

Abstract
CD30 is a cell-surface receptor that can augment lymphocyte activation and survival through its ability to induce the transcription factor NF-κB. CD30, however, has also been implicated in the induction of apoptotic cell death of lymphocytes. Here we show that one of the effects of CD30 signal transduction is to render cells sensitive to apoptosis induced by the type 1 tumor necrosis factor receptor (TNFR1). This sensitization is dependent on the TRAF-binding sites within the CD30 cytoplasmic domain. One of the proteins that binds to these sites is TRAF2, a signal transduction molecule that is also utilized by TNFR1 to mediate the activation of several downstream kinases and transcription factors. During CD30 signal transduction, we found that binding of TRAF2 to the cytoplasmic domain of CD30 results in the rapid depletion of TRAF2 and the associated protein TRAF1 by proteolysis. These data suggest a model in which CD30 limits its own ability to transduce cell survival signals through signal-coupled depletion of TRAF2. Depletion of intracellular TRAF2 and its coassociated proteins also increased the sensitivity of the cell to undergoing apoptosis during activation of death-inducing receptors such as TNFR1. Consistent with this hypothesis, expression of a dominant-negative form of TRAF2 was found to potentiate TNFR1-mediated death. These studies provide a potential mechanism through which CD30, as well as other TRAF-binding members of the TNFR superfamily, can negatively regulate cell survival.
Keywords: Apoptosis, NF-κB, CD30, TRAF, TNF, proteases
An essential feature of the immune system is the ability to tolerate large fluctuations in cell number. Homeostasis is thought to be achieved through the integration of intracellular and extracellular signals that function to balance the degree of cell proliferation with the rate of cell death (Oltvai and Korsmeyer 1994; Allison and Krummel 1995). Lymphocyte activation and proliferation is initiated by the engagement of antigen with its clonotypic cell-surface receptor (Weiss and Littman 1994). Additional costimulatory signals, however, are also required. For example, costimulatory signals can be mediated in T cells by signaling through the CD28 receptor (Allison and Krummel 1995) and in B cells through the CD40 receptor, a member of the tumor necrosis factor/nerve growth factor (TNF/NGF) receptor superfamily (Choi et al. 1995).
The TNF/NGF receptor superfamily is a group of related cell-surface receptors that include the types 1 and 2 TNF receptors (TNFR1 and TNFR2), Fas, CD27, 4-1BB, and CD30 (Vassalli 1992; Armitage 1994; Beyaert and Fiers 1994; Gruss and Dower 1995). Many of these receptors have been found to play costimulatory roles in cell proliferation, whereas others have been shown to induce apoptosis. Interestingly, several members of this family, such as TNFR2 (Tartaglia et al. 1993; Zheng et al. 1995), CD40 (Foy et al. 1996; Rathmell et al. 1996), and CD30 (see below), have been shown to play dual roles, although the mechanism(s) underlying this apparent ambiguity is unknown. To understand this phenomenon, we have examined the signaling properties of one of these receptors, CD30.
CD30 is a lymphoid cell-restricted receptor (Dürkop et al. 1992) that is normally expressed on activated and memory T cells (Ellis et al. 1993). CD30 was originally identified on the surface of Hodgkin’s lymphoma cells (Schwab et al. 1982; Stein et al. 1985b; Falini et al. 1995), and high levels of CD30 expression have also been found to correlate with disease progression in anaplastic large cell lymphoma (Nadali et al. 1995). Signal transduction through CD30 has been found to augment T-cell receptor-mediated proliferation under some circumstances (Smith et al. 1993), but in other cases to potentiate apoptosis (Lee et al. 1996). Activation of CD30 has also been shown to stimulate HIV expression (Maggi et al. 1995), probably through its ability to activate the transcription factor NF-κB (Siebenlist et al. 1994; Biswas et al. 1995). Targeted disruption of the murine CD30 gene, however, has implicated CD30 in thymic negative selection (Amakawa et al. 1996).
The signaling properties of TNFR-related receptors are being rapidly elucidated. Several TNF receptor family members, including Fas, TNFR1, and DR3/Wsl/Apo-3, have been found to contain an ∼80-residue motif designated the death domain (Tartaglia et al. 1993; Takahashi et al. 1994; Chinnaiyan et al. 1996; Kitson et al. 1996; Marsters et al. 1996; Pan et al. 1997), and signaling through these receptors has been shown to induce apoptosis in a manner that is dependent on dimerization of the death domain of the receptor with similar death domains of cytoplasmic factors such as FADD, TRADD, and RIP (Chinnaiyan et al. 1995; Hsu et al. 1995; Stanger et al. 1995). In contrast, the cytoplasmic domains of a number of other TNF receptor family members, including TNFR2 and CD40, have been shown to interact with members of the recently described TNF receptor-associated factor (TRAF) family of signaling molecules (Baker and Reddy 1996). We and others have recently identified TRAF1, TRAF2, TRAF3, and TRAF5 as factors that can be recruited to the cytoplasmic domain of CD30 and have shown that these molecules play central roles in signal transduction through this receptor (Ansieau et al. 1996; Gedrich et al. 1996; Lee et al. 1996; Aizawa et al. 1997; Duckett et al. 1997; Tsitsikov et al. 1997).
Six mammalian TRAF proteins have been identified to date (Baker and Reddy 1996). These factors have been shown to possess distinct affinities for different members of the TNF receptor superfamily. The TRAF proteins have been suggested to be important in promoting cell survival because of their role in promoting NF-κB activation through receptors of the TNF receptor family (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996). In addition, animals with a targeted TRAF3 disruption (Xu et al. 1996) and transgenic mice expressing TRAF1 (Speiser et al. 1997) have both been reported to display defects in lymphocyte development and survival.
In this report we describe a mechanism by which signaling through CD30 can both promote initial cell survival while at the same time setting in motion a signaling-dependent mechanism to leave the cell subsequently more susceptible to apoptosis through death domain-containing receptors. The ability of CD30 to promote cell survival is dependent on its ability to recruit a TRAF-dependent signal transduction complex (Duckett et al. 1997). Now, we show that the recruitment of TRAF2 into this complex results in the signal-coupled depletion of TRAF2 and its coassociated signaling molecule TRAF1. This depletion in TRAF2 correlates with increased cellular sensitivity to cell death in response to TNFR1 signal transduction. This phenomenon is not unique to CD30 signal transduction, but also occurs following TNFR2 signal transduction. Consistent with the central role of the TRAF2 signaling complex in modulating the apoptotic sensitivity of cells in response to TNFR1 engagement, dominant-negative TRAF2 was found to directly mimic the ability of CD30 signal transduction to sensitize cells to TNFR1-induced apoptosis.
Results
CD30 signaling potentiates TNFR1-mediated apoptosis
Recent papers have shown an important role for NF-κB activation in the regulation of cell survival (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996), and we have shown that CD30 is a potent activator of NF-κB (Duckett et al. 1997). The human embryonic kidney cell line 293 previously has been shown to express TNFR1 but not TNFR2 (Liu et al. 1996; C.S. Duckett and C.B. Thompson, unpubl.). 293 cells were transfected with expression vectors encoding green fluorescent protein (GFP) as a marker of transfection, together with a plasmid encoding a chimeric protein comprised of the cytoplasmic domain of CD30 fused to the extracellular and transmembrane domains of CD28. This chimeric protein is expressed on the cell surface as a constitutive homodimer, and this dimerization is sufficient to initiate CD30 signal transduction in a ligand-independent manner (Duckett et al. 1997). Expression of this protein on the cell surface results in potent NF-κB activation. In contrast, a CD28 deletion mutant lacking a cytoplasmic domain (CD28ΔTail) has no effect on NF-κB activation (Duckett et al. 1997). The present experiments were initiated to determine whether CD30 signal transduction could inhibit the ability of death domain-containing proteins such as TNFR1 from inducing apoptosis. In our initial experiments, cells were stimulated with recombinant TNF-α 12 hr after transfection and subsequently evaluated for viability by nuclear staining with 4′,6′-diamidino-2-phenylindole (DAPI) and fluorescence microscopy. Contrary to our expectations, TNF-α had no effect on the viability of parental 293 cells or the cells transfected with the tailless CD28 construct. In contrast, 70% of the CD30 transfectants died in response to 18 hr of TNF-α treatment (Fig. 1A). The nuclear condensation and morphological changes as observed by DAPI staining and analysis by fluorescence microscopy were indicative of apoptosis (data not shown). This cell death was dependent on TNF-α, as the CD30 chimera in the absence of TNF-α had no significant effect on viability. Flow cytometric analysis of cells transfected with the CD28–CD30 and the CD28ΔTail vectors surface expression revealed very similar levels of CD28 surface expression (data not shown).
Figure 1.


Potentiation of TNFR1-mediated cell death by constitutively activated chimeric CD30. (A) 293 cells were transfected with a vector (100 ng) encoding GFP together with expression vectors (1 μg) encoding a fusion between CD28 and CD30, a mutated CD28–CD30 vector lacking the carboxy-terminal TRAF-binding sites (ΔTraf), or a control CD28 mutant lacking a cytoplasmic tail (ΔTail), as indicated. Twelve hours following transfection, cells were stimulated with human TNF-α (200 U/ml) or a medium control for a further 24 hr, and viability of GFP-expressing cells was determined by fluorescence microscopy as described in Materials and Methods. (B) CD30-enhanced death is blocked by apoptosis inhibitors. 293 cells were transfected either with the CD28 ΔTail control vector (Control) or with the CD28–CD30 chimera (CD30), together with the indicated expression vectors. Transfectants were stimulated with recombinant TNF-α and viability determined as described above and in Materials and Methods.
Previous work has shown that CD30 signal transduction is dependent on a TRAF-binding region located within the carboxy-terminal 36 amino acids of the cytoplasmic domain (Duckett et al. 1997). To determine the role of the TRAF-binding domain of CD30 in the potentiation of TNF-α-induced death, we examined the ability of a mutant of CD28–CD30 chimera that lacks the TRAF-binding motifs (ΔTraf) to potentiate TNF-α-mediated death. In contrast to the vector containing the full-length CD30 tail, this mutant was unable to induce the sensitivity to TNF-α (Fig. 1A). Thus, contrary to our expectations, these data suggest that CD30 can potentiate TNF-mediated apoptosis. This potentiation is dependent on the TRAF-binding sites in CD30.
To characterize the mechanism of cell death induced by the combination of CD30 signaling and TNF-α, expression vectors were cotransfected encoding viral proteins that are known to block apoptotic cell death induced by members of the caspase family of cysteine proteases. Expression of either the baculoviral apoptotic inhibitor, P35 (Clem et al. 1991; Bump et al. 1995), or the cowpox virus inhibitor, CrmA (Gagliardini et al. 1994; Komiyama et al. 1994), completely abrogated the cell death induced by TNF-α plus CD30 signaling (Fig. 1B), indicating the involvement of caspases and confirming that the cell death was caused by apoptosis.
TRAF2 is degraded upon recruitment to CD30
The experiments described above suggested a role for the TRAF proteins in the potentiation of TNFR1-mediated death. Because TRAF2 has also been found to be involved in TNFR1-mediated signaling (Hsu et al. 1996), we sought to characterize cellular TRAF2. Endogenous TRAF2 protein in 293 cells was not detectable by immunoblot analysis with commercially available TRAF2-specific antibodies (Fig. 2A; data not shown), indicating that TRAF2 is normally expressed at levels below the threshold of detection in this cell type. Therefore, a mammalian expression vector encoding human TRAF2 was transfected into 293 cells together with either the chimeric CD28 vector containing the CD30 cytoplasmic tail, or the CD28 vector lacking a cytoplasmic tail. Surprisingly, whereas TRAF2 protein was easily detected in lysates from cells cotransfected with the TRAF2 plus control CD28 tailless vectors, TRAF2 was virtually undetectable when cotransfected with the CD28–CD30 chimera (Fig. 2A). This effect was not observed when TRAF2 was coexpressed with a CD30 vector lacking the TRAF-binding domains. High levels of TRAF2 protein were also detected when the TRAF2 vector was transfected alone or with a variety of additional control vectors (data not shown).
Figure 2.

TRAF2 is destabilized by CD30. 293 cells were cotransfected with TRAF2 and CD28 chimera expression vectors as indicated. (A) Cells were lysed 18 hr following transfection, standardized for protein levels, and TRAF2 levels were detected by immunoblot analysis as described in Materials and Methods. (B) Total RNA was prepared from an equivalent aliquot of cells used for A, and TRAF2 mRNA was evaluated by Northern analysis with a TRAF2 cDNA probe. (Bottom) An ethidium bromide-stained agarose gel to standardize the RNA samples; (top) an autoradiograph of the TRAF2-probed blot.
To investigate why we failed to detect TRAF2 protein in cells transfected with TRAF2 in the presence of the chimeric CD28–CD30 receptor, the levels of TRAF2 RNA were examined by Northern analysis. The amounts of TRAF2 RNA were similar in cells cotransfected with the CD28ΔTail, CD28–CD30, and CD28–CD30ΔTraf vectors (Fig. 2B). These data suggest that TRAF2 is being transcribed at equivalent rates in the presence or absence of the CD28–CD30 receptor. The failure of TRAF2 protein to accumulate appears to be mediated by post-transcriptional mechanisms.
CD30-induced destabilization of TRAF2 can be blocked by protease inhibitors
To further characterize the process of CD30-induced TRAF2 destabilization, a panel of protease inhibitors were tested for their ability to inhibit CD30-induced TRAF2 degradation (Fig. 3). Lactacystin, a specific inhibitor of the ubiquitin-proteasome pathway (Krappmann et al. 1996), was unable to rescue the degradation of TRAF2 (Fig. 3), although a number of less specific compounds such as E64 and TLCK were able to inhibit TRAF2 degradation. In addition, several inhibitors of the calpain family of cysteine proteases were found to efficiently block CD30-induced TRAF2 degradation. The calpains are a family of cytoplasmic, calcium-activated cysteine proteases that have been implicated in the induction of apoptosis (Squier and Cohen 1997). These data suggest that the binding of TRAF2 to the cytoplasmic tail of CD30 activates a protease or proteases that result in the degradation of TRAF2.
Figure 3.

TRAF2 destabilization can be blocked by protease inhibitors. 293 cells were transfected with TRAF2 and chimeric CD28–CD30 expression vectors as indicated. Parallel wells were stimulated 36 hr following transfection with the indicated protease inhibitors each at a final concentration of 50 μm or with a DMSO solvent control at a final concentration of 0.001%, and lysates were prepared 48 hr after transfection. Standardized extracts were examined for TRAF2 by immunoblot analysis as described in Materials and Methods.
Heteromeric complexes containing TRAF2 are also degraded upon recruitment to CD30
It has previously been shown that TRAF1, TRAF2, and TRAF3 can be directly recruited to the cytoplasmic domain of CD30 (Ansieau et al. 1996; Gedrich et al. 1996; Lee et al. 1996). More recently, three additional members of the TRAF family have also been identified, TRAF4, TRAF5, and TRAF6 (Tomasetto et al. 1995; Cao et al. 1996; Ishida et al. 1996a,b; Nakano et al. 1996; Aizawa et al. 1997). Therefore, the six known TRAF proteins were assessed for their abilities to be degraded upon CD30 signaling (Table 1). Interestingly, TRAF2, TRAF3, and TRAF5 were degraded, presumably upon binding to CD30, whereas the levels of TRAF1, TRAF4, and TRAF6 were not evidently affected by coexpression of CD30 (Table 1). TRAF1 is able to bind to the CD30 tail, but was not degraded, suggesting that additional determinants may be present in TRAFs 2, 3, and 5 which confer binding-specific degradation.
Table 1.
Comparison of the ability of CD30 to bind the six known TRAF proteins with its degradative properties
|
|
Binding to CD30
|
Degradation by CD30
|
|---|---|---|
| TRAF1 | +++ | − |
| TRAF2 | +++ | +++ |
| TRAF3 | +++ | + |
| TRAF4 | − | − |
| TRAF5 | +++ | + |
| TRAF6 | − | − |
The Cd30 binding column has been amalgamated from current published data (Ansieau et al. 1996; Gedrich et al. 1996; Lee et al. 1996) and unpublished data. The degradation by CD30 column summarizes the data obtained by coexpression of CD28–CD30 chimeras with expression vectors encoding wild-type TRAF1, TRAF2, or TRAF3, with Myc epitope-tagged TRAF4 and TRAF5, or with FLAG epitope-tagged TRAF6, followed by immunoblot analysis, as indicated. (+++) The respective TRAF protein was reduced >80% by coexpression with the CD30 chimera, as determined by immunoblot analysis, (+) a reduction of 50% or less; (−) no change.
As shown in Table 1, TRAF1 was not degraded by CD30 signal transduction. Because TRAF1 and TRAF2 can form heterodimers (Rothe et al. 1994, 1995b), we tested the possibility that TRAF1 might function to stabilize TRAF2. Expression vectors encoding TRAF1 and TRAF2 were cotransfected into 293 cells together with either the CD28–CD30 vector or the control vector lacking a cytoplasmic tail. Cotransfection of TRAF1 had no stabilizing effect on TRAF2 (Fig. 4A). In fact, whereas expression of TRAF1 alone was not affected by the presence of CD30, coexpression of TRAF1 with TRAF2 resulted in the almost complete disappearance of TRAF1, suggesting that recruitment by TRAF2 of TRAF1 into a heteromeric complex with CD30 resulted in degradation of both TRAF proteins.
Figure 4.


Determinants of TRAF degradation. (A) 293 cells were transfected with expression vectors encoding the indicated proteins. Lysates were prepared 48 hr following transfection and standardized aliquots were subjected to immunoblot analysis with a polyclonal antibody to TRAF1 (top) or TRAF2 (bottom). (B) 293 cells were transfected with the indicated plasmids, including either wild-type TRAF2 (1-501) or a dominant-negative mutant (87–501) lacking a functional RING finger, as indicated. Cells were prepared 24 hr following transfection and analyzed as described in A with a TRAF2 polyclonal antibody.
The RING finger of TRAF2 is required for CD30-mediated degradation
Both TRAF1 and TRAF2 possess similar characteristics of recruitment to the two TRAF-binding sites in CD30 (Gedrich et al. 1996), but as shown in Figure 4A, TRAF1 was not degraded directly by CD30 signal transduction. The principal structural difference between TRAF1 and TRAF2 is the presence of a RING finger domain in TRAF2. Therefore, the properties of a mutant version of TRAF2 that lacks the RING finger domain were examined. Deletion of the amino-terminal 86 residues of TRAF2 has previously been reported to disrupt the RING finger domain of TRAF2 without diminishing its ability to bind to CD30 (Gedrich et al. 1996). This mutant, TRAF2 (87–501), functions in a dominant-negative manner to inhibit CD30-mediated signaling (Duckett et al. 1997). As shown in Figure 4B, TRAF2 (87–501) was not degraded by coexpression with CD30, whereas in control transfections, the level of full-length TRAF2 was dramatically reduced. These findings suggest that the amino terminus of TRAF2, which contains a RING finger, is required for degradation following recruitment to the cytoplasmic tail of CD30.
Previous studies have shown that TRAF2 is able to form homodimers, as well as heterodimers with TRAF1 (Rothe et al. 1995b). Because TRAF1 was degraded when coexpressed with TRAF2 and CD30, we reasoned that the dominant-negative TRAF2 (87–501), which is not degraded when coexpressed with CD30 (Fig. 4B), might become subject to degradation if coexpressed with full-length TRAF2. As shown in Figure 4B, levels of cotransfected full-length TRAF2 and truncated TRAF2 (87–501) were dramatically reduced on coexpression with the CD28–CD30 chimera. This suggests that a complex containing both the full-length and the truncated (87–501) forms of TRAF2 is degraded upon recruitment to CD30.
Both TRAF-binding sites in CD30 contribute to TRAF degradation
Previously we have shown that TRAF2 can bind independently to either of two 5–7 residue elements in CD30 (domains 2A and 2B), and that each of these elements are independently capable of recruiting TRAF proteins and inducing NF-κB activation (Gedrich et al. 1996; Duckett et al. 1997). Therefore, additional mutants of the CD30 cytoplasmic tail were tested to determine which of the two TRAF-binding elements was required to produce destabilization of TRAF2. The TRAF2 expression vector was cotransfected into 293 cells together with chimeric CD30 variants lacking the TRAF-binding domain 2A only, 2B only, or both 2A and 2B. Individual disruption of either TRAF-binding domain 2A or 2B resulted in mutants that induced intermediate levels of TRAF2 degradation (Fig. 5). In control transfections, wild-type CD30 resulted in the almost complete loss of TRAF2. These data suggest that whereas either TRAF-binding site is capable of partially destabilizing TRAF2, both sites are required for maximal destabilization.
Figure 5.

Both TRAF-binding sites in CD30 contribute to TRAF degradation. 293 cells were cotransfected with TRAF2 and CD28 chimera expression vectors encoding wild-type CD30, tailless CD28 (ΔTail), or mutated proteins lacking both TRAF-binding motifs (Δ dom 2) or each of the individual TRAF-binding domains (Δ dom 2A or Δ dom 2B), as indicated. Cells were lysed 18 hr following transfection and standardized for protein levels, and TRAF2 levels were detected by immunoblot analysis as described in Materials and Methods.
TNFR2 can also enhance TNFR1 sensitivity and induce TRAF2 degradation
To determine whether the ability to heighten sensitivity to TNFR1 signals and to induce the degradation of TRAF2 is a unique property of CD30 or is shared by other TNF receptor family members, we examined the properties of TNFR2. A chimeric vector was constructed encoding the cytoplasmic domain of TNFR2 fused to the extracellular and transmembrane domains of CD28. Transfection of this vector into 293 cells resulted in comparable levels of both surface CD28 expression and NF-κB induction to those observed after transfection of the CD28–CD30 chimera or the control CD28Δtail vector (data not shown). Similar to the results found with the CD28–CD30 protein, cells transfected with the CD28–TNFR2 chimera were also sensitized to TNFR1-mediated death. The degree of sensitization was almost identical to that found for the CD28–CD30 chimera (Fig. 6A). In addition, cotransfection of CD28–TNFR2 with TRAF2 also induced the degradation of TRAF2 (Fig. 6B), revealing that the enhancement of TNFR1 sensitivity, as well as the TRAF2 degradation following recruitment to the receptor, is not restricted to CD30, but can occur with other members of the TNF receptor superfamily.
Figure 6.


TNFR2 can enhance TNFR1 sensitivity and induces TRAF2 degradation. (A) 293 cells were transfected with the indicated CD28 chimeric vectors (1 μg), along with a GFP vector (100 ng), exactly as described in the legend to Fig. 1. Transfectants were stimulated with TNF-α (200 U/ml) and cell viability was determined by DAPI staining and fluorescence microscopy as described in Materials and Methods. (B) 293 cells were transfected with the TRAF2 expression vector along with the indicated chimeric vectors. Twelve hours following transfection, cells were either mock treated or treated with human TNF-α (200 U/ml) as indicated. Cells were harvested 24 hr following transfection, standardized by protein content and TRAF2 was detected by immunoblot analysis as described in Materials and Methods.
Blockade of TRAF2 signaling can potentiate TNFR1-mediated death
The experiments described above show that the recruitment of TRAF2 to CD30 results in its destabilization by a mechanism that involves proteolysis. Activation of TNFR1 has also been shown to effect the recruitment of TRAF2 to the TNFR1 cytoplasmic tail, by virtue of a TNFR1–TRADD–TRAF2 interaction (Hsu et al. 1996), and this recruitment has been implicated in the activation of downstream signals, notably NF-κB (Rothe et al. 1995b) and JNK (Natoli et al. 1997) activation. One consequence of TRAF2 degradation by CD30 might be to inhibit TRAF-mediated signaling by TNFR1. To test this possibility, 293 cells were transfected with the dominant-negative TRAF2 (87–501) vector, treated with either recombinant TNF-α or a media control for 24 hr, and assayed for viability. As shown in Figure 7, similar to the effect of the constitutively active CD30 chimera, expression of dominant-negative TRAF2 greatly sensitized the cells TNF-α-mediated death, suggesting that blocking the ability of TRAF proteins to participate in TNFR1-dependent signal transduction can enhance the sensitivity to TNF-α.
Figure 7.

Dominant-negative TRAF2 can potentiate TNFR1-mediated death. 293 cells were transfected with 100 ng of GFP plasmid, together with 1 μg of the indicated plasmids, and either mock treated or treated with human TNF-α (200 U/ml), and cell viability was determined as described in the legend to Fig. 1 and in Materials and Methods.
CD30 signaling abrogates the induction of NF-κB by TNF-α
Previous studies have found that the induction of NF-κB can confer protection from TNF-α-mediated death (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996). Because TRAF2 has been reported to play an integral role in the activation of NF-κB by TNFR1 (Hsu et al. 1996), the ability of cells to respond to TNF-α following prolonged CD30 signal transduction was examined. Cells were transfected with the chimeric CD28–CD30 plasmid or with the tailless control plasmid. Forty-eight hours later, cells were stimulated with TNF-α and then evaluated for NF-κB levels by luciferase reporter gene assays. Cells transfected with the CD28–CD30 plasmid were unable to induce NF-κB in response to TNF-α treatment (Fig. 8), whereas cells transfected with the control plasmid induced NF-κB-dependent reporter gene activity in response to TNF-α.
Figure 8.
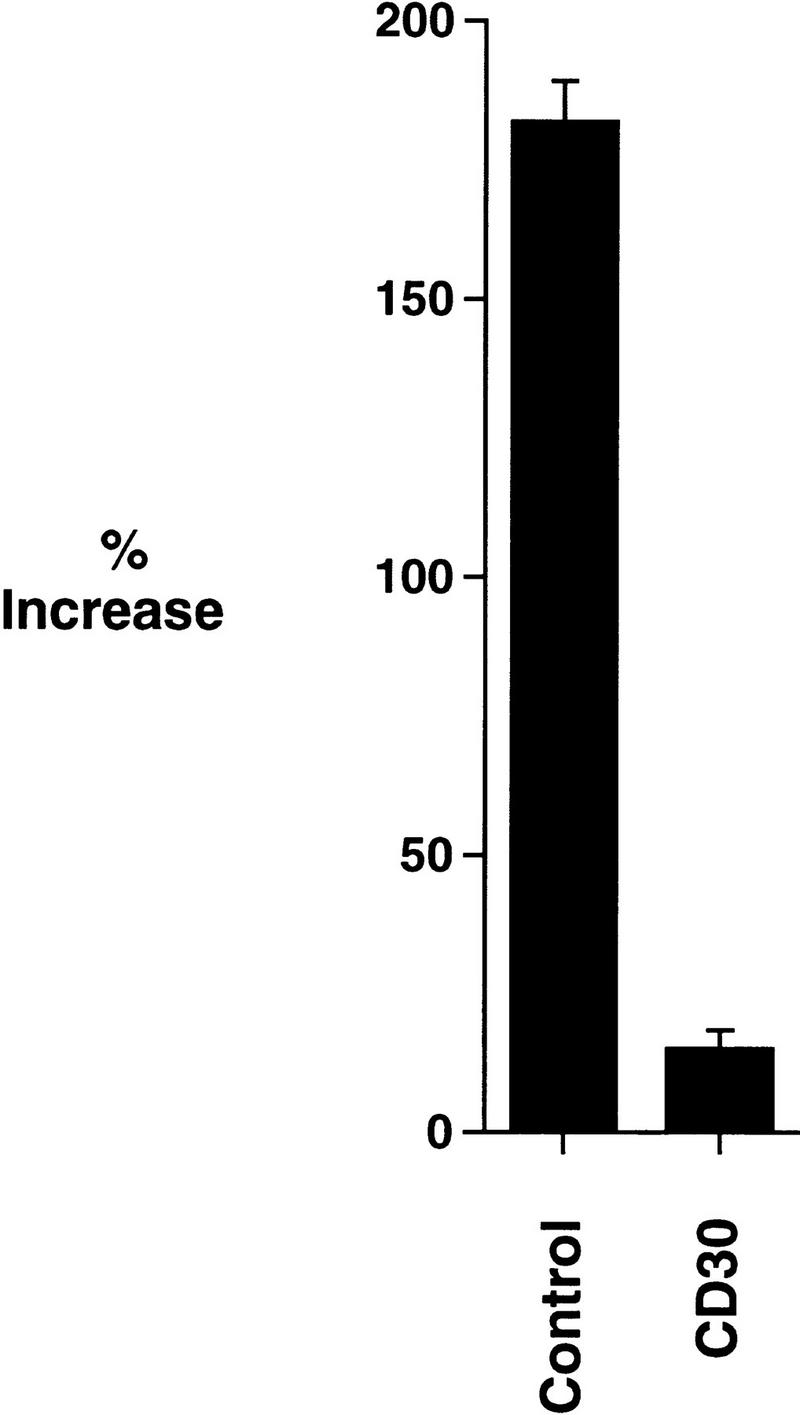
CD30 signaling abrogates NF-κB induction by TNF-α. 293 cells were transfected with 50 ng each of κB-luciferase reporter plasmid and β-galactosidase plasmid, along with 100 ng of either CD28–CD30 vector or tailless CD28 as indicated. Cells were stimulated with TNF-α 48 hr following transfection, and incubated further for 10 hr prior to harvest. The induction of NF-κB is presented as the percentage increase in NF-κB of TNF-α-treated samples over the activity of the reporter plasmid in mock-treated cells. Luciferase assays were performed as described in Materials and Methods.
Discussion
A subfamily of TNF receptor superfamily members has recently been defined that shares the common ability to initiate signal transduction through TRAF proteins, leading to the induction of NF-κB. In most cases, this promotes cell survival. These same receptors, however, have been implicated in cell death, under some circumstances. Unlike TNFR1, Fas and DR3/Wsl/Apo-3 (Tartaglia et al. 1993; Takahashi et al. 1994; Chinnaiyan et al. 1996; Kitson et al. 1996; Marsters et al. 1996; Pan et al. 1997), which contain death domains capable of inducing apoptosis through the recruitment and activation of caspases (Nagata 1996), it is not known how these receptors can induce apoptosis.
As an example, CD30 appears to play dual roles in the cell. CD30 has been reported to promote the development and differentiation of T helper type 2 (Th2) cells (Del Prete et al. 1995), and, therefore, has been associated with cellular survival and proliferation. In contrast, other findings have implicated CD30 in the activation of cell death (Lee et al. 1996). Disruption of murine CD30 by homologous recombination (Amakawa et al. 1996) has implicated a role for this receptor in thymic negative selection.
Similar to CD30, TNFR2 has been shown to exert both pro-survival and pro-death effects on the cell. TNFR2 can confer protection from cell death, and cellular proliferation (Rothe et al. 1994). In contrast, TNFR2 has been reported to play a role in apoptosis of mature T cells (Zheng et al. 1995). Several models have been proposed to account for the opposing properties of TNFR2. The rapid association/dissociation kinetics of TNFR2 for its ligand have led to the ligand passing hypothesis (Tartaglia et al. 1993), in which TNFR2 is postulated to recruit TNF-α and provide a source of ligand for TNFR1, thus suggesting a passive role for TNFR2 in signaling. Grell and coworkers have suggested that TNFR2 specifically binds membrane-associated TNF-α and thereby induces cell death (Grell et al. 1995), although this does not necessarily exclude the ligand-passing model. More recently, it has been shown that the cytoplasmic domain of TNFR2 is required for the potentiation of TNFR1-mediated death (Weiss et al. 1997), suggesting that cooperative signaling through these receptors, perhaps by ligand-induced oligomerization, may play a role in this cooperative response.
The experiments described here may provide an explanation for the dual effects of CD30 and TNFR2. Stimulation of 293 cells with TNF-α alone did not induce a significant degree of cell death (Fig. 1). Although 293 cells contain the product of the antiapoptotic adenovirus gene E1B 19K, which may function to raise the cellular apoptotic threshold, constitutive signal transduction mediated either by a CD28–CD30 or a CD28–TNFR2 chimera potentiated TNFR1-induced cell death (Figs. 1A and 6A). The TRAF-binding domain in CD30 was required for potentiation (Fig. 1A), and other reports have shown that the TRAF-binding element in TNFR2 is required for sensitization by this receptor (Weiss et al. 1997). This suggests that CD30 signaling functions by the same mechanism as TNFR2 to potentiate TNFR1-mediated death. Because CD30 and TNFR2 are normally activated by independent ligands, this raises the possibility that other members of the TNF receptor superfamily that utilize the TRAF signaling pathway may also function to potentiate TNFR1-mediated death by a similar mechanism. It is formally possible that the apoptotic properties of CD30 and TNFR2 are mediated by recruitment and activation of caspases, for example, through the formation of a TRAF2–TRADD–FADD–caspase complex. Because CD30 was unable to induce cell death in the absence of a TNFR1 signal, however, it is more likely that CD30 functions as a cofactor to potentiate the cytotoxic effects of TNFR1.
As shown in Figure 2A, we found that TRAF2 protein was almost completely degraded by activation of CD30-mediated, TRAF-dependent signals. This destabilization did not occur at the transcriptional level (Fig. 2B), but could be inhibited by several protease inhibitors (Fig. 3), suggesting the involvement of a proteolytic program that specifically degrades TRAF2.
Degradation of TRAF2 could be induced by recruitment to either of the two TRAF-binding domains in CD30 (Fig. 5) that have previously been identified (Gedrich et al. 1996). Degradation could also be induced by recruitment to TNFR2 (Fig. 6B), suggesting a shared pathway. Signal transduction mediated by CD30 and TNFR2 utilizes TRAF2, which subsequently leads to NF-κB activation. This activation has been previously shown to involve the degradation of IκB by the ubiquitin–proteasome pathway (Palombella et al. 1994; Chen et al. 1995, 1996; Henkel et al. 1995; DiDonato et al. 1996; Hateboer et al. 1996). Signaling through endogenous TNFR1, however, which also results in the TRAF2-mediated activation of NF-κB (Hsu et al. 1996) and induces the degradation of IκB (Chen et al. 1995; DiDonato et al. 1995; Henkel et al. 1995), did not result in the destabilization of TRAF2 (Fig. 6B). Although it is possible that ectopically expressed, activated TNFR1 might also induce TRAF2 degradation, these findings suggest that the determinants of TRAF2 degradation are distinct from those of IκB degradation.
Signaling through CD30 does not induce a general degradation of all TRAF proteins (Table 1). Moreover, binding of TRAF proteins to CD30 is not sufficient to induce degradation, because TRAF1 binds directly to CD30 (Gedrich et al. 1996) but is not degraded. One difference that may account for this is the absence of a RING finger domain in TRAF1. As shown in Figure 4B, the disruption of the RING finger of TRAF2 abrogated degradation, suggesting that the RING finger of TRAF2 may play a role in destabilization. Interestingly, the RING finger has also been reported to be required for NF-κB activation by TRAF2 (Rothe et al. 1995b), suggesting that the RING finger may be required both for protein–protein interactions with downstream signaling components as well as for the targeting of TRAF2 for degradation. Thus, the TRAF2 (87–501) mutant is not subject to degradation and so might block NF-κB activation by competitively binding to CD30, thereby preventing recruitment of endogenous TRAF2 to CD30. The construction of fusion proteins between TRAF1 and TRAF2, however, have so far failed to generate a chimeric TRAF1–TRAF2 molecule that could be degraded by CD30 signaling (C.S. Duckett and C.B. Thompson, unpubl.), suggesting that additional determinants in TRAF2 may also be required. Also noteworthy is the observation that TRAF1 can be degraded when coexpressed with TRAF2. This finding suggests that the degradation of TRAF2 can extend to additional molecules associated with TRAF2, and this might also be the case for factors unrelated to the TRAFs such as the c-IAPs, TANK/I-TRAF, Nik, A20 and TRIP, all of which have been shown to associate directly with TRAF2 (Rothe et al. 1995a, 1996; Cheng and Baltimore 1996; Song et al. 1996; Lee et al. 1997; Malinin et al. 1997).
TRAF2 is a signaling intermediate that is also utilized by TNFR1 (Hsu et al. 1996). Whereas TRAF2 can bind directly to the cytoplasmic domains of TNFR2 and CD30, it is recruited indirectly to TNFR1 through a TNFR1–TRADD–TRAF2 interaction (Hsu et al. 1996), where it is thought to mediate NF-κB activation. Several groups have reported that NF-κB induction can confer antiapoptotic properties on the cell (Beg and Baltimore 1996; Van Antwerp et al. 1996; Wang et al. 1996). Expression of a dominant-negative mutant of TRAF2, which blocks NF-κB induction by TNFR1, potentiated TNFR1-mediated death (Fig. 7). Therefore, the inhibition of TRAF2-induced signaling, either by expression of a dominant-negative TRAF2 or by the CD30-induced degradation of TRAF2, can sensitize cells to TNFR1-mediated death. Consistent with this hypothesis, CD30 signal transduction and concomitant TRAF2 degradation abrogated the ability of TNF-α to induce NF-κB (Fig. 8). A further prediction of this model is that the treatment of cells with protease inhibitors that can block TRAF2 degradation might confer protection to death induced by TNFR1 plus CD30 signal transduction. Preliminary studies suggest that this is the case (C.S. Duckett and C.B. Thompson, unpubl.). The compounds that inhibit TRAF2 degradation, however, also cross-react with the ubiquitin–proteasome pathway and have been shown to block NF-κB induction by TNF-α, and the combined treatment of TNF-α plus protease inhibitors was found to be toxic to the untransfected and control transfected populations of cells (C.S. Duckett and C.B. Thompson, unpubl.).
These studies describe a mechanism by which members of the tumor necrosis receptor superfamily may cooperate to modulate cell survival. The recruitment of TRAF2 to the cytoplasmic tails of CD30 or to TNFR2 is followed by its degradation. There are at least two consequences of this event. First, the removal of TRAF2 would be predicted to limit the duration of the receptor-induced signal, thus ensuring that the signal is self regulating. Second, several TNFR2-associated proteins, such as TRAF2 and the c-IAPs, have been found to possess antiapoptotic properties (Liston et al. 1996; Liu et al. 1996; Uren et al. 1996), and because TRAF2-associated proteins were also found to be degraded by CD30, elimination of these factors might be predicted to sensitize the cell to additional signals such as those mediated by TNFR1 (Fig. 9). The observation that CD30 and TNFR2 signaling can sensitize cells to death in the context of TNFR1 signals might also explain how signaling through CD30 or through TNFR2 can induce disparate effects ranging from cellular proliferation to apoptosis (Smith et al. 1993; Stein et al. 1985a,b). The removal of TRAF2 by degradation might therefore be expected to interfere with the induction by TNFR1 of antiapoptotic signals, resulting in a shift to the remaining TRADD–FADD–caspase pathway (Fig. 9).
Figure 9.

Model: Control of TRAF2 levels may modulate the sensitivity of cells to TNFR1 signals (see text for details).
The induction of proteolytic degradation of TRAF2 following recruitment to CD30 and TNFR2 will result in the termination of signal transduction through these receptors. Following decay of the NF-κB-mediated survival signal, cells will be subsequently more susceptible to dying if activated by death domain-containing receptors such as TNFR1, unless TRAF2 and its coassociated signaling molecules can be regenerated during this interval. Factors that increase the expression of these proteins are, therefore, likely to help maintain the duration of survival signaling through CD30 and TNFR2. These data suggest that the rate of receptor-mediated TRAF2 consumption and TRAF2 translation are likely to play a dynamic role in the regulation of cell survival.
Materials and methods
Plasmids
The chimeric CD28–CD30 vectors, the Δtail control plasmid, and the expression vectors encoding TRAF1, TRAF2, and TRAF3 have been described previously (Duckett et al. 1997). The ΔTraf vector encodes the CD28 extracellular domain fused to residues 410–531 of CD30, a deletion that previously has been shown to abrogate TRAF binding (Gedrich et al. 1996; Duckett et al. 1997). The CD28–TNFR2 chimera was constructed by PCR amplification of the TNFR2 cytoplasmic tail from a human T-cell cDNA library with the following primers: 5′-ATACTCGAGACAGGTGAAAAAGAAGCCCTTGTGCC-3′ and 5′-AATTCTAGACCACCTTGGCTACGACACAGCCCAC-3′. The PCR product was subcloned in-frame into the modified CD28 expression vector described previously (Duckett et al. 1997). The GFP expression vector was obtained from Clontech laboratories. The Myc epitope-tagged human TRAF4/CART1 expression vector was kindly provided by C. Rudin and J. Van Dongen (both at University of Chicago, IL). The murine TRAF5 vector was kindly provided by R. Arch (University of Chicago, IL); the amino terminus was modified to incorporate the Myc epitope tag (Duckett et al. 1996) prior to subcloning into the pcDNA3 mammalian expression vector (Invitrogen). Human TRAF6 was obtained by PCR amplification from a human T-cell cDNA library followed by subcloning into the pFLAG-CMV-2 expression vector (Kodak).
Cells and transfections
Human embryonic kidney 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, 2 mm glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin, and grown at 37°C in 5% CO2. Six-well plates were seeded with 2 × 105 cells per well in 2 ml of medium. The medium was replaced the following day, and cells were transfected by the calcium phosphate procedure as described previously (Duckett et al. 1997).
Viability assays
Cells were cotransfected with 100 ng of GFP plasmid and 1 μg of CD28 chimeric plasmid as indicated in the text. Unless stated otherwise, the medium was removed 12 hr post-transfection and replaced either with fresh medium containing human recombinant TNF-α (Boehringer Mannheim) at 200 U/ml or with medium alone, and cells were maintained for a further 18 to 24 hr as indicated, before harvest. Medium from transfected cells was collected, cells were washed once with phosphate buffered saline (PBS) supplemented with 1 mm ethylenediaminetetraacetic acid (EDTA), and cells were detached from the plate in the same buffer. Cells, washes, and medium were pooled, and cells were pelleted by centrifugation at 1500 rpm at 4°C. Pellets were washed once in PBS/EDTA and fixed in PBS containing 1.25% glutaraldehyde (1 ml). Fixed cells were washed once with PBS, resuspended in 1 ml PBS containing 4′,6′-diamidino-2-phenylindole (DAPI) at 20 ng/ml, and incubated for 10 min at room temperature. Stained cells were washed once in PBS and resuspended in 1 ml PBS. Aliquots (typically 50 μl) were spun onto microscope slides with a Cytopro cytocentrifuge (Wescor), and cells were examined by fluorescence microscopy by use of a Leitz DM-RB microscope.
Analysis of TRAF protein levels
Cells were cotransfected with 100 ng of the indicated CD28 chimeric vector and 1 μg of TRAF expression vector. Lysates were prepared 12–48 hr after transfection as indicated in the figure legends, as follows: Cells were washed once in PBS and lysed in 0.5 ml of lysis buffer [25 mm HEPES at pH 7.9, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, 10% glycerol, 1 mm dithiothreitol, 0.1 mm phenylmethanesulfonyl fluoride, and a cocktail of protease inhibitors (Boehringer Mannheim)]. Protein levels were standardized with the Bradford reagent (Bio-Rad). Samples were electrophoresed on 9.5% SDS–polyacrylamide gels and immunoblotted with commercial rabbit polyclonal antibodies to human TRAF1, TRAF2, or TRAF3 (Santa Cruz Biotechnologies), or with murine monoclonal antibodies to the Myc epitope (Pharmingen; clone 9E10) or FLAG (Kodak; M2) as indicated in the text. Blots were subsequently probed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse monoclonal antibodies (Amersham) as required and developed with an enhanced chemiluminescence detection system (Amersham).
RNA extraction and Northern blot analysis
Total cellular RNA was isolated with the TRIzol protocol (Life Technologies). RNA quantitation, electrophoresis, and transfer to nitrocellulose were performed as described (Yang et al. 1995), and blots were probed with a nick-translated 32P-labeled DNA fragment encompassing residues 87–501 of human TRAF2.
Reporter gene analysis
Six-well plates were seeded with 293 cells at a density of 5 × 105 cells per well in 2 ml of medium, and triplicate wells were transfected with 50 ng each of a κB-responsive luciferase reporter plasmid containing two canonical κB sites (Klug et al. 1994) and a β-galactosidase transfection efficiency control plasmid, together with 100 ng of either the CD28–CD30 or the Δtail control plasmid, as indicated. Transfectants were maintained for 48 hr and stimulated where indicated with recombinant human TNF-α (200 U/ml; Boehringer Mannheim) or a medium control for 10 hr. Cells were harvested and luciferase reporter gene activity assayed as described previously (Duckett et al. 1997).
Acknowledgments
We thank R. Arch, R. Gedrich, M. Gilfillan, A. Minn, C. Rudin, and members of the Thompson lab for thoughtful discussions and critical review of the manuscript; R. Arch, C. Rudin, and J. Van Dongen for providing TRAF4 and TRAF5 cDNAs; H. Singh for providing luciferase plasmids; and T. Lindsten for assistance with Northern blots. This work was supported by research grant P01 DK49799 (to C.B.T.) from the National Institutes of Health. C.S.D. is a Special Fellow of the Leukemia Society of America.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Craig@Knapp.uchicago.edu; FAX (773) 702-1576.
References
- Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, Yagita H, Okumura K, Inoue J, Watanabe T. Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NF-κB activation. J Biol Chem. 1997;272:2042–2045. doi: 10.1074/jbc.272.4.2042. [DOI] [PubMed] [Google Scholar]
- Allison J, Krummel MF. The yin and yan of T cell costimulation. Science. 1995;270:932–933. doi: 10.1126/science.270.5238.932. [DOI] [PubMed] [Google Scholar]
- Amakawa R, Hakem A, Kundig TM, Matsuyama T, Simard JJL, Timms E, Wakeham A, Mittruecker H-W, Griesser H, Takimoto H, Schmits R, Shahinian A, Ohashi PS, Penninger JM, Mak TW. Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell. 1996;84:551–562. doi: 10.1016/s0092-8674(00)81031-4. [DOI] [PubMed] [Google Scholar]
- Ansieau S, Scheffrahn I, Mosialos G, Brand H, Duyster J, Kaye K, Harada J, Dougall B, Hübinger G, Kieff E, Hermann F, Leutz A, Gruss H-J. Tumor necrosis factor receptor-associated factor (TRAF)-1, TRAF-2, and TRAF-3 interact in vivo with the CD30 cytoplasmic domain; TRAF-2 mediates CD30-induced nuclear factor kappa B activation. Proc Natl Acad Sci. 1996;93:14053–14058. doi: 10.1073/pnas.93.24.14053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Armitage RJ. Tumor necrosis factor receptor superfamily members and their ligands. Curr Opin Immunol. 1994;6:407–413. doi: 10.1016/0952-7915(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene. 1996;12:1–9. [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Fiers W. Molecular mechanisms of tumor necrosis factor-induced cytotoxicity. What we do understand and what we do not. FEBS Lett. 1994;340:9–16. doi: 10.1016/0014-5793(94)80163-0. [DOI] [PubMed] [Google Scholar]
- Biswas P, Smith CA, Goletti D, Hardy EC, Jackson RW, Fauci AS. Cross-linking of CD30 induces HIV expression in chronically infected T cells. Immunity. 1995;2:587–596. doi: 10.1016/1074-7613(95)90003-9. [DOI] [PubMed] [Google Scholar]
- Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, Licari P, Mankovich J, Shi L, Greenberg AH, Miller LK, Wong WW. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes & Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- Chen Z, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Cheng G, Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD40L-mediated NF-κB activation. Genes & Dev. 1996;10:963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O’Rourke K, Yu G-L, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- Choi MS, Boise LH, Gottschalk AR, Quintans J, Thompson CB, Klaus GG. The role of bcl-xL in CD40-mediated rescue from anti-μ-induced apoptosis. Eur J Immunol. 1995;25:1352–1357. doi: 10.1002/eji.1830250533. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- Del Prete G, De Carli M, D’Elios MM, Daniel KC, Almerigogna F, Alderson M, Smith CA, Thomas E, Romagnani S. CD30-mediated signaling promotes the development of human T helper type 2-like cells. J Exp Med. 1995;182:1655–1661. doi: 10.1084/jem.182.6.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. Phophorylation of IκBα precedes but is not sufficient for its dissociation from NF-κB. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett CS, Nava VE, Gedrich RW, Clem RJ, Van Dongen JL, Gilfillan MC, Shiels H, Hardwick JM, Thompson CB. A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J. 1996;15:2685–2694. [PMC free article] [PubMed] [Google Scholar]
- Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. Induction of nuclear factor κB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol. 1997;17:1535–1542. doi: 10.1128/mcb.17.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell. 1992;68:421–427. doi: 10.1016/0092-8674(92)90180-k. [DOI] [PubMed] [Google Scholar]
- Ellis TM, Simms PE, Slivnick DJ, Jäck H-M, Fisher RI. CD30 is a signal-transducing molecule that defines a subset of human activated CD45RO+ T cells. J Immunol. 1993;151:2380–2389. [PubMed] [Google Scholar]
- Falini B, Pileri S, Pizzolo G, Dürkop H, Flenghi L, Stirpe F, Martelli MF, Stein H. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85:1–14. [PubMed] [Google Scholar]
- Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand gp39. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- Gagliardini V, Fernandez PA, Lee RK, Drexler HC, Rotello RJ, Fishman MC, Yuan J. Prevention of vertebrate neuronal death by the crmA gene. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen JL, Thompson CB. CD30 contains two binding sites with different specificities for members of the TRAF family of signal transducing proteins. J Biol Chem. 1996;271:12852–12858. doi: 10.1074/jbc.271.22.12852. [DOI] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Gruss H-J, Dower SK. Tumor necrosis factor ligand superfamily: Involvement in the pathology of malignant lymphomas. Blood. 1995;85:3378–3404. [PubMed] [Google Scholar]
- Hateboer G, Kerkhoven RM, Shvarts A, Bernards R, Beijersbergen RL. Degradation of E2F by the ubiquitin-proteasome pathway: Regulation by retinoblastoma family proteins and adenovirus transforming proteins. Genes & Dev. 1996;10:2960–2970. doi: 10.1101/gad.10.23.2960. [DOI] [PubMed] [Google Scholar]
- Henkel T, Machleidt T, Alkalay I, Krönke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Nature. 1995;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu H-B, Pan M-G, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Ishida T, Mizushima S-I, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J-I. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996a;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- Ishida T, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J-I. TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci. 1996b;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson J, Raven T, Jiang Y-P, Goeddel DV, Giles KM, Pun K-T, Grinham CJ, Brown R, Farrow SN. A death-domain containing receptor that mediates apoptosis. Nature. 1996;384:372–375. doi: 10.1038/384372a0. [DOI] [PubMed] [Google Scholar]
- Klug CA, Gerety SJ, Shah PC, Chen Y-Y, Rice NR, Rosenberg N, Singh H. The v-abl tyrosine kinase negatively regulates NF-κB/Rel factors and blocks κ gene transcription in pre-B lymphocytes. Genes & Dev. 1994;8:678–687. doi: 10.1101/gad.8.6.678. [DOI] [PubMed] [Google Scholar]
- Komiyama T, Ray CA, Pickup DJ, Howard AD, Thornberry NA, Peterson EP, Salvesen G. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J Biol Chem. 1994;269:19331–19337. [PubMed] [Google Scholar]
- Krappmann D, Wulczyn FG, Scheidereit C. Different mechanisms control signal-induced degradation and basal turnover of the NF-κB inhibitor IκBα in vivo. EMBO J. 1996;15:6716–6726. [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Park CG, Choi Y. T cell receptor-dependent cell death of T cell hybridomas mediated by the CD30 cytoplasmic domain in association with tumor necrosis factor receptor-associated factors. J Exp Med. 1996;183:669–674. doi: 10.1084/jem.183.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee SY, Choi Y. TRAF-interacting protein (TRIP): A novel component of the tumor necrosis factor receptor (TNFR)- and CD30-TRAF signaling complexes that inhibits TRAF2-mediated NF-κB activation. J Exp Med. 1997;185:1275–1285. doi: 10.1084/jem.185.7.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda J-E, MacKenzie A, Korneluk RG. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis, while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Maggi E, Annunziato F, Manetti R, Biagiotti R, Giudizi MG, Ravina A, Almerigogna F, Boiani N, Alderson M, Romagnani S. Activation of HIV expression by CD30 triggering in CD4+ cells from HIV-infected individuals. Immunity. 1995;3:251–255. doi: 10.1016/1074-7613(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Malinin NL, Boldin MP, Kovalenko A, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- Marsters SA, Sheridan JP, Donahue CJ, Pitti RM, Gray CL, Goddard AD, Bauer KD, Ashkenazi A. Apo-3, a new member of the tumor necrosis factor receptor family, contains a death domain and activates apoptosis and NF-κB. Curr Biol. 1996;6:1669–1676. doi: 10.1016/s0960-9822(02)70791-4. [DOI] [PubMed] [Google Scholar]
- Nadali G, Vinante F, Stein H, Todeschini G, Tecchio C, Morosato L, Chilosi M, Menestrina F, Kinney MC, Greer JP, Latza U, Perona G, Pizzolo G. Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large-cell lymphoma. J Clin Oncol. 1995;13:1355–1360. doi: 10.1200/JCO.1995.13.6.1355. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis: Telling cells their time is up. Curr Biol. 1996;6:1241–1243. doi: 10.1016/s0960-9822(02)70706-9. [DOI] [PubMed] [Google Scholar]
- Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware CF, Yagita H, Okumura K. TRAF5, an activator of NF-κB and putative signal transducer for the lymphotoxin-β receptor. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, Levrero M. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: Dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319–329. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995a;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science. 1995b;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- Rothe M, Xiong J, Shu H-B, Williamson K, Goddard A, Goeddel DV. I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc Natl Acad Sci. 1996;93:8241–8246. doi: 10.1073/pnas.93.16.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M, Diehl V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymhoid cells. Nature. 1982;299:65–67. doi: 10.1038/299065a0. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA, Grabstein KH, Gliniak B, McAlister IB, Fanslow W, Alderson M, Falk B, Gimpel S, Gillis S, Din WS, Goodwin RG, Armitage RJ. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993;73:1349–1360. doi: 10.1016/0092-8674(93)90361-s. [DOI] [PubMed] [Google Scholar]
- Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc Natl Acad Sci. 1996;93:6721–6725. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Lee SY, Wong B, Arron J, Santana A, Kong Y-Y, Ohashi PS, Choi Y. A regulatory role for TRAF1 in antigen-induced apoptosis of T cells. J Exp Med. 1997;185:1777–1783. doi: 10.1084/jem.185.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier MK, Cohen JJ. Calpain, an upstream regulator of thymocyte apoptosis. J Immunol. 1997;158:3690–3697. [PubMed] [Google Scholar]
- Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- Stein H, Gerdes J, Lemke H, Mason DY. Evidence of Sternberg-Reed cells being derived from activated lymphocytes. In: Gallo RC, Neth R, Greaves MF, Janka G, editors. Modern trends in human leukemia VI. Heidelberg, Germany: Springer-Verlag; 1985a. pp. 441–444. [DOI] [PubMed] [Google Scholar]
- Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, Gatter K, Falini B, Delsol G, Lemke H, Schwarting R, Lennert K. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985b;66:848–858. [PubMed] [Google Scholar]
- Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice caused by a point mutation in Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV, Reynolds C, Figari IS, Weber RF, Fendly BM, Palladino MA., Jr Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. J Immunol. 1993;151:4637–4641. [PubMed] [Google Scholar]
- Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: The 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268:18542–18548. [PubMed] [Google Scholar]
- Tomasetto C, Régnier CH, Moog-Lutz C, Mattei MG, Chenard MP, Lidereau R, Basset P, Rio MC. Identification of four novel human genes amplified and overexpressed in breast carcinoma and localized to the q11–q21.3 region of chromosome 17. Genomics. 1995;28:367–376. doi: 10.1006/geno.1995.1163. [DOI] [PubMed] [Google Scholar]
- Tsitsikov E, Wright DA, Geha RS. CD30 induction of human immunodeficiency virus gene transcription is mediated by TRAF2. Proc Natl Acad Sci. 1997;94:1390–1395. doi: 10.1073/pnas.94.4.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A, Pakusch M, Hawkins C, Puls KL, Vaux DL. Cloning and expression of apoptosis inhibitory proteins homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc Natl Acad Sci. 1996;93:4974–4978. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green D, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Mayo MW, Baldwin ASJ. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Weiss T, Grell M, Hessabi B, Bourteele S, Müller G, Scheurich P, Wajant H. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80. J Immunol. 1997;158:2398–2404. [PubMed] [Google Scholar]
- Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- Yang H, Duckett CS, Lindsten T. iPABP, an inducible poly(A)-binding protein detected in activated human T cells. Mol Cell Biol. 1995;15:6770–6776. doi: 10.1128/mcb.15.12.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]