3-kb band representative of the mutant CPP32ex3 allele (mt) are indicated with arrows. (C) Western blot analysis of CPP32 expression. Cell extracts from wild-type (wt), two homozygous CPP32ex3 (mt) ES cells, and Jurkat cells were electrophoresed on a 10% SDS–polyacrylamide gel, transferred to nitrocellulose, and hybridized with anti-human CPP32 antiserum. The arrow indicates that the CPP32 band is present in the wild-type ES and Jurkat cells but not in the two CPP32ex3−/− ES cells.
Chimeric mice derived from two independent heterozygous ES clones were backcrossed to C57BL/6J mice, and heterozygous mice were crossed to produce homozygous mutant offspring. CPP32ex3−/− mice derived from the two CPP32 heterozygous ES cell clones were indistinguishable in phenotype and responses to apoptotic stimuli. The genotypes of the mice were confirmed by Southern blot analysis (Fig. 1B).
In anticipation of a potential early lethality of CPP32 mutant mice, and to study the role of CPP32 during ES cell apoptosis, we derived CPP32ex3−/− ES cells by selecting two CPP32ex3+/− clones at a higher G418 concentration and screening for loss of the wild-type allele (Nishina et al. 1997). One homozygous CPP32ex3 clone was obtained from each CPP32ex3 heterozygous ES cell line (Fig. 1B). Western blot analysis with anti-human CPP32 antiserum indicated that the 32-kD CPP32 proenzyme was expressed in wild-type ES cells but not in CPP32ex3 homozygous ES cells (Fig. 1C).
To assess the role of CPP32 in thymocyte and peripheral T cell development and apoptosis, the two CPP32ex3−/− ES clones were injected into rag2 null blastocysts to generate somatic chimeras. rag2−/− mice reconstituted with either of the CPP32ex3−/− ES cell clones showed similar results. CPP32ex3−/− and control MEFs from day 12.5 of gestation were also derived to study the role of CPP32 in the apoptosis of both normal and transformed fibroblasts.
CPP32ex3 mutation affects embryonic development and reduces mouse life span
CPP32ex3 mutant mice were born at lower than the expected Mendelian frequency (9%). Most survived until only 4–5 weeks of age. Consistent with the previously reported CPP32 mutant mice, our CPP32ex3−/− mice were smaller than their littermates, showed neurological abnormalities, and had visible masses in their heads that represent ectopic masses of supernumerary cells in place of pyknotic clusters that represent normal apoptosis that occur during brain development (Kuida et al. 1996).
Lack of expression of CPP32 does not affect immature T and B cell development
Thymi and spleens from CPP32ex3−/− mice were smaller than in the wild type in proportion to their decreased overall body size. Thymi and spleens from CPP32ex3−/− mice contained normal distributions of the expected cell populations. Consistent with a previous report, apoptosis of thymic T cells induced by CD3ε crosslinking, anti-CD95 (Fas), or dexamethasone was unaltered in the absence of CPP32, as was apoptosis in response to TNFα (data not shown).
To address the effect of CPP32 on immature B cell apoptosis, bone marrow cells from mutant and wild-type mice were treated with anti-IgM for 24 or 48 hr and the apoptosis of B220+CD43+ immature B cells was monitored with 7-Aminoactinomycin D (7-AAD) staining (Schmid et al. 1994). No apoptotic differences were observed in the treated immature B cells in the absence of CPP32 expression (data not shown). Thus, CPP32 is not required during T and B cell differentiation.
CPP32 deficient peripheral T cells are less susceptible to AICD-, CD3ε-, and CD95 (Fas)-induced apoptosis
Activated T cells must be removed by apoptosis at the end of an immune response to maintain cellular homeostasis (Akbar et al. 1997). To determine the function of CPP32 in the activation-induced cell death (AICD) of peripheral T cells, the responses of mature T cells to several stimuli were examined. In response to CDε-cross-linking, with either anti-CD28 mAb or lL2, wild-type as well as CPP32ex3−/− T cells showed significant [3H]thymidine incorporation by 3 days after stimulation. The level of [3H]thymidine incorporation in the wild type, however, dropped by day 4, but remained high in the mutant CPP32ex3−/− cells (Fig. 2A).
Figure 2.


Reduced activation induced cell death in the absence of CPP32. (A,B) [3H]Thymidine uptake and cell death of stimulated CPP32wt and CPP32ex3−/− lymphocytes. (C) [3H]Thymidine uptake of stimulated T cells from rag2−/− chimeric mice reconstituted with CPP32wt and CPP32ex3−/− ES cells. (D) [3H]Thymidine uptake of SEA-stimulated lymphocytes from wild-type and CPP32ex3−/− mice. Purified lymph node responder T cells (1 × 105 cells/well) were activated with plate-bound anti-CD3ε alone or with soluble anti-CD28 mAb or IL-2 (A,C), and SEA (D). After 3, 4, or 5 days of stimulation, [3H]thymidine uptake was determined. Similar experiments were performed with 1 × 106 lymph node cells cultured in 24-well tissue culture plates. At day 4 of stimulation, cells were harvested and the percentage of cell death was determined as indicated in Materials and Methods (B). Each panel shows results representative of at least four independent experiments. Solid bars represent CPP32wt cells; open bars represent CPP32ex3−/− cells.
To determine whether the increased [3H]thymidine incorporation observed in CPP32ex3−/− T cells was caused by a defect in cell death, the wild-type and mutant CPP32ex3−/− activated cells were stained and assessed for viability by trypan blue exclusion and by costaining with annexin V-FITC and propidium iodide (PI). Annexin V binds phophatidyl serine (PS) on the plasma membrane, allowing detection of the membrane disintegration, which is an early signature event in apoptosis (Fadok et al. 1992).
Activated CPP32ex3−/− lymphocytes showed low percentages and numbers of dying cells compared with controls when analyzed with 7-AAD staining and cell count by hemocytometer by trypan blue exclusion, respectively (Fig. 2B and data not shown). Similar results were obtained when activated T lymphocytes from rag2−/− mice reconstituted with CPP32ex3−/− ES cells were compared with activated T lymphocytes derived from rag2−/− mice reconstituted with wild-type ES cells (Fig. 2C). Thus, CPP32ex3−/− activated peripheral T cells showed higher and more persistent [3H]thymidine incorporation, likely the result of decreased cell death. Similarly, in response to superantigen staphylococcal enterotoxin A (SEA), a potent activator of T cells, CPP32ex3−/− peripheral T lymphocytes showed greater and more persistent [3H]thymidine incorporation associated with reduced cell death compared with wild-type controls (Fig. 2B,D). These results are in keeping with the recent report that shows cystein inhibitors prevent activation induced peripheral T cell apoptosis (Yang et al. 1997).
Peripheral T cells were also examined for their susceptibility to cell death by CD3ε and CD95 antibody. Activated lymphocytes were treated for 24 and 48 hr with CD3ε or CD95 antibody. Significant differences in viability were seen 24 hr after CD3ε crosslinking; (27% and 46% viable cells in CPP32ex3+/− and CPP32ex3−/− T cells, respectively). Similar differences were seen following induction of cell death with anti-CD95; (29% vs 42% viable cells in CPP32ex3+/− and CPP32ex3−/− T cells, respectively). Morphological changes were seen also that contribute to the importance of CPP32 in these cells (discussed in detail in another section of this paper). These results indicate that CPP32 makes an essential contribution to apoptosis in peripheral T cells.
CPP32 expression is not required for cytotoxic killing of target cells
Cytotoxic T lymphocytes (CTLs) lyse target cells in a contact-dependent manner with two independent mechanisms. The first involves the secretion of the pore-forming protein perforin onto the target cell, whereas the second requires the interaction of Fas ligand on the effector cell with Fas on the surface of the target cell. It is thought that secreted granzymes, a family of serine proteases colocalizing with perforin in the cytoplasmic granules of CTLs, participate in the first pathway in inducing target cell death. Granzyme B is the main candidate for this function because of its ability to process and activate several caspases in vitro, including CPP32, ICE–LAP3 (caspase 7), and ICE–LAP6 (caspase 9) (Darmon et al. 1995, 1996; Chinnaiyan et al. 1996; Duan et al. 1996; Quan et al. 1996).
To determine whether the expression of CPP32 in the target cell is required for sensitivity to the perforindependent pathway of T cell-mediated cytotoxicity, CPP32ex3−/− and heterozygous control MEFs were labeled with a LCMV–GP-derived epitope peptide and used as target cells for LCMV-specific CTLs. Both types of target cells showed a similar level of 51Cr release when incubated with CTLs from C57BL/6 mice (Fig 3). These results indicate that CPP32 is not required for target cell lysis with CTLs.
Figure 3.

Cytotoxic killing of target cells in the absence of CPP32. CPP32ex3−/− (□) and CPP32ex3 heterozygous MEFs (▪), as well as control MC57G cells (▵) were labeled with a LCMV-GP derived epitope peptide and used as target cells for LCMV-specific CTLs derived from C57BL/6 mice. Different effector/target cell ratios were tested. 51Cr release was determined after 5 hr incubation of target cells with effector cells.
CPP32 involvement in bone marrow neutrophil apoptosis
Neutrophils play a major role in inflammatory responses, and their clearance by apoptosis is required to prevent inflammatory damage of tissues (Liles 1997). To determine whether CPP32 is required for neutrophil apoptosis, bone marrow (BM) neutrophils from mutant and wild-type mice were treated with cycloheximide (CHX). CHX has been shown previously to induce neutrophil apoptosis (Tsuchida et al. 1995). CPP32ex3−/− BM neutrophils showed a significant resistance to cell death induced with CHX treatment (Fig. 4). Therefore, CPP32 expression is required for CHX-induced apoptosis in BM neutrophils.
Figure 4.

BM neutrophil apoptosis in the absence of CPP32. BM neutophils were incubated with medium alone (Con), or with CHX (1 or 10 μg/ml). After 3 hr incubation, cells were harvested and the viability of BM neutrophils was assessed by analysis of 7-AAD+ GR-1+ cells. Solid bars represent wild-type mice; open bars represent CPP32-deficient mice. Data show mean ± s.e.m. for 10 animals in each group, analyzed in three separate experiments. CPP32-deficient BM neutophils display significantly less cell death after incubation with 10 μg/ml of CHX than do wild-type cells.
CPP32-deficient ES cells are resistant to UV-irradiation and osmotic shock-induced apoptosis
The role of CPP32 in mediating apoptosis in ES cells was addressed by use of two independent CPP32ex3−/− ES clones. CPP32ex3−/− and wild-type ES cells were treated with several apoptotic stimuli, including UV-irradiation, γ-irradiation, sorbitol, and heat shock. Cell viability was monitored by either staining with annexin V and PI, or by trypan blue exclusion. No differences in apoptosis were observed between the mutant and wild-type ES cells in response to γ-irradiation (Fig. 5A) or heat shock (data not shown). In response to UV-irradiation or osmotic shock following sorbitol treatment, however, CPP32ex3−/− ES cells showed high resistance to apoptosis compared with wild-type controls (Fig. 5B,C). Western blot analysis showed that in ES cells, CPP32 is processed in response to UV-irradiation but not γ-irradiation (data not shown). These results indicate that, in ES cells, CPP32 is necessary for UV-irradiation and sorbitol induced apoptosis, but not apoptosis induced by heat shock or γ-irradiation.
Figure 5.
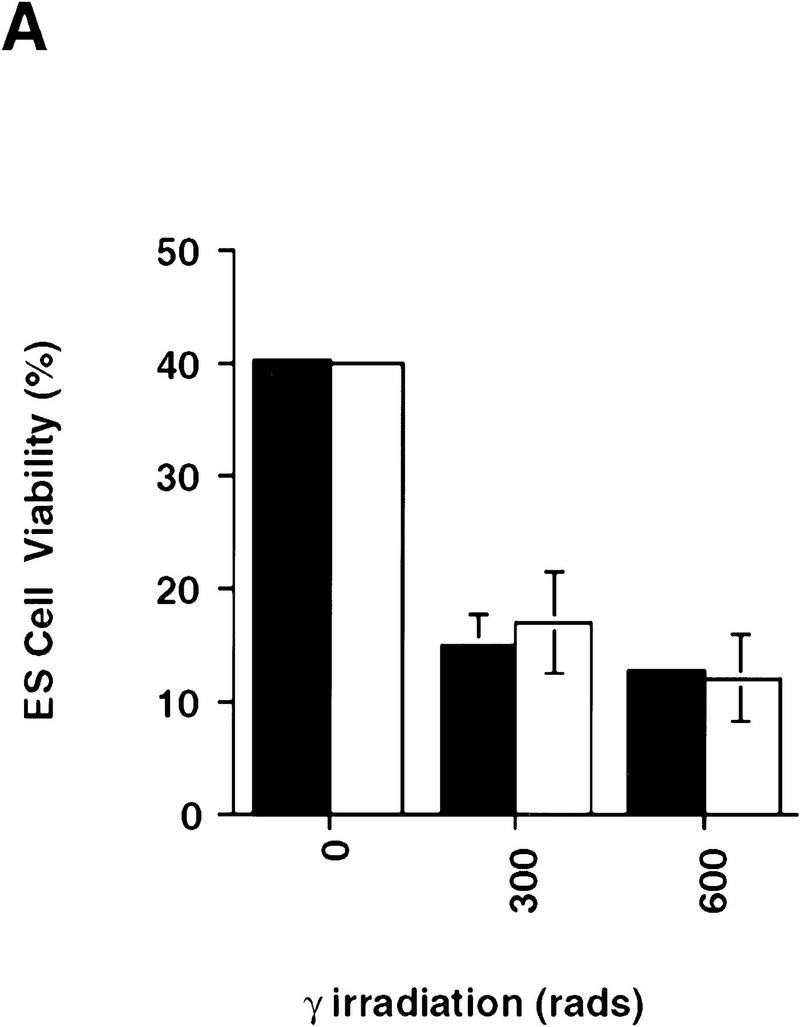
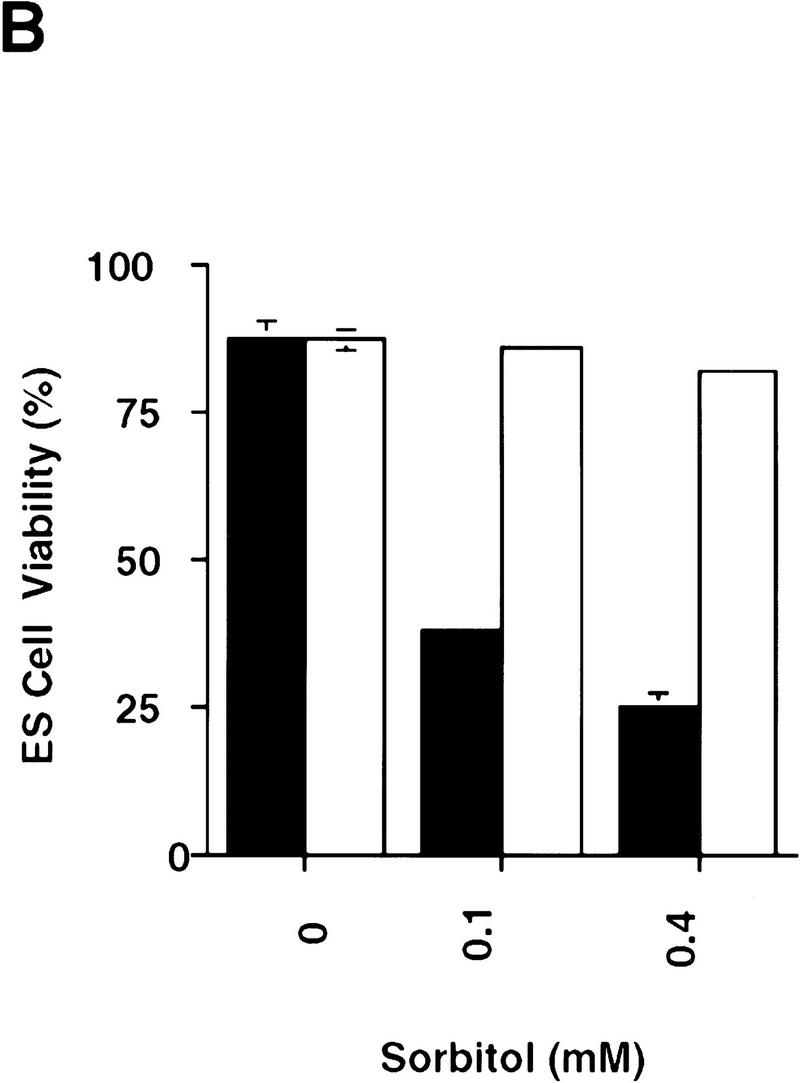
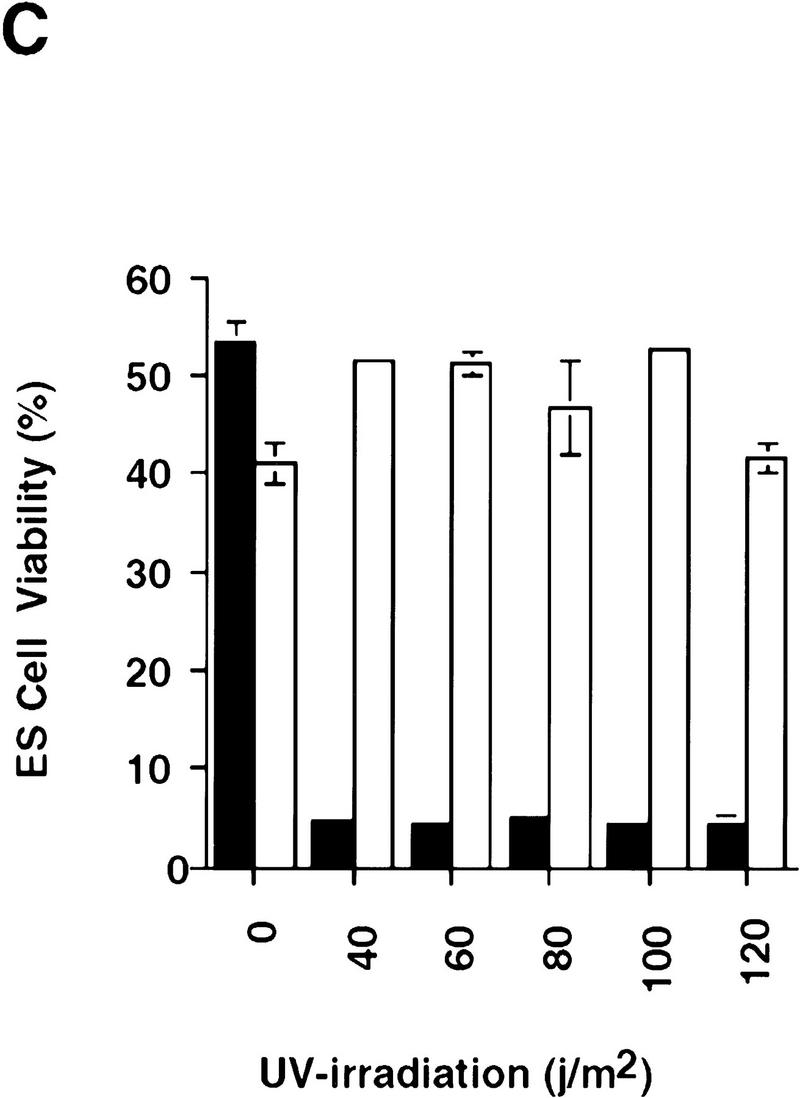
Impaired apoptosis in mutant CPP32ex3−/− ES cells. Induction of cell death in CPP32ex3+/− and two different CPP32ex3−/− ES cell clones by γ-irradiation (A), sorbitol (B), and UV irradiation (C). Unstimulated controls are also shown. Cells were harvested after the indicated stimulus and analyzed for cell viability with flow cytometry PI and Annexin V-FITC costaining. Similar results were obtained in three different experiments. (Solid bars) CPP32wt; (open bars) CPP32ex3−/−.
Reduced cell death in oncogenically transformed MEFs lacking CPP32
Many anticancer drugs exert their effects by inducing tumor cell apoptosis (Mesner et al. 1997). Therefore, the chemosensitivity of tumor cells and consequently, the ability of a genotoxic agent influence to tumor development, are controlled by a precisely organized sequence of caspase activation. To examine the requirement of CPP32 for programmed death of oncogenically transformed cells, we used an experimental system on the basis of the proapoptotic activity of E1A and ras oncogenes. Primary MEFs transformed with E1A and Ras oncoproteins become sensitive to several apoptosis-inducing stimuli, including γ-irradiation, serum withdrawal, mTNFα and several chemotherapeutic drugs (Lowe et al. 1993; Lanni et al. 1997; McCurrach et al. 1997; Samuelson et al. 1997). Therefore, by introducing E1A and Ras into MEFs obtained from control and knockout mutant mice, it is possible to examine the requirement for the deleted gene during apoptosis induced by different agents.
With this objective, early passage MEFs from wild-type and CPP32ex3−/− knockout mice were transformed with E1A and ras oncogenes by retroviral gene transfer, and the resulting cell populations were examined for viability following treatment with the apoptotic stimuli described above. As can be seen in Figure 6, both the time response (A–C) and the sensitivity (D–F) to these agents were compromised in E1A/Ras-transformed CPP32ex3−/− MEFs.
Figure 6.
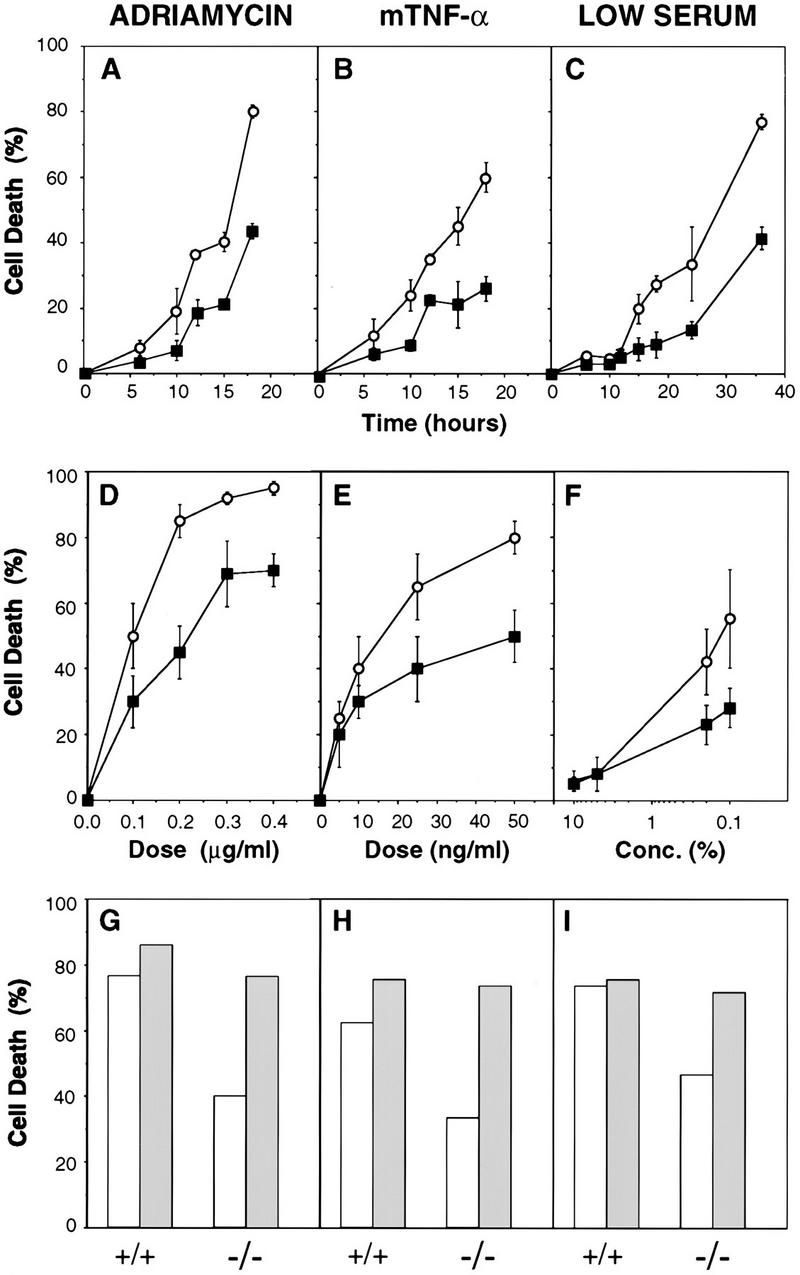
Reduced chemosensitivity of E1A/Ras-transformed MEFs in the absence of CPP32 and its restoration by reintroduction of wild-type CPP32. (A,C) Wild-type (○) and CPP32ex3−/− (▪) MEFs were treated with a single dose of adriamycin (0.2 μg/ml) (A), mTNFα (50 ng/ml) (B), or serum withdrawal (0.2% FCS) (C), and collected at the indicated times. Cell death was determined by a trypan blue exclusion assay. (D–F) Response of 13S/Ras MEFs to 24 hr treatment with increasing amounts of adriamycin (D), mTNF-α (E), and with decreasing amounts of serum (F). (○) Wild type; (▪) CPP32ex3−/−. Each point represents the mean ± s.d. from three independent experiments. (G–I) Effect of introduction of wild-type CPP32 (shaded bars) on the cell death response of the 13S/Ras MEFs. The effect of the pWZLHygro vector (open bars) on both wild-type and CPP32ex3−/− cells is also shown as a control. Data correspond to the percentage of dead cells scored at 18 hr after treatment with 0.2 μg/ml adriamycin (G), 50 ng/ml mTNFα (H), or serum withdrawal (0.2% FCS) (I).
To demonstrate that the apoptotic defects of the CPP32ex3−/− MEFs were a direct consequence of the CPP32 deficiency rather than the result of variations in the genetic background, a wild-type CPP32 gene was reintroduced into E1A/ras MEFs by retroviral gene transfer. Overexpression of CPP32 was not toxic itself, because it did not induce spontaneous cell death or any of the hallmarks of apoptosis (data not shown). On treatment with apoptotic stimuli, however, CPP32ex3−/− cells expressing exogenously transfected CPP32 died at levels similar to cells harboring only endogenous CPP32 (Fig. 6G–I). Hence, exogenous CPP32 rescued the cell death defect of CPP32ex3−/− MEFs.
Aberrant apoptosis select cell types lacking CPP32
CPP32 was initially considered the major caspase responsible for the proteolytic processing of poly(ADP–ribase) polymerase (PARP) (Nicholson et al. 1995; Tewari et al. 1995; for review, see Thornberry et al. 1997). CPP32 also has been implicated in other aspects of apoptosis, including externalization of PS, various morphological changes, and oligonucleosomal DNA degradation (Liu et al. 1997; Rudel et al. 1997). To examine the requirement for CPP32 in these processes, E1A/Ras wild type and CPP32ex3−/− MEFs were treated as indicated above, and dying cells were examined for caspase activation, PARP cleavage, changes in PS distribution, chromatin structure and DNA degradation. As a control, the same cells were treated with bleach to induce necrosis.
In wild-type MEFs expressing E1A and Ras, adriamycin treatment induced CPP32 processing (Fig. 7A) and proteolysis of PARP into 89- and 24-kD fragments (Fig. 7B). Similar results were obtained upon mTNFα and serum withdrawal (Fig. 7C; data not shown). By use of an affinity probe (biotin–DEVD–amk) known to recognize active caspases (Thornberry 1994; Takahashi et al. 1996), a dramatic decrease in total caspase activation in CPP32ex3−/− cells was observed (not shown). PARP, however, was still processed in CPP32ex3−/− cells treated with adriamycin, mTNFα, or serum deprivation to an extent proportional to the number of dead cells (Fig. 7B,C). In contrast, CPP32 was not activated on bleach treatment (Fig. 7A) and PARP was completely absent (Fig. 7B), presumably owing to general protein degradation. Thus, CPP32 is not required for PARP processing in E1A/Ras-transformed cells.
Figure 7.
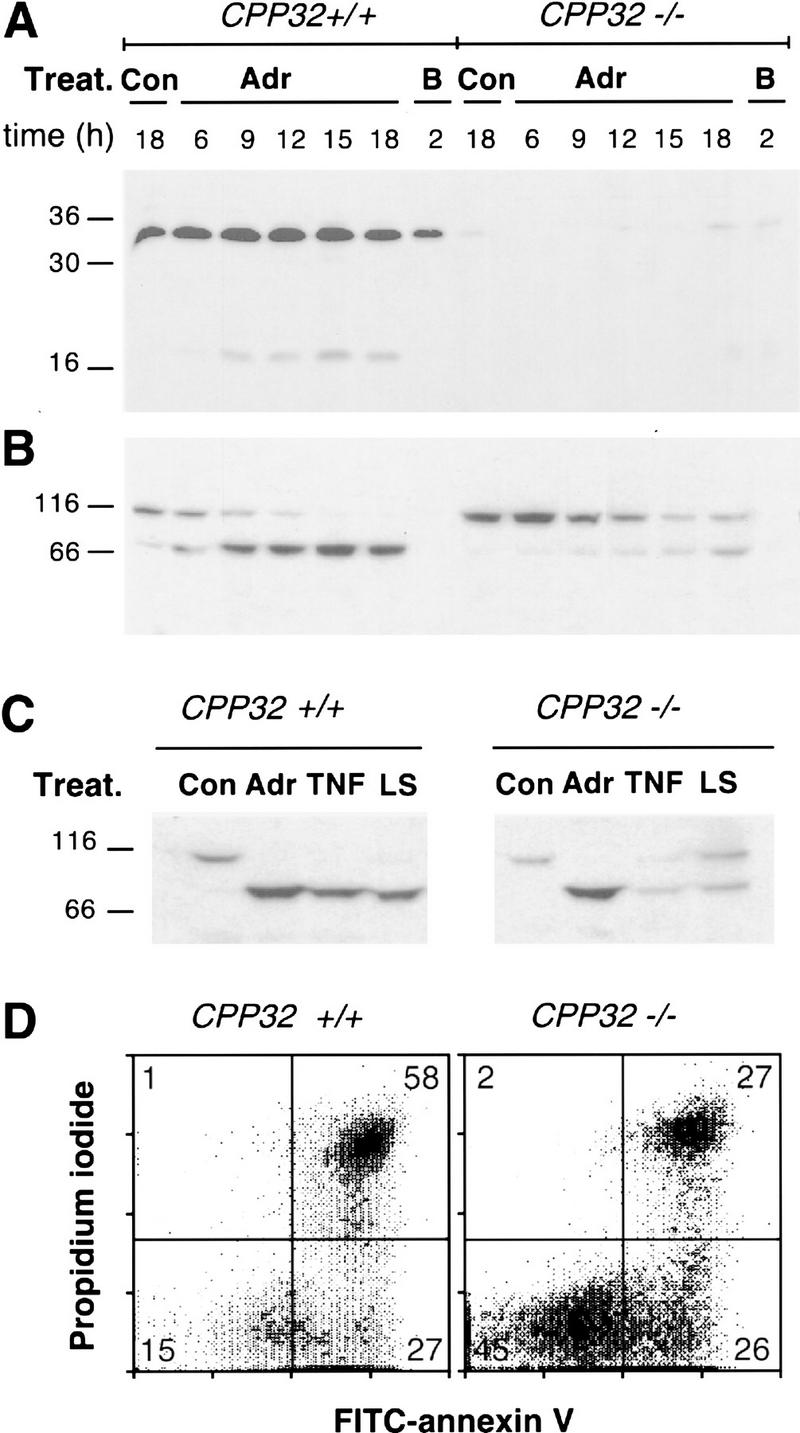
Apoptotic-like processing of PARP and loss-of-membrane phospholipid asymmetry in cell death in the absence of CPP32. (A,B) 13S/Ras MEFs of the indicated genotype were not treated as control (Con) or treated with adriamycin 0.2 μg/ml (Adr). The time courses of CPP32 processing (A) and PARP (B) are shown. As a control for events occurring in necrotic cells, both wild-type and CPP32ex3−/− MEFs were placed for 2 hr in 0.5% bleach (B). (C) CPP32 is not essential for PARP cleavage following adriamycin (0.2 μg/ml, 24 hr), mTNF-α (50 ng/ml, 24 hr) or low serum (0.2%, 36 hr) treatments of 13S/Ras MEFs. (D) Fluorescence dot blots of FITC–Annexin V and PI stained 13S/Ras wild-type and CPP32ex3−/− MEFs incubated for 24 hr in medium with 0.2% serum. In both cell types, cell death occurs with changes in the plasma membrane that lead to the exposure of phosphotydil serine, allowing interaction (and therefore, staining) with Annexin V (right). PI staining (top right) is indicative of late stages of cell death.
To assess the contribution of CPP32 to membrane changes during apoptosis, the distribution of PS was analyzed by Annexin V–FITC and PI staining. Consistent with the cell viability data, CPP32ex3−/− cells displayed an increased annexin V-negative population compared with their wild-type counterparts, (45% vs. 15 % respectively; cf. bottom left quadrants in Fig. 7D), indicating a reduction in cell death. A substantial number of CPP32ex3−/− cells, however, were Annexin V-positive but PI-negative (bottom right quadrants of Fig. 7D), indicating binding of Annexin V to cells with intact plasma membranes. This staining pattern is indicative of PS relocalization to the outer plasma membrane, a fundamental characteristic that distinguishes apoptosis from necrosis.
To examine nuclear changes during apoptosis following various apoptotic stimuli, oncogenically transformed MEFs, ES cells, and peripheral T cells were analyzed. E1A/Ras-transformed MEFs deficient for CPP32 showed marked defects in chromatin condensation and DNA degradation following treatment with various apoptotic stimuli. DAPI staining revealed that dying E1A/Ras-transformed CPP32ex3−/− cells did not display the collapsed chromatin pattern characteristic of apoptosis that was clearly evident in wild-type cells (Fig. 8A). Furthermore, dying CPP32ex3−/− MEFs displayed a complete absence of oligonucleosomal DNA fragments, or ladders when analyzed by agarose gel electrophoresis. Reintroduction of exogenous CPP32 restored normal patterns of chromatin condensation and DNA degradation in CPP32ex3−/− cells treated with apoptotic agents (Fig. 8B; data not shown), indicating that CPP32-deficiency was directly responsible for these defects.
Figure 8.

Abnormal chromatin condensation and absence of oligonucleosomal DNA degradation in dying CPP32ex3−/− 13S/Ras MEFs. (A) Fluorescence microscopy visualization of DAPI staining of DNA from wild-type (+/+), and CPP32ex3−/− (−/−) MEFs untreated control (Con) or treated with 0.2 μg/ml adriamycin (Adr) and 50 ng/ml mTNF-α for 18 hr and 0.2% serum (LS) for 36 hr. Chromatin condensation in CPP32ex3−/− did not progress to a fully collapsed state, and no apoptotic bodies were observed on any of the treatments tested. (B) DNA extracted from untreated control (Con) and adriamycin-treated (0.2 μg/ml) wild-type and CPP32ex3−/− 13S/Ras MEFs. After treatment, wild-type MEFs display the classical laddering degradation of DNA; the laddering does not occur in the CPP32ex3−/− cells.
Similar observation of nuclear changes were seen in peripheral T cells following UV irradiation and following death induction with CD3ε or Fas antibody (Fig. 9A). Both chromatin condensation and DNA laddering were not seen in untreated T cells, but following apoptosis induction with anti-CD3ε and anti-CD95, distinct chromatin condensation was seen in T cells from wild-type mice but incomplete chromatin condensation was seen in the mutant T cells (Fig. 9A,B).
Figure 9.
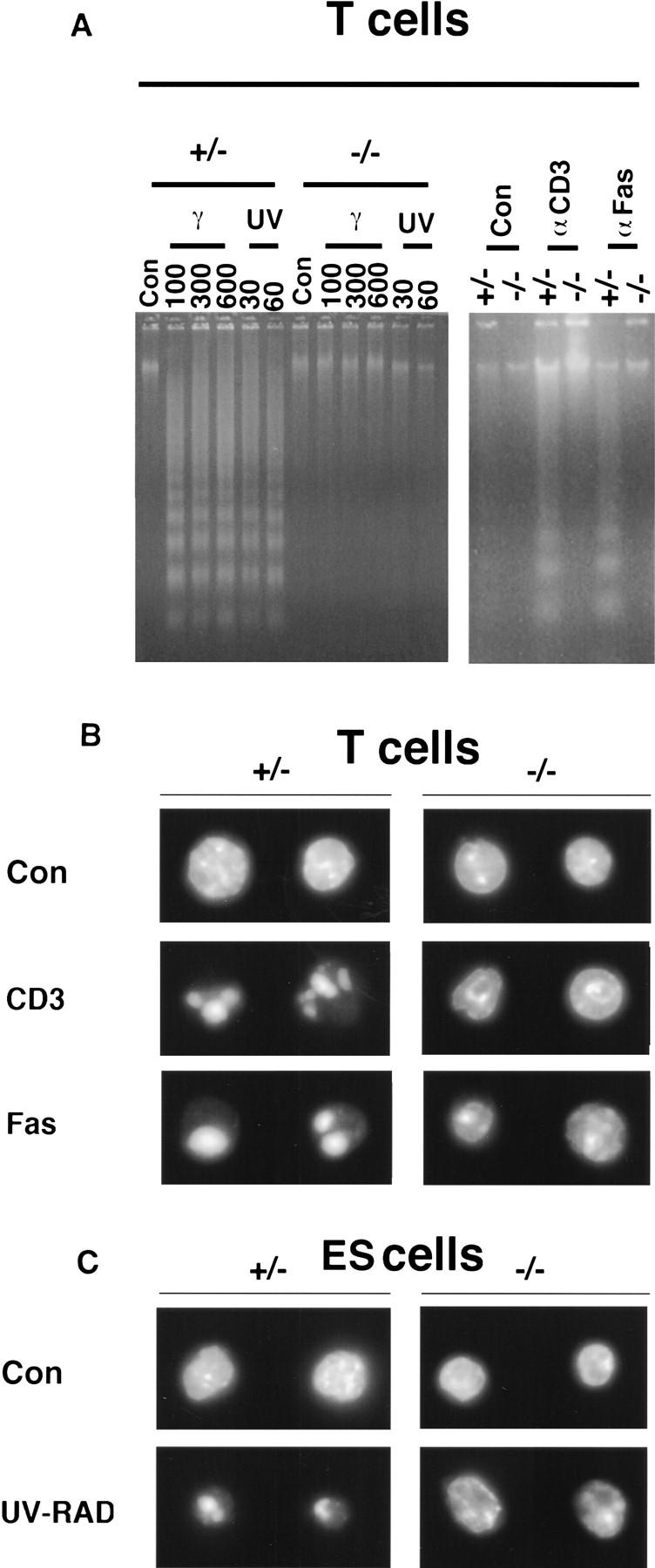
Absence of DNA laddering and abnormal chromatin condensation in dying peripheral activated CPP32ex3−/− T cells and ES cells. (A) DNA was extracted from CPP32ex3+/− and CPP32ex3−/− peripheral T cells untreated control (Con), and treated with UV- and γ-irradiation at 100, 300, and 600 rads and 30 and 60 J/m2, respectively. DNA laddering is seen in the wild-type group but not in CPP32ex3−/− T cells. Activated peripheral T cells were also treated for 48 hr with anti-CD3ε and anti-Fas antibodies. Mutant cells showed absence of DNA ladder formation following apoptotic stimuli. (B) Fluorescence microscopy visualization of DAPI staining of DNA from peripheral T cells and ES cells. CPP32ex3+/− and CPP32ex3−/− activated peripheral T cells were treated with anti-CD3ε, and anti-Fas for 48 hr and fixed in 5% paraformaldehyde and stained with DAPI. In wild-type T cells, normal chromatin condensation is seen, but in CPP32ex3−/− T cells, clear and complete chromatin condensation is not seen. CPP32ex3+/− and CPP32ex3−/− ES cells were also stained with DAPI following no treatment control (Con) and 24 hr after UV-irradiation (UV-RAD) at 60 J/m2. In untreated control (Con) section, no chromatin condensation is seen in both CPP32ex3+/− and CPP32ex3−/− group. Following UV-irradiation, chromatin is condensed in CPP32ex3+/− ES cells, but clear and complete chromatin condensation is not seen in CPP32ex3−/− ES cells.
In ES cells, CPP32 was selectively dispensable for γ- but not UV- irradiated cells as mentioned in the previous section. In keeping with this observation, chromatin condensation was seen only following γ- (data not shown) but not following UV-irradiation in CPP32ex3−/− ES cells (Fig. 9C). In CPP32ex3+/− ES cells, chromatin showed clear condensation following UV-irradiation, but in CPP32ex3−/− ES cells, there was abnormal chromatin condensation in which chromatin remained diffuse in the nucleus. As well, DNA laddering was seen only in the CPP32ex3+/− ES cells and absent in the CPP32ex3−/− ES cells (data not shown).
Electron microscopy of apoptotic MEFs and peripheral T cells confirmed that the absence of CPP32 resulted in aberrant chromatin condensation (Fig. 10 and data not shown). CPP32ex3−/− cells were highly vacuolized, contained swollen mitochondria, and appeared incapable of completely breaking down various cellular organelles. Although some of these characteristics are suggestive of necrotic cell death, the ability of CPP32ex3−/− cells to cleave PARP and redistribute PS strongly suggests that, in select cell types, CPP32 deficiency results in an aberrant form of apoptosis.
Figure 10.

Electron microscopy of dead wild-type (A) and CPP32ex3−/− (B) 13S/Ras MEFs. Cells were treated with adriamycin (0.2 μg/ml) for 24 hr and processed for electron microscopy visualization as described in Materials and Methods. Shown are thin sections of nonadherent (presumably dying) cells that become detached (dead) after treatment. Note the aberrant chromatin (ch) condensation and vacuolization of the mutant CPP32ex3−/− MEFs.
These observations seen in multiple cell systems enforce the specific role of CPP32 in specific cell systems in a stimulus-dependent manner.
Discussion
A great deal of our understanding of the regulation of apoptosis comes from studies of lower order organisms such as worms (C. elegans) and fruit flies (Drosophila melanogaster). Whereas apoptosis in these species may be induced by many different signals (White 1993), there is evidence that distinct pathways can lead to a common apoptotic program. For example, apoptosis in response to many different death-inducing signals is inhibited by the expression of Bcl2 or baculovirus p35 (Clem et al. 1991; Sugimoto et al. 1994; Armstrong et al. 1996). In addition, the Drosophila gene reaper encodes a protein capable of integrating information from different signaling pathways and converging them in the activation of what appears to be a single apoptotic program. On the basis of these and other observations, it has been proposed that the machinery for PCD is mediated by one common mechanism (Hale et al. 1996).
In the mammalian system, the complexity of the organism has required the evolution of a large number of ICE homologs as the effectors of cell death. Therefore, it is not a simple task to understand how the death machinery is regulated in higher species. Different apoptotic stimuli can have very different effects on cell growth and viability depending on the tissue origin (Kastan et al. 1992; Clarke et al. 1993; Lowe et al. 1993). Furthermore, even within the same cell type, the sensitivity to a given apoptotic stimulus can be altered by developmental stage or by oncogenic mutation (Lowe et al. 1993; Clayton et al. 1997). Despite this diversity, the morphological features of apoptotic cell death are remarkably constant, suggesting that a similar convergence of death signals on a single apoptotic program might apply equally well to mammalian systems.
CPP32 is considered to be a prototypical caspase and a key effector of PCD (Fernandes-Alnemri et al. 1994; Nicholson et al. 1995; Tewari et al. 1995; Casciola-Rosen et al. 1996). Given this view, it was surprising that an earlier report on the effects of CPP32 mutation in mice cited only defects in neuronal apoptosis (Kuida et al. 1996). In this study, we used mice, ES cells, and MEFs deficient for CPP32 to assess the role of CPP32 in apoptosis in a wide variety of biological and physiological settings. By taking a comprehensive approach, we have demonstrated the fundamental contribution of CPP32 to many forms of apoptosis. We have also demonstrated that CPP32 is involved in several other systems besides the nervous system, and that it influences the PCD of peripheral lymphoid cells, neutrophils, ES cells and embryonic fibroblasts.
The consequences of CPP32 deficiency are remarkably context-dependent, with apoptotic defects being both tissue- and stimulus-specific. Moreover, in some settings, CPP32 loss affects some hallmarks of apoptosis but not others. These findings lead us to conclude that the notion of common apoptotic machinery is too simplistic. The apoptotic machinery may be as complex and diverse as the stimuli that activate cell death.
Tissue-specific consequences of CPP32 inactivation
The expression of CPP32 in thymocytes and immature B cells, and its processing in response to several stimuli, suggested a role for this caspase in the PCD of these cells. Apoptosis of immature T and B cells has a major role in building the mature T and B cell repertoires (Smith et al. 1989; Hartley et al. 1993). Studies of the role of CPP32 in immature thymocyte apoptosis (this study and Kuida et al. 1996) as well as in immature B cell apoptosis have indicated that CPP32 is dispensable for PCD in these cells. In contrast, more differentiated cells in the T cell lineage, such as peripheral T cells, showed dependence on CPP32 expression for normal AICD. This tissue-specific requirement for CPP32 in apoptosis was also observed in ES cells, and in oncogenically transformed MEFs. Thus, in contrast to thymocytes, absence of CPP32 expression resulted in ES cells resistant to UV-irradiation-induced apoptosis. These results suggest that, in most tissues and in response to most apoptotic stimuli, there are multiple caspases present that are either redundant in function or acting in concert with one another. In certain cases, however, such as in ES cells following UV-irradiation or osmotic shock, no other caspase can compensate for the absence of CPP32, making it an indispensable caspase.
Stimuli-specific involvement of CPP32 within the same cell type
It has been demonstrated that the apoptotic machinery can be activated by increased levels of p53. DNA breaks caused by γ-irradiation, the presence of DNA repair intermediates from UV-irradiation, and other stress signals such as hypoxia or drugs have all been shown to increase levels of p53 (Lowe et al. 1993; Lee et al. 1995; Martin et al. 1995; Graeber et al. 1996). The upstream pathways leading to CPP32 processing and activation have not been well characterized, but the above findings have led to the postulation that p53 is upstream of ICE/ced3 family of proteases. In this study we show that CPP32 responds differentially to several different cell-damaging stimuli. CPP32ex3−/− ES cells were resistant to apoptosis induced by UV-irradiation but susceptible to cell death induced by γ-irradiation.
The stimuli-specific involvement of CPP32 within the same cell type was also well established by use of oncogenically transformed MEFs. In these cells, CPP32 is required for normal apoptosis following treatment with TNFα, adriamycin, or serum deprivation, but not in response to CTL killing. These results indicate that apoptosis regulation is a complex process and that upstream pathways leading to CPP32 activation can be surprisingly distinct, even in response to similar treatments.
Aberrant apoptosis select cell types
CPP32 appears to have specific tasks in apoptosis. CPP32 deficiency delays apoptosis in E1A/ras-transformed MEFs following treatment with several stimuli, and, when they did die, CPP32ex3−/−-transformed MEFs displayed some hallmarks of apoptosis but not others. In particular, PARP cleavage and PS relocalization to the outer leaflet of the plasma membrane occurred as expected, but the chromatin was neither properly condensed nor the DNA degraded. In addition, these cells appeared to be incapable of completely breaking down cellular organelles. These defects were also seen in CPP32ex3−/− peripheral T cells and ES cells following various apoptotic stimuli highlighting a consistent pattern of the important role that CPP32 has in nuclear events in apoptosis. Two proteins, DNA fragmentation factor (DFF) and p21-activated kinase 2 (PAK2), were reported recently to be targets for activated CPP32 (Liu et al. 1997; Rudel et al. 1997). These proteins are both activated in response to apoptotic stimuli and have been shown to induce DNA fragmentation. A possible defect in the expression or the activation of either DFF or PAK2 might explain the absence of nuclear condensation and DNA fragmentation in the mutant CPP32ex3 cells, and this possibility is under investigation. Other activated caspases were detected with biotinylated tetrapeptide caspase inhibitors in CPP32ex3−/− MEFs following treatment with apoptotic stimuli. Moreover, at least one other known caspase (caspase 7) is processed in CPP32-deficient cells undergoing cell death (data not shown). Taken together, these results provide the first direct evidence that, in some situations, multiple caspases work in concert to bring about the complete biochemical and morphological changes characteristic of apoptotic cell death.
Redundancy vs. specificity
CPP32 has been shown to be activated in many forms of cell death, but the fact that apoptosis can proceed in the absence of CPP32 argues that either other caspases can compensate for CPP32, or that the caspases are redundant in function. Inhibitors of the ICE–CED3 family have been shown to inhibit thymocyte apoptosis (Clayton et al. 1997), but the absence of CPP32 expression does not lead to deleterious effects in thymocytes, indicating that in these cells, other caspases can play compensatory roles. This study has revealed a large number of circumstances, however, in which a loss of CPP32 leads to defective apoptosis that cannot be relieved by other caspases, showing that the role of CPP32 in apoptosis is remarkably context-dependent. Therefore, despite the fact that most apoptotic programs produce similar morphological and biochemical endpoints, the route to these endpoints can be highly variable, even at the level of the apoptotic machinery.
Much investigation has focused on the consequences of CPP32 activation during apoptosis. Our results imply that it is not possible to generalize the role of specific caspases to all cell types or all stimuli. This specificity may have important implications for antiapoptotic therapies. In particular, because apoptosis is essential for many mammalian processes, global inactivation of the apoptotic machinery might have deleterious consequences for the entire organism. Our data argue, however, that inhibition of a central caspase can have context-dependent effects on apoptosis, raising the possibility that inhibitors of specific caspases will show specificity for certain forms of cell death.
In this study, we have shown for the first time that CPP32 is required in multiple systems and cell types in a context-specific manner. We have also demonstrated that CPP32 is specifically required for selective morphological changes during apoptosis. This CPP32 mutant animal model allows a detailed assessment of the complex cell death process and more importantly, will lead to more focused examinations and possible therapeutic studies of human diseases such as AIDS and cancer.
Materials and methods
Cells
E14K ES cells from 129/Ola mice were maintained on a layer of mitomycin C-treated embryonic fibroblasts in Dulbecco’s modified Eagle culture medium (DMEM), supplemented with leukemia inhibitory factor, 15% fetal calf serum (FCS), l-glutamine, and β-mercaptoethanol.
Generation of CPP32ex3 mutant mice
A 129/J mouse genomic library was screened with a CPP32 mouse cDNA probe. Three overlapping phage genomic clones were isolated and included CPP32 exons 2–4. A targeting vector was designed to replace a 910-bp genomic fragment containing CPP32 exons 3 with the PGKneo resistance expression cassette. The targeting vector (20 μg) was linearized with KpnI and electroporated into E14K ES cells (5 × 106) (Bio-Rad Gene Pulser, 0.34 kV, 0.25 mF). The cells were subsequently cultured in the presence of 300 μg/ml G418 (Sigma) for 10 days. Homologous recombinants were identified by PCR. An external primer specific for the CPP32 gene upstream of the targeting construct (5′-TCTATTTGTTCAGTGTTGGAT-3′) and a primer specific for the neo cassette (5′-TCATTCTCAGTATTGTTTTGC-3′) were used in the PCR analysis. Colonies positive by PCR were genotyped by Southern blotting of HindIII-digested DNA and hybridized with random hexamer [32P]dCTP-labeled DNA probes (Amersham). An EcoR1 600-bp CPP32 external probe, and a neo specific probe, were used for hybridization. Three correctly targeted colonies were identified.
Chimeric mice were produced by microinjection of targeted ES cells into 3.5-day C57BL/6J blastocysts that were transferred to CD1 pseudopregnant foster mothers. Chimeric males were mated with C57BL/6J females (Jackson Laboratories), and germ-line transmission of the mutant allele was verified by PCR and Southern blot analysis of tail DNA from F1 offspring with agouti coat color. Two injected ES colonies contributed to the germ line of mice. F2 offspring from heterozygous intercrosses were genotyped by Southern blotting. Mutant mice derived from the two targeted ES cells showed the same phenotype.
Generation of CPP32ex3 Homozygous ES cells and rag2 mutant chimera
Two different CPP32ex3+/− ES clones were cultured at an increased concentration of G418. At 0.6 mg/ml G418, >90% of the colonies died. Colonies that were resistant to 0.6 mg/ml G418 were analyzed for homologous recombination of the second allele by PCR with primers to detect the deleted portion of the CPP32 gene (5′-CCTCAGGTGCCAGTCATCCAT-3′ and 5′-CCATACATGGGAGCAAGTCAG-3′). Colonies that were homozygous by PCR for the CPP32ex3 mutation were expanded and their genotype confirmed by Southern blot analysis. The second allele was inactivated in ∼4% of ES cells resistant to 0.6 mg/ml G418. The two homozygous CPP32ex3 ES clones were injected into rag2−/− blastocysts and chimeric mice were generated as described previously.
Apoptotic induction of peripheral T cells
Activation-induced cell death
Lymph node cells and splenocytes (1 × 105) from wild-type or CPP32ex3 mutant mice were incubated in 96-well round bottom plates (Nunc, Kamptrop, Denmark) in 200 μl of HL-1 medium (Hycor Biomedical Inc, Irvine, CA) supplemented with 2 mm glutamine, 50 m β-mercaptoethanol, penicillin, streptomycin, and 2% FCS. Cells were stimulated by the addition of superantigen SEA (5 μg/ml) or 50 U/ml of recombinant mouse IL2 (Genzyme, Cambridge, MA). For stimulation with cross linked anti-CD3ε antibodies, and plates were coated with polyclonal rabbit anti-hamster antibodies (Jackson Laboratories) at 10 μg/ml in PBS at 4°C overnight. Plates were washed with PBS and incubated for 3 hr with anti-CD3ε mAb (clone: 145-2C11; PharMingen) at 1 μg/ml in PBS and washed again with PBS. After the indicated time of culture, 1 μCi/well of [3H]thymidine (NEN, Boston, MA) was added to the cell cultures. After a subsequent 12–18 hr of culture, cells were harvested (Filtermate-196 harvester; Canberra Packard, Mississauga, Canada) and [3H]thymidine incorporation was determined (Matrix-96 direct β counter, Canberra Packard).
Parallel experiments were performed with 1 × 106 lymph node cells cultured in 24 well tissue culture plates. At day 4 of stimulation, cells were harvested and percentage of cell death was determined by Annexin V and PI staining with flow cytometry. Cells were also counted with hemocytometer by trypan blue exclusion.
CD3ε- and CD95-mediated apoptosis
Lymphocytes from CPP32ex3+/− and CPP32ex3−/− mice were isolated and cultured in plate bound anti-CD3ε (10 μg/ml) for 24 hr and then cultured in Con A (10%) for 4 days. Then, the activated lymphocytes were treated with either plate bound anti-CD3ε (10 μg/ml) or anti-CD95 (1 μg/ml) for 48 hr. Cells were then analyzed for Annexin V and PI staining as well as prepared for DAPI staining and for DNA laddering as described below.
BM neutrophil apoptosis
BM cells were harvested from the femurs of wild-type or CPP32ex3−/− mutant mice by flushing with 2 ml of RPMI containing 10% FCS. Cells from each mouse were pooled, washed, and resuspended at 10 × 106 cells/ml in RPMI containing 10% FCS. BM cells (2 × 106) were incubated at 37°C in a final volume of 1 ml in round-bottomed 5-ml polypropylene tubes containing medium alone, or medium containing mTNF-α (10 ng/ml) or cycloheximide (1 μg/ml or 10 μg/ml). After 3 hr incubation, cells were harvested and the viability of BM neutrophils was assessed by analysis of 7-AAD+ GR-1+ cells. Monoclonal antibodies used for FACS analysis were Mac-1 FITC and GR-1 PE (both PharMingen). By use of a gate on Mac-1+/GR-1+ cells, the viability of BM neutrophils was determined by analysis of 7-AAD-positive cells specifically in this population.
ES cell death induction
One heterozygous and two homozygous CPP32ex3 ES cell clones (1 × 105) were plated on 1% gelatinized plates (24 well plates; Costar) in ES cell media. Twenty-four hours after the initial plating at 37°C, the following cell death stimuli were given: UV-irradiation at 40, 60, 80, 100, and 120 J/m2 with DNA Stratalinker (Stratagene), γ-irradiation at 300, 600, and 1000 rads (Gammacell 40 Exactor); 0.1 mm and 0.4 m sorbito; low serum at 0.1% FCS; and heat shock (43°C for 30 min). The same number of cells was also grown at 37°C as a control. Cell counts were determined on days 1, 2, 3, and 4 after the initial induction of cell death, by use of hemocytometer and trypan blue exclusion.
ES cells were also harvested 24 and 48 hr after the induction of cell death for FACS analysis to detect apoptotic and viable cells. ES cells were resuspended in 200 μl of PBS-1% BSA and incubated with 10 μl of Annexin V–FITC and 10 μl of PI at room temperature for 20 min. Four hundred microliters of buffer, provided by the manufacturer (R & D Systems), was added and viable cells were quantitated by flow cytometry.
Generation of CPP32ex3−/− primary MEFs
Primary MEFs derived from wild-type and CPP32ex3−/− day 12.5 embryos were prepared as described previously (Serrano et al. 1997). All cultures were maintained in DMEM (GIBCO) supplemented with 10% FCS and 1% penicillin G/streptomycin sulfate (Sigma).
Retrovirus-mediated gene transfer
Ecotropic retroviruses were produced by the Phoenix packaging line (generously provided by G. Nolan, Stanford University, CA) according to a previously described calcium phosphate precipitation method (McCurrach et al. 1997; Serrano et al. 1997). Transformation of wild-type and CPP32ex3−/− MEFs was achieved by sequential infections (Serrano et al. 1997) with the following retroviral vectors: pLPC-13S that coexpresses 13S E1A cDNA with puromycin phosphotransferase, and pWZL-Ras/CD8, coexpressing human H-ras V12 cDNA with the CD8 cell surface marker. Infected pLPC-13S cells were selected in medium containing 2.5 μg/ml puromycin (Sigma) and after infection with pWZL–Ras/CD8, cells were recovered with magnetic beads conjugated to anti-CD8 (Dynal).
For analyzing the effects of ectopic expression of CPP32, CPP32 cDNA was subcloned into pWZLHygro, a retroviral vector expressing hygromycin phosphotransferase. Selection of infected cells was performed in medium containing 100 μg/ml hygromycin B (Boehringer Mannheim).
Programmed cell death in 13S/Ras MEFs
Induction of cell death
Cells (3 × 106 to 4 × 106) were plated in 10-cm plates (2 plates per treatment). After 8 hr, cells were washed twice with PBS and treated with the indicated amounts of adriamycin (Sigma) or mTNFα (Boehringer Mannheim) in DMEM–10% FCS. The effect of serum withdrawal was analyzed by placing the cells in DMEM supplemented with different amounts of FCS. At the indicated times, adherent and nonadherent cells were pooled and prepared for the various cell death analyses (see below). Wild-type and CPP32ex3−/− E1A/Ras-transformed MEFs were treated with 0.5% bleach for the indicated time (2 hr), collected and processed for caspase labeling and Western blot analysis as indicated for adriamycin, TNF, and low serum treatments. The amount of dead cells on bleach treatment, as estimated by trypan blue exclusion, was 90 ±5% for both cell types. Nuclei analysis of bleach-treated MEFs by DAPI staining did not show apoptotic-like chromatin condensation.
Cell viability
Approximately 1 × 105 cells were collected for trypan blue exclusion analysis and scored within 1 hr after stopping treatment. At least 200 cells were counted for each point. The percentage of dead cells was also determined by flow cytometric analysis by use of PI and Annexin V–FITC costaining according to the manufacturer’s (R & D Systems) instructions.
PARP processing
Approximately 2 × 106 cells were washed with PBS, pelleted, and frozen at 70°C. Cells were lysed in NP-40% lysis buffer (1% NP-40, 150 mm NaCl, 50 mm Tris-HCl at pH 8.0, 1 mm sodium vanadate) supplemented with protease inhibitor cocktail (0.1 mm PMSF, 10 μg/ml of cytochalasin B, 2 μg/ml of chymostatin, leupeptin, antipain, and pepstatin) for 60 min on ice. Lysates were centrifuged at 10,000 rpm for 5 min at 4°C, and protein concentration was estimated by the Bio-Rad protein assay with BSA as the standard. Thirty micrograms of total protein was loaded onto 10% SDS-PAGE and transferred to an Immobilon-P membrane (Millipore). Western blot analysis was carried out according to standard procedures by ECL detection. PARP antibody and the secondary antibody, horseradish peroxidase-conjugated sheep anti-mouse, were both used at a 1:1000 dilution.
DNA fragmentation analysis
Approximately 3 × 106 treated cells were washed in cold PBS, pelleted and frozen at −70°C. Total DNA was extracted by overnight incubation at 4°C in 0.5 ml of lysis buffer (50 mm Tris-HCl at pH 7.5, 10 mm EDTA at pH 8.0, 200 mm NaCl, 1% NP-40, 50 ng/ml of freshly added proteinase K). Lysates were centrifuged at 5000 rpm for 10 min at 4°C. DNA was recovered by standard phenol–chloroform extraction and ethanol precipitation. Dry pellets were resuspended in 50 μg/ml TE–RNase and incubated at 37°C for 30 min. Samples were loaded on to a 1.5% native agarose gel and visualized by EtBr staining.
Visualization of chromatin condensation
Approximately 1 × 105 cells were fixed in 5% paraformaldehyde (Mallinckrodt) and DNA was stained with DAPI (1 μg/ml). Images of the condensation state of the chromatin were taken with a fluorescence microscope coupled to a Photometrics PXL CCD camera (Photometrics Ltd.)
Analysis of cell death by electron microscopy
Adherent cells were rinsed twice in 1× PBS, and fixed in 0.2 m cacodylate buffer/3% glutaraldehyde for 30 min. Nonadherent cells were lightly pelleted and embedded in 3% agar following fixation. The fixation was removed with three rinses in 0.2 m cacodylate. Samples were then postfixed in 1% OsO4 in 0.1 m cacodylate for 1 hr, rinsed once in dH2O, dehydrated through a graded ethanol series, and embedded in epon–araldite. Thin sections were cut on a Reichert Ultracut E, picked up on copper grids, and poststained with uranyl acetate and Reynold’s lead citrate. Grids were examined and photographed in a Hitachi H-7000 TEM.
Acknowledgments
We thank the technical support of Julia Potter, Drew Wakeham, Annick Itie, Christine Mirtsos, Antonio Ruiz de la Vera, Denis Bouchard, Suzanne Plyte, and Mary Saunders for scientific editing. Electron microscopy was done with help of D. Spector. We also thank J. Rodrigues for the CPP32 retroviral vector, Raffick Sekaly for CPP32 antibody and Y. Lazebnik for helpful advice. M.S. is supported by a postdoctoral fellowship from the Spanish Ministry of Education and Human Frontiers Science Foundation. S.W.L. is a Kimmel Scholar. This research was supported by grants CA13106 from the National Cancer Institute (S.W.L.) and by the National Cancer Institute of Canada (T.W.M).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tmak@uci.utoronto.ca; FAX (416) 204-5300.
References
- Akbar AN, Salmon M. Cellular environments and apoptosis: Tissue microenvironments control activated T-cell death. Immunol Today. 1997;18:72–76. doi: 10.1016/s0167-5699(97)01003-7. [DOI] [PubMed] [Google Scholar]
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: A fundamental principle of apoptotic. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan AM, Hanna WL, Orth K, Duan H, Poirier GG, Froelich CJ, Dixit VM. Cytotoxic T-cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr Biol. 1996;6:897–899. doi: 10.1016/s0960-9822(02)00614-0. [DOI] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Clayton LK, Ghendler Y, Mizoguchi E, Patch RJ, Ocain TD, Orth K, Bhan AK, Dixit VM, Reinherz EL. T-cell receptor ligation by peptide/MHC induces activation of a caspase in immature thymocytes: The molecular basis of negative selection. EMBO J. 1997;16:2282–2293. doi: 10.1093/emboj/16.9.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- Darmon AJ, Ley TJ, Nicholson DW, Bleackley RC. Cleavage of CPP32 by granzyme B represents a critical role for granzyme B in the induction of target cell DNA fragmentation. J Biol Chem. 1996;271:21709–21712. doi: 10.1074/jbc.271.36.21709. [DOI] [PubMed] [Google Scholar]
- Duan H, Orth K, Chinnaiyan AM, Poirier GG, Froelich CJ, He WW, Dixit VM. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem. 1996;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 β- converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nature Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Green DR, Martin SJ. The killer and the executioner: How apoptosis controls malignancy. Curr Opin Immunol. 1995;7:694–703. doi: 10.1016/0952-7915(95)80079-4. [DOI] [PubMed] [Google Scholar]
- Hale AJ, Smith CA, Sutherland LC, Stoneman VE, Longthorne V, Culhane AC, Williams GT. Apoptosis: Molecular regulation of cell death. Eur J Biochem. 1996;237:884. [PubMed] [Google Scholar]
- Hartley SB, Cooke MP, Fulcher DA, Harris AW, Cory S, Basten A, Goodnow CC. Elimination of self-reactive B lymphocytes proceeds in two stages: Arrested development and cell death. Cell. 1993;72:325–335. doi: 10.1016/0092-8674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. Programmed cell death in invertebrates. Curr Opin Genet Dev. 1996;6:34–38. doi: 10.1016/s0959-437x(96)90007-6. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994a;4:581–586. doi: 10.1016/0959-437x(94)90076-f. [DOI] [PubMed] [Google Scholar]
- ————— C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994b;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32- deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Lanni JS, Lowe SW, Licitra EJ, Liu JO, Jacks T. p53-independent apoptosis induced by paclitaxel through an indirect mechanism. Proc Natl Acad Sci. 1997;94:9679–9683. doi: 10.1073/pnas.94.18.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell. 1995;81:1013–1020. doi: 10.1016/s0092-8674(05)80006-6. [DOI] [PubMed] [Google Scholar]
- Liles CW. Apoptosis—role in infection and inflammation. Curr Opin Infect Dis. 1997;10:165–170. [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Newmeyer DD, Mathias S, Farschon DM, Wang HG, Reed JC, Kolesnick RN, Green DR. Cell-free reconstitution of Fas-, UV radiation- and ceramide-induced apoptosis. EMBO J. 1995;14:5191–5200. doi: 10.1002/j.1460-2075.1995.tb00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ, Lowe SW. Bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci. 1997;94:2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner PW, Jr, Budihardjo II, Kaufmann SH. Chemotherapy-induced apoptosis. Adv Pharmacol. 1997;41:461–499. doi: 10.1016/s1054-3589(08)61069-8. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nishina H, Fischer KD, Radvanyi L, Shahinian A, Hakem R, Rubie EA, Bernstein A, Mak TW, Woodgett JR, Penninger JM. Stress-signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- Quan LT, Tewari M, O’Rourke K, Dixit V, Snipas SJ, Poirier GG, Ray C, Pickup DJ, Salvesen GS. Proteolytic activation of the cell death protease Yama/CPP32 by granzyme B. Proc Natl Acad Sci. 1996;93:1972–1976. doi: 10.1073/pnas.93.5.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- Samuelson AV, Lowe SW. Selective induction of p53 and chemosensitivity in Rb-deficient cells by E1A mutants unable to bind the retinoblastsoma-related proteins. Proc Natl Acad Sci. 1997;94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid I, Uittenbogaart CH, Giorgi JV. Sensitive method for measuring apoptosis and cell surface phenotype in human thymocytes by flow cytometry. Cytometry. 1994;1:12–20. doi: 10.1002/cyto.990150104. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Smith CA, Williams GT, Kingston R, Jenkinson EJ, Owen JJ. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature. 1989;337:181–184. doi: 10.1038/337181a0. [DOI] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Sugimoto A, Friesen PD, Rothman JH. Baculovirus p35 prevents developmentally programmed cell death and rescues a ced-9 mutant in the nematode Caenorhabditis elegans. EMBO J. 1994;13:2023–2028. doi: 10.1002/j.1460-2075.1994.tb06475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Earnshaw WC. ICE-related proteases in apoptosis. Curr Opin Genet Dev. 1996;6:50–55. doi: 10.1016/s0959-437x(96)90010-6. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thornberry NA. Interleukin-1 beta converting enzyme. Methods Enzymol. 1994;244:615–631. doi: 10.1016/0076-6879(94)44045-x. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Tsuchida H, Takeda Y, Takei H, Shinzawa H, Takahashi T, Sendo F. In vivo regulation of rat neutrophil apoptosis occurring spontaneously or induced with TNF-alpha or cycloheximide. J Immunol. 1995;154:2403–2412. [PubMed] [Google Scholar]
- Vaux DL, Strasser A. The molecular biology of apoptosis. Proc Natl Acad Sci. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Death-defying acts: A meeting review on apoptosis. Genes & Dev. 1993;7:2277–2284. doi: 10.1101/gad.7.12a.2277. [DOI] [PubMed] [Google Scholar]
- Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes & Dev. 1996;10:1073–1083. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- Yang Y, Liu ZH, Ware CF, Ashwell JD. A cysteine protease inhibitor prevents activation-induced T-cell apoptosis and death of peripheral blood cells from human immunodeficiency virus-infected individuals by inhibiting upregulation of Fas ligand. Blood. 1997;89:550–557. [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zhou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]