

Abstract
Although reductions in the expression of the calcium-buffering proteins calbindin D-28K (CB) and parvalbumin (PV) have been observed in the aging brain, it is unknown whether these changes contribute to age-related hippocampal dysfunction. To address this issue, we measured basal hippocampal metabolism and hippocampal structure across the lifespan of C57BL/6J, calbindin D-28k knockout (CBKO) and parvalbumin knockout (PVKO) mice. Basal metabolism was estimated using steady state relative cerebral blood volume (rCBV), which is a variant of fMRI that provides the highest spatial resolution, optimal for the analysis of individual subregions of the hippocampal formation. We found that like primates, normal aging in C57BL/6J mice is characterized by an age-dependent decline in rCBV-estimated dentate gyrus metabolism. Although abnormal hippocampal fMRI signals were observed in CBKO and PVKO mice, only CBKO mice showed accelerated age-dependent decline of rCBV-estimated metabolism in the dentate gyrus. We also found age-independent structural changes in CBKO mice, which included an enlarged hippocampus and neocortex as well as global brain hypertrophy. These metabolic and structural changes in CBKO mice correlated with a deficit in hippocampus-dependent learning in the active place avoidance task. Our results suggest that the decrease in CB that occurs during normal aging is involved in age-related hippocampal metabolic decline. Our findings also illustrate the value of using multiple MRI techniques in transgenic mice to investigate mechanisms involved in the functional and structural changes that occur during aging.
Keywords: fMRI (functional magnetic resonance imaging), Parvalbumin, CBV, aging, CBKO
INTRODUCTION
The hippocampal formation, a structure vital for memory, has been shown to be particularly vulnerable to dysfunction during the aging process in humans, non-human primates and rodents (Gazzaley et al., 1996; Geinisman et al., 1995; Small et al., 2004; West, 1993; West et al., 1994). Changes in the regulation of intracellular calcium levels have been proposed to contribute to age-related hippocampal dysfunction (Khachaturian, 1994; Ouanounou et al., 1999). Physiological Ca2+ entry into neurons plays an important role in multiple cellular functions, such as metabolism, gene regulation and synaptic transmission, including its short- and long-term modulation. However prolonged increases of intracellular calcium levels [Ca2+]i can cause neuronal dysfunction (Hara and Snyder, 2007). To avoid excessive [Ca2+]i elevations, neurons are equipped with machinery that continuously modulates the temporal and spatial patterns of Ca2+ signaling. These regulatory systems include sequestration into the endoplasmic reticulum and mitochondria, activation of plasma membrane Ca2+ pumps, and buffering by Ca2+-binding proteins (CaBPs).
Among the many CaBPs, calbindin D-28k (CB) and parvalbumin (PV) are two that have been found to be downregulated in the aging brain (Armbrecht et al., 1999; Bu et al., 2003; de Jong et al., 1996; Geula et al., 2003; Kishimoto et al., 1998). They are both abundant cytosolic proteins that typically segregate into separate neuronal populations (Andressen et al., 1993; Schwaller, 2003). Calbindin D-28k knockout (CBKO) (Airaksinen et al., 1997) and parvalbumin knockout (PVKO) mice (Schwaller et al., 1999) have been generated and specific yet mild phenotypes have been described (Schwaller, 2003; Schwaller, 2010). Interestingly, in single KO mice, the CaBP that remains is neither upregulated nor expressed ectopically in neuronal populations not seen in wild-type (WT) mice. It has been hypothesized that since the biophysical properties (Ca2+-binding affinities, kinetics, diffusion) of these two buffers are considerably different (Faas et al., 2007; Schwaller, 2003), they cannot substitute for one another when one is absent.
In the hippocampus, CB is primarily found in dentate granule cells, a subpopulation of CA1 pyramidal cells, and some interneurons (Baimbridge, 1992; Celio, 1990; Freund and Buzsaki, 1996; Kosaka et al., 1987). The expression of CB, which is modulated by the functional state of the hippocampal circuit (Nagerl et al., 2000), is required for normal short-term synaptic plasticity between granule cells of the dentate gyrus and CA3 neurons (Blatow et al., 2003), and modulates the excitability of CA1 pyramidal cells (Arabadzisz et al., 2002). In contrast, PV is mainly expressed in a population of fast-spiking interneurons that are thought to play a key role in the generation of network oscillations in the gamma frequency range (30–100Hz) (Fuchs et al., 2007; Vreugdenhil et al., 2003).
Despite what is known about the function of CB and PV in the adult hippocampus, the role of these CaBPs in the aging hippocampus is not well defined. We set out to investigate the relationship between CB and PV and age-related decline in hippocampal function by measuring changes in basal hippocampal function that occurred during aging in CBKO and PVKO mice. For purposes of this study, we operationally define aging as the physical changes that occur with increases in life span, which are typically associated with declining function(s). The function of interest here was basal metabolism in the hippocampus estimated using steady state cerebral blood volume (CBV) values. CBV is a variant of functional magnetic resonance imaging (fMRI) that provides an MRI correlate of metabolism and yields the highest spatial resolution in humans, non-human primates and rodents (Moreno et al., 2007; Small et al., 2004; Wu et al., 2008). Such high-resolution is particularly important when investigating function of the hippocampal formation, since it allows for the analysis of the individual subregions. These subregions, which include the entorhinal cortex, the dentate gyrus, the CA1 and CA3 subfields, and the subiculum, each house a molecularly-distinct population of neurons that are differentially vulnerable to mechanisms of dysfunction. Steady-state contrast enhanced CBV mapping has been successfully used by our laboratories as well as others to pinpoint subregions of the hippocampal formation that are differentially affected in Alzheimer’s disease (Moreno et al., 2007), schizophrenia (Gaisler-Salomon et al., 2009; Schobel et al., 2009), aging (Small et al., 2004), diabetes (Wu et al., 2008), and by acute pharmacological interventions of the serotonergic pathways (Mueggler et al., 2010).
We began our study by first establishing how hippocampus metabolism changes in vivo during the course of normal aging. This involved acquiring rCBV measures of each hippocampal subregion in wild-type (WT) C57BL/6J mice at different ages. Then we generated rCBV maps of the hippocampal formation across the lifespan of CBKO and PVKO mice and evaluated how a lack of each protein affected the rate of rCBV decline that occurred with age in each hippocampal subregion. To evaluate whether age-related rCBV changes were accompanied by structural changes, we used MRI to measure anatomical volume in CBKO and PVKO mice of different ages. Finally, we used hippocampus-dependent learning in the active place avoidance task to assess the functional significance of the MRI findings.
MATERIALS AND METHODS
Animals
Calbindin D-28k (CBKO) and parvalbumin knockout (PVKO) mice have been described previously (Airaksinen et al., 1997, Schwaller et al., 1999). CBKO and PVKO mice used in this study were congenic with C57BL/6J. Mating of heterozygous transgenic mice was carried out with C57BL/6J for at least 10 generations. The absence of CB in CBKO mice was confirmed with immunocytochemistry using the well-characterized antiserum CB38 (1:25k; SWant, Bellinzona, Switzerland) against calbindin D-28K. The absence of PV in PVKO mouse was confirmed with the anti-parvalbumin antiserum PV28 (1:5k; SWant, Bellinzona, Switzerland) (Supplementary Fig. 3).
MRI experiments were performed in CBKO and PVKO mice across their life span. Specifically, fifty-two MRI experiments were performed with CBKO mice and age- and sex-matched wild-type (WT) controls of the following ages: 1.5 months (N=6), 6 months (N=5), 9 months (N=6), 12 months (N=4) and 16 months of age (N=5). Twenty MRI experiments were performed on a total of 20 PVKO mice, which were 1.5 months (N=5), 6 months (N=5), 9 months (N=5) and 16 months (N=5) of age. The same age- and sex-matched WT mice that were used as control groups for the CBKO experiment were also used as controls for the PVKO experiment, since all mutants have the same C57BL/6J background. The weights of the animals ranged from 28 to 42 grams for genetically modified mice and 26 to 41 grams for controls. All experiments were performed in compliance with National Institutes of Health regulations and approved by the Institutional Animal Care and Use Committees of Columbia University and SUNY Downstate Medical Center following the NIH Guide for the Care and Use of Laboratory Animals.
Experimental Design
Images from CBKO and WT mice were acquired for a cross-sectional study and a longitudinal study. We used 26 WT and 26 CBKO mice ranging from 1.5 to 16 months of age in the cross sectional study, in which each animal is imaged once. A subset of these mice (11 CBKO and 13 WT mice) was also used in a longitudinal study, in which each mouse was imaged at 1.5, 6, and 9 months of age. A smaller subgroup of these mice (5 CBKO and 5 WT) was imaged at both 4 and 12 months of age. PVKO mice were all imaged once for a cross-sectional study.
An additional group of WT C57BL/6J mice (rCBV- MRI experiments retrieved from our MRI-archives) was analyzed at the following ages: 1.5 months (N=6), 6 months (N=6), 9 months (N=6), 12 months (N=6), 14 months (N=6), 16 months (N=4) 18 months (N=8), 21 months (N=12) and 24 months of age (N=4). Males and females were counterbalanced across groups and all mice were imaged once for a cross-sectional study.
Functional Imaging
Cerebral blood volume (CBV) maps were acquired using methods previously described (Moreno et al., 2007). Subjects were imaged with a Brucker AVANCE 400WB spectrometer (Brucker NMR, Inc., Billerica, MA) outfitted with an 89 mm bore 9.4 Tesla vertical Brucker magnet (Oxford Instruments Ltd., UK), a 30 mm-i.d. birdcage RF probe, and a shielded gradient system (100 G/cm). Anesthesia was used to immobilize the animals and minimize fear and anxiety induced by the loud environment of the scanner. Like other fMRI studies in rodents, we used isoflurane for anesthesia (induction phase 3 vol. % and maintenance 1.1–1.5 vol. % at 1 l/min air flow, via a nose cone), under conditions that do not significantly affect heart rate, respiratory rate or SaO2 in C57BL/6J mice (Moreno et al., 2006). Although isoflurane causes a decrease in global glucose uptake, it does not significantly uncouple the hemodynamic response at 1 minimal alveolar concentration (MAC) (Lenz et al., 1998).
At high-fields, T2-weighted scans generate images with superior contrast compared to T1-weighted scans. Therefore, we relied on T2-weighted scans for CBV mapping in mice. Three scout scans were first acquired so we could position the subsequent T2-weighted images along the standard anatomical orientations in a reproducible manner. Optimal horizontal images were determined empirically by repositioning the animal to ensure the animal’s brain was located at the center of the MRI coils. Shimming was performed in order to optimize B0 field homogeneity. By running the auto shim procedure, the three linear shim channels (x,y,z) were automatically adjusted by means of an iterative software routine that maximized the area of the free inductive decay (FID). T2-weighted images were obtained with a fast spin echo (FSE) sequence with TR/TEeff = 2000ms/70ms, rapid acquisition with relaxation enhancement (RARE) factor = 16, FOV= 20 mm, acquisition matrix = 256 × 256, 16 slices, with a slice thickness =0.6 mm, slice gap = 0.1 mm and number of excitations (NEX) =28. The resulting voxel size was 0.00365404 mm3. Each set of images required 15 minutes, and 5 sets of images were acquired sequentially. The first 2 sets were acquired pre-contrast and the subsequent 3 were acquired post-contrast. Gadodiamide (Omniscan, Amersham, Oslo, Norway) was injected (10 mmol/kg) via a 0.5 mm catheter placed intraperitonealy (IP) and saline (2 ml) was injected IP at the end of imaging to improve the clearance of the contrast agent.
Signal to noise ratio (SNR) was calculated as follows: First in a pre contrast baseline T2 weighted image, mean signal intensity (SI) of an appropriate region of interest (ROI) was measured. Then the average standard deviation (stdev) of an identical sized ROI of the surrounding air was defined as noise. Subsequently, the SNR value was obtained using the following equation: SNR= mean SI-ROI (brain)/stdev ROI (air) {eq.1}. Given the effect of gadolinium on T2 signal, contrast to noise ratio (CNR) was calculated as CNR= (Mean ROI-SI pre – Mean ROI-SI post)/stdev (air) ROI pre {eq.2}.
ROI-CBV data had SNR with values between 17–25 and CNR values between 5–7. During imaging, acquisition CNR was also assessed by measuring the contrast-induced percent change in signal in the thalamus (comparing the last post-contrast image to the pre-contrast image). If contrast-induced percentile change was less than 20%, an extra dose of 3 mmol/kg gadodiamide was administered and a new post contrast series was obtained 15 minutes later. If a 20% signal change was still not achieved, the images were rejected prior to analysis. Heart rate, respiratory rate and SaO2 were continuously monitored using pulse oximetry (Model V33304, Sergivet). The probe was attached to the lower abdomen and normality of physiological parameters was used as criterion to continue with the imaging session.
An investigator blinded to subject grouping performed all image processing. Although anesthesia and soft head fixation effectively minimized head motion, the AIR program was used to co-register the images. A Gnu plot was generated to assess the quality of the co-registration, and an individual study was rejected if a shift greater than 1 pixel dimension was detected. Relative CBV was mapped as changes of transverse relaxation rate (ΔR2) induced by the contrast agent (CA). Note that R2 = 1/T2. When the contrast agent reaches uniform distribution, then CBV maps can be measured from steady state T2-weighted images as: CBV α ΔR2= ln (Spre/Spost)/TE {eq.3}. Where TE is the effective echo time, Spre is the T2 weighted signal before CA administration, and Spost is the T2-weighted signal after CA reaches steady state. We have previously studied the kinetics of gadolinium (IP) generated CBV maps in C57BL/6J mice and identified the 37.5 minute time-point as the optimum post-contrast time interval (Moreno et al., 2006). The derived ΔR2 maps were then divided by 4 pixels with the highest ΔR2 measured from the posterior cerebral vein, yielding relative CBV maps (rCBV). We have published a detailed discussion on the necessity for normalization of CBV maps (Moreno et al., 2006, Moreno et al., 2007).
Modeling and empirical data have demonstrated that gradient echo (GE) sequences are more sensitive to large vessels than to microvessels (Boxerman et al., 1995, Simonsen et al., 2000). We took advantage of this fact in a separate group of mice (N=6) injected with gadodiamide, where we used a three-dimensional gradient echo (GE) sequence with TR/TE=40ms/5.5ms, flip angle= 5o, acquisition matrix 192×192×16, and NEX= 8. This provides S*pre and S*post that are pre-contrast and post-contrast signal intensities for GE images, which better illustrate the vascular structure used to normalize CBV maps, as shown in Fig. 1C&D.
Figure 1. Mapping of hippocampal cerebral blood volumes.
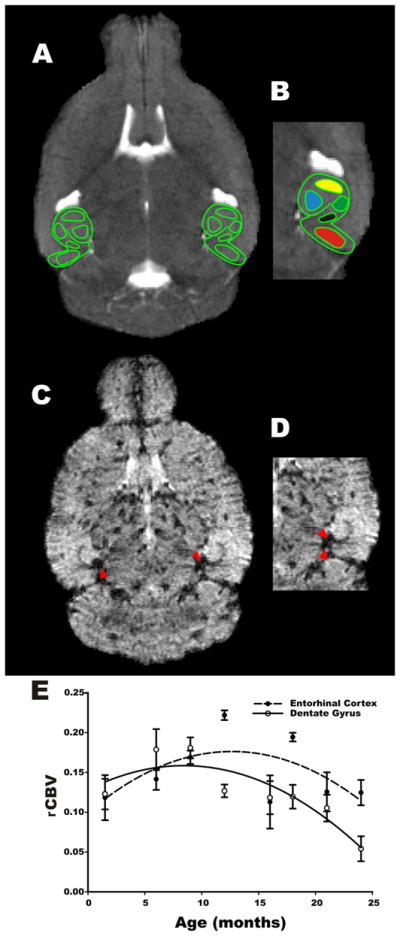
A) Horizontal section of a T2-weighted image from a 6-month-old WT C57BL/6J mouse at the mid-body of the hippocampal formation (outlined in green). B) Close up view of A, in which hippocampal formation external morphology appearance and internal architecture (outlined in green) is best visualized. Regions of interest were identified within the entorhinal cortex [red], subiculum [black], CA1 subfield [green], CA3 subfield [yellow] and dentate gyrus [blue]. C) T2* weighted image, axial slice at the level of the mid-body of the hippocampal formation, 37.5 minutes after 10 mmol/kg (IP) gadodiamide was injected. Arrowheads identify the posterior cerebral vein (PCV). D) Close up view of the hippocampal formation and its relation to PCV identified by arrowheads. E) Temporal analysis of metabolic brain changes during normal aging showed a significant age-related decline in DG rCBV, while no significant change was observed in the EC of WT mice. The curve illustrates a quadratic fit of the data points.
Hippocampus Subregional Analysis
Although MRI sections were acquired throughout the brain, the images used for data analysis were restricted to the mid-body of the hippocampal formation, unless otherwise specified. We have consistently found that a horizontal slice acquired through the mid-body of the hippocampal formation provides optimal visualization of hippocampal morphology and internal architecture. The external morphology of the hippocampus was manually traced, as was the internal architecture that follows the hippocampal sulcus and the internal white matter tracts (Fig. 1A&B). Regions-of-interest (ROIs) of five hippocampal subregions were then identified based on the following anatomical criteria: a) Entorhinal cortex [EC]. The lateral boundary follows the grey/white junction of the perirhinal cortex; the medial boundary is the medial aspect of the temporal lobe; the superior boundary is the beginning of the collateral sulcus; the inferior boundary is the lateral tip of the brain. b) Subiculum [SC]. The medial boundary is the medial extent of the hippocampal sulcus and/or the horizontal inflection of the hippocampus; the inferior boundary is the white matter of the underlying perirhinal cortex; the superior boundary is the hippocampal sulcus; the lateral boundary is a few pixels medial to the vertical inflection of the hippocampus. c) CA1 subfield. The lateral boundary is the lateral aspect of the hippocampal proper; the medial boundary is the hippocampal fissure; the superior boundary is the superior curvature of the hippocampus proper; the inferior boundary is the inferior curvature of the hippocampus proper. d) CA3 subfield. The lateral boundary is the beginning of the superior curvature of the hippocampus proper; the medial boundary is the end of this curvature; the superior boundary is the superior aspect of the hippocampus proper; the inferior boundary is the superior tip of the hippocampal fissure. e) Dentate gyrus [DG]. The lateral boundary is the lateral aspect of the hippocampus proper; the medial boundary is the hippocampal fissure; the superior boundary is the superior curvature of the hippocampus proper; the inferior boundary is the inferior aspect of the hippocampus proper. Of note, the border zones between any two subregions cannot be identified without histological landmarks and were therefore excluded from the ROIs (Fig. 1B). For experiments involving a comparison of CA1 in dorsal vs. ventral hippocampus, an additional horizontal slice corresponding to the stereotaxic coordinates in mm: interarural = 6.16, bregma = −2.88, lateral =2.95 and DV level = 2.12 was selected to identify a dorsal CA1 ROI. A second slice corresponding to the stereotactic coordinates in mm: interaural = 2.56, bregma = −2.88, lateral = 3.2 and dorsoventral (DV) level = 4.8, was selected to identify a ventral CA1 ROI (supplementary Fig 2). These coordinates were chosen based on the MBL C57BL/6J atlas (www.mbl.org). The minimum size of the ROIs was 26 pixels. Right and left hippocampal subregions were measured and averaged.
In order to estimate measurement reliability for rCBV, normalized CBV values for the same mouse were compared across two imaging sessions that were separated by a brief period, during which it is reasonable to expect minimal or no changes. Eight 9-month-old mice (4 CBKO and 4 controls) were imaged within 48 hours. The interclass correlation between values paired by mouse, but randomly assigned to test or retest were: r[CA3]=0.97, r[DG]=0.96, r[CA1]=0.89, r[EC]=0.93, r[SC]= 0.912.
Structural Imaging
The MRIs generated for functional analysis were used to obtain anatomical volumes as well. Both 2D-FSE (all mice) and 3D-GE data (18 mice) were used to generate brain volumes utilizing the ITK-SNAP program (Yushkevich et al., 2006), which allows for active contour generation. For the present work manual boundary drawing was used, which involved slice-by-slice boundary drawing on available orthogonal views. This method has a coupling cursor between 2D slices and the 3D display, which significantly helps to reduce slice-by-slice jitter that occurs in this type of segmentation. Manual segmentation was performed by three highly trained raters using a standard atlas. Although there was a high correlation between 2D and 3D MRI (r=0.87), 2D images consistently provided slightly higher volumes. The volumes measured in 2D images were obtained by multiplying the pixel size by the inter-slice distance (SI=0.7mm). A slight overestimation may have resulted because the volume from where the signal is obtained reflects a smaller slice thickness (ST=0.6mm). Therefore we calculated the average height in the slice dimension for a given ROI (Hs) and multiplied the 2D volumes by the following correction factor: (1-(1-ST/SI)/Hs)). For instance Hs=1.2: (1-(0.6/0.7)/1.2=0.88; Hs = 2.34: (1-(1–0.6/0.7)/2.34)=0.94. Volume based reliability analysis, comparing 3D and corrected 2D images was performed using one-way random effects on intra-class correlation statistics. We found excellent agreement between 3D and corrected 2D images by the three raters in all areas except the thalamus, which was subsequently removed from the structural volumetric analysis. Mean values of the correlations were: neocortex=0.972, ventricles=0.934, striatum= 0.965, whole brain=0.938, thalamus 0.712. Both rCBV-ROIs and anatomical ROI-segmentations were compared between the three raters using common intra-class correlation statistics. The analysis showed high agreement between the raters for all the ROIs analyzed with values between 0.8912 and 0.9710. Right and left structures were measured and the total volume was reported.
Statistical Analyses
Cross-Sectional Data
Standard statistical methods were used for the cross-sectional data. These included generalized multivariate linear models, bivariate correlations, intra-class correlations, ANOVA, repeated measures ANOVA and curve estimation. The specific selection of variables and methods is specified for each experiment in the results section. Outcome variables were normally distributed.
Longitudinal Data
Longitudinal data were analyzed by applying generalized estimating equations (GEE) (Zeger et al., 1988), with repeated measures. This statistical method takes into account that there were multiple observations per subject, which are likely to be correlated, and treats them as clusters. The dependent variables were the normalized rCBV values, the independent variable was genotype and the interaction parameter was genotype and time. Sex and specific anatomical regions were included as covariates in subsequent analyses.
The GEE analysis yields coefficient values, which represent the associations between a factor score and variables included in the model. There were three main coefficients of interest in each model: one comparing the rCBV values at baseline, one relating the change in rCBV with time, and an interaction term for time and genotype. The main variable of interest was the interaction coefficient, which indicates whether there was a significant difference between genotypes in the rate of rCBV change over time. To corroborate the results of the GEE analysis, longitudinal data were also analyzed with a mixed effect model with random intercept.
Behavioral Testing
Hippocampus-dependent learning was tested in young (3–6 months of age) and middle-aged/old (9–21 months of age) CBKO and age-matched WT C57BL/6J mice using an active place avoidance task (Cimadevilla et al., 2001a). This spatial task involves placing the animal on a circular (40 cm diameter) platform that rotates clockwise at a speed of 1 rpm. The rotating platform is open to the room environment, within which a 60° region is designated as a shock zone. In this task, entrance into the shock zone results in a brief constant current foot shock (500 ms, 60 Hz, 0.2 mA) that is scrambled across pairs of the platform floor’s parallel rods. Mice receive additional shocks of the same intensity and duration every 1.5 seconds until they leave the shock zone. Animals quickly learn to actively avoid the shock zone whenever the arena rotation brings them within its vicinity, by running into the non-shock areas of the room (Fig. 4A). PC-based software tracks the position of the mouse by analyzing images from an overhead camera and delivers shocks appropriately (Tracker, Bio-Signal Group Corp., Brooklyn, NY). The number of times the animal entered the shock zone, the distance it actively moved across the platform, and other parameters of behavior were computed offline by Track Analysis software (Bio-Signal Group Corp., Brooklyn, NY).
Figure 4. Spatial learning deficit in CBKO mice.

A) Schematic representation of the active place avoidance task: The active place avoidance task involves placing the mouse on a rotating platform, open to visual cues within the room. A stationary 60° region of the room is designated as the shock zone. An overhead camera is used to track the movement of the mouse during the trial session. B) The average number of times animals from each group entered the shock zone averaged across all 9 training trials. Both young and middle aged/old CBKO mice entered the shock zone significantly more often than WT mice. (p<0.05) (means ± SEM). C) Distance traveled by each group averaged across habituation and all 9 training trials. Individual t tests confirmed that middle-aged/old CBKO mice walked a significantly shorter distance on the rotating platform than middle-aged/old WT controls. Young CBKO and WT mice did not differ on this measure (p<0.05) (means ± SEM). D) To control for movement differences between the groups, the number of shock zone entrances each animal made per meter traveled was calculated and averaged across all 9 training trials. The results confirm our finding that both young and middle-age/old CBKO mice entered the shock zone significantly more often than WT mice. (p<0.05) (means ± SEM).
At the start of the experiment, mice were habituated to handling and the training environment for 10 minutes, during which time the shock was turned off and they walked freely on the rotating platform. Then the shock was turned on and mice received three 10-minute training trials every day for three days, with an inter-trial interval of 50 minutes. Active place avoidance was measured by averaging the number of times mice entered the shock zone across all 9 trials. Speed of movement and total path length were also compared across groups. A total of ten young (5 CBKO and 5 WT) and 8 middle-aged/old (4 CBKO and 4 WT) sex-matched mice were tested. Data were analyzed by two-way ANOVA and Student’s t test for independent samples. Significance was accepted for p<0.05.
RESULTS
Hippocampal rCBV Values During Normal Aging
We evaluated a large number of WT mice across their lifespan in a cross-sectional study to establish how rCBV measures change in the hippocampus during the normal aging process. Newly acquired rCBV data were combined with data from previous studies for a total of 59 C57BL/6J mice, ranging from 1.5 to 24 months of age (see material and methods for details). The data were fit with a quadratic curve (Fig. 1E) and a curve estimation analysis was performed to evaluate the temporal profile of rCBV changes in different subregions of the hippocampus. In this analysis, rCBV measures from different regions of interest (ROIs) were included as the dependent variables and age was included as the independent variable. In agreement with a previous primate study (Small et al., 2004), we found an age-dependent decline in rCBV values specifically in the DG (F(1,56)=12.28 p=0.0003). In contrast, no significant changes were observed in other subregions of the hippocampal formation, including the entorhinal cortex (F(1,56) =0.087; p=0.764) (Fig. 1E). These data confirm that fMRI-rCBV is sensitive enough to detect age-related changes in subregions of the hippocampal formation in C57BL/6J mice.
Hippocampal rCBV Values in CBKO mice
To gain insight into the role of CB in the aging hippocampus, rCBV maps were generated in 26 CBKO and 26 WT controls ranging from 1.5 to 16 months of age. Each subject was imaged once in this cross-sectional study. Data were initially analyzed with an analysis-of-variance (ANOVA) model, in which genotype (CBKO vs. WT) was included as the fixed factor, rCBV values measured from individual hippocampal subregions were included as the dependent variables, and sex was included as a covariate. Results revealed that compared to WT mice, CBKO mice had significantly lower rCBV values in the DG (F(1,50)=6.94, p=0.010) and CA1 (F(1,50)=6.22, p=0.016) subregions of the hippocampus. Values in the entorhinal cortex, subiculum, and CA3 subregions of the hippocampal formation did not differ significantly between the two groups (Fig. 2A) and there were no significant sex differences.
Figure 2. Spatial and temporal pattern of hippocampal rCBV measures in CaBP KO mice.

Mean normalized relative cerebral blood volumes (rCBV) were measured from each hippocampal subregion in KO and WT mice. A) Significantly lower values were observed in the DG and CA1 of CBKO mice. B) Comparison of rCBV values in PVKO mice demonstrated a similar spatial pattern, where lower rCBV measures were also observed in DG and CA1. C) KO and WT mice were assigned to a young group or middle-aged group (see text for details). The estimated marginal means of rCBV in CBKO (squares), PVKO (triangles) and WT controls (circles) in DG. D) The estimated marginal means of rCBV for each group in EC. Note that the only age-dependent decline is in the DG of CBKO mice. E-H). Representative horizontal T2-weigthted images (left) and close up views of the hippocampal formation (right) using pseudo color rCBV maps in CBKO, PVKO, and WT mice at 9-months of age. Warmer colors represent larger rCBV values.
EC=entorhinal cortex, SC=subiculum, CA1=CA1subfield, CA3=CA3 subfield, DG=dentate gyrus, CBKO=calbindin D-28k knockout, PVKO= parvalbumin knockout, WT= wild-type * denotes p value ≤ 0.05, and ** p value ≤ 0.01.
Because there are known dorsal-ventral differences in CB expression in rodent CA1, we obtained rCBV values from dorsal and ventral CA1 in the same group of CBKO and WT mice. We found that CBKO mice had significantly lower CA1 rCBV values in ventral hippocampus (F(1,50)= 5.49, p= 0.029) but not dorsal hippocampus (F(1,50)= 0.702, p= 0.411) when compared to WT controls (supplementary Figure 2). These findings are consistent with the anatomical distribution of CB in CA1, which is more abundant in ventral than dorsal hippocampus (Baimbridge et al., 1991).
To evaluate the degree to which rCBV measures in the hippocampus decreased with age, mice were assigned to a young group (1.5 and 6 months of age) or a middle-aged group (9, 12 and 16 months of age) and a generalized linear model (GLM) was used to evaluate the genotype × age interaction, (fixed factors) controlling for sex and multiple comparisons. This analysis showed a significant genotype × age interaction in the DG (F(4)= 4.89, p=0.031) but not in CA1 (F(4)=2.037, p=0.108). Consistent with the 1-way ANOVA results above, the genotype × age interaction was not significant, in the entorhinal cortex (F(4)=1.02, p=0.782) (Fig. 2C & D) or the other hippocampal subregions (subiculum, CA3, dorsal CA1 or ventral CA1). These results indicate that the DG was the only region in which the rCBV decline was clearly age-related in CBKO mice.
To further verify this effect, we performed a longitudinal analysis on a subset of mice used in the cross-sectional study (16 CBKO and 18 WT mice). These mice were imaged repeatedly from 1.5 to 12 months of age. The longitudinal rCBV dataset was analyzed using a generalized estimating equations (GEE) model that accommodated the age-range and repeated measures (Zeger et al., 1988). The main term of interest was the genotype × time interaction. Consistent with the results of the cross-sectional study, the results of this analysis (Table 1) showed: 1) a significant effect of genotype in CA1 (Wald chi square= 5.16, p=0.023), but no genotype × time interaction (Wald chi square=1.26, p= 0.261), indicating that at the earliest time point (1.5 months) CA1 values were already significantly lower in CBKO mice compared to WT controls. 2) No significant effect of genotype in the DG (Wald chi square= 1.44, p=0.229), but a significant genotype × time interaction (Wald chi square= 6.15, p=0.013), indicating an age-dependent decline in DG rCBV values in CBKO mice. These effects were confirmed using a mixed effect model with random intercept.
Table 1.
Longitudinal rCBV Dataset in CBKO Mice.
| ROI | Variable | Estimated β | Standard error | Wald Chi-Square | Significance |
|---|---|---|---|---|---|
| Entorhinal Cortex | Genotype | 0.061 | 0.032 | 3.66 | 0.063 |
| Time | 0.001 | 0.002 | 0.25 | 0.616 | |
| Genotype × Time | −0.010 | 0.003 | 3.86 | 0.053 | |
| CA1 subfield | Genotype | 0.004 | 0.023 | 5.16 | 0.023* |
| Time | 0.002 | 0.026 | 0.02 | 0.877 | |
| Genotype × Time | −0.004 | 0.003 | 1.26 | 0.261 | |
| CA3 subfield | Genotype | 0.037 | 0.030 | 1.48 | 0.223 |
| Time | 0.000 | 0.002 | 2.92 | 0.087 | |
| Genotype × Time | −0.006 | 0.003 | 3.47 | 0.062 | |
| Dentate gyrus | Genotype | 0.027 | 0.022 | 1.44 | 0.229 |
| Time | 0.001 | 0.002 | 2.10 | 0.147 | |
| Genotype × Time | −0.007 | 0.002 | 6.15 | 0.013* | |
| Subiculum | Genotype | 0.064 | 0.031 | 4.13 | 0.052 |
| Time | 0.004 | 0.003 | 0.05 | 0.824 | |
| Genotype ×Time | −0.010 | 0.005 | 3.70 | 0.054 |
Genotype represents CBKO mice vs. age matched controls
Time represents the repeated measurements
Genotype × Time is the interaction term representing age dependency of the changes.
denotes statically significant difference p ≤ 0.05.
Hippocampal rCBV Values in PVKO mice
Parvalbumin (PV) is also a CaBP that is prominently expressed in the hippocampus, although in a separate subpopulation of cells that do not contain CB (Celio, 1990). We investigated whether the absence of PV in PVKO mice also affects hippocampal rCBV values in an age-related fashion. rCBV maps were generated in 20 PVKO and age-matched controls ranging from 1.5 to 16 months of age, each of which was imaged once in a cross-sectional study. Similar to what was described above for CBKO mice, PVKO mice were separated into a young group (1.5 and 6 months of age) and a middle-aged group (9 and 16 months of age) and results were analyzed with a generalized linear model (GLM). The only difference between this analysis and that described above is that data were unavailable for 12-month-old PVKO mice. Results revealed that like CBKO mice, PVKO mice had lower rCBV values in the DG (F(1,38) =6.81 p=0.015) and CA1 (F(1,38) =4.12, p=0.049) (Fig. 2B) compared to WT controls. However, unlike CBKO mice there was no significant genotype × age interaction in any hippocampal subregion, indicating that the lower rCBV values detected in PVKO mice were not age-dependent (Fig. 2C&D).
Structural Changes in CBKO and PVKO Mice
To investigate whether a lack of CB or PV affects age-related changes in brain structure, we used axial 2D-T2 and 3D-T2*- weighted images to perform a volumetric analysis in CBKO, PVKO and WT mice. The following cerebral structures were included in the analysis: ventricles, hippocampus, neocortex, striatum and whole brain (Supplementary Fig. 1A-C). Results were normalized by generating a ratio of each specific structure to the volume of the whole brain. Because the results of the statistical analyses using these normalized values did not vary significantly from those using raw values, only the raw values are reported. The same CBKO, PVKO and WT groups used to generate rCBV maps were used here.
Data were analyzed with an ANOVA in which genotype was included as the fixed factor, volume of each structure was included as the dependent variable, and age and sex were included as covariates. Results revealed that compared to WT controls, CBKO mice had a significantly enlarged hippocampus (24.65 ± 1.12 vs. 21.06 ± 0.67; F(1,56)= 6.873, p=0.02) neocortex (116 ± 2.98 vs. 108 ± 4.36; F(1,56)=5.021, p=0.034) and whole brain (474.45 ± 8.4 vs. 452.68 ± 5.3; F(1,56)=11.082, p=0.002; volumes in mm3) (Fig. 3A). When young and middle-aged groups were compared, no significant differences were observed, indicating that the volumetric changes in CBKO mice were not age-dependent. The brain volumes of PVKO mice did not differ significantly from WT mice in any of the analyzed regions (Fig. 3B) and no age-dependent differences were detected.
Figure 3. Volumetric analysis of CBKO and PVKO mice.
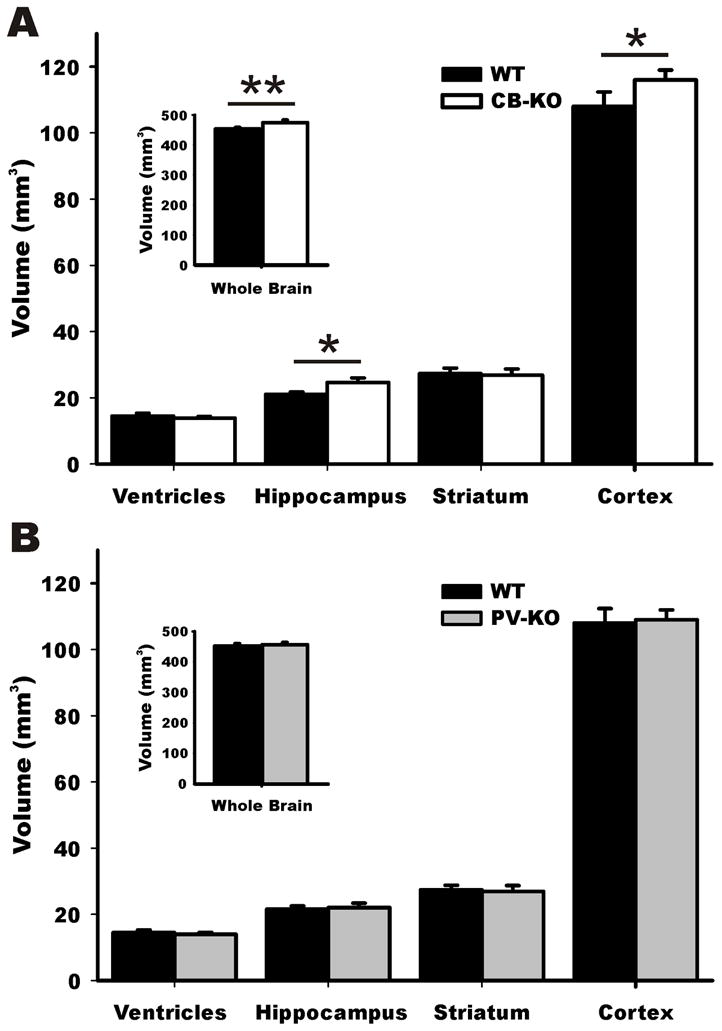
A) The volume of the hippocampus and cortex was significantly larger in CBKO mice compared to WT controls. Whole brain volumes were also significantly larger in this group (inset). B) The volumes of PVKO mice were not significantly different from WT controls in any of the brain regions analyzed.
Mean volumes in mm3 and SD for the different ROIs. WT= wild-type, * denotes p value ≤ 0.05, and ** p value ≤ 0.01
To evaluate whether there was a relationship between anatomical volume and rCBV, a bivariate correlation analysis was performed. However, no significant correlation was observed between these parameters in any of the brain regions analyzed.
Hippocampus-Dependent Learning in CBKO Mice
We evaluated whether altered hippocampal rCBV and structure are associated with impaired hippocampus-dependent learning by testing CBKO and WT mice in an active place avoidance task. In this task, animals placed on a rotating platform are trained to avoid a region of the stationary room that has been designated as a shock zone (see material and methods for details). Optimal performance in this task depends upon normal hippocampal functioning (Cimadevilla et al., 2001; Wesierska et al., 2005) and persistent long-term potentiation in the hippocampus, which is a proposed physiological mechanism of memory storage (Pastalkova et al., 2006). We tested eight middle aged/old (4 CBKO; 4 WT) and ten young (5 CBKO; 5 WT) mice and evaluated their acquisition of active place avoidance by measuring the number of times mice entered the shock zone. On average, young and middle-aged/old WT mice only entered the shock zone a few times, indicating both age groups successfully learned the task. In contrast, CBKO mice of both age groups entered the shock zone more often than their respective WT controls, indicating they were impaired in learning the location of the shock zone (Fig. 4B). We confirmed these effects with a two-way ANOVA with factors: age (young vs. middle-aged/old) and genotype (CBKO vs. WT), which revealed a significant effect of genotype (F(1,14)=5.60, p<0.05), no significant effect of age (F(1,14)=0.16, p=0.69) and no significant age × genotype interaction (F(1,14)=0.07, p=0.79). Given previous reports that CBKO mice display mild ataxia (Airaksinen et al., 1997), we compared the movement of CBKO mice with their respective control groups during habituation and across all 9 training trials. Indeed, we found that on average, the middle-aged/old CBKO mice walked more slowly (speed: 2.99 + 0.27 cm/s) and covered a shorter distance on the rotating platform (17.92 + 1.63 m) than middle-aged/old WT mice (speed: 3.81 ± 0.17 cm/s; distance 22.83 ± 1.02 m). These differences were not seen with young CBKO (speed: 3.42 ± 0.11 cm/s; distance: 20.54 ± 0.66 m) and young WT mice (speed: 3.53 ± 0.20 cm/s; distance: 21.19 ± 1.21 m). To confirm these findings, we first compared average distance traveled across groups by a two-way ANOVA with factors: age (young vs. middle-aged) and genotype (CBKO vs. WT). We found a significant effect of genotype (F(1,14)=5.87, p<0.05), no significant effect of age (F(1,14)=0.18, p=0.67), and no significant age × genotype interaction (F(1,14)=3.43, p=0.085). Student’s t tests supported the impression (Fig. 4C) that the middle-aged/old CBKO mice walked a shorter distance than middle-aged/old WT mice (t(6)=2.55, p<0.05), while the young CBKO and WT mice walked similar distances (t(8)=0.48, p=0.65). We controlled for these movement differences by calculating the number of shock zone entrances each animal made per distance traveled (Fig. 4D) and compared the groups by a two-way ANOVA with factors: age (young vs. middle-aged) and genotype (CBKO vs. WT). The analysis confirmed that active place avoidance was impaired in both young and middle-aged/old CBKO mice, because the effect of genotype was significant (F(1,14)=5.98, p<0.05), the effect of age (F(1,14)=0.15, p=0.71) was not significant and the age × genotype interaction (F(1,14)=0.02, p=0.88) was not significant. This behavioral impairment demonstrates that the changes detected with fMRI in both young and middle-aged CBKO mice correlate with a deficit in hippocampus-dependent learning.
DISCUSSION
Aging in different species, including humans, is accompanied by altered CNS Ca2+ homeostasis (Kirischuk and Verkhratsky, 1996). According to the “Ca2+ aging hypothesis,” increased [Ca2+]i is the main cause of dysfunction during the aging process (Ouanounou et al., 1999). This increase in [Ca2+]i could result from reduced buffering by CaBPs, such as CB and PV, both of which decrease with age (de Jong et al., 1996). Age-related loss of CB has been described in the hippocampus as well as the cerebellum, nucleus basalis of Meynert, olfactory bulb, cerebral cortex and retina (Bu et al., 2003; de Jong et al., 1996; Geula et al., 2003; Iacopino and Christakos, 1990; Kishimoto et al., 1998). An age-related decrease in PV has also been described in the hippocampus, although the decrease is not as pronounced as that found with CB (de Jong et al., 1996).
The aim of the present study was to explore the role of CB and PV in age-related changes in the hippocampus. We hypothesized that if a decrease in either CaBP is involved with aging, then the complete lack of that protein in KO mice (Supplementary Fig. 3) would accelerate age-related changes in hippocampal function. The hippocampal function we focused on was basal metabolism, which was estimated with fMRI-rCBV. The first step in our study was to establish an fMRI-marker of normal aging, so that accelerated aging could be identified. This involved using rCBV to measure basal hippocampal metabolism in WT mice across their life span. Then we compared these measures to rCBV values in CBKO and PVKO mice at different ages. An earlier age-related decline in rCBV in any hippocampal subregion was taken as correlational evidence of accelerated aging in that region. In addition, we evaluated whether age-related metabolic changes were accompanied by structural changes or impairments in hippocampus-dependent learning in KO mice.
Using fMRI-rCBV to Measure Metabolic Changes in the Brain During Aging
The use of CBV as an estimate of brain metabolism has been validated in several studies using a range of MRI approaches across a variety of species (van Zijl et al., 1998; Wu et al., 2003; Wu et al., 2002). Although there are a number of other correlates of brain metabolism, such as cerebral blood flow, deoxyhemoglobin and glucose uptake, we used fMRI- rCBV maps because they provide superior spatial resolution (80 μm in plane resolution). The specific protocol used here involved basal (steady-state) imaging, which allows for the visualization of small hippocampal subregions. This is much more difficult to do with stimulus-induced fMRI responses (BOLD signals), which require rapid imaging and consequently provide lower spatial resolution. Previous studies have used steady-state contrast enhanced-CBV mapping to identify specific patterns of metabolic changes in the hippocampus in AD (Moreno et al., 2007), schizophrenia (Gaisler-Salomon et al., 2009; Schobel et al., 2009), aging (Small et al., 2004), diabetes (Wu et al., 2008) and following acute pharmacological interventions of serotonergic pathways (Mueggler et al., 2010).
A Decline in Dentate Gyrus Metabolism During Normal Aging
We found that normal aging in C57BL/6J mice involves a decline in metabolic function that differentially affects the DG. This finding is consistent with previous studies utilizing different techniques that also found that changes in the DG are associated with the aging process (Gazzaley et al., 1996; Geinisman et al., 1995; Small et al., 2004; West, 1993; West et al., 1994). It is also consistent with data presented in our previous publication (Moreno et al., 2007), which used the same fMRI-rCBV technique to image the DG in C57BL/6J mice. Together, these results demonstrate that fMRI-CBV is sensitive enough to detect statistically significant age-related changes in the DG of mice, although using advanced ages (>18 months) in the analysis is required. Based on these results, we propose that an age-dependent decline in DG rCBV is a marker of normal aging and an increase in the rate of this decline is indicative of accelerated hippocampal aging. Although this conjecture remains to be proven, we adopt this viewpoint for interpreting the results of KO mice.
The Absence of CB But Not PV Is Associated With Accelerated Age-related Decline in DG Metabolism
The fMRI-rCBV analysis of KO mice revealed the following: a) Compared to WT controls, CBKO mice have lower rCBV values in the DG and CA1 hippocampal subregions. b) Only the decline in DG rCBV is age-dependent in CBKO mice. c) Compared to WT controls, PVKO mice have lower rCBV values in the DG and CA1 hippocampal subregions. d) None of the changes in rCBV were age-dependent in PVKO mice. Together, these findings demonstrate that the absence of CB and PV correlate with metabolic dysfunction in the DG and CA1 subfields of the hippocampus. However, only the absence of CB is associated with accelerated age-related metabolic decline in the hippocampus. We therefore interpret this as evidence supporting a specific role for CB during aging in this brain region.
Our finding that metabolic abnormalities in CBKO and PVKO mice was limited to the DG and CA1 subregions of the hippocampus, identifies these areas as the most sensitive to altered Ca2+ homeostasis. This finding is in line with the expression patterns of CB and PV, both of which are found in the DG and CA1, although in different cell types. CB is strongly expressed in DG granule cells and CA1 pyramidal cells and is weakly expressed in the CA3 subfield in a few perikarya from interneurons (Celio, 1990), In contrast, PV is only expressed in interneurons of the hippocampus, specifically in the granule cell layer and hilus of the DG, and in stratum pyramidale and stratum oriens of the CA1 and CA3 subregions (Kosaka et al., 1987). Interestingly, there are pronounced age-related changes in the expression of CB throughout the hippocampus (Lee et al., 2009) and only a small age-related decrease in PV in the DG (de Jong et al., 1996). Such differences in the types of neurons where these proteins are expressed and their precise expression pattern during aging may account for why we found evidence in support of a specific role of CB and not PV in the aging hippocampus.
The specific role of DG metabolism in normal aging and in aging CBKO mice may be attributable to qualities that are specific to this brain region. DG activity is sparsely organized (Fricke and Prince, 1984; Santhakumar et al., 2005), compared to CA1 (Church and McLennan, 1989) and CA3 (Leutgeb et al., 2007; Traub et al., 1991). Perhaps sparse cell activity in DG allows the absence of CB to be better tolerated initially, but leads to accumulating neuronal dysfunction with time. An additional mechanism that may account for the selective role of the DG in aging is adult neurogenesis, which also declines with age in rodents (Kuhn et al., 1996), non-human primates (Leuner et al., 2007) and possibly humans (Manganas et al., 2007). Adult hippocampal neurogenesis originates in the subgranular zone of the DG and leads to the formation of new DG granule cells. If the growth of new dentate granule cells serves as a selective protective mechanism in this region, then it would account for why we detected metabolic changes in the CA1 subregion across all age groups, but changes in the DG with age, as the rate of the neurogenesis declines. However, further studies are necessary before we can determine what role, if any, neurogenesis plays in mediating the effects of CB on normal DG function. Alternatively, it is possible that young CBKO mice have compensatory mechanisms to cope with the absence of CB in the DG or that the alterations in young CBKO mice do not manifest at the level of rCBV.
Although CB-immunoreactive (-ir) neurons are present in all layers of the entorhinal cortex (EC), we found no differences between CBKO and WT mice in EC rCBV levels. These findings suggest that the entorhinal cortex is less sensitive than the DG and CA1 subregions to alterations caused by the absence of CB. Furthermore, metabolic changes in the DG and CA1 subregions cannot be attributed to dysfunction in their entorhinal inputs. Given that the hippocampal circuit is heavily interconnected, a primary dysfunction in any given subregion could lead to secondary dysfunctions in neighboring subregions. However, our results demonstrate that this is not necessarily the case.
Structural changes associated with lack of CB and PV expression
We found structural changes in CBKO but not PVKO mice. These included significant volumetric increases in the hippocampus and neocortex as well as global brain hypertrophy (large brain volumes, a tendency to smaller ventricles). Morphological changes previously reported in CBKO but not PVKO mice include a significant increase in the length and volume of dendritic spines in cerebellar Purkinje cells. (Vecellio et al., 2000). Our structural MRI data indicate that such changes may also take place in the hippocampus. Given that spine morphology can shape Ca2+ signaling in dendrites and spines (Schmidt et al., 2003), changes in spine morphology could underlie the metabolic changes we detected. We addressed this possibility by exploring whether changes in brain volume correlated with the normalized CBV (rCBV) values in CBKO mice and found no significant correlation. Therefore, the structural changes in CBKO mice do not themselves account for the functional differences we found. Interestingly, the structural and functional values tended to change in the opposite directions (a decrease in rCBV corresponded to an increase in volume). Although a positive correlation between volume and function is often assumed, a negative correlation similar to the one we found here has been reported in autism (Williams and Minshew, 2007).
Impaired Hippocampus Dependent Learning in CBKO mice
Using an active place avoidance task, we found impaired hippocampus-dependent learning in both young and middle-age/old CBKO mice. Since the task is sensitive to overall hippocampal dysfunction (Cimadevilla et al., 2001), this learning deficit can be attributed to the increase in hippocampus volume and/or the reduction in CA1 metabolism detected in both age groups. Evidence that CA1 dysfunction alone is sufficient to underlie the impairment comes from ongoing work in our lab, in which global cerebral ischemia is induced in rats, consequent damage to CA1 neurons is quantified, and rats are trained in the active place avoidance task (Popp et al., 2008). These animals, which have damage largely restricted to CA1, are like CBKO mice in that they are impaired in learning this task, as measured by shock zone entrances. The important role of CA1 neurons in this task is further indicated by the significant negative correlation we find between intact CA1 neurons and the number of entrances into the shock zone in these rats. Our finding that there is no behavioral difference between young and middle-aged/old CBKO mice indicates that age-related changes in the DG are not sufficient to either exacerbate or compensate for the other structural and functional deficits found in these mice. Together, our behavioral results verify that the metabolic and structural changes detected with MRI techniques in the hippocampus of CBKO mice are of functional relevance.
Do CB and PV Play a Role in Neuroprotection?
The functional and structural data presented here implicate CB and PV in a neuroprotective role. Although there is a vast literature that supports such a role, the evidence is often correlational (Schwaller, 2009), with a focus on CB rather than PV. For example, aged mammals that have a significant decrease in the expression of CB in the DG also have decreased synaptic connections (Geinisman et al., 1995) and NMDA receptors (Gazzaley et al., 1996). In several amyloidogenic Alzheimer’s disease (AD) mouse models, there is a loss of CB in DG granule cells and the development of hippocampus based electrographic seizures (Berridge, 2010; Palop and Mucke, 2009). In one mouse model of AD, the loss of CB in DG granule cells correlates strongly with hippocampus-dependent learning deficits and a decrease in c-Fos in DG granule cells (Palop et al., 2003). Unfortunately, these studies do not rule out the possibility that a decrease in CB simply reflects altered intracellular Ca2+ homeostasis and a different component of the Ca2+ signalosome (Berridge et al., 2003) is responsible for dysfunction.
Additional evidence in favor of a neuroprotective role of CB comes from a study showing that cultured hippocampal CB-ir neurons are more resistant to neurotoxicity than CB-ir negative ones (Mattson et al., 1991). Similarly, ectopic expression of CB by herpes simplex virus (HSV)-mediated transfer of the CB gene also results in increased survival of hippocampal neurons exposed to excitotoxic insults (Phillips et al., 1999). Although both studies indicate that CB-ir neurons are less affected by neurotoxicity, they did not explore the precise role of CB by specifically down-regulating CB or using CBKO mice, which limits interpretation of the data.
The results of several studies challenge a role for CB in neuroprotection. For example, in human medial temporal lobe epilepsy (mTLE), CB expression is decreased or even completely absent in DG granule cells and Ca2+-dependent inactivation of voltage-gated Ca2+ channels is increased, which consequently reduces Ca2+ influx during repetitive stimuli (Nagerl et al., 2000). Under these circumstances, the loss or decrease of CB is beneficial, since it is thought to increase survival of DG granule cells. In rabbits, CB is absent in CA1 pyramidal cells, yet there is an age-dependent functional decline in these cells (de Jong et al., 1996), indicating that CB is not involved in this process. In a mouse model of kainite-mediated temporal lobe epilepsy and excitotoxicity, hippocampal damage was unaffected by the absence of CB and PV in KO mice (Bouilleret et al., 2000). In a different study, there was actually less hippocampal damage in CBKO mice than controls after carotid artery occlusion (Klapstein et al., 1998). Because all of these cell injury models involve considerably different procedures, it is not known if they share common mechanisms leading to cell death. Importantly, it is not known if these mechanisms play a role in age-mediated neurodegeneration, which is a much slower process than cell death resulting from one of the above procedures.
Our interpretation of the above data is that downregulation of CB may serve as a protective mechanism under specific circumstances. However, in the context of aging, downregulation of CB correlates with dysfunction. Our finding that constitutive absence of CB is associated with earlier age-related metabolic decline in the DG supports this hypothesis. While it is often assumed that an age-related neuroprotective function of CB is mediated through its direct modulation of Ca2+, it is possible that other mechanisms of action are involved, such as inhibition of caspase activity. It has been shown that CB acts as an anti-apoptotic signal by inhibiting caspase-3 in osteocytic and osteoblastic cells (Bellido et al., 2000; Christakos and Liu, 2004), and does so independent of its Ca2+-binding properties. CB may perform a similar function in neurons, although it is unclear if this is also independent of its role in Ca2+ buffering (Fan et al., 2007; Lema Tome et al., 2006). It is therefore possible that the metabolic changes we detected in CBKO mice are mediated by enhanced apoptosis, resulting from a lack of inhibition of caspase activity, and not a change in Ca2+ buffering.
Like CB, the case for a neuroprotective role of PV is also debated. While a neuroprotective effect has been consistently documented in motor neurons (Dekkers et al., 2004; Van Den Bosch et al., 2002), the findings are less clear in other cell types in vivo (Bouilleret et al., 2000; Maetzler et al., 2004) and in vitro (D’Orlando et al., 2002). The decline in rCBV-estimated metabolism we found in CA1 and DG of PVKO mice supports a neuroprotective role for PV in the hippocampus, although unlike CB, this appears to be unrelated to aging.
Conclusions
The present study used novel MRI techniques to identify functional and structural abnormalities in specific hippocampal subregions of mice lacking the Ca2+-binding proteins, CB or PV. Analyzing CBKO and PVKO mice over their life span allowed for the identification of metabolic and volumetric changes associated with advanced age and the absence of a CaBP. We found that DG metabolism declines during normal aging and that the constitutive absence of CB but not PV is associated with an earlier decline in DG metabolism. These results suggest that the normal decrease in CB that occurs with age is involved in age-related hippocampal metabolic decline. Understanding the relationship between the absence of CB and accelerated decline in hippocampal function may lead to the identification of general molecular pathways that are a fundamental part of the aging process.
Supplementary Material
Acknowledgments
We would like to thank: Mr. K Hess at Columbia University for his excellent technical help with the mouse lines, P. Flom, PhD at SUNY Downstate Medical Center for his help with the statistical methods and K. Todkar at University of Fribourg for the help of the immunohistochemical staining. This research was supported by NIH grants AG027476 to HM and AG025161 to SS and Swiss National Science Foundation grant 31003A-130680 to BS.
Grant sponsors: NIH; Grant number AG027476 to HM and AG025161 to SS. Swiss National Science Foundation; Grant number: 31003A_130680 to BS
Contributor Information
Herman Moreno, Email: herman.moreno@downstate.edu, SUNY Downstate Medical Center, The Robert F. Furchgott Center for Neural and Behavioral Science, Department of Neurology and Pharmacology &Physiology, 450 Clarkson Avenue Box 29, Brooklyn NY USA 11203, Phone: (718) 270-4660.
Nesha S. Burghardt, Email: nsb2113@columbia.edu, Columbia University, Departments of Neuroscience, Pharmacology & Psychiatry, 1051 Riverside Drive, Unit 87, New York, NY USA 10032.
Daniel Vela-Duarte, Email: Daniel.Vela@downstate.edu, SUNY Downstate Medical Center, Department of Pharmacology & Physiology, 450 Clarkson Avenue Box 29, Brooklyn NY USA 11203.
James Masciotti, Email: jmm2014@columbia.edu, Columbia University, Department of Biomedical Engineering, 630 West 168th Street, PH # 19, New York, NY USA 10032, Columbia University.
Fan Hua, Email: fh2107@columbia.edu, Columbia University, Department of Radiology, 630 West 168th Street, PH # 19, New York, NY USA 10032.
André A. Fenton, Email: afenton@nyu.edu, New York University, Center for Neural Science, New York University, 4 Washington Place, New York, NY USA 10003-6621 And SUNY Downstate Medical Center, The Robert F. Furchgott Center for Neural and Behavioral Science, Department of Pharmacology & Physiology, 450 Clarkson Avenue, Brooklyn NY USA 11203
Beat Schwaller, Email: Beat.Schwaller@unifr.ch, University of Fribourg, Unit of Anatomy, Department of Medicine, 1, route Albert-Gockel, CH-1700 Fribourg Switzerland.
Scott A. Small, Email: sas68@columbia.edu, Columbia University, School of Physicians and Surgeons, 630 West 168th Street, PH # 19, New York, NY USA 10032
References
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci U S A. 1997;94(4):1488–93. doi: 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andressen C, Blumcke I, Celio MR. Calcium-binding proteins: selective markers of nerve cells. Cell Tissue Res. 1993;271(2):181–208. doi: 10.1007/BF00318606. [DOI] [PubMed] [Google Scholar]
- Arabadzisz D, Ylinen A, Emri Z. Increased inter-spike intervals and fast after-hyperpolarization of action potentials in rat hippocampal pyramidal cells accompanied with altered calbindin immunoreactivity 10–12 months after global forebrain ischemia. Neurosci Lett. 2002;331(2):103–6. doi: 10.1016/s0304-3940(02)00864-9. [DOI] [PubMed] [Google Scholar]
- Armbrecht HJ, Boltz MA, Kumar VB, Flood JF, Morley JE. Effect of age on calcium-dependent proteins in hippocampus of senescence-accelerated mice. Brain Res. 1999;842(2):287–93. doi: 10.1016/s0006-8993(99)01802-8. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG. Calcium-binding proteins in the dentate gyrus. Epilepsy Res Suppl. 1992;7:211–20. [PubMed] [Google Scholar]
- Baimbridge KG, Peet MJ, McLennan H, Church J. Bursting response to current-evoked depolarization in rat CA1 pyramidal neurons is correlated with lucifer yellow dye coupling but not with the presence of calbindin-D28k. Synapse. 1991;7(4):269–77. doi: 10.1002/syn.890070404. [DOI] [PubMed] [Google Scholar]
- Bellido T, Huening M, Raval-Pandya M, Manolagas SC, Christakos S. Calbindin-D28k is expressed in osteoblastic cells and suppresses their apoptosis by inhibiting caspase-3 activity. J Biol Chem. 2000;275(34):26328–32. doi: 10.1074/jbc.M003600200. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 2010;459(3):441–9. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38(1):79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Bouilleret V, Schwaller B, Schurmans S, Celio MR, Fritschy JM. Neurodegenerative and morphogenic changes in a mouse model of temporal lobe epilepsy do not depend on the expression of the calcium-binding proteins parvalbumin, calbindin, or calretinin. Neuroscience. 2000;97(1):47–58. doi: 10.1016/s0306-4522(00)00017-8. [DOI] [PubMed] [Google Scholar]
- Bu J, Sathyendra V, Nagykery N, Geula C. Age-related changes in calbindin-D28k, calretinin, and parvalbumin-immunoreactive neurons in the human cerebral cortex. Exp Neurol. 2003;182(1):220–31. doi: 10.1016/s0014-4886(03)00094-3. [DOI] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35(2):375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- Christakos S, Liu Y. Biological actions and mechanism of action of calbindin in the process of apoptosis. J Steroid Biochem Mol Biol. 2004;89–90(1–5):401–4. doi: 10.1016/j.jsbmb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Church J, McLennan H. Electrophysiological properties of rat CA1 pyramidal neurones in vitro modified by changes in extracellular bicarbonate. J Physiol. 1989;415:85–108. doi: 10.1113/jphysiol.1989.sp017713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimadevilla JM, Wesierska M, Fenton AA, Bures J. Inactivating one hippocampus impairs avoidance of a stable room-defined place during dissociation of arena cues from room cues by rotation of the arena. Proc Natl Acad Sci U S A. 2001;98(6):3531–6. doi: 10.1073/pnas.051628398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orlando C, Celio MR, Schwaller B. Calretinin and calbindin D-28k, but not parvalbumin protect against glutamate-induced delayed excitotoxicity in transfected N18-RE 105 neuroblastoma-retina hybrid cells. Brain Res. 2002;945(2):181–90. doi: 10.1016/s0006-8993(02)02753-1. [DOI] [PubMed] [Google Scholar]
- de Jong GI, Naber PA, Van der Zee EA, Thompson LT, Disterhoft JF, Luiten PG. Age-related loss of calcium binding proteins in rabbit hippocampus. Neurobiol Aging. 1996;17(3):459–65. doi: 10.1016/0197-4580(96)00030-9. [DOI] [PubMed] [Google Scholar]
- Dekkers J, Bayley P, Dick JR, Schwaller B, Berchtold MW, Greensmith L. Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death. Neuroscience. 2004;123(2):459–66. doi: 10.1016/j.neuroscience.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Faas GC, Schwaller B, Vergara JL, Mody I. Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 2007;5(11):e311. doi: 10.1371/journal.pbio.0050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Shi L, Gu Y, Zhao Y, Xie J, Qiao J, Yang GY, Wang Y, Lu CZ. Pretreatment with PTD-calbindin D 28k alleviates rat brain injury induced by ischemia and reperfusion. J Cereb Blood Flow Metab. 2007;27(4):719–28. doi: 10.1038/sj.jcbfm.9600373. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6(4):347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fricke RA, Prince DA. Electrophysiology of dentate gyrus granule cells. J Neurophysiol. 1984;51(2):195–209. doi: 10.1152/jn.1984.51.2.195. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53(4):591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, Lewandowski N, Fairhurst S, Wang Y, Conjard-Duplany A, et al. Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacology. 2009;34(10):2305–22. doi: 10.1038/npp.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzaley AH, Siegel SJ, Kordower JH, Mufson EJ, Morrison JH. Circuit-specific alterations of N-methyl-D-aspartate receptor subunit 1 in the dentate gyrus of aged monkeys. Proc Natl Acad Sci U S A. 1996;93(7):3121–5. doi: 10.1073/pnas.93.7.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y, Detoledo-Morrell L, Morrell F, Heller RE. Hippocampal markers of age-related memory dysfunction: behavioral, electrophysiological and morphological perspectives. Prog Neurobiol. 1995;45(3):223–52. doi: 10.1016/0301-0082(94)00047-l. [DOI] [PubMed] [Google Scholar]
- Geula C, Bu J, Nagykery N, Scinto LF, Chan J, Joseph J, Parker R, Wu CK. Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss. J Comp Neurol. 2003;455(2):249–59. doi: 10.1002/cne.10475. [DOI] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–41. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Iacopino AM, Christakos S. Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc Natl Acad Sci U S A. 1990;87(11):4078–82. doi: 10.1073/pnas.87.11.4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Verkhratsky A. Calcium homeostasis in aged neurones. Life Sci. 1996;59(5–6):451–9. doi: 10.1016/0024-3205(96)00324-4. [DOI] [PubMed] [Google Scholar]
- Kishimoto J, Tsuchiya T, Cox H, Emson PC, Nakayama Y. Age-related changes of calbindin-D28k, calretinin, and parvalbumin mRNAs in the hamster brain. Neurobiol Aging. 1998;19(1):77–82. doi: 10.1016/s0197-4580(97)00166-8. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Vietla S, Lieberman DN, Gray PA, Airaksinen MS, Thoenen H, Meyer M, Mody I. Calbindin-D28k fails to protect hippocampal neurons against ischemia in spite of its cytoplasmic calcium buffering properties: evidence from calbindin-D28k knockout mice. Neuroscience. 1998;85(2):361–73. doi: 10.1016/s0306-4522(97)00632-5. [DOI] [PubMed] [Google Scholar]
- Kosaka T, Katsumaru H, Hama K, Wu JY, Heizmann CW. GABAergic neurons containing the Ca2+-binding protein parvalbumin in the rat hippocampus and dentate gyrus. Brain Res. 1987;419(1–2):119–30. doi: 10.1016/0006-8993(87)90575-0. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16(6):2027–33. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Hwang IK, Yoo KY, Choi JH, Park OK, Lee JC, Jeong YG, Lee IS, Won MH. Calbindin d-28k immunoreactivity and its protein level in hippocampal subregions during normal aging in gerbils. Cell Mol Neurobiol. 2009;29(5):665–72. doi: 10.1007/s10571-009-9360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema Tome CM, Bauer C, Nottingham C, Smith C, Blackstone K, Brown L, Hlavaty C, Nelson C, Daker R, Sola R, et al. MK801-induced caspase-3 in the postnatal brain: inverse relationship with calcium binding proteins. Neuroscience. 2006;141(3):1351–63. doi: 10.1016/j.neuroscience.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Leuner B, Kozorovitskiy Y, Gross CG, Gould E. Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci U S A. 2007;104(43):17169–73. doi: 10.1073/pnas.0708228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315(5814):961–6. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Maetzler W, Nitsch C, Bendfeldt K, Racay P, Vollenweider F, Schwaller B. Ectopic parvalbumin expression in mouse forebrain neurons increases excitotoxic injury provoked by ibotenic acid injection into the striatum. Exp Neurol. 2004;186(1):78–88. doi: 10.1016/j.expneurol.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Manganas LN, Zhang X, Li Y, Hazel RD, Smith SD, Wagshul ME, Henn F, Benveniste H, Djuric PM, Enikolopov G, et al. Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science. 2007;318(5852):980–5. doi: 10.1126/science.1147851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Rychlik B, Chu C, Christakos S. Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron. 1991;6(1):41–51. doi: 10.1016/0896-6273(91)90120-o. [DOI] [PubMed] [Google Scholar]
- Moreno H, Wu WE, Lee T, Brickman A, Mayeux R, Brown TR, Small SA. Imaging the Abeta-related neurotoxicity of Alzheimer disease. Arch Neurol. 2007;64(10):1467–77. doi: 10.1001/archneur.64.10.1467. [DOI] [PubMed] [Google Scholar]
- Mueggler T, Razoux F, Russig H, Buehler A, Franklin TB, Baltes C, Mansuy IM, Rudin M. Mapping of CBV changes in 5-HT(1A) terminal fields by functional MRI in the mouse brain. Eur Neuropsychopharmacol. 2010 doi: 10.1016/j.euroneuro.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20(5):1831–6. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouanounou A, Zhang L, Charlton MP, Carlen PL. Differential modulation of synaptic transmission by calcium chelators in young and aged hippocampal CA1 neurons: evidence for altered calcium homeostasis in aging. J Neurosci. 1999;19(3):906–15. doi: 10.1523/JNEUROSCI.19-03-00906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100(16):9572–7. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66(4):435–40. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313(5790):1141–4. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Phillips RG, Meier TJ, Giuli LC, McLaughlin JR, Ho DY, Sapolsky RM. Calbindin D28K gene transfer via herpes simplex virus amplicon vector decreases hippocampal damage in vivo following neurotoxic insults. J Neurochem. 1999;73(3):1200–5. doi: 10.1046/j.1471-4159.1999.0731200.x. [DOI] [PubMed] [Google Scholar]
- Popp S, Kelemen E, Fenton AA, Cottrell E, Kass I. Effects of low-dose lidocaine administration on transient global cerebral ischemia in rats. Society for Neuroscience. 2008 doi: 10.1016/j.neuroscience.2011.06.086. Abstract 152.14/AA8. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Aradi I, Soltesz I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. J Neurophysiol. 2005;93(1):437–53. doi: 10.1152/jn.00777.2004. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Stiefel KM, Racay P, Schwaller B, Eilers J. Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin D28k. J Physiol. 2003;551(Pt 1):13–32. doi: 10.1113/jphysiol.2002.035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66(9):938–46. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. Calcium Buffers. Elsevier Sciences; 2003. [Google Scholar]
- Schwaller B. The continuing disappearance of “pure” Ca2+ buffers. Cell Mol Life Sci. 2009;66(2):275–300. doi: 10.1007/s00018-008-8564-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol. 2010;2(11):a004051. doi: 10.1101/cshperspect.a004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Dick J, Dhoot G, Carroll S, Vrbova G, Nicotera P, Pette D, Wyss A, Bluethmann H, Hunziker W, et al. Prolonged contraction-relaxation cycle of fast-twitch muscles in parvalbumin knockout mice. Am J Physiol. 1999;276(2 Pt 1):C395–403. doi: 10.1152/ajpcell.1999.276.2.C395. [DOI] [PubMed] [Google Scholar]
- Small SA, Chawla MK, Buonocore M, Rapp PR, Barnes CA. Imaging correlates of brain function in monkeys and rats isolates a hippocampal subregion differentially vulnerable to aging. Proc Natl Acad Sci U S A. 2004;101(18):7181–6. doi: 10.1073/pnas.0400285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66(2):635–50. doi: 10.1152/jn.1991.66.2.635. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Schwaller B, Vleminckx V, Meijers B, Stork S, Ruehlicke T, Van Houtte E, Klaassen H, Celio MR, Missiaen L, et al. Protective effect of parvalbumin on excitotoxic motor neuron death. Exp Neurol. 2002;174(2):150–61. doi: 10.1006/exnr.2001.7858. [DOI] [PubMed] [Google Scholar]
- van Zijl PC, Eleff SM, Ulatowski JA, Oja JM, Ulug AM, Traystman RJ, Kauppinen RA. Quantitative assessment of blood flow, blood volume and blood oxygenation effects in functional magnetic resonance imaging. Nat Med. 1998;4(2):159–67. doi: 10.1038/nm0298-159. [DOI] [PubMed] [Google Scholar]
- Vecellio M, Schwaller B, Meyer M, Hunziker W, Celio MR. Alterations in Purkinje cell spines of calbindin D-28 k and parvalbumin knock-out mice. Eur J Neurosci. 2000;12(3):945–54. doi: 10.1046/j.1460-9568.2000.00986.x. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B. Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol. 2003;89(3):1414–22. doi: 10.1152/jn.00576.2002. [DOI] [PubMed] [Google Scholar]
- Wesierska M, Dockery C, Fenton AA. Beyond memory, navigation, and inhibition: behavioral evidence for hippocampus-dependent cognitive coordination in the rat. J Neurosci. 2005;25(9):2413–9. doi: 10.1523/JNEUROSCI.3962-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging. 1993;14(4):287–93. doi: 10.1016/0197-4580(93)90113-p. [DOI] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344(8925):769–72. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- Williams DL, Minshew NJ. Understanding autism and related disorders: what has imaging taught us? Neuroimaging Clin N Am. 2007;17(4):495–509. ix. doi: 10.1016/j.nic.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EX, Wong KK, Andrassy M, Tang H. High-resolution in vivo CBV mapping with MRI in wild-type mice. Magn Reson Med. 2003;49(4):765–70. doi: 10.1002/mrm.10425. [DOI] [PubMed] [Google Scholar]
- Wu G, Luo F, Li Z, Zhao X, Li SJ. Transient relationships among BOLD, CBV, and CBF changes in rat brain as detected by functional MRI. Magn Reson Med. 2002;48(6):987–93. doi: 10.1002/mrm.10317. [DOI] [PubMed] [Google Scholar]
- Wu W, Brickman AM, Luchsinger J, Ferrazzano P, Pichiule P, Yoshita M, Brown T, DeCarli C, Barnes CA, Mayeux R, et al. The brain in the age of old: the hippocampal formation is targeted differentially by diseases of late life. Ann Neurol. 2008;64(6):698–706. doi: 10.1002/ana.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44(4):1049–60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.