

Abstract
We have designed a pair of biotinylated peptide nucleic acid (PNA) probes targeting two sequences in 18S rRNA (from the parasite Trypanosoma brucei) at a distance of 191 nt (corresponding to maximum distance of ca. 60 nm) from each other. The PNA probes were individually bound to (strept)avidin-coated fluorescent beads, differing in size and color [green beads (1 µm) and red beads (5.9 µm)], thereby allowing distinct detection of each PNA probe by conventional fluorescence microscopy. These two PNA beads showed easily detectable co-localization when simultaneously hybridizing to a target nucleic acid. The assay detected the parasite 18S rRNA down to 1.6 fmol while there was no such co-localization visible with human 18S rRNA not containing the PNA targets. Furthermore, the assay showed positive detection with 1.6 ng of total RNA (corresponding to RNA from ca. 300 parasites). Upon further optimization this method may provide a new tool for a diagnosis of Human African Trypanosomiasis (HAT) and it may more generally have applications within diagnostics for (neglected) infectious diseases.
Key words: diagnostics, fluorescence microscopy, fluorescent bead, PNA, ribosomal RNA, Trypanosome
Introduction
Sensitive and sequence specific detection of nucleic acids is a prerequisite for genetic analyses in basic, preclinical and clinical research. A large variety of methods are available, most of which rely on PCR amplification of the target nucleic acid prior to actual detection. In many diagnostic applications, in particular within infectious (bacterial, viral, parasitic) diseases, a quick “yes or no” answer is desired. Sequence specific detection of nucleic acids based on amplification (using e.g., PCR,1 PCR-ELISA,2 mass spectrometry,3 proximity ligation4) is extremely sensitive and may in principle detect single molecules. However, they also require well-equipped laboratories together with well-skilled technicians to obtain reliable results. Therefore, simplified amplification-independent methods that do not require advanced instrumentation, are highly warranted, particularly for diagnosis of neglected infectious diseases occurring in developing countries such as Human African Trypanosomiasis (HAT).5
Artificial DNA, such as locked nucleic acid (LNA),6 peptide nucleic acid (PNA)7 and morpholino oligomers,8 provide biologically stable, high affinity probes exhibiting high sequence selectivity for detection of target nucleic acids (DNA and RNA) via hybridization. For instance, LNA arrays for microRNA analyses are widely used,9 and fluorescent in situ hybridization (FISH) detection of telomere sequence by PNA probes10 and identification of specific bacteria using a PNA-FISH method11 have gained wide acceptance.
Fluorescence detection methods are extremely sensitive and in principle (using advanced instruments for e.g., total internal reflection fluorescence (TIRF)12 or two-photon-excited fluorescence13 analysis) allow detection of single molecules, but typically this is seldom achieved due to high background. Single molecule detection is also possible by microscopy (AFM, electron microscopy, fluorescence) techniques using nucleic acid probes conjugated to (nano)particles, and by combining two gold nanoparticles (of different size) hybridizing to the same nucleic acid target, the specificity is dramatically improved.14
In order to combine the high sensitivity of fluorescence detection with the excellent specificity of two bead simultaneous hybridization, we decided to use (strept)avidin coated fluorescent polystyrene beads to which biotinylated PNA oligomer probes can be coupled to create a hybridization directed co-localization detection method for nucleic acids. Two different (size and color) beads, which are easily visualized and discriminated by conventional fluorescence microscopy techniques, were chosen and each coupled to a biotin PNA targeting a unique site on the analyte nucleic acid. When binding the same nucleic acid molecule, the two beads will co-locate as an indicator for the presence of analyte nucleic acid.
To test the concept of this technique, we chose two unique sequences within the 18S rRNA from the parasite Trypanosoma brucei as probe target because of a significant demand for detection (of the parasite) as well as the high rRNA content in the parasite (ca.1 million copies/parasite),15 and we show that by simple fluorescence microscopy analysis we are able to specifically detect the presence of 18S rRNA in a total RNA extract from less than 300 parasites.
Results and Discussion
For the detection of Trypanosoma brucei 18S RNA, we made a pair of biotinylated peptide nucleic acid (PNA) probes which were attached on the surface of fluorescent beads for visualization (Fig. 1A). Two fluorescent (strept)avidin coated beads of different size and color [green beads (GB, 1 µm) and red bead (RB, 5.9 µm)] that can easily and simultaneously be located and distinguished from each other by conventional fluorescence microscopy (Fig. 1A) were chosen. The two biotinylated 18-mer PNA oligomers complementary to and specific for two targets on the Trypanosoma brucei 18S rRNA separated by 191 nucleotides (ca. 60 nm) were bound to the red (PNA3534) and green (PNA3533) beads, respectively (Fig. 1B).
Figure 1.

Fluorescence microscopy detection of beads and co-localization principle. Red fluorescent beads (RBs) are 5.9 µm in size, surface modified with streptavidin and including magnetite for magnetic separation. Green fluorescent beads (GBs) are 1 µm in size and surface modified with avidin. (A) Fluorescent microscopic images of GB, RB and a mixture of the two. (B) Illustration of target sequence detection by two bead/PNA complexes through target hybridization via the PNA probes. Fluorescent beads were complexed with biotin-labeled PNA 3533 and PNA 3534 respectively for GB and RB. These two bead/PNA complexes were incubated with target nucleic acids. Two beads show proximity location upon the hybridization of two PNAs to their target sequences.
In a preliminary characterization of this system we chose a simple, short synthetic oligodeoxynucleotide target (instead of RNA). For synthetic reasons the DNA sequence was shortened by129 nt in the middle of the two PNA binding sites (relative to the original parasite 18S RNA target) resulting in a 62 nt DNA. This DNA (97 nt) was sequentially incubated with the pair of fluorescent PNA-beads (Fig. 2A) for 1 h at room temperature and subjected to fluorescence microscopy analysis. Using varying amounts of DNA (0–1,000 fmol), we consistently found that it is possible to detect co-localization of two beads even at the lowest (40 fmol) concentration, and there was an increase in the number of red and green beads showing co-localization in a dose-dependent manner, although this was not quantitatively addressed in these preliminary experiments (Fig. 2A). Furthermore, red beads exhibiting co-localization with more than one green bead were observed at all DNA concentrations, but (as would be expected) appeared more pronounced at higher DNA doses (Figs. 2B and 3A). To rule out nonspecific complex formation, we performed a number of control experiments (Fig. 2B), clearly demonstrating that co-localization requires the presence of DNA and only takes place with PNA coated beads. Furthermore, selective enrichment of the red (magnetic) beads through a washing step makes identification of the double bound target (co-localization) much more effective (Fig. 2B). Identification of genuinely hybridization connected (co-localized) green and red beads may be distinguished from coincidental co-localization by difference in vertical location by varying the microscopic focus level, since out of focus beads appear blurred and enlarged (Fig. 3B). Nonetheless, we found it advantageous to include a washing step in further experiments for easy detection.
Figure 2.

Detection of a 97 nt target DNA with a pair of PNA-Beads. Two PNA beads (red beads (RB) with PNA3534 and green beads (GB) with PNA3533), prepared as illustrated in Figure 1, were incubated with the target DNA and subsequently analyzed by fluorescence microscopy. RBs, showing a co-localization with GB(s) are circled with dotted red line, and the co-localized GBs are indicated by yellow arrows. (A) Dose dependent detection of target DNA (0–1,000 fmol). (B) The complex formation was tested in the presence or absence of target DNA (40 fmol) with or without PNA probe. The samples were subjected to fluorescence microscopy before and after magnetic enrichment. A two step hybridization was performed to assure Red beads/DNA complexation first as the Red beads have less DNA binding sites than GBs (<500) and less free-movement than GBs Red beads are six times bigger than GBs (i.e., much heavier) although we are not sure if it is necessary.
Figure 3.
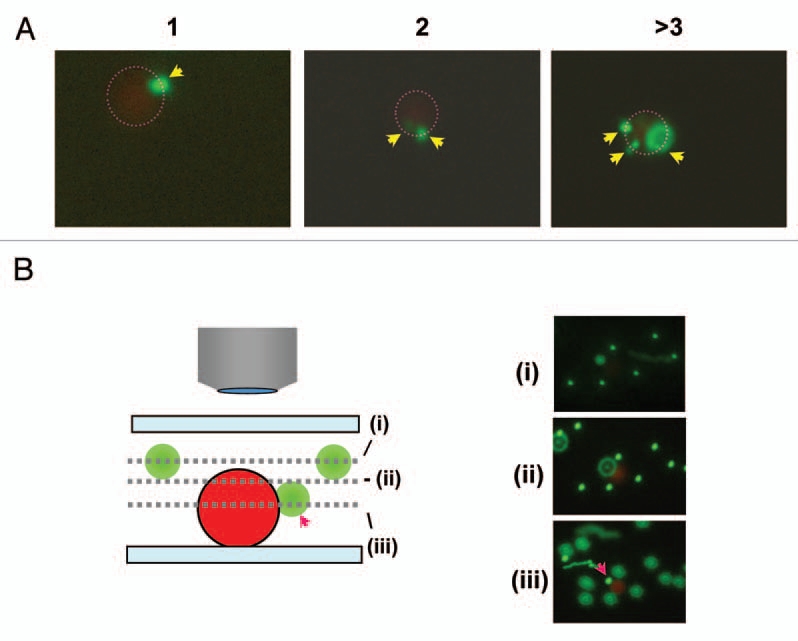
Example of two bead co-localization. (A) Typical examples of co-localization of GB and RB with different GB numbers (e.g., 1, 2 and >3). (B) Detection of GBs at different depth and proximity from RB. Three pictures were taken with different focal depth (i, ii and iii) (including schematic drawing) from the same area. The red arrow indicates the GB exhibiting co-localization with RB. Some GBs appear to be larger due to location in a different focal plane (depth) (blurring).
To explore the detection sensitivity, we tested this assay with lower DNA doses (200, 40, 8 and 1.6 fmol) (Fig. 4). In analogy with the experiment in Figure 2, we observed complex formation in a dose-dependent manner, although rigorous quantification was not performed. The detection limit for the target DNA under these conditions was 8 fmol, and as a control for specificity no co-localization was observed using human 18S rRNA, which does not contain targets for the PNAs. In contrast, when we used in vitro transcribed 18S rRNA from the parasite Trypanosoma brucei (Fig. 5A), co-localization of red beads with at least one green bead was observed down to 1.6 fmol RNA, and at concentrations higher than 8 fmol, more than 20% of the red beads showed co-localization with green bead(s) while this number was only around 5% in case of 1.6 fmol RNA (ca 200 beads counted for each). For diagnostic purposes a lower detection limit of 1.6 fmol target RNA is relevant, although it is still one order of magnitude higher than a desired detection limit of 0.16 fmol corresponding to 100 parasites17 given that one parasite contains approximately 1 million copies of 18S-rRNA/parasite.15
Figure 4.

Dose dependent detection of target nucleic acids with human 18S rRNA negative control. Experiment analogous to the one presented in Figure 2.
Figure 5.

Dose dependent detection of trypanosoma18S rRNA. The PNA beads (red beads (RB) with PNA3534 and green beads (GB) with PNA3533), were incubated with the indicated amounts of (A) in vitro transcribed target 18S RNA or (B) total trypanosome RNA, and subjected to fluorescence microscopy analysis (×250 for top raw and ×400 for other raws) after magnetic enrichment. RBs in co-localization with GB(s) are circled with a dotted red line where GBs with co-localization are indicated by yellow arrows.
To further validate this assay, we decided to test a total RNA preparation from Trypanosoma brucei procyclic cultures (Fig. 5B) in stead of pure target RNA prepared by in vitro transcription. By using this RNA preparation, we were still able to detect co-localization of beads (as indicator for specific hybridization of both PNA probes) down to 1.6 ng of RNA per sample giving >5% of the red beads exhibiting co-localization with at least one green bead. The total RNA amount of 1.6 ng corresponds to ca 0.5 fmol of 18S rRNA (assuming that the 18S rRNA constitutes 20% of the total RNA) and this amount of target rRNA may be obtained from 300 parasites (based on the assumption of 1 million copies of 18S rRNA/cell). For comparison, other methods, including amplification protocols such as real time PCR and loop-mediated isothermal amplification (LAMP)18,19 e.g., in a dipstick format,20 claim to detect infections with 5–10 parasites/ml blood (clinical samples). Although, this sensitivity level has not yet been reached by the presently described bead co-localization principle, a detection limit of 0.5 fmol rRNA is quite encouraging as it is accomplished with a very simple method not requiring amplification. The bead co-localization methodology is indeed a single molecule detection principle and therefore we are confident that it may be significantly optimized in terms of sample preparation and analysis, and should be useful for specific rRNA (or other nucleic acid) detection in general in other contexts, not least within diagnostics of other infectious diseases. The presently used PNAs are specitic for Trypanosoma brucei, but it remains to be seen to which extent the assay is capable of distinguishing different Trypanosoma species. Furthermore, improved detection limits should be possible if combined with computerized image analysis or by flow cytometric (FACS) or microfluidics sorting for counting co-localized beads.
Materials and Methods
Synthesis of PNA.
PNA synthesis was carried out as reported elsewhere in reference 16. Biotin (Bio) was covalently linked to the PNA at the N-terminal through three ethylene glycol type linkers (eg2, 11-amino-3,6,9-trioxaundecanoic acid). PNAs were HPLC-purified and characterized by mass spectrometry. Purified PNAs were lyophilized and stored at 4°C until use. The sequences of the PNAs are as following: PNA3533, Bio-(eg2)3-CCG CTC CCG TGT TTC TTG-Lys-NH2; PNA3534, Bio-(eg2)3-GAA ACA CCG ACC CAA GGC-Lys-NH2.
Target nucleic acids.
Sequence of chemically synthesized DNA (97 nt) was as follows where PNA target sequences were underlined: 5′-CAA GAA ACA CGG GAG CGG TTC CTC CTC ACT TTC ACG CAT GTC ATG CAT GCG AAG TCC TTG GGA GAT TAT GGG GCC GCG TGC CTT GGG TCG GTG TTT C-3′.
Trypanosome (accession #, M12676) and human (accession #: K03432) 18S rRNA samples were prepared by in vitro transcription. The Trypanosoma brucei 18S rRNA fragment of the following sequence, where PNA target sequences are underlined and the truncated region for the above mentioned DNA is shown in italic, was used: 5′-CAA GAA ACA CGG GAG CGG UUC CUC CUC ACU UUC ACG CAU GUC AUG CAU GCG AGG GGG CGU CCG UGA AUU UUA CUG UGA CCA AAA AAG UGC GAC CAA AGC AGU CUG CCG ACU UGA AUU ACA AAG CAU GGG AUA ACG AAG CAU CAG CCC UGG GGC CAC CGU UUC GGC UUU UGU UGG UUU UAG AAG UCC UUG GGA GAU UAU GGG GCC GCG UGC CUU GGG UCG GUG UUU C-3′. Total RNA was extracted from Trypansoma brucei procyclic cell culture (500,000 parasites/each extraction) using the RNaqueous-micro kit (Ambion).
Beads-PNA complex preparation.
Two distinct beads (different in color and size) were used for complex preparation. The red fluorescent beads (RB) were magnetic (for capturing), streptavidin surface modified and 5.9 µm in size (QuantumPlexM SP Streptavidin (Bangs laboratories)). The green fluorescent (GB) were surface modified with avidin and 1.01 µm in size (Neutral avidin coated Dragon Green polystyrene beads (Bangs laboratories)). A pair of two PNA/bead complexes was prepared by incubating each PNA with one of the beads (PNA3353 (10 nmol) was incubated with the GB (eq. to 1.4 nmol in biotin binding capacity) and PNA3354 (500 pmol) was incubated with RB (eq. to 2.5 pmol in biotin binding capacity)) for 30 min in PBS at room temperature. After 30 min incubation, the complexes were extensively washed with PBS using a centrifugation (10,000x g for 5 min) to remove unbound PNA and used for further analysis.
Hybridization of target nucleic acid with bead/PNA complexes and detection by fluorescence microscopy.
The target nucleic acid was incubated with the PNA3353/GB complex for 1 h at room temperature in PBS and subsequently with the PNA3354/RB complex (biotin binding capacity of 93 pmol and 0.1 pmol respectively for RB and GB) for 1 h at room temperature in PBS. Then the nucleic acid/beads complexes were washed once with PBS following manufacture instructions using a strong magnet to remove the excess GB and unbound target nucleic acids. Finally the resultant nucleic acid/beads complexes were resuspended in PBS and analyzed by conventional fluorescence microscopy (Diaplan, Leitz) with x500 magnification using a proper filter block [XF70 (Omega Optical) for only red, H3 (Leitz) for both green and red] unless otherwise stated. Images were captured with an installed CCD camera INFINITY2 (Lumenera) using the accompanying software for image acquisition and processing.
Acknowledgments
This work was supported by the European Commission, 6. Framework EU project TRYLEIDIAG (contract no. INCO-CT-2005-015379). Deborggraeve is a postdoctoral fellow of the Research Foundation Flanders (FWO).
References
- 1.Becker S, Franco JR, Simarro PP, Stich A, Abel PM, Steverding D. Real-time PCR for detection of Trypanosoma brucei in human blood samples. Diagn Microbiol Infect Dis. 2004;50:193–199. doi: 10.1016/j.diagmicrobio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Sanchez J, Pineda JA, Andreu-Lopez M, Delgado J, Macias J, De La Rosa R, Morillas-Marquez F. The high sensitivity of a PCR-ELISA in the diagnosis of cutaneous and visceral leishmaniasis caused by Leishmania infantum. Ann Trop Med Parasitol. 2002;96:669–677. doi: 10.1179/000349802125001906. [DOI] [PubMed] [Google Scholar]
- 3.Sampath R, Hofstadler SA, Blyn LB, Eshoo MW, Hall TA, Massire C, et al. Rapid identification of emerging pathogens: coronavirus. Emerg Infect Dis. 2005;11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landegren U, Schallmeiner E, Nilsson M, Fredriksson S, Baner J, Gullberg M, et al. Molecular tools for a molecular medicine: analyzing genes, transcripts and proteins using padlock and proximity probes. J Mol Recognit. 2004;17:194–197. doi: 10.1002/jmr.664. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy PG. The continuing problem of human African trypanosomiasis (sleeping sickness) Ann Neurol. 2008;64:116–126. doi: 10.1002/ana.21429. [DOI] [PubMed] [Google Scholar]
- 6.Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci USA. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 8.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 9.Stenvang J, Silahtaroglu AN, Lindow M, Elmen J, Kauppinen S. The utility of LNA in microRNA-based cancer diagnostics and therapeutics. Semin Cancer Biol. 2008;18:89–102. doi: 10.1016/j.semcancer.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Taneja KL. Localization of trinucleotide repeat sequences in myotonic dystrophy cells using a single fluorochrome-labeled PNA probe. Biotechniques. 1998;24:472–476. doi: 10.2144/98243rr02. [DOI] [PubMed] [Google Scholar]
- 11.Malic S, Hill KE, Hayes A, Percival SL, Thomas DW, Williams DW. Detection and identification of specific bacteria in wound biofilms using peptide nucleic acid fluorescent in situ hybridization (PNA FISH) Microbiology. 2009;155:2603–2611. doi: 10.1099/mic.0.028712-0. [DOI] [PubMed] [Google Scholar]
- 12.Truskey GA, Burmeister JS, Grapa E, Reichert WM. Total internal reflection fluorescence microscopy (TIRFM). II. Topographical mapping of relative cell/substratum separation distances. J Cell Sci. 1992;103:491–499. doi: 10.1242/jcs.103.2.491. [DOI] [PubMed] [Google Scholar]
- 13.Mertz J, Xu C, Webb WW. Single-molecule detection by two-photon-excited fluorescence. Optics letters. 1995;20:2532. doi: 10.1364/ol.20.002532. [DOI] [PubMed] [Google Scholar]
- 14.Stadler AL, Sun D, Maye MM, van der Lelie D, Gang O. Site-Selective Binding of Nanoparticles to Double-Stranded DNA via Peptide Nucleic Acid “Invasion”. ACS nano. 2011;5:2467–2474. doi: 10.1021/nn101355n. [DOI] [PubMed] [Google Scholar]
- 15.van Eys GJ, Schoone GJ, Kroon NC, Ebeling SB. Sequence analysis of small subunit ribosomal RNA genes and its use for detection and identification of Leishmania parasites. Mol Biochem Parasitol. 1992;51:133–142. doi: 10.1016/0166-6851(92)90208-2. [DOI] [PubMed] [Google Scholar]
- 16.Christensen L, Fitzpatrick R, Gildea B, Petersen KH, Hansen HF, Koch T, et al. Solid-phase synthesis of peptide nucleic acids. J Pept Sci. 1995;1:175–183. doi: 10.1002/psc.310010304. [DOI] [PubMed] [Google Scholar]
- 17.Robays J, Bilengue MM, Van der Stuyft P, Boelaert M. The effectiveness of active population screening and treatment for sleeping sickness control in the Democratic Republic of Congo. Trop Med Int Health. 2004;9:542–550. doi: 10.1111/j.1365-3156.2004.01240.x. [DOI] [PubMed] [Google Scholar]
- 18.Njiru ZK, Mikosza AS, Armstrong T, Enyaru JC, Ndung'u JM, Thompson AR. Loop-mediated isothermal amplification (LAMP) method for rapid detection of Trypanosoma brucei rhodesiense. PLoS neglected tropical diseases. 2008;2:147. doi: 10.1371/journal.pntd.0000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mugasa CM, Laurent T, Schoone GJ, Kager PA, Lubega GW, Schallig HD. Nucleic acid sequence-based amplification with oligochromatography for detection of Trypanosoma brucei in clinical samples. J Clin Microbiol. 2009;47:630–635. doi: 10.1128/JCM.01430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deborggraeve S, Claes F, Laurent T, Mertens P, Leclipteux T, Dujardin JC, et al. Molecular dipstick test for diagnosis of sleeping sickness. J Clin Microbiol. 2006;44:2884–2889. doi: 10.1128/JCM.02594-05. [DOI] [PMC free article] [PubMed] [Google Scholar]