

Abstract
The molecular mechanisms controlling bone extracellular matrix (ECM) deposition by differentiated osteoblasts in postnatal life, called hereafter bone formation, are unknown. This contrasts with the growing knowledge about the genetic control of osteoblast differentiation during embryonic development. Cbfa1, a transcriptional activator of osteoblast differentiation during embryonic development, is also expressed in differentiated osteoblasts postnatally. The perinatal lethality occurring in Cbfa1-deficient mice has prevented so far the study of its function after birth. To determine if Cbfa1 plays a role during bone formation we generated transgenic mice overexpressing Cbfa1 DNA-binding domain (ΔCbfa1) in differentiated osteoblasts only postnatally. ΔCbfa1 has a higher affinity for DNA than Cbfa1 itself, has no transcriptional activity on its own, and can act in a dominant-negative manner in DNA cotransfection assays. ΔCbfa1-expressing mice have a normal skeleton at birth but develop an osteopenic phenotype thereafter. Dynamic histomorphometric studies show that this phenotype is caused by a major decrease in the bone formation rate in the face of a normal number of osteoblasts thus indicating that once osteoblasts are differentiated Cbfa1 regulates their function. Molecular analyses reveal that the expression of the genes expressed in osteoblasts and encoding bone ECM proteins is nearly abolished in transgenic mice, and ex vivo assays demonstrated that ΔCbfa1-expressing osteoblasts were less active than wild-type osteoblasts. We also show that Cbfa1 regulates positively the activity of its own promoter, which has the highest affinity Cbfa1-binding sites characterized. This study demonstrates that beyond its differentiation function Cbfa1 is the first transcriptional activator of bone formation identified to date and illustrates that developmentally important genes control physiological processes postnatally.
Keywords: Cbfa1, osteoblast function, bone formation, dominant negative, Cbfa1 autoregulation
Bone extracellular matrix (ECM) deposition or bone formation by differentiated osteoblasts starts late during development and lasts throughout life. This is a totally different process than bone development, which is characterized by cell commitment and differentiation. The bone ECM contains two types of proteins: the collagens, mostly type I collagen, which account for 90% of the bone matrix proteins (Gehron-Robey 1996), and the noncollagenous proteins, including osteocalcin, osteopontin, and bone sialo protein (Hauschka et al. 1989; Bianco et al. 1991; Denhardt and Guo 1993). Postnatally the production of bone ECM by osteoblasts must be regulated tightly to allow skeleton growth to occur and to replace bone resorbed by the osteoclasts throughout life (Ott 1996). Defective bone formation by the osteoblasts is at the origin of crippling and frequent genetic diseases such as osteogenesis imperfecta (Prockop and Kivirikko 1984; Byers 1990; Rowe and Shapiro 1998), and of acquired diseases like osteoporosis, the most frequent metabolic bone disease (Kleerekoper and Avioli 1998). In the United States only, >1 million osteoporotic fractures are diagnosed each year, resulting in substantial morbidity and mortality. This rate, which is expected to accelerate because of the unprecedented increase in life expectancy, exemplifies the importance of identifying molecular regulators of bone formation.
Cbfa1 is the only osteoblast-specific transcription factor identified to date. Molecular and genetic evidence have demonstrated that it acts as an activator of osteoblast differentiation during embryonic development in mouse and human. Indeed, Cbfa1 is expressed in cells of the osteoblastic lineage during development, it regulates osteoblast-specific expression of Osteocalcin and Osteopontin, can induce osteoblastic differentiation of nonosteoblastic cells (Ducy et al. 1997), and patients heterozygous for mutations or deletions of CBFA1 develop cleidocranial dysplasia (CCD) (Lee et al. 1997; Mundlos et al. 1997). Likewise, inactivation of Cbfa1 in mice leads to a total absence of osteoblasts in homozygous mutant animals, and to a CCD phenotype in heterozygous mutant animals (Komori et al. 1997; Otto et al. 1997). Thus Cbfa1 is an indispensable regulator of osteoblast differentiation that fullfills a function dominant to and nonredundant with the function of any other gene product.
This progress in our understanding of osteoblast differentiation during development has left unanswered a major question of skeleton biology, namely: What are the molecules controlling bone formation once osteoblast differentiation has occured? This lack of knowledge about the regulation of bone formation contrasts also with the growing body of information regarding the control of osteoclastic bone resorption (Soriano et al. 1991; Simonet et al. 1997; Bucay et al. 1998; Lacey et al. 1998; Yasuda et al. 1998).
Two correlative arguments suggest that Cbfa1 may be involved in postnatal bone formation. First, Cbfa1 is expressed at high levels in osteoblasts after birth (Ducy et al. 1997). Second, it regulates the in vitro and in vivo expression of Osteocalcin (Ducy et al. 1997; Frendo et al. 1998), a gene virtually not expressed before birth and that is the hallmark of the differentiated osteoblast phenotype (Owen et al. 1990; Stein et al. 1990; Aubin and Liu 1996). For these reasons, we decided to test whether Cbfa1 is a determinant of bone formation by differentiated osteoblasts postnatally. This question could not be answered in vivo until now because the deletion of Cbfa1 leads to perinatal lethality in mice (Komori et al. 1997; Otto et al. 1997), and no juvenile or more severe osteoporosis has been described in CCD patients. The observation that haploinsufficiency at the Cbfa1 locus causes CCD suggests that if Cbfa1 controls bone formation postnatally it should be possible to demonstrate this by altering its level of expression and/or its function postnatally. The availability of a cell-specific and stage-specific promoter, such as the Osteocalcin promoter that is not active before birth, provides us with an excellent tool to address this question.
We show here, through a time-specific, cell-specific, and stage-specific inhibition-of-function experiment that Cbfa1 is required for bone formation by differentiated osteoblasts after birth. By controlling its own expression positively Cbfa1 is at the top of a genetic cascade regulating bone ECM deposition. Inhibition of this autoregulatory loop in differentiated osteoblasts results in an osteopenic phenotype caused by the near abolition of expression of ECM-related genes, including type I collagen-encoding genes, without any overt effect on osteoblast differentiation. These results uncover a transcriptional pathway governing bone formation by differentiated osteoblasts and identify Cbfa1 as the first transcriptional activator of this process.
Results
ΔCbfa1 inhibits Cbfa1 transactivation function in a dominant-negative manner
Cbfa1 is one of the three known mouse Cbfa genes (Bae et al. 1992, 1995; Ogawa et al. 1993; Ducy et al. 1997). In Western blot analysis an antiserum that recognizes all three Cbfa proteins detected only Cbfa1 in calvaria osteoblasts (Fig. 1A, lane 5), thus indicating that Cbfa1 is the only Cbfa protein detectable in osteoblasts, which is consistent with the phenotype observed when the gene is deleted.
Figure 1.

Characterization of ΔCbfa1 activity in vitro. (A) Cbfa1 is the only detectable Cbfa expressed in osteoblasts. Western blot analysis of nuclear extracts from COS cells transfected with the empty vector (−, lane 1), a Cbfa3 (lane 2), a Cbfa2 (lane 3), or a Cbfa1 (lane 3) expression vector or from calvaria (lane 5). The antiserum used recognize all three Cbfa proteins (indicated by white asteriks). (B) DNA cotransfections in COS cells. Overexpression of ΔCbfa1 does not transactivate an OSE2–luc chimeric promoter construct, whereas overexpression of Cbfa1 does. (−) cotransfection with the empty vector. (C) Osteocalcin 160-bp promoter–luc activation by endogenous Cbfa1 is inhibited by cotransfection of increasing amounts of ΔCbfa1 in ROS 17/2.8 osteoblastic cells. (DBD) DNA-binding domain. (D) Different affinity of Cbfa1 and ΔCbfa1 for OSE2. DNA competition in EMSA were performed with identical amount of GST–Cbfa1 or GST–ΔCbfa1, labeled OSE2, and increasing amounts of unlabeled OSE2 oligonucleotide (comp.). Incubation time was 5 min. (E) High stability of the ΔCbfa1–OSE2 complex. EMSA was performed with a fixed quantity of protein (GST–Cbfa1 or GST–ΔCbfa1), labeled OSE2, and a 100-fold molar excess of cold OSE2. Incubation times in presence of competitor were as indicated.
It has been shown that the DNA-binding domain of runt-related proteins has no detectable transactivation function on its own (Bae et al. 1994; Aranson et al. 1997; Tracey et al. 1998). Consistent with these observations, Cbfa1 DNA-binding domain (ΔCbfa1) failed to transactivate a reporter vector containing six OSE2 elements, the Cbfa1-binding sites (Ducy and Karsenty 1995) in COS cells that do not express any Cbfa genes (Kurokawa et al. 1996) (Fig. 1B). In DNA cotransfection experiments performed in ROS 17/2.8 osteoblastic cells that do express Cbfa1 (Ducy and Karsenty 1995), increasing amount of ΔCbfa1 led to a threefold decrease of activity of a reporter vector containing a 160-bp Osteocalcin promoter fragment fused to the luciferase (luc) reporter gene (Fig. 1C). The activity of this Osteocalcin promoter fragment is partly dependent on the presence of one OSE2 element (Ducy and Karsenty 1995; Frendo et al. 1998). These results demonstrate that ΔCbfa1 has no transactivation ability on its own but can impair Cbfa1 transactivation function.
To define the molecular basis of this inhibition of transcription we performed DNA-binding assays with wild-type and truncated recombinant proteins. In the condition of electrophoretic mobility shift assay (EMSA) GST–ΔCbfa1 had a higher affinity for OSE2 than the full-length GST–Cbfa1 protein (Fig. 1D). Moreover the ΔCbfa1–DNA complex was far more stable than the Cbfa1–DNA complex (Fig. 1E). These data indicate that the dominant-negative function of ΔCbfa1 is because of its higher affinity for OSE2, and the higher stability of the ΔCbfa1–DNA complex.
Absence of skeletal abnormalities in newborn mice expressing ΔCbfa1 in a differentiated osteoblast-specific manner
Osteocalcin is the most osteoblast-specific gene known (Hauschka et al. 1989). Its expression is not only cell-specific but also time- and stage-specific. Indeed, its expression is virtually absent before birth, is restricted to differentiated osteoblasts able to produce a bone ECM, and is absent in osteoblast progenitors (Owen et al. 1990; Stein et al. 1990; Aubin and Liu 1996). Recently, we have shown that a 1.3-kb fragment of the mouse Osteocalcin (OG2) promoter contains all the regulatory elements necessary to confer differentiated osteoblast- and postnatal-specific expression to a reporter gene in vivo (Frendo et al. 1998). This Osteocalcin promoter fragment constitutes a unique resource to address osteoblast function without affecting osteoblast differentiation.
To determine whether ΔCbfa1 affects osteoblastic bone formation in vivo postnatally we generated a construct containing ΔCbfa1 coding sequence under the control of the 1.3-kb OG2 promoter (Fig. 2A) and used it to generate transgenic mice. In these transgenic animals, called thereafter ΔCbfa1-expressing mice, ΔCbfa1 was expressed only in bone (Fig. 2B). Two different lines of transgenic mice were generated, both lines had a similar level of expression of the transgene (Fig. 2C) and identical results were obtained with their progenies. The ΔCbfa1-expressing mice were phenotypically normal at birth; had a normally mineralized skeleton, an indication that their osteoblasts are functional; had no features characteristic of a CCD phenotype; and there was no perinatal lethality (Fig. 2D). The bone histology of these mice was also normal at birth (data not shown).
Figure 2.

Generation of ΔCbfa1-expressing mice. (A) Representation of the ΔCbfa1 transgene. (DBD) DNA-binding domain. (B) RT–PCR showing ΔCbfa1 bone-specific expression in transgenic mice (arrow). Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane (bottom). (C) RT–PCR analysis comparing ΔCbfa1 level of expression in the two independent transgenic lines analyzed in this study (F21 and F28, top). RNAs were extracted from long bones of 1-month-old mice. Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane (bottom). PCR reactions were performed in presence of [32P]dCTP. (D) Skeletal preparation of newborn wild-type and ΔCbfa1-expressing mice. Note the presence of normally developed skull (arrowhead) and clavicles (arrow), excluding a CCD phenotype. No delayed or impaired mineralization is visible at that stage unlike the case in the Cbfa1-deficient mice (Komori et al. 1997; Otto et al. 1997).
Postnatal osteopenia without osteoblast depletion in ΔCbfa1-expressing mice
Within 2 weeks after birth the ΔCbfa1-expressing mice developed a short stature phenotype (Fig. 3A). X-ray analysis showed shorter and more lucent long bones with thinner cortices, three features indicative of bone loss (Fig. 3B,C). The short stature phenotype was probably not caused by chondrocyte abnormalities as the growth plate cartilage appeared histologically and histomorphometrically indistinguishable between wild-type and ΔCbfa1-expressing animals (Fig. 3D and growth plate thickness: wild type = 228.26 ± 11.86 μm; ΔCbfa1 = 232.97 ± 26.76 μm). Conventional histology in both long bones and vertebrae showed a decreased amount of trabecular bone and thinner cortices, confirming the existence of an osteopenia affecting the whole bone in the ΔCbfa1-expressing mice (Fig. 3E–G). Inspection of the cells at high magnification following toluidine blue staining showed that the osteoblasts had the same morphology in wild-type and ΔCbfa1-expressing mice. However, the thickness of the osteoid layer was significantly thinner in ΔCbfa1-expressing mice (Fig. 3H).
Figure 3.
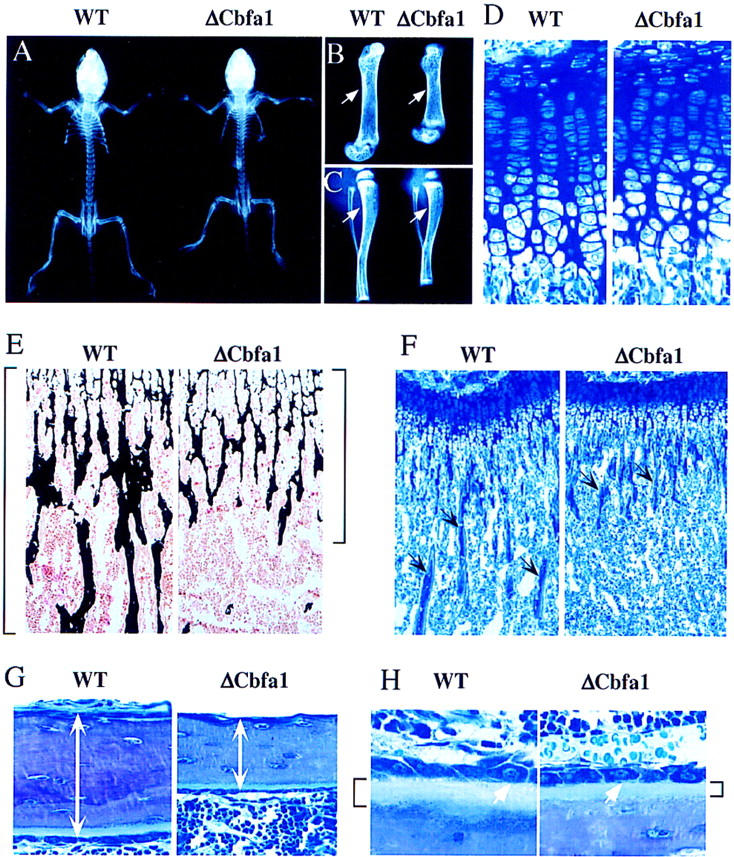
Radiological and histological analysis of ΔCbfa1-expressing mice. (A–C) X-ray analysis of 2-week-old wild-type (WT) and ΔCbfa1-expressing mice (ΔCbfa1). ΔCbfa1-expressing mice are smaller and have shorter long bones with decreased cortical thickness (arrows). (D) Histologic analysis of the growth plate cartilage in tibia of 3-week-old wild-type and ΔCbfa1 littermates. (E–H) Histologic analysis of cancellous (E,F,H) and cortical (G) bone in 3-week-old wild-type and ΔCbfa1-expressing littermates. (E) Longitudinal sections through the tibia at the metaphysis showing mineralized trabecular bone stained in black by von Kossa and osteoid and cartilage counterstained in pink with Kernechtrot. Brackets indicate the extend of calcified bone matrix. (F) Longitudinal sections through the tibia at the metaphysis stained with toluidine blue showing the decreased amount of bone trabeculae (arrows) present in the ΔCbfa1-expressing mice. (G) Longitudinal sections through the tibia stained by toluidine blue. Cortical thickness are indicated by white arrows. (H) High magnification visualization of the osteoblasts (white arrows) present at the surface of the tibia trabeculae showing that they appear morphologically identical in 3-week-old wild-type and ΔCbfa1-expressing littermates. Note that the osteoid layer present below the osteoblasts and staining light blue (brackets) is significantly thinner in ΔCbfa1-expressing mice compared to wild-type littermates.
To determine if this osteopenic phenotype was attributable to a functional defect of differentiated osteoblasts or to a decrease of the osteoblast number, static and dynamic histomorphometric analyses were performed over a 10-day period (day 10 to 20 postnatal) using tetracyclin/calcein, a marker of bone formation (Parfitt et al. 1987). This dynamic analysis aims at recording all the histological abnormalities existing during this period. As shown by the distance between the tetracyclin and calcein labels there was a major decrease (three- to fourfold) in the amount of newly formed bone in the ΔCbfa1-expressing mice compared to wild-type animals (Fig. 4A). The rate of bone formation was indeed 70% reduced in ΔCbfa1-expressing mice (Fig. 4B) and the osteoid thickness, an indicator of bone matrix deposition in the presence of an unchanged mineralization rate, was decreased significantly (Fig. 4C). Likewise, the bone volume was reduced in the ΔCbfa1-expressing mice (Fig. 4D).
Figure 4.

Bone formation and cell parameters in ΔCbfa1-expressing mice. (A) Fluorescent micrographs of the two labeled mineralization fronts at the mid-diaphysis of the tibiae of 3-week-old wild-type (WT) and ΔCbfa1-expressing mice. The brackets between the two labelings, tetracyclin at the top and calcein at the bottom, indicate the amount of newly formed bone. Note the three- to four-fold decrease in the distance between the two labeled areas in ΔCbfa1-expressing mice (B–D) Comparison of bone formation parameters in 3-week-old wild-type and ΔCbfa1-expressing mice. Measurement of bone formation rate (B), osteoid thickness (C), and trabecular bone volume (bone volume over total volume, BV/TV) (D). (E–I) Comparison of cell surfaces and numbers in 3-week-old wild-type and ΔCbfa1-expressing mice. (ObS) osteoblast surface; (BS) bone surface; (NOb) number of osteoblasts; (Bpm) bone perimeter (mm2); (OcS) osteoclast surface; (NOc) number of osteoclasts. Bars represent means ± s.e.m. (*) Statistically significant difference between wild-type and transgenic mice groups (*) P < 0.05; n = 6.
More importantly, this decrease in the bone formation rate occurred while there was no decrease in osteoblast and osteocyte numbers in the ΔCbfa1-expressing mice (Fig. 4E–G). This indicates that the osteopenia induced by the osteoblast-specific expression of ΔCbfa1 was caused by a functional failure of the osteoblasts, not by an absence or a depletion of the osteoblast population as in the Cbfa1-deficient mice. The number of osteoclasts was also identical in wild-type and transgenic animals (Fig. 4H,I).
Taken together these results indicate that ΔCbfa1 was interfering with a second function of Cbfa1, namely the maintenance of bone formation by differentiated osteoblasts.
ΔCbfa1-expressing osteoblasts are less active than wild-type osteoblasts
When put in culture osteoblastic cells synthesize type I collagen and alkaline phosphatase whose presence can be detected by colorimetric assays (Bancroft and Stevens 1996). Furthermore, following the synthesis of this collagen-rich matrix these cells have the unique ability to form compact nodules that become mineralized eventually (Aubin and Liu 1996; Stein et al. 1996). Thus, the size and the very existence of nodules is a reflection of the amount of extracellular matrix surrounding the cells. To determine whether the cells identified in ΔCbfa1-expressing mice as osteoblasts by morphologic criteria were bona fide osteoblasts we established osteoblast culture from newborn calvaria of wild-type and ΔCbfa1-expressing mice. Cells from the transgenic animals did form a collagen-rich matrix and mineralized nodules indicating that they were functional osteoblasts (Fig. 5A–C). However these nodules appeared smaller and less compact than the one observed when using cells from wild-type animals (Fig. 5A,B). The ECM synthetized by the ΔCbfa1-expressing cells stained positively for collagen and cellular alkaline phosphatase was detected but again these stainings were weaker than the one observed in wild-type nodules (Fig. 5B–D). Finally, von Kossa’s staining for mineral showed that the ECM surrounding the nodules generated using ΔCbfa1-expressing cells was mineralized poorly compared to the one surrounding the wild-type nodules (Fig. 5E,F). Thus, the results of these functional ex vivo assays indicate that the bones of the ΔCbfa1-expressing mice did contain functional osteoblasts but that these cells were less active than wild-type osteoblasts. This is in full agreement with the result of the histomorphometric analysis showing a normal number of osteoblasts but a decreased bone formation rate.
Figure 5.

Functional analysis of the osteoblasts derived from ΔCbfa1 mice. Ex vivo culture of osteoblasts derived from calvaria of newborn wild-type and ΔCbfa1-expressing mice were analyzed after 20 days in presence of mineralization medium (see Materials and Methods). (A) Goldner trichrome staining showing that the ΔCbfa1-expressing osteoblasts form smaller and fewer nodules (arrows). (B) van Gieson staining for collagen (pink/purple color). Note the lighter aspect of the matrix secreted by the ΔCbfa1-expressing osteoblasts. (C) von Kossa staining showing that the bone matrix secreted by the ΔCbfa1-expressing osteoblasts is mineralized poorly compared to wild-type cultures. (D) Higher magnification of the culture presented in B. There are fewer and smaller collagen-secreting nodules in cultures derived from ΔCbfa1-expressing mice than from their wild-type littermates. (E) High magnification of cells stained for alkaline phosphatase. ΔCbfa1-expressing osteoblasts are synthetizing alkaline phosphatase (blue) but at a lower level than wild-type mice. (F,G) von Kossa staining for mineralized bone matrix. (F) Less mineralized nodules (arrows) are present in the ΔCbfa1-derived cultures. (G) Higher magnification of the culture presented in F, photographied in polarized light. The mineralized nodules (black) present in cultures derived from ΔCbfa1-expressing mice are smaller than the one derived from their wild-type littermates.
Decreased expression of the genes encoding the main bone matrix proteins in ΔCbfa1-expressing mice
In the presence of a normal number of osteoblasts an osteopenia could develop only if each osteoblast is producing less of the various proteins required to form the bone ECM. Accordingly, the expression of the two genes encoding osteocalcin (OG1 and OG2) (Desbois et al. 1994), the most abundant noncollagenous protein (Hauschka et al. 1989), of Bone sialo protein, another osteoblast-specific gene (Bianco et al. 1991), and of Osteopontin (Denhardt and Guo 1993), were all decreased severely in 2- and 4-week-old ΔCbfa1-expressing mice (Figs. 6A and 7C, below). These proteins account for only 10% of the protein content of the bone ECM, therefore the decreased expression of these genes alone could not explain the osteopenic phenotype of the ΔCbfa1-expressing mice. Type I collagen is the most abundant constituent of the bone matrix, accounting for 90% of the bone protein content. It has been largely demonstrated that its integrity is required for proper bone formation and for skeleton growth postnatally (Prockop and Kivirikko 1984; Byers 1990; Rowe and Shapiro 1998). The expression of α1(I) and α2(I) collagen, the two genes encoding type I collagen, was decreased markedly in ΔCbfa1-expressing mice compared to wild-type littermates (Fig. 6A). The expression of Hprt was not affected in the ΔCbfa1-expressing mice indicating that the effects we observed were specific. The expression of α1(I) collagen was not reduced in skin fibroblasts of the ΔCbfa1-expressing mice (Fig. 6B) further demonstrating the specificity of the abnormalities observed in the bones of these animals.
Figure 6.

Osteoblast gene expression in ΔCbfa1-expressing mice. (A) RT–PCR analysis of gene expression in long bones of 2-week-old wild-type (wt) and ΔCbfa1-expressing mice. Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane. Expression of Osteocalcin, Bsp, and Osteopontin is virtually abolished, whereas expression of α1(I)collagen is reduced markedly, and the expresssion of α2(I)collagen is reduced moderately. (B) Northern blot analysis of α1(I)collagen expression in skin of 2-week-old wild-type and ΔCbfa1-expressing mice. The Gapdh cDNA was used to reprobe this Northern blot to assess equal loadings in the different lanes (bottom).
Figure 7.
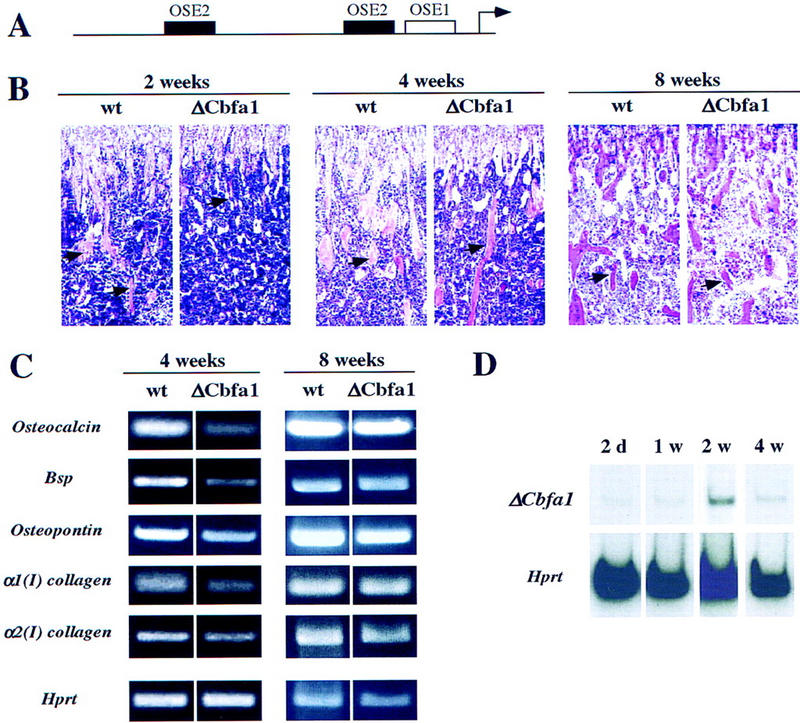
Auto-down-regulation of the ΔCbfa1 transgene and transient aspect of the osteopenic phenotype in ΔCbfa1-expressing mice. (A) Functional organization of the 1.3-kb OG2 promoter. The OSE1 site and the two OSE2 sites are indicated by open and solid boxes, respectively. (B) Histological analysis of long bones (tibiae) of 2-, 4-, and 8-week-old wild-type and ΔCbfa1-expressing mice. Sections through the metaphyses stained with hematoxylin/eosin. The amount of trabecular bone (pink, arrows) is severely decreased in 2-week-old trangenic mice and return to normal in 8-week-old animals. (C) RT–PCR analysis of ECM protein-encoding gene expression in long bones of 4- and 8-week-old wild-type and ΔCbfa1-expressing mice. Expression of Osteocalcin, Bone sialo protein, Osteopontin, α1(I)collagen, and α2(I)collagen in the transgenic mice is still decreased in 4-week-old animals, but less than that in 2-week-old mice (cf. with Fig. 6A), and returns to normal in 8-week-old mice. Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane. (D) Temporal analysis of ΔCbfa1 expression in transgenic animals. Expression of the transgene peaks at 2 weeks of age and is decreased severely thereafter. Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane.
Evolution of the phenotype of ΔCbfa1-expressing mice
The activity of the OG2 promoter fragment used to drive ΔCbfa1 is dependent partly for its expression on two OSE2 elements (Ducy and Karsenty 1995; Frendo et al. 1998). Given the higher affinity of ΔCbfa1 than Cbfa1 for DNA, one would predict that at one point the level of expression of the transgene should decrease. Its expression should not be abolished because of the presence in this promoter fragment of OSE1, another osteoblast-specific element (Fig. 7A). Consistent with this model we could observe a histological phenotype for the first 4 weeks of life of the ΔCbfa1-expressing animals (Fig. 7B). Beyond 4 weeks the phenotype faded and the mice had a normal life span (Fig. 7B). Likewise, the expression of the bone matrix genes was nearly abolished at 2 weeks of age, decreased at 4 weeks of age, and was normal in older animals (Figs. 6A and 7C). As expected, the expression of the transgene peaked at 2 weeks and decreased thereafter (Fig. 7D).
Down-regulation of Cbfa1 expression in ΔCbfa1-expressing mice
The near complete abolition of osteoblast-specific gene expression observed in the ΔCbfa1-expressing mice could be explained by a combination of several mechanisms: The level of expression of the transgene could be extremely high, ΔCbfa1 could bind to DNA with a higher affinity than full-length Cbfa1, and/or Cbfa1 could regulate its own expression. Comparison of the transgene and endogenous gene levels of expression revealed that they were similar (Fig. 8A), thus ruling out the first mechanism. As shown in Figure 1, D and E, ΔCbfa1 bound DNA with a higher affinity for OSE2 than the full-length Cbfa1, and the ΔCbfa1–DNA complex was more stable than the Cbfa1–DNA complex. This provided one molecular explanation for the progression of the phenotype.
Figure 8.

Characterization of Cbfa1 autoregulation. (A) RT–PCR analysis comparing the ΔCbfa1 transgene (top) and endogenous Cbfa1 (bottom) levels of expression in long bones of 2-day-old transgenic mice. (B) Down-regulation of Cbfa1 expression in 2-week-old ΔCbfa1 compared to wild-type (WT) mice. RT–PCR analysis of gene expression in long bones of 2-week-old wild-type and ΔCbfa1-expressing mice. Amplification of exon 2 of Hprt was used as an internal control for cDNA quality and PCR efficiency in each lane. (C) Presence of three identical OSE2 elements (shaded boxes) in the promoter of the mouse and human Cbfa1 genes. (D) Binding in EMSA of ROS 17/2.8 nuclear extracts to the OSE2 elements present in Cbfa1. The specific protein–DNA complex (arrow) is supershifted by preincubation with an antiserum against Cbfa1. (E) Binding in EMSA of GST–Cbfa1 to the wild-type (wt) but not the mutated (mut) versions of the OSE2s present in Cbfa1. (F) Functional characterization of the Cbfa1 OSE2 elements. Activation of the Cbfa1 promoter containing wild-type OSE2 elements upon cotransfection with a Cbfa1 expression vector in COS cells. Mutation of the OSE2 elements inhibits this effect.
To search for additional explanations we studied endogenous Cbfa1 expression in ΔCbfa1-expressing mice. Endogenous Cbfa1 expression was nearly abolished in every transgenic mouse analyzed at 2 weeks of age (Fig. 8B), implying that Cbfa1 controls its own expression. Sequence inspection of the 5′ region of the Cbfa1 mouse and human genes showed the presence of three conserved consensus OSE2 sites that were analyzed further (Fig. 8C). In EMSA, osteoblast nuclear extracts bound to oligonucleotides containing these OSE2 elements, an antibody against Cbfa1 caused an upward shift of the protein–DNA complex (Fig. 8D), and recombinant Cbfa1 bound to the wild-type oligonucleotides but not to their mutated counterparts (Fig. 8E). Finally, in DNA cotransfection experiments exogenous Cbfa1 transactivated a Cbfa1 promoter–luc chimeric gene containing wild-type OSE2 elements but not a similar construct containing mutant OSE2 elements (Fig. 8F). Taken together these experiments, along with the near abolition of Cbfa1 expression in ΔCbfa1-expressing mice, demonstrate that those elements are bona fide OSE2s, and that Cbfa1 is a positive regulator of its own expression.
Remarkably, quantitative DNA-binding assays performed with decreasing amounts of recombinant Cbfa1 and a fixed quantity of probes demonstrated that Cbfa1 had a 10- to 100-fold higher affinity for the OSE2 elements in Cbfa1 than for any other known OSE2 element (Fig. 9). It is interesting to note that the OSE2 site with the lowest affinity for Cbfa1 is located in α2(I) collagen, the gene whose expression was the least affected in the ΔCbfa1-expressing mice (Fig. 6A). This 100:1 order of magnitude higher affinity of Cbfa1 for the sites present in the Cbfa1 promoter than for any other sites most likely contributed to the severity of the phenotype observed in the ΔCbfa1-expressing mice and emphasizes the importance of this autoregulatory loop as a critical means through which Cbfa1 controls structural gene expression and thereby bone formation by differentiated osteoblasts.
Figure 9.

Different affinities of various OSE2s for GST–Cbfa1. Labeled OSE2s present in the Cbfa1, Osteocalcin, and type I collagen promoters were used as probes in EMSA with decreasing amount of recombinant protein as expressed by arbitrary units. A binding unit is defined as the quantity of GST–Cbfa1 required to shift 50% of the Osteocalcin promoter OSE2a probe, the initial OSE2 element described (Ducy and Karsenty 1995). GST–Cbfa1 has a 10- and 100-fold higher affinity for Cbfa1 OSE2s than for the Osteocalcin and type I collagen genes OSE2s, respectively.
Discussion
Our study uncovers a genetic cascade with Cbfa1 at its origin that controls bone formation after osteoblast differentiation is achieved. It illustrates that developmentally important genes may have additional and critical roles beyond embryonic development.
Bone formation is a different process than bone development
Bone ECM deposition by differentiated osteoblasts, or bone formation, occurs throughout life and fulfills several physiological functions after birth. During postnatal development, bone formation is required during endochondral ossification to replace the cartilagenous templates near the growth plate cartilage thereby ensuring proper longitudinal bone growth. During adulthood bone formation in the context of bone remodeling must be regulated tightly to replace the bone resorbed by the osteoclasts to maintain a near constant bone mass. Thus, any qualitative or quantitative defect of the osteoblasts, inherited or acquired, could result in short stature, bone fragility, and/or osteoporosis, the most frequent metabolic bone disease. These multiple requirements underscore the need to understand the molecular basis of bone formation by the osteoblasts and to identify molecule(s) regulating this function.
Mouse and human genetic studies have demonstrated that the integrity of structural proteins of the skeleton is critical for a proper bone formation. Mutations in either one of the two genes encoding type I collagen cause osteogenesis imperfecta (OI), a disease characterized by a decrease in bone formation and whose severity can vary from lethal forms to osteoporosis-like phenotypes (Prockop and Kivirikko 1984; Byers 1990). The potential severity of the OI phenotype is consistent with the abundance of type I collagen in bone matrix. Likewise, genetic analyses have uncovered the role of noncollagenous proteins such as alkaline phosphatase, Bone sialo protein, Osteocalcin, Osteopontin, and Fibrillin, among others, as regulators of bone matrix deposition, matrix mineralization, osteoclast function, and tensile strength of the bones (J.E. Aubin, pers. comm.; Ducy et al. 1996; Pereira et al. 1997; Rittling et al. 1998). Taken together, these molecules are the effectors of the osteoblasts’ function. However, what remains to be identified is/are regulator(s) of osteoblast function.
Cbfa1 is a regulator of osteoblast function
Because of its expression in differentiated osteoblasts postnatally and its role in regulating Osteocalcin (Ducy et al. 1997), the latest gene to be expressed in osteoblasts (Owen et al. 1990; Stein et al. 1990; Aubin and Liu 1996), we hypothesized that Cbfa1 could regulate bone formation and used a dominant-negative approach to test this hypothesis. Three lines of evidence indicate that Cbfa1 is the only Cbfa protein present in osteoblasts in vivo. First, in situ hybridization and Western analyses failed to detect Cbfa2 or Cbfa3 in osteoblasts (Simeone et al. 1995; Wijmenga et al. 1995; Ducy et al. 1997; this study). Second, we and others have failed to identify new Cbfa genes expressed in the skeleton. Third and more importantly, deletion of the Cbfa1 gene led to a complete absence of osteoblast in homozygous mutant mice indicating that no other transcription factor can fulfill Cbfa1’s regulatory function during osteoblast differentiation (Komori et al. 1997; Otto et al. 1997) Thus, the phenotype observed in the ΔCbfa1-expressing mice is caused by a specific inhibition of Cbfa1 function.
To demonstrate a second function for Cbfa1 in differentiated osteoblasts, distinct from its function during osteoblast differentiation, we took advantage of the Osteocalcin promoter which is active only in differentiated osteoblasts able to produce a matrix (Owen et al. 1990; Stein et al. 1990; Aubin and Liu 1996; Frendo et al. 1998). The results of this study demonstrate that Cbfa1 controls bone formation by differentiated osteoblasts. Cbfa1 is at the top of a genetic cascade controlling positively its own expression and the expression of the major osteoblast-specific and osteoblast-enriched genes through its binding to multiple OSE2 elements present in these genes (Fig. 9). This autoregulatory loop of Cbfa1 expression is so critical for proper bone formation because Cbfa1 has a much higher affinity for its binding sites in Cbfa1 than for any other known binding sites. It is possible that other OSE2 elements present elsewhere in Cbfa1 could play an additional role in the autoregulation of Cbfa1 expression and in the severity of the phenotype of the ΔCbfa1-expressing mice. This autoregulatory loop of Cbfa1 expression could also be important during development to maintain osteoblast differentiation once it is initiated. Our findings distinguish Cbfa1 from other molecules such as the bone morphogenetic proteins (BMPs) (Hogan 1996) that can induce bone formation by recapitulating all the cell differentiation events occurring during skeleton development. Instead, Cbfa1 acts as a maintenance factor of the differentiated osteoblasts by simply regulating the rate of bone matrix deposition by already differentiated cells.
In light of the absence of reported juvenile or more severe osteoporosis in CCD patients the phenotype we observed is unexpected. This is likely because of the more severe decrease of expression of the genes encoding bone ECM proteins, notably type I collagen, in the ΔCbfa1-expressing mice compared to the heterozygous Cbfa1-deficient mice. Figure 10 summarizes the two aspects of Cbfa1 function: During embryonic development Cbfa1 controls cell differentiation along the osteoblastic pathway, postnatally Cbfa1 has an additional function, it controls directly bone matrix deposition by differentiated osteoblasts.
Figure 10.

Schematic representation of Cbfa1 function during and after embryonic development. (A) During embryogenesis Cbfa1 controls differentiation of a mesenchymal progenitor cell toward the osteoblastic lineage. (B) After birth, in fully differentiated osteoblasts, Cbfa1 controls the rate of bone formation by regulating directly the expression of the bone ECM-encoding genes. Moreover, Cbfa1 regulates its own gene expression.
Cbfa1 as a tool to study bone remodeling
This second function of Cbfa1 has important biological implications. First, some skeletal features observed in CCD patients like short stature, growth retardation, and abnormally shaped epiphyses and metaphyses (Marie and Santon 1898; Soule 1946) have long been viewed as proof that the CCD gene has a role during postnatal skeletal growth (Mundlos and Olsen 1997). These features can now be explained by the fact that Cbfa1 control bone formation by differentiated osteoblasts postnatally. Second, this study raises the possibility that mutations in the Cbfa1 gene that leave intact the DNA-binding domain of Cbfa1 but affect its transactivation function may result in slightly more severe postnatal manifestations in CCD patients. More generally, mutations affecting one of the transactivation domains of Cbfa1 (Thirunavukkarasu et al. 1998) could be at the origin of some form of genetically inherited low or high bone mass diseases. The ability of Cbfa1 to control the function of already differentiated osteoblasts, together with the fact that haploinsufficiency at the Cbfa1 locus is at the origin of CCD (Lee et al. 1997; Mundlos et al. 1997; Otto et al. 1997), raises the hypothesis that increasing Cbfa1 rate of transcription in elder individuals may be a way to prevent or to treat osteopenic diseases. This warrants further investigation of the already established regulation of Cbfa1 expression by growth factors and hormones (Ducy et al. 1997) (Fig. 10). Lastly, this study along with the recent characterization of the role of osteoprotegerin and its ligand during bone resorption (Simonet et al. 1997; Lacey et al. 1998; Yasuda et al. 1998) provides some of the molecular tools that are necessary to understand the mechanisms controlling the maintenance of a near constant bone mass through bone remodeling during adulthood.
Beyond the involvment of Cbfa1 in bone formation a more general implication of these results is that other genes that play critical roles during embryonic development and that are also expressed after birth could also be involved during physiological processes postnatally. For instance, it is tempting to speculate that this function of Cbfa1 in cell physiology may be a feature shared by many transcriptional activators of cell differentiation; the easy-to-score nature of bone formation providing a favorable setting to uncover this aspect of their biology.
Materials and methods
DNA construction and generation of transgenic mice
For DNA transfections the 396-bp NcoI–HindIII fragment of Cbfa1 encoding the DNA-binding domain was cloned into the pEFBOS expression vector (Mizumisha and Nagata 1990). ΔCbfa1 transgenic mice bear a fusion gene composed of a 1.3-kb fragment of the mouse OG2 promoter (Ducy and Karsenty 1995) fused to Cbfa1 DNA-binding domain and the 3′ untranslated and polyA signal of the human G-CSF cDNA (Nagata et al. 1986). Transgenic founders were generated by pronuclear injection according to standard techniques (Bonnerot and Nicolas 1993). Genotype of transgenic animals was determined by PCR using as primers 5′-CGGAGCGGACGAGGCAAGAGTTTC-3′ and 5′-ACGGTGGGGAAGACTGTCCTGCCTG-3′. To generate prokaryotic expression vectors the AcyI–HincII fragment and the NcoI–HindIII fragment of the Cbfa1 cDNA, encoding the full-length protein and the DNA-binding domain, respectively, were ligated in-frame with the GST-coding sequence of the pGEX-4T3 vector (Pharmacia). Luc chimeric constructs were generated by cloning multimerized oligonucleotides or promoter fragments in the pA3luc vector (Goldberg et al. 1992). Mutagenesis of the Cbfa1 promoter was performed by PCR amplification using as primers: 5′-TTCTGTGAGGTCACAAAGAACATGA-3′ and 5′-ATACTTGTTCTTCTGTTCTTGTTTGTGAGGCGA-3′.
Western blot analysis
The pCMV5 expression vector (Andersson et al. 1989) empty or containing the Osf2/Cbfa1 (Ducy et al. 1997), Cbfa2 (Bae et al. 1992), or Cbfa3 (Bae et al. 1995) cDNAs was transfected in COS cells. Nuclear extracts were prepared 24 hr later as described (Ducy and Karsenty 1995). Nuclear extracts from mouse calvaria were prepared according Deryckere and Gannon (1994). SDS-PAGE and immunoblotting were performed according standard procedures (Ausubel et al. 1995) using an antiserum against the peptide SFFWDPSTSRRFSPPS and the ECL detection kit (Amersham).
DNA-binding assays
Labeling of oligonucleotide probes, EMSA, and supershift assays were done as described (Ducy and Karsenty 1995; Ducy et al. 1997). Each experiment was repeated at least three times. GST-tagged proteins were enriched according to standard protocols (Ausubel et al. 1995). Nuclear extracts were prepared as described previously (Ducy and Karsenty 1995). Top-strand sequences of the double-stranded oligonucleotides used in EMSA were α2(I)OSE2, 5′-CTTTGTGGATACGCGGACTTTGA-3′; α2(I)mutOSE2, 5′-CTTTGTTCATACGCGGACTTTGA-3′; Cbfa1–OSE2a, 5′-ATTCGCCTCACAAACAACCACAGAACCACAAGT-3′; Cbfa1–mutOSE2a, 5′-ATTCGCCTCACAAACAAGAACAGAACCACAAGT-3′; Cbfa1–OSE2b, 5′-TTCTGTGAGGTCACAAACCACATGA-3′; Cbfa1–mutOSE2b, 5′-TTCTGTGAGGTCACAAAGAACATGA-3′. Osteocalcin OSE2a, and α1(I)OSE2 oligonucleotides have been described elsewhere (Ducy and Karsenty 1995; Ducy et al. 1997; Frendo et al. 1998).
Morphological and histological analyses
Skeletons from newborn pups were prepared as described (Kochhar 1973) and stained with alcian blue 8GX and alizarin red S. For histological analysis mice were sacrificed at 2, 3, 4, and 8 weeks of age. For assessment of dynamic histomorphometric indices, mice of the 3-week group were injected with tetracyclin and calcein 10 and 2 days, respectively prior to sacrifices, according to standard tetracyclin/calcein double-labeling procedure (Vignery and Baron 1980). After radiological analysis of the skeleton (Faxitron, Munich, Germany), long bones and vertebrae were dissected out and fixed in 4% formaldehyde for 18 hr at 4°C. Undecalcified bones were embedded in methylmethacrylate, and 5-μm sections were prepared on a rotation microtome (Jung, Heidelberg, Germany) as described previously (Hahn et al. 1991; Amling et al. 1997). Sections were stained with 1% toluidine blue, or von Kossa reagent (3% silver nitrate counterstained with Kernechtrot), or hematoxylin/eosin, and evaluated using a Zeiss microscope (Carl Zeiss, Jena, Germany). Histomorphometrical analysis was performed on tibiae and vertebrae according to the American Society for Bone and Mineral Research (ASBMR) standards (Parfitt et al. 1987) using the OsteoMeasure Analysis System (Osteometrix, Atlanta, GA). Statistical differences between groups were assessed by Student’s t-test. Histology and histomorphometry data shown in Figures 3 and 4 were obtained from tibia metaphyses.
Ex vivo experiments
Osteoblasts from individual calvaria of newborn wild-type (n = 4) or transgenic (n = 4) pups were isolated according to the following protocol. After incubation for 40 min in αMEM-0.1 mg/ml collagenase P–2.5% trypsin at 37°C, shaking, calvaria were washed in αMEM, cut in pieces and transfered in αMEM-0.1 mg/ml collagenase P-10% trypsin for 1 hr at 37°C and shook. Digestion was stopped by addition of 10 volumes of αMEM/10% FBS. The cells were allowed to attach for 48 hr and were then replated at a density of 12,000 cells per cm2 in αMEM/10% FBS for 2 days. Thereafter the medium was supplemented with 5 mm β-glycerophosphate and 100 μg/ml ascorbic acid (mineralization medium) and replaced every 2 days. Cultures were maintained for 20 days before analysis for alkaline phosphatase and collagen synthesis (van Gieson staining), and for presence of a mineralized matrix (Goldner trichrome and von Kossa staining) according to standard protocols (Bancroft and Stevens 1996).
RT–PCR analysis
For RNA preparation long bones were dissected free of surrounding tissues. Epiphyses were cut out and the bone marrow flushed. Three to four animals were analyzed independently. We used RT–PCR to analyze variations of gene expression between individual wild-type and transgenic animals because the amount of RNA obtained per animal prevented us to perform Northern blot analyses. RNA extraction, cDNA synthesis, and PCR amplification were performed using standard protocols (Ausubel et al. 1995). Exon 2 amplification of the Hprt gene was used as internal control for the quantity and quality of cDNAs. The following sets of primer were used: ΔCbfa1, 5′-CAGCAGTGTTCCCCATCTGGGTCCT-3′ and 5′-GGGGGGTGAAGAGGTGGAGGGTGAC-3′; Hprt, 5′-GTTGAGAGATCATCTCCACC-3′ and 5′-AGCGATGATGAACCAGGTTA-3′; Osteocalcin, 5′-GTTCAGGGTGTGTCGTCGAAC-3′ and 5′-TTTCGGCTCGACGGTCTCAAA-3′; BSP, 5′-GAGCCAGGACTGCCGAAAGGAA-3′ and 5′-CCGTTGTCTCCTCCGCTGCTGC-3′; Osteopontin, 5′-CATTGCCTCCTCCCTCCCGGTG-3′ and 5′-GTCATCACCTCGGCCGTTGGGG-3′; α1(I) collagen, 5′-CCTGGTAAAGATGGTGCC-3′ and 5′-CACCAGGTTCACCTTTCGCACC-3′; α2(I) collagen, 5′-TGGTCCTCTGGGCATCTCAGGC-3′ and 5′-GGTGAACCTGCTGTTGCCCTCA-3′; Cbfa1, 5′-GAGGCCGCCGCACGACAACCGCA-3′ and 5′-ACGGTGGGGAAGACTGTCCTGCCTG-3′.
DNA transfections and luc assays
ROS 17/2.8 osteoblastic cells were transfected with increasing amount of expression vector, 5 μg of luc reporter vector and 2 μg of pSVβgal plasmid. COS cells were transfected with 5 μg of expression vector, 5 μg of luc reporter vector and 2 μg of pSVβgal plasmid. Transfection, luc assays, and β-galactosidase assays were performed as described (Ducy and Karsenty 1995). Data represent ratios of luc/β-galactosidase activities and values are means of six to eight independent transfection experiments; error bars represent standard deviation of the means.
Acknowledgments
G.K. is indebted to M. Sato for his generous help and advice. P.D. is grateful to M. Machado for her help in analyzing the ex vivo cultures. We thank Y. Ito for the PEBP2αB and PEBP2αC cDNAs and H. Bellen for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health and the March of Dimes Foundation (G.K.) and an Arthritis Investigator Award (P.D.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL karsenty@bcm.tmc.edu; FAX (713) 798-1465.
References
- Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick W M, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone related peptide in a signalling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranson BD, Fisher AL, Blechman K, Caudy M, Gergen JP. Groucho-dependent and -independent repression activities of Runt domain proteins. Mol Cell Biol. 1997;17:5581–5587. doi: 10.1128/mcb.17.9.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin JE, Liu F. The osteoblast lineage. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. San Diego, CA: Academic Press; 1996. pp. 51–67. [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: John Wiley & Sons; 1995. [Google Scholar]
- Bancroft JD, Stevens A. Theory and practice of histological techniques. New York, NY: Churchil Livingston; 1996. [Google Scholar]
- Bae S-C, Yamaguchi-Iwai Y, Ogawa E, Maruyama M, Inuzuka M, Kagoshima H, Shigesada K, Satake M, Ito Y. Isolation of PEBP2αB cDNA representing the mouse homolog of human acute myeloid leukemia gene. Oncogene. 1992;8:809–814. [PubMed] [Google Scholar]
- Bae S-C, Ogawa E, Maruyama M, Oka H, Satake M, Shigesada K, Jenkins NA, Gilbert DJ, Copeland NG, Ito Y. PEBP2αB/mouse AML1 consists of multiple isoforms that possess differential transactivation potentials. Mol Cell Biol. 1994;14:3242–3252. doi: 10.1128/mcb.14.5.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S-C, Takahashi E-I, Zhang YW, Ogawa E, Shigesada K, Namba Y, Satake M, Ito Y. Cloning, mapping and expression of PEBP2αC, a third gene encoding the mammalian runt domain. Gene. 1995;159:245–248. doi: 10.1016/0378-1119(95)00060-j. [DOI] [PubMed] [Google Scholar]
- Bianco P, Fisher LW, Young MF, Termine JD, Gehron Robey P. Expression of bone sialo protein (BSP) in developing human tissues. Calcif Tissue Int. 1991;49:421–426. doi: 10.1007/BF02555854. [DOI] [PubMed] [Google Scholar]
- Bonnerot C, Nicolas JF. Guide to techniques in mouse development. Methods Enzymol. 1993;225:747–799. doi: 10.1016/0076-6879(93)25003-k. [DOI] [PubMed] [Google Scholar]
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Lin Tan H, Xu W, Lacey DL, Boyle WJ, Simonet S W. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes & Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers PH. Brittle bones—fragile molecules: Disorders of collagen gene structure and expression. Trends Genet. 1990;6:293–300. doi: 10.1016/0168-9525(90)90235-x. [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Guo X. Osteopontin: A protein with diverse functions. FASEB J. 1993;7:1475–1482. [PubMed] [Google Scholar]
- Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. BioTechniques. 1994;16:405. [PubMed] [Google Scholar]
- Desbois C, Hogue DA, Karsenty G. The mouse osteocalcin gene cluster contains three genes with two separate spatial and temporal patterns of expression. J Biol Chem. 1994;269:1183–1190. [PubMed] [Google Scholar]
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–1869. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, Bradley A, Karsenty G. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall A, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Frendo J-L, Xiao G, Franceschi RT, Karsenty G, Ducy P. Functional hierarchy between two OSE2 elements in the control of Osteocalcin gene expression in vivo. J Biol Chem. 1998;273:30509–30516. doi: 10.1074/jbc.273.46.30509. [DOI] [PubMed] [Google Scholar]
- Gehron-Robey P. Bone matrix proteoglycans and glycoproteins. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. San Diego, CA: Academic Press; 1996. pp. 155–166. [Google Scholar]
- Goldberg H, Helaakoski T, Garrett LA, Karsenty G, Pellegrino A, Lozano G, Maity S, de Crombrugghe B. Tissue-specific expression of the mouse α2(I) collagen promoter: Studies in transgenic mice and in tissue cultured cells. J Biol Chem. 1992;267:19622–19630. [PubMed] [Google Scholar]
- Hahn M, Vogel M, Delling G. Undecalcified preparation of bone tissue: Report of technical experience and development of new methods. Virchows Archiv A Pathol Anat. 1991;418:1–7. doi: 10.1007/BF01600238. [DOI] [PubMed] [Google Scholar]
- Hauschka P, Lian J, Cole D, Gundberg C. Osteocalcin and matrix Gla protein: Vitamin K-dependent proteins in bone. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- Hogan BLM. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes & Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- Kleerekoper M, Avioli LV. Osteoporosis pathogenesis and therapy. In: Avioli LV, Krane SM, editors. Metabolic bone disease. San Diego, CA: Academic Press; 1998. pp. 387–389. [Google Scholar]
- Kochhar DM. Limb development in mouse embryos. Analysis of teratogenic effects of retinoic acid. Teratology. 1973;7:289–298. doi: 10.1002/tera.1420070310. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kurokawa M, Tanaka T, Tanaka K, Hirano N, Ogawa S, Mitani K, Yazaki Y, Hirai H. A conserved cysteine residue in the runt homology domain of AML1 is required for the DNA-binding ability and the transforming activity on fibroblasts. J Biol Chem. 1996;271:16870–16876. doi: 10.1074/jbc.271.28.16870. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan H-L, Kelley MJ, Dunstan CR, Burgess T, Elliot R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian Y-X, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding OSF2/CBFA1 in patients affected with cleidocranial dysplasia. Nat Genet. 1997;16:307–311. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- Marie P, Santon P. Sur la dysostose cleidocrannienne hereditaire. Rev Neurol. 1898;6:835–838. [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mamalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundlos S, Olsen BR. Heritable diseases of the skeleton. Part I: Molecular insights into skeletal development-transcription factors and signaling pathways. FASEB J. 1997;11:125–132. doi: 10.1096/fasebj.11.2.9039954. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JHM, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- Nagata S, Tsuchiya M, Yamazaki T, Yamamoto O, Hirata Y, Kubota N, Oheda M, Nomura H, Ono M. Molecular cloning and expression of cDNA for human granulocyte colony-stimulating factor. Nature. 1986;319:415–418. doi: 10.1038/319415a0. [DOI] [PubMed] [Google Scholar]
- Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott SM. Theoritical and methodoligical approach. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. San Diego, CA: Academic Press; 1996. pp. 231–234. [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GWH, Beddington RSP, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Owen TA, Aronow M, Shalhoub V, Barone LM, Wilming L, Tassinari MS, Kennedy MB, Pockwinse S, Lian JB, Stein GS. Progressive development of the rat osteoblast phenotype in vitro: Reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol. 1990;143:420–430. doi: 10.1002/jcp.1041430304. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott S M, Recker RR. Bone histomorphometry. Standardization of nomenclature, symbols, and units. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- Prockop DJ, Kivirikko KI. Heritable diseases of collagen. New Engl J Med. 1984;311:376–386. doi: 10.1056/NEJM198408093110606. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Matsumoto HN, McKee MD, Nanci A, An X-R, Novick KE, Kowalski AJ, Noda M, Denhardt DT. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13:1101–1111. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- Rowe DW, Shapiro JR. Osteogenesis imperfecta. In: Avioli LV, Krane SM, editors. Metabolic bone disease. San Diego, CA: Academic Press; 1998. pp. 651–678. [Google Scholar]
- Simeone A, Daga A, Calabi F. Expression of runt in the mouse embryo. Devel Dyn. 1995;203:61–70. doi: 10.1002/aja.1002030107. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ Amgen EST Program. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the C-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Soule AB. Mutational dysostosis (Cleidocranial dysostosis) J Bone Joint Surg. 1946;1:81–102. [PubMed] [Google Scholar]
- Stein GS, Lian JB, Owen TA. Bone cell differentiation: A functionally coupled relationship between expression of cell-growth- and tissue-specific genes. Curr Opin Cell Biol. 1990;2:1018–1027. doi: 10.1016/0955-0674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- Stein GS, Lian JB, Stein JL, van Wijnen AJ, Frenkel B, Montecino M. Mechanisms regulating osteoblast proliferation and differentiation. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. San Diego, CA: Academic Press; 1996. pp. 69–86. [Google Scholar]
- Thirunavukkarasu K, Mahajan M, McLarren KW, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transciption factor Cbfa1/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfb. Mol Cell Biol. 1998;18:4197–4208. doi: 10.1128/mcb.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey WD, Jr, Pepling ME, Horb ME, Thomsen GH, Gergen JP. A Xenopus homologue of aml-1 reveals unexpected patterning mechanisms leading to the formation of embryonic blood. Development. 1998;125:1371–1380. doi: 10.1242/dev.125.8.1371. [DOI] [PubMed] [Google Scholar]
- Vignery A, Baron R. Dynamic histomorphometry of alveolar bone remodeling in the adult rat. Anat Rec. 1980;196:191–200. doi: 10.1002/ar.1091960210. [DOI] [PubMed] [Google Scholar]
- Wijmenga C, Speck NA, Dracopoli NC, Hofker MH, Liu P, Collins FS. Identification of a new murine runt domain-containing gene, Cbf3a, and localization of the human homolog, CBF3A, to chromosome 1p35-pter. Genomics. 1995;26:611–614. doi: 10.1016/0888-7543(95)80185-o. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]