

Abstract
Bone resorption and remodeling is an intricately controlled, physiological process that requires the function of osteoclasts. The processes governing both the differentiation and activation of osteoclasts involve signals induced by osteoprotegerin ligand (OPGL), a member of tumor necrosis factor (TNF) superfamily, and its cognate receptor RANK. The molecular mechanisms of the intracellular signal transduction remain to be elucidated. Here we report that mice deficient in TNF receptor-associated factor 6 (TRAF6) are osteopetrotic with defects in bone remodeling and tooth eruption due to impaired osteoclast function. Using in vitro assays, we demonstrate that TRAF6 is crucial not only in IL-1 and CD40 signaling but also, surprisingly, in LPS signaling. Furthermore, like TRAF2 and TRAF3, TRAF6 is essential for perinatal and postnatal survival. These findings establish unexpectedly diverse and critical roles for TRAF6 in perinatal and postnatal survival, bone metabolism, LPS, and cytokine signaling.
Keywords: TRAF6, osteopetrosis, lipopolysaccharide, interleukin-1, CD40
Members of the tumor necrosis factor receptor (TNFR) superfamily are known to mediate important and diverse physiological functions, including apoptosis, osteoclastogenesis, and immune system regulation (Liu et al. 1996; Anderson et al. 1997; Lacey et al. 1998). The specific biological outcomes governed by these cell surface receptors are dependent not only on stimulation by their cognate ligands but also on various cytoplasmic signal transducing proteins (Liu et al. 1996; Arch et al. 1998). TNFR-associated factors (TRAFs) have been implicated in mediating signals induced by a subset of TNFR family members, including TNFR1, TNFR2, LTβR, CD30, and CD40 (Arch et al. 1998). Members of the TRAF family of proteins are characterized by a conserved carboxy-terminal TRAF-C domain, a coiled–coil TRAF-N domain and, excluding TRAF1, an amino-terminal ring finger domain (Rothe et al. 1994; Cao et al. 1996b). The TRAF-C domain mediates interactions among TRAF proteins as well as their association with members of the TNFR superfamily. The ring finger domain is thought to be essential for downstream signaling (Rothe et al. 1995; Cao et al. 1996a; Takeuchi et al. 1996).
Several members of the TRAF family, including TRAF2, TRAF5, and TRAF6 have been implicated in regulating signals from various TNFR family members, leading to the activation of nuclear factor-κ B (NF-κB) (Cheng et al. 1995; Rothe et al. 1995; Ishida et al. 1996a,b; Nakano et al. 1996; Hsu et al. 1997; Marsters et al. 1997; Song et al. 1997; Muzio et al. 1998). NF-κB is a ubiquitously expressed transcription factor that controls a plethora of genes, including those involved in regulating apoptosis, immune responses, and embryonic development. For example, signaling of CD40, a receptor expressed on B lymphocytes and certain accessory cells such as dendritic cells (Banchereau et al. 1994), involves the recruitment of TRAF1, TRAF2, TRAF3, TRAF5, and TRAF6 (Pullen et al. 1998). Although the physiological roles of individual TRAF proteins in CD40 signaling remain to be clarified, ex vivo data have suggested a potentially distinguished role for TRAF6. Of the six TRAFs described to date, TRAF6 shows the least homology to the prototypical TRAF domain sequence (Cao et al. 1996b). TRAF6 binds to a cytoplasmic domain of CD40, which is distinct from that containing the binding sites for TRAF1, TRAF2, TRAF3, and TRAF 5 (Ishida et al. 1996a). Furthermore, the TRAF6-binding region is thought to be required for CD40-mediated NF-κB activation (Ishida et al. 1996a).
Another unique feature of TRAF6 is that unlike other TRAFs, it has also been implicated in interleukin-1 (IL-1) signaling (Cao et al. 1996b), leading to the activation of NF-κB. Overexpression of a dominant-negative mutant of TRAF6 in human 293 cells was found to inhibit IL-1-induced NF-κB activation (Cao et al. 1996b). IL-1 is a multifunctional, pro-inflammatory cytokine, which can signal through the IL-1 type 1 receptor (IL-1R1) (Dinarello 1996). Key players involved in this transduction pathway have been identified recently (Huang et al. 1997; Malinin et al. 1997; Muzio et al. 1997; Wesche et al. 1997a), and TRAF6 mediates the signal by interacting with the IL-1-receptor-associated kinase (IRAK) (Cao et al. 1996a; Muzio et al. 1997; Wesche et al. 1997b).
To date, the physiologic roles of two TRAF family members have been determined by gene targeting (Xu et al. 1996; Yeh et al. 1997). Both TRAF2- and TRAF3-deficient mice appear normal at birth but become progressively runted and die prematurely. TRAF2-deficient mice have elevated levels of serum TNF. Ex vivo assays demonstrated that TRAF2−/− cells were highly sensitive to TNF-induced cell death and, in the absence of TRAF2, TNF-mediated JNK/SAPK activation was greatly reduced (Yeh et al. 1997). On the other hand, reconstitution of mice with TRAF3−/− fetal liver cells revealed a requirement for TRAF3 in T-dependent immune responses (Xu et al. 1996).
To investigate the physiologic role of TRAF6, we have generated TRAF6-deficient mice. Analyses of these mutant mice indicate that TRAF6 is essential for both perinatal and postnatal survival. Surprisingly, TRAF6 mutant mice exhibit osteopetrosis. Furthermore, TRAF6-deficient cells showed defective responses when treated with lipopolysaccharide (LPS). Our results not only confirm the crucial role of TRAF6 in IL-1 and CD40 signaling but also reveal an unexpected requirement for TRAF6 in both LPS signaling and bone metabolism.
Results
Generation of TRAF6-deficient mice
The murine traf6 gene was disrupted by introducing a targeted mutation into embryonic stem (ES) cells of 129J background (Fig. 1A). Two independent heterozygous (TRAF6+/−) mutant ES cell lines were microinjected into C57BL/6 (B6) blastocysts, which were subsequently implanted into pseudopregnant CD-1 (ICR) recipients. Male chimeras generated from both cell lines were used to generate TRAF6+/− mice, which were intercrossed to produce TRAF6−/− mice. Homozygous mutants derived from either ES cell line had identical phenotypes. Homologous recombination was confirmed by Southern blot analysis (Fig. 1B). It has been shown previously by Northern blot analysis that TRAF6 is highly expressed in murine kidney (Ishida et al. 1996a). Western blot analysis using a TRAF6-specific antibody showed that TRAF6 protein levels were undetectable in lysates of TRAF6−/− kidney, indicative of a null mutation (Fig. 1C).
Figure 1.

Disruption of the murine traf6 gene by homologous recombination. (A). Restriction enzyme maps of the 5′ region of the endogenous traf6 locus (top) and the targeting construct (middle) are shown. The targeting vector was engineered to replace the exon containing the ATG translation initiation codon with PGK-neo in the opposite orientation (bottom). This exon encodes the first 100 amino acids of TRAF6, representing a portion of the amino-terminal ring-finger domain. EcoRI digestion generates a 4.3-kb wild-type band and a 2.6-kb mutant band. The flanking region (FR) probe used for Southern blot analysis is represented by an oval. (E) EcoRI; (N) NotI. (B) Southern blot analysis of genomic DNA from primary embryonic fibroblasts harvested from E14.5 wild-type (+/+), heterozygous (+/−) and homozygous (−/−) TRAF6 embryos. DNA was digested with EcoRI and hybridized with radiolabeled FR. Arrows indicate the 4.3-kb wild-type (WT) and the 2.6-kb mutant (MT) bands. (C) Immunoprecipitation and Western blot analysis of TRAF6 protein. TRAF6 was immunoprecipitated from total kidney lysate of TRAF6+/+, TRAF6+/−, and TRAF6−/− mice using polyclonal anti-TRAF6 antibody raised against the full-length TRAF6 protein. Western blots of both total lysates (left) and immunoprecipitates (right) were probed with the same antibody. The arrow indicates the presence of the 60-kD TRAF6 protein in +/+ lysates, a reduced level of TRAF6 in +/− lysates, and the absence of TRAF6 in −/− lysates.
TRAF6 is required for perinatal and postnatal survival
When the first 327 live pups of heterozygous intercrosses were examined, only 11% were genotyped as TRAF6−/− (Table 1). However, a normal Mendelian ratio of TRAF6−/− mutants prevailed in embryos examined at day 14.5 to day 17.5 postconception (Table 1). Viable TRAF6−/− mice appeared normal at birth but failed to thrive normally and died prematurely. The few TRAF6−/− mice that survived >2 weeks exhibited 20%–30% deficits in both body mass and length (not shown). Although the mutant heart and liver were moderately enlarged (1.3-fold), the spleen was two to six times larger than that of control littermates. TRAF6−/− mice also exhibited moderate normocytic, hypochromic anemia (not shown).
Table 1.
Genotypic analysis of offspring derived from TRAF6 heterozygous intercrosses
| Stage
|
No. per genotype
|
||||
|---|---|---|---|---|---|
| +/+
|
+/−
|
−/−
|
Total
|
Percentages −/−
|
|
| E14.5 | 5 | 6 | 4 | 15 | 27 |
| E17.5 | 8 | 17 | 7 | 32 | 22 |
| 2 weeks old | 104 | 187 | 36 | 327 | 11 |
Embryos of various stages of gestation were obtained by setting up timed breedings between TRAF6+/− males and females (129 × B6 background). E14.5 was defined as 14.5 days after conception. Viable pups (n = 327) were genotyped ∼2 weeks after birth. Genotypes were determined primarily by PCR analysis. To confirm the validity of the PCR results, genomic Southern blots were performed in parallel for the first several litters.
TRAF6-deficient mice are osteopetrotic
A surprising phenotypic finding was that TRAF6−/− mice were osteopetrotic. Radiographic analysis of TRAF6−/− mice showed that their long bones and vertebral bodies were radio-opaque. The long bones, especially the femur, were reduced in length and exhibited a distinct broadening at the ends attributable to a failure in bone modeling (Fig. 2A, arrow). Molars and incisors of all TRAF6−/− mice examined had failed to erupt (Fig. 2A, arrowheads), a typical finding in osteopetrosis, as bone resorption is required to open an avenue through the jawbone for teeth to erupt. Peripheral quantitative computed tomography (pQCT) measurement of the proximal tibial bone metaphysis showed elevations in total [266 ± 18 mg/cm3 (TRAF6−/−, n = 6) vs. 234 ± 7 mg/cm3 (TRAF6+/−, n = 4)] and trabecular (269 ± 24 vs. 200 ± 23 mg/cm3) bone mineral density. The marrow space in the femoral shaft was primarily filled with bony trabeculae and large amounts of hypertrophic cartilage (Fig. 2B), also characteristic of osteopetrosis and failed bone resorption. In contrast to the long bones, bones that develop by intramembranous formation, such as the skull, appeared radiographically normal in TRAF6−/− mice. Biochemical analysis of serum revealed normal levels of calcium but reduced levels of phosphate compared to control littermates (data not shown).
Figure 2.
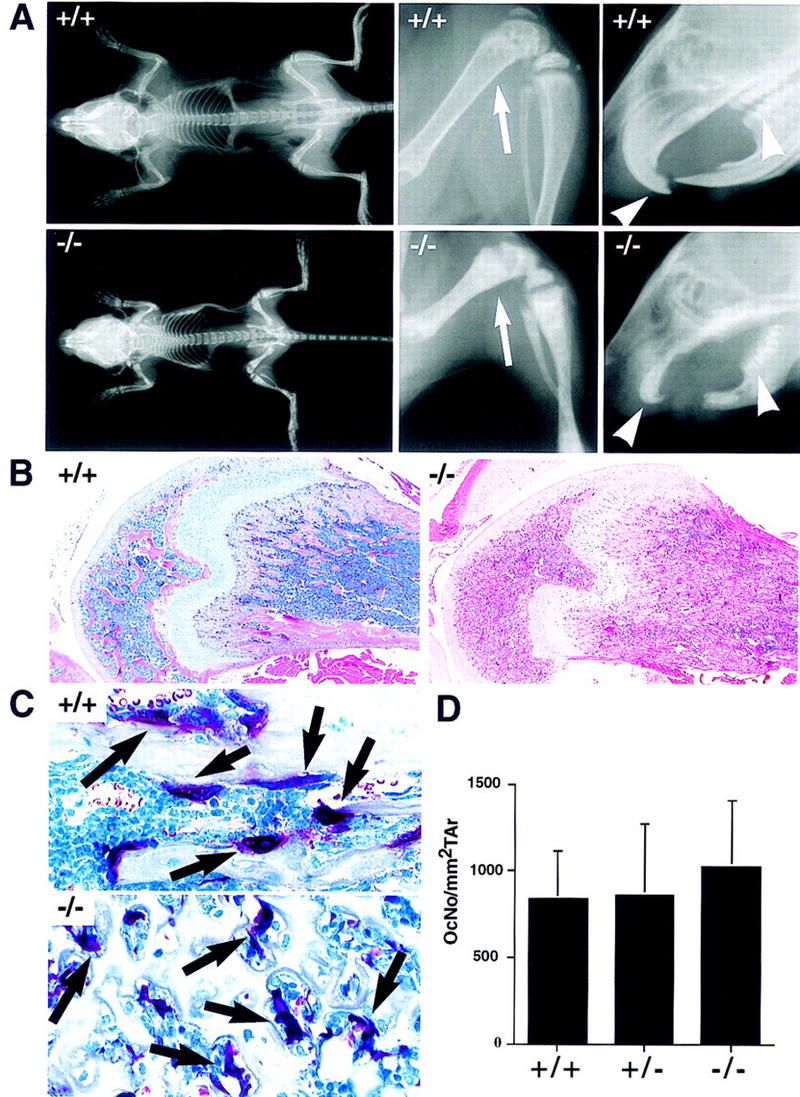
Severe osteopetrosis in the presence of TRAP+ osteoclasts in TRAF6−/− mice. (A) X-ray analysis of the bone density of a 4-week-old TRAF6 knockout (−/−) and a wild-type littermate (+/+) mouse. A complete scan of the skeletons of both mice, including their femurs, tibia/fibulas, and facial regions was performed. Note the increase in overall bone density in the TRAF6−/− mouse, the shortening of its long bones, the enlargement of its metaphyses (arrow), as well as the absence of incisors and molars (arrowheads) in its oral cavity. Skeletal structure and morphogenesis of flat bones appears normal. (B) H&E staining depicting histological changes in bone. The shaft of the femur in TRAF6−/− mice is filled with cartilage and bone. There is some evidence of periosteal bone modeling occurring adjacent to the growth plates. Magnification, 4×. (C) TRAP staining. Presence of multinucleate TRAP+ osteoclasts (arrows) in TRAF6−/− mice. Most TRAF6−/− osteoclasts are withdrawn from the bone surface, whereas osteoclasts in wild type mice are attached to the bone. Magnification 60×. (D) The number of TRAP+ osteoclasts per mm2 tissue area in TRAF6−/−, TRAF6+/−, and TRAF6+/+ mice is comparable.
Examination of osteoclasts in TRAF6−/− mice showed that the number of tartrate-resistant acid phosphatase (TRAP)-positive cells per square millimeter of tissue area was comparable in wild-type, heterozygous, and knockout mice (Fig. 2D). Interestingly, TRAP+ TRAF6−/− osteoclasts lacked contact with bone surfaces (Fig. 2C, arrows). This observation was confirmed by electron microscopy, which showed that most TRAF6−/− osteoclasts were withdrawn from the bone surface (Fig. 3, cf. A and B). A few cells appeared to form structures reminiscent of attachment zones (Fig. 3C, asterisk) and disorganized ruffled borders (Fig. 3C, large arrowhead), resulting in small areas of bone resorption (Fig. 3C, arrow). [Ruffled borders are specialized membranous areas of the osteoclast, which allow it to attach to the bone matrix and secrete hydrogen ions and digestive enzymes that resorb bone (Baron et al. 1993).] However, even the attached osteoclasts appeared unable to resorb significant amounts of bone (Fig. 3C, row of arrowheads), suggesting that a defect in osteoclast function, rather than differentiation, was responsible for the osteopetrosis observed in TRAF6-deficient animals. These results point to an unexpected role for TRAF6 in bone metabolism.
Figure 3.
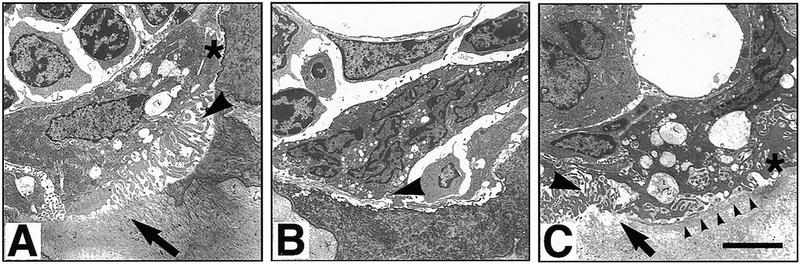
Electron microscopy of osteoclasts in TRAF6−/− and TRAF6+/− mice. (A) TRAF6+/− osteoclast seen in a resorption lacuna. The depicted cell exhibits an attachment zone (asterisk), cytoplasmic vacuolization, and ruffled border (arrowhead), features of a normally activated, mineral-resorbing (arrow) osteoclast. (B) This cell illustrates the typical osteoclast in TRAF6−/− mice. There is no evidence of activation or mineral resorption, the cell forms no attachment zone or ruffled border (arrowhead), and is in limited contact with the underlying bone. (C) One of the few activated osteoclasts in a TRAF6−/− mouse. The cell is in contact with the mineralized bone, forms an adhesion zone (asterisk), and has a partial and disorganized ruffled border (large arrowhead). Some resorption is occurring as evidenced by the dissolved bone material (arrow) and the cytoplasmic vacuolization of the cell. However, on most of the bone surface covered, there is no ruffled border formation and no bone resorption (line of arrowheads). Size bar, 5 μm.
Impaired CD40 and LPS-induced proliferation of TRAF6-deficient B lymphocytes
To assess the physiologic role of TRAF6 in CD40 signaling, we first examined the proliferation of splenocytes enriched for B cells stimulated with an agonistic rat anti-mouse CD40 monoclonal antibody, recombinant murine IL-4, or LPS for 2, 3, or 4 days. In the absence of TRAF6, anti-CD40-stimulated B cells failed to proliferate (Fig. 4A). The original experimental design included LPS as a positive control for B cell proliferation. Quite unexpectedly, LPS-stimulated proliferation was inhibited dramatically in TRAF6−/− B cells (Fig. 4A). In contrast, TRAF6 does not appear to be involved in T cell responses, as no significant differences from the wild type in the proliferation of TRAF6−/− T cells stimulated with anti-CD3ε, anti-CD3ε plus anti-CD28, staphylococcal enterotoxin A (SEA), or concanavalin A were observed (data not shown).
Figure 4.

Impaired proliferation, induction of iNOS, and LPS signaling in TRAF6-deficient cells. (A) Proliferation index of enriched (see Materials and Methods) splenic B cells from wild-type (█) or TRAF6−/− (□) mice (A–C), determined by calculating the ratio of [3H]thymidine uptake (cpm.) in treated cells vs. that in untreated cells. Cells were stimulated with anti-CD40, IL-4, anti-CD40 plus IL-4 or LPS for 72 hr. [3H]Thymidine uptake in both wild-type and TRAF6−/− cells treated with an isotype control was not significantly different from that in untreated cells (not shown). Because of the young age of the mice, proliferation in response to IgM cross-linking could not be determined. Data shown are representative of three independent experiments. (B) Induction of iNOS in thioglycollate-elicited PMφ macrophages treated for 24 hr with 50 U/ml mIFNγ, 50 U/ml mIFNγ plus 10 ng/ml mIL-1β, or 50 U/ml mIFNγ plus 10 ng/ml mTNFα. Supernatants were assayed for nitrite (see Materials and Methods). Data shown represent the mean nitrite production ± s.e.m. for TRAF6−/− (n = 8) and wild-type littermates (n = 10) analyzed in five independent experiments. (C) Induction of iNOS in BMMφ macrophages treated for 48 hr with increasing concentrations of LPS, 50 U/ml mIFNγ, or 50 U/ml mIFNγ plus 10 ng/ml mTNFα. Supernatants were assayed for nitrite, as in B. Data show the mean ± s.e.m of triplicate samples from one experiment representative of three independent trials. (N.D.) Nondetectable.
Impaired activation of inducible nitric oxide synthase in TRAF6-deficient macrophages
To further investigate the role of TRAF6 in LPS and cytokine signaling, thioglycollate-elicited peritoneal (PMφ) and naive bone marrow (BMMφ) macrophages derived from TRAF6−/− and wild-type littermates were harvested and treated with either IL-1β, IFNγ, or TNFα in various combinations or with increasing doses of LPS. Treatment of wild-type PMφ macrophages with TNFα (not shown), IL-1β, or IFNγ alone does not induce iNOS (inducible nitrous oxide synthase) significantly; however a dramatic synergistic induction of iNOS activity occurred when the cells were treated with either IL-1β or TNFα in combination with IFNγ (Fig. 4B). PMφ macrophages derived from TRAF6−/− mice were unresponsive to IL-1β but responded like the wild type to TNFα plus IFNγ (Fig. 4B). iNOS was induced in a dose-dependent manner in wild-type BMMφ macrophages following treatment with increasing concentrations of LPS (Fig. 4C, left). In contrast, TRAF6−/− BMMφ macrophages were impaired in their ability to induce iNOS, even at high doses of LPS (Fig. 4C, left). This defect was specific to the LPS signaling pathway, because the combination of TNFα plus IFNγ was able to activate iNOS in TRAF6−/− BMMφ macrophages (Fig. 4C, right). Furthermore, TRAF6−/− peripheral blood leukocytes (PBLs) were impaired in their ability to secrete TNFα in response to LPS treatment (not shown). Taken together, these results clearly demonstrate an unexpected requirement for TRAF6 in LPS signaling.
TRAF6 is required for IL-1, CD40, and LPS-induced activation of NF-κB and IL-1-mediated JNK/SAPK activation
Because signaling through IL-1, CD40, or LPS ultimately leads to NF-κB activation, we examined the activation of this transcription factor in various cell types. NF-κB activation was determined in primary embryonic fibroblasts (EFs), splenocytes, and Abelson-transformed pre-B cell lines derived from TRAF6−/− and wild-type mice. NF-κB activation in EF cells was induced by treatment with either IL-1β or TNFα, whereas splenocytes and Abelson-transformed pre-B cells were stimulated with anti-CD40 or LPS. Electrophoretic mobility shift assays (EMSAs) demonstrated that NF-κB activation was impaired in TRAF6−/− EF cells or splenocytes treated with either IL-1β or CD40 but not in mutant cells treated with TNFα (Fig. 5A,B). LPS-induced NF-κB activation in TRAF6−/− Abelson pre-B cells was reduced significantly compared to the wild type (Fig. 5C).
Figure 5.

NF-κB and JNK/SAPK activation in TRAF6-deficient fibroblasts and Abelson-transformed pre-B cells. (A) TRAF6−/− and wild-type EF cells were incubated with 10 ng/ml mIL-1β or mTNFα for the indicated time periods. Equivalent amounts of nuclear extract protein were incubated with a radiolabeled probe containing NF-κB binding sites. Activation of NF-κB was determined using EMSA as described in Materials and Methods. (B) NF-κB activation in response to anti-CD40. Naive splenocytes from TRAF6−/− and wild-type mice were incubated with an agonistic rat anti-mouse CD40 monoclonal antibody (5 μg/ml) or an isotype control (5 μg/ml, for 30 min) (denoted i) for the indicated time periods. Activation of NF-κB was determined as in A. Similar results were obtained using two independent TRAF6−/− and TRAF6+/+ Abelson-transformed pre-B cell lines (not shown). (C) TRAF6-deficient and wild-type Abelson-transformed pre-B cells were incubated with 20 μg/ml LPS for the indicated time points. Activation of NF-κB was determined as in A. (D) JNK/SAPK activation. TRAF6-deficient and wild-type EF cells were incubated with 10 ng/ml IL-1β or 10 μg/ml anisomycin for 30 min (denoted A) for the indicated time points. Activation of SAPK/JNK was determined using a kinase assay as described in Materials and Methods.
Although overexpression of TRAF6 was shown previously to activate JNK/SAPK (Song et al. 1997), a possible role for TRAF6 in IL-1-mediated JNK/SAPK activation has not been demonstrated clearly (O’Neill and Greene 1998). Here we show an absence of JNK/SAPK activation in TRAF6−/− EF cells treated with IL-1β (Fig. 5D). Because another stress stimulus, anisomycin, was still able to activate JNK/SAPK in TRAF6−/− cells (Fig. 5D), TRAF6 appears to be specifically required for the IL-1 pathway of JNK/SAPK activation. Together, these results define crucial roles for TRAF6 in signaling directly related to the activation of both NF-κB and JNK/SAPK.
Discussion
A common feature of all TRAF knockout mice generated to date is their failure to thrive normally and survive until adulthood. Apart from this similarity, each TRAF knockout mouse is phenotypically distinct, as demonstrated by the marked differences between TRAF6-, TRAF2- and TRAF3-deficient mice (Xu et al. 1996; Yeh et al. 1997). The results of this study demonstrate that TRAF6 is an important transducer in not only IL-1 and CD40 signaling (as had been suggested by previous ex vivo work) but also in LPS signaling. In addition, we have discovered a novel physiologic role for TRAF6 in the regulation of bone metabolism.
Osteopetrosis in TRAF6-deficient mice
Unlike the reported TRAF2 or TRAF3 knockout mice, TRAF6-deficient mice exhibited osteopetrosis. In most other osteopetrotic mice, such as the p50/p52 double mutant (Franzoso et al. 1997), the c-Fos mutant (Wang et al. 1992; Grigoriades et al. 1994), and PU.1-deficient mice (Tondravi et al. 1997), osteopetrosis is attributed to defects in the differentiation of hematopoietic precursors into mature osteoclasts. In contrast, mice deficient for the tyrosine kinase c-src (Soriano et al. 1991) are osteopetrotic because of a defect in osteoclast function rather than development. The osteopetrotic phenotype of TRAF6−/− mice is similar to that of c-src-deficient mice. Both mutants have normal numbers of mature osteoclasts that are impaired in the formation of ruffled borders, and hence, do not resorb bone properly (Boyce et al. 1992).
c-src has been implicated in signaling by colony-stimulating factor-1 (CSF-1) (Insogna et al. 1997). Treatment of normal osteoclasts with CSF-1 induces cytoplasmic spreading, c-src kinase activity, and protein tyrosine phosphorylation (Insogna et al. 1997). Although CSF-1 fails to induce spreading in c-src−/− osteoclasts (Insogna et al. 1997), whether CSF-1 signaling is required to trigger bone resorption by osteoclasts remains to be clarified. Interestingly, CSF-1−/− mice are severely deficient in mature macrophages and osteoclasts (Yoshida et al. 1990), unlike TRAF6−/− mice. These data argue against a role for TRAF6 in CSF-1 signaling. Furthermore, it is unlikely that disruption of IL-1 or CD40 signaling in TRAF6−/− mice is the cause of the osteopetrosis, as mice deficient in IL-1R1, IL-1β (Zheng et al. 1995; Leon et al. 1996; Glaccum et al. 1997; Labow et al. 1997), MyD88 (Adachi et al. 1998), CD40 (Kawabe et al. 1994), or CD40L (Xu et al. 1994) do not exhibit osteopetrosis.
The role of TRAF6 during osteoclast maturation and activation processes is not known but is likely to involve signal transduction from an osteoclast surface receptor. On the basis of what is known about osteoclast biology, a candidate signaling receptor is RANK (receptor activator of NF-κB), a novel TNFR family member related most closely to CD40 (Anderson et al. 1997). RANK has recently been localized to the surface of osteoclast progenitor cells (Hsu et al. 1999). The ligand for RANK, also known as osteoprotegerin ligand (OPGL), is essential for both osteoclast differentiation and activation (Lacey et al. 1998; Kong et al. 1999). Most recently, it was demonstrated that multiple TRAF proteins, including TRAF1, TRAF2, TRAF3, TRAF5, and TRAF6 can interact with the carboxyl terminus of RANK (Darnay et al. 1998; Galibert et al. 1998; Wong et al. 1998; Hsu et al. 1999). However, the physiological contribution of each TRAF protein to RANK signaling remains to be elucidated.
Given that TRAF6 and OPGL are believed to act via the same signaling receptor, namely RANK, one would expect both mutant mice to be phenotypically similar. On the contrary, mice deficient in OPGL differ from TRAF6−/− mice, in that the former are completely devoid of osteoclasts (Kong et al. 1999). There are two possible mechanisms that may account for this discrepancy. First, in addition to acting on RANK, OPGL may act on some heretofore uncharacterized receptor that specifically mediates the generation of osteoclasts independently of TRAF6. Future studies examining the physiological functions of RANK using gene targeting techniques are required to address this issue. Alternatively, it is also possible that RANK is the only receptor for OPGL, but its downstream signaling diverges. In this scenario, the signal(s) leading to osteoclast generation may be compensated by other molecules such as TRAF2 and/or TRAF5, where TRAF6 is indispensable in transducing signals mediating mature osteoclast functions. Future studies aimed at generating mutant mice deficient in multiple TRAF proteins will help answer these questions.
The osteoclast-activating role of OPGL has been examined previously in vitro (Fuller et al. 1998; Lacey et al. 1998). Treatment of isolated osteoclasts with OPGL led to a rapid increase in pseudopodial motility and stimulated bone resorption, effects that were specifically inhibited following the addition of the decoy receptor osteoprotegerin (OPG) (Fuller et al. 1998). Therefore, OPGL–RANK signaling, in addition to playing a role in osteoclast differentiation, is also important for the activation of mature osteoclasts. Our results suggest that TRAF6 could be a signal transducer downstream of RANK that is specifically required for the pathway leading to osteoclast activation.
Requirement for TRAF6 in LPS signaling
Several exciting findings implicate TLR2 (Toll-like receptor 2) and TLR4 in LPS signaling. Both are type 1 transmembrane proteins with cytoplasmic domains sharing similarities with the intracellular region of the IL-1R1 (Gay and Keith 1991; Medzhitov et al. 1997). TLR2 is activated by LPS in a response that depends on LPS-binding protein and is enhanced by CD14 (Yang et al. 1998). Overexpression of TLR2 confers LPS inducibility of NF-κB activation in mammalian 293 cells, and a dominant-negative mutant of TRAF6 suppresses NF-κB activation in LPS-treated 293 cells transiently expressing TLR2 (Kirschning et al. 1998). Recently, the resistance of two mutant strains of mice (C3H/HeJ and C57BL/10ScCr) to endotoxin has been attributed to destructive mutations of TLR4 (Poltorak et al. 1998). Overexpression of dominant-negative TRAF6 also impairs TLR4-induced NF-κB activity (Muzio et al. 1998). Taken together, these results strongly suggest a physiologically important role for TRAF6 in transducing LPS-mediated signals downstream of TLR2 and/or TLR4. Annually, a considerable number of human fatalities occur due to endotoxic shock, the result of activation of the immune system by endotoxin/LPS (Fenton and Golenbock 1998). The elucidation of the signaling molecules in the LPS pathway(s) may lead to the discovery of potential targets for the pharmacological modulation of endotoxic shock.
In conclusion, this study has revealed important new information on the physiologic functions of TRAF6. Through the generation and analysis of TRAF6-deficient mice, we have demonstrated critical roles for TRAF6 in perinatal and postnatal survival and in IL-1 and CD40 signal transduction. We have also identified a novel role for TRAF6 in bone metabolism, in that its presence is required to prevent osteopetrosis. Most importantly, these studies have uncovered an unexpected and prominent role for TRAF6 in LPS signal transduction. Our findings should make a significant contribution to advancing the heretofore limited knowledge of the pathophysiologic mechanisms underlying endotoxic shock.
Materials and methods
Animal husbandry
Mice were housed in a specific-pathogen-free facility according to the ethical and institutional guidelines of the Ontario Cancer Institute. Pathogen status was monitored by standard microbiological and parasitological examinations as well as by histopathological staining for specific pathogens. All mice analyzed in this investigation appeared healthy at the time of use.
Gross anatomical and histological analyses
Groups of TRAF6+/+, TRAF6+/−, and TRAF6−/− mice were necropsied on day 21 or 28 after birth. Radiography was performed using a Faxitron X-ray system (model 43855A, Faxitron X-ray Corp., Buffalo Grove, IL). Bone mineral density was determined in two 0.5-mm cross sections of bone taken at 1.5 and 2.0 mm from the proximal end of the tibia. Both total and trabecular bone mineral density (defined as the innermost 20% of the bone cross section) in the metaphysis were determined (XMICE 5.2, Stratec, Germany). Bone tissue was decalcified using formic acid and embedded in paraffin. Osteoclasts were identified by their expression of the specific marker TRAP. TRAP activity was determined using a method of enzyme histochemistry that results in the specific red staining of TRAP+ cells, that is, osteoclasts (Simonet et al. 1997). Osteoclasts were counted in TRAP-stained sections with the aid of a graded eyepiece. Measurements were made in an area of ∼0.4 mm2 just distal to the proximal tibial growth plate in the primary spongiosa. The osteoclast number was determined relative to the tissue area measured.
Immunoprecipitation and Western blot analysis
Kidneys from TRAF6+/+, TRAF6+/−, and TRAF6−/− littermates were homogenized and lysed in ice-cold lysis buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Triton X-100, 20 mm EDTA, and the protease inhibitors phenylmethylsulfonyl fluoride (PMSF, 10 μg/ml), leupeptin (1μg/ml), aprotinin (2 μg/ml), and sodium orthovanadate (1 mm). TRAF6 was immunoprecipitated from 1.5 mg of total protein using 25 μl of protein A beads (Pharmacia) and 1 μl of a polyclonal anti-human TRAF6 antiserum that was raised against the full-length TRAF6. Immunoprecipitates and total lysates were fractionated by gel electrophoresis on an 8% Tris-glycine polyacrylamide gel (Novex, San Diego, CA) and transferred onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The Western blots were probed with a 1:1000 dilution of the anti-TRAF6 antibody and developed with enhanced chemiluminescence (ECL, Amersham) according to the manufacturer’s instructions. To verify that equal amounts of protein were loaded, blots were reprobed with a 1:100 dilution of an anti-β-actin antibody (Sigma).
Electron microscopy
The lumbar vertebrae were removed at necropsy, split lengthwise, and immersion-fixed in cold 0.1 m sodium cacodylate buffer containing 2.5% glutaraldehyde and 1.6% paraformaldehyde at pH 7.4. After 24 hr at 4°C, the specimens were transferred to a decalcification solution consisting of 5% EDTA and 1% glutaraldehyde in 0.1 m sodium cacodylate buffer. The decalcification was considered complete after 7 days, and the vertebrae were rinsed in buffer, postfixed in 1% aqueous osmium tetroxide, dehydrated in ethanol, and embedded in epoxy resin.
Each of the blocks was initially sectioned at 1–2 μm, stained with toluidine blue, and examined by light microscopy. Selected blocks were then trimmed and subjected to ultrathin sectioning. The collected sections were contrast-enhanced with uranyl acetate and lead citrate prior to examination at 100 kV on a Philips CM120 transmission electron microscope.
Cellular proliferation assays
Splenocytes from two healthy, 10-day-old TRAF6−/− or wild-type mice were pooled. Cell suspensions were prepared by passing the spleens through a fine wire mesh. Erythrocytes were lysed using ACK (0.155 m ammonium chloride, 0.1 m disodium EDTA, 0.01 m potassium bicarbonate) for 3 min on ice. T cells were depleted by incubating the suspension for 1 hr at 37°C with a combination of three monoclonal antibodies (Maroun and Julius 1994), anti-Thy1.2 (H013.4.9-2), anti-CD4 (RL-172-4H), and anti-CD8 (3.168), in conjunction with guinea pig complement (Cedarlane). Mac-1+ cells were depleted by panning using Optilux petri dishes (Falcon) coated with 5μg/ml mouse anti-rat IgG (Jackson Immunoresearch Laboratories) and rat anti-mouse Mac-1 (M1/70, ATCC). The cell suspensions were incubated in the coated plates for 1 hr at 4°C, followed by the recovery of Mac-1− cells (enriched B cells) in three washes.
For proliferation assays, 1 × 105 viable cells per well (96-well, flat-bottom plate) were cultured in quadruplicate in OptiMEM (GIBCO) supplemented with 5 × 10−5 m 2-mercaptoethanol, NaHCO3 (2.4 grams/liter), streptomycin (5 μg/ml) and 5% heat-inactivated fetal bovine serum (GIBCO). Cells were treated with 10 μg/ml rat IgG2a anti-mouse CD40 mAb (Pharmingen, 3/23), 10 μg/ml rat IgG2a isotype control (Pharmingen, 11020D), 10 μg/ml LPS (Difco), or 5 ng/ml recombinant murine IL-4 (Genzyme) for 2, 3, or 4 days. Plates were pulsed with 1 μCi [3H]thymidine for 6 hr and harvested onto glass fiber filters. [3H]Thymidine uptake was measured using a scintillation counter (Topcount, Canberra Packard).
EMSA
Primary embryonic fibroblasts (2 × 106), splenocytes (1 × 107), and Abelson-transformed pre-B cells (1 × 107) were either untreated or stimulated with 10 ng/ml mIL-1β (Genzyme), 10 ng/ml mTNFα (Genzyme), 20 μg/ml LPS (Sigma), 5 μg/ml anti-mouse CD40 (3/23) mAb (IgG2a κ) (PharMingen), or 5 μg/ml rat anti-human creatinine kinase mAb (IgG2a κ) (isotype control) for various times. Nuclear extracts were harvested and EMSAs were performed as described previously (Yeh et al. 1997).
JNK/SAPK kinase assay
Primary embryonic fibroblasts (1 × 106) were serum-starved for 3 hr in DMEM containing 10 mm HEPES and 0.1% fatty acid-free BSA. The cells were then treated with either 10 ng/ml recombinant murine IL-1β for various times (Genzyme) or 10 μg/ml anisomycin for 30 min. JNK/SAPK was immunoprecipitated using anti-JNK1 C-17 rabbit polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA), and the kinase assay was performed as described previously (Yeh et al. 1997).
NO production assay
BMMφ macrophages were derived from bone marrow cells after 6 days culture in 50 ng/ml recombinant murine CSF (R&D Systems, Minneapolis, MN). Elicited PMφ macrophages were obtained from peritoneal lavage of mice injected 5 days previously with 1 ml of 4% thioglycollate broth (Sigma). Macrophages (1 × 105) were incubated with various concentrations of LPS, (50 U/ml) mIFNγ, (10 ng/ml) mIL-1β, or (10 ng/ml) mTNFα for 48 hr (BMMφ) or 24 hr (PMφ) in 96-well flat-bottomed microtiter plates. The culture supernatants were assayed for nitrite, the stable end product of NO production, by the Greiss method (Green et al. 1982). To adjust for any discrepancies in adherent cell numbers, nitrite concentration was calculated as nmoles of nitrite per milligram of protein after determination of protein content in representative control wells.
Acknowledgments
We thank Louis-Martin Boucher, Young-Yun Kong, Arda Shahinian, Hiroki Yoshida, Razquallah Hakem, David Kägi, Andrew Hessel, Hiroshi Nishina, Paul Waterhouse, Seiji Kondo, Denis Bouchard, Michelle Ng, Betty Hum, Diane Duryea, Carol Burgh, Ennis Julian, and Yan Cheng for reagents, instructive discussions, and/or technical assistance, as well as other members of the Mak laboratory for helpful discussions and advice. We also thank Irene Ng for excellent administrative support and Mary Saunders for scientific editing. This work was supported in part by an Ontario Graduate Scholarship. Mark A. Lomaga is a doctoral student at the Faculty of Pharmacy, University of Toronto.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tmak@oci.utoronto.ca; FAX (416) 204-5300.
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1 and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Maraskovsky E, Billingley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes & Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Bazan F, Blanchard F, Briere JP, Galizzi C, van Kooten C, Liu YL, Rousset F, Saeland S. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- Baron R, Revesloot J-H, Neff L, Chakraborty M, Chatterjee A, Lomri A, Horne W. Biology of the osteoclast. In: Noda M, editor. Cellular and molecular biology of bone. New York, NY: Springer Publishing; 1993. pp. 445–495. [Google Scholar]
- Boyce B, Yoneda T, Lowe C, Soriano P, Mundy G. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and to resorb bone in mice. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Henzel W, Gao X. IRAK: A kinase associated with the interleukin-1 receptor. Science. 1996a;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996b;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, Baltimore D. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- Darnay BG, Haridas V, Ni J, Moore PA, Aggarwal BB. Characterization of the intracellular domain of receptor activator of NF-κB (RANK) J Biol Chem. 1998;273:20551–20555. doi: 10.1074/jbc.273.32.20551. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leuk Biol. 1998;64:25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-κB in osteoclast and B-cell development. Genes & Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller K, Wong B, Fox S, Choi Y, Chambers TJ. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. 1998;188:997–1001. doi: 10.1084/jem.188.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert L, Tometsko ME, Anderson DM, Cosman D, Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-κB, a member of the TNFR superfamily. J Biol Chem. 1998;273:34120–34127. doi: 10.1074/jbc.273.51.34120. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Keith FJ. Drosophila Toll and IL-1 receptor. Nature. 1991;351:355–356. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type 1 receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15-N] nitrate in biological fluids. Ann Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- Hsu H, Solovyev I, Colombero A, Elliott R, Kelley M, Boyle WJ. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J Biol Chem. 1997;272:13471–13474. doi: 10.1074/jbc.272.21.13471. [DOI] [PubMed] [Google Scholar]
- Hsu, H., D.L. Lacey, C.R. Dunstan, I. Solovyev, A. Colombero, E. Timms, H.-L. Tan, G. Elliott, M.J. Kelley, I. Sarosi, L. Wang, X.-Z. Xia, R. Elliott, L. Chiu, T. Black, S. Scully, C. Capparelli, S. Morony, G. Shimamoto, M.B. Bass, and W.J. Boyle. 1999. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci 99: (in press). [DOI] [PMC free article] [PubMed]
- Huang J, Gao X, Li S, Cao Z. Recruitment of IRAK to the interleukin-1 receptor complex requires interleukin-1 receptor accessory protein. Proc Natl Acad Sci. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insogna KL, Sahni M, Grey AB, Tanaka S, Horne WC, Neff L, Mitnick M, Levy JB, Baron R. Colony-stimulating factor-1 induces cytoskeletal reorganization and c-src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest. 1997;100:2476–2485. doi: 10.1172/JCI119790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. Identification of TRAF6, a novel tumor-necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996a;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- Ishida TK, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J. TRAF5, a novel tumor necrosis factor receptor-associated factor protein, mediates CD40 signaling. Proc Natl Acad Sci. 1996b;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal centre formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Kirschning CJ, Wesche H, Ayres TM, Rothe M. Human Toll-like receptor 2 confers responsiveness to bacterial LPS. J Exp Med. 1998;188:2091–2097. doi: 10.1084/jem.188.11.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y-Y, Yoshida H, Sarosi I, Tan H-L, Timms E, Capparelli C, Morony S, Van G, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, T cell activation, and lymph node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB, Bartfai T, Solorzano C, Moldawer LL, Chizzonite R, McIntyre KW. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type 1 receptor-deficient mice. J Immunol. 1997;159:2452–2461. [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan H-L, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian Y-X, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Leon LR, Conn CA, Glaccum M, Kluger MJ. IL-1 type 1 receptor mediates acute phase response to terpentine, but not lipopolysaccharide, in mice. Am J Physiol Regul Integr Comp Physiol. 1996;40:R1668–R1675. doi: 10.1152/ajpregu.1996.271.6.R1668. [DOI] [PubMed] [Google Scholar]
- Liu Z-G, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector junctions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Malinin N, Boldin MP, Kovalenko A, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- Maroun CR, Julius M. Distinct roles for CD4 and CD8 as co-receptors in T cell receptor signalling. Eur J Immunol. 1994;24:959–966. doi: 10.1002/eji.1830240427. [DOI] [PubMed] [Google Scholar]
- Marsters SA, Ayres TM, Skubatch M, Gray CL, Rothe M, Ashkenazi A. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-κB and AP-1. J Biol Chem. 1997;272:14029–14032. doi: 10.1074/jbc.272.22.14029. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signalling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human Toll signaling pathway: Divergence of nuclear factor κ B and JNK/SAPK activation upstream of tumor necrosis receptor-associated factor 6 (TRAF6) J Exp Med. 1998;187:2097–2101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware CF, Yagita H, Okumura K. TRAF5, an activator of NF-kappa B and putative signal transducer for the lymphotoxin-beta receptor. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, Greene C. Signal transduction pathways activated by the IL-1 receptor family: Ancient signaling machinery in mammals, insects, and plants. J Leukoc Biol. 1998;63:650–657. [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in TLR4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Pullen SS, Miller HG, Everdeen DS, Dang TTA, Crute JJ, Kehry MR. CD40-Tumor necrosis factor receptor associated factor (TRAF) interactions: Regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–11845. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan H-L, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Boyle WJ. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Song HY, Regnier CH, Kirschning C, Goeddel D, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: Bifurcation of nuclear factor-κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Rothe M, Goeddel DV. Anatomy of TRAF2. Distinct domains for nuclear factor-kappa B activation and association with tumor necrosis signaling proteins. J Biol Chem. 1996;271:19935–19942. doi: 10.1074/jbc.271.33.19935. [DOI] [PubMed] [Google Scholar]
- Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-Fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Wesche H, Henzel W, Schillinglaw W, Li S, Cao Z. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997a;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases) J Biol Chem. 1997b;272:7727–7731. doi: 10.1074/jbc.272.12.7727. [DOI] [PubMed] [Google Scholar]
- Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–28359. doi: 10.1074/jbc.273.43.28355. [DOI] [PubMed] [Google Scholar]
- Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle R, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- Yang R-B, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–288. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- Yeh W-C, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Early lethality, fuctional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;71:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S-I, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S-I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–443. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A. Resistance to fever induction and impaired acute phase response in interleukin-1β-deficient mice. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]