

Abstract
Acetaminophen (APAP) overdose is one of the most frequent causes of acute liver failure in the United States and is primarily mediated by toxic metabolites which accumulate in the liver upon depletion of glutathione stores. However, cells of the innate immune system, including NK cells, neutrophils, and Kupffer cells, have also been implicated in the centrilobular liver necrosis associated with APAP. We have recently shown that dendritic cells (DC) regulate intra-hepatic inflammation in chronic liver disease and, therefore, postulated that DC may also modulate the hepatotoxic effects of APAP. We found that DC immune-phenotype was markedly altered after APAP challenge. In particular, liver DC expressed higher MHC II, co-stimulatory molecules, and Toll-like Receptors, and produced higher IL-6, MCP-1, and TNF-α. Conversely, spleen DC were unaltered. However, APAP-induced centrilobular necrosis, and its associated mortality, was markedly exacerbated upon DC depletion. Conversely, endogenous DC expansion using FMS-like tyrosine kinase 3 ligand (Flt3L) protected mice from APAP injury. Our mechanistic studies showed that APAP liver DC had the particular capacity to prevent NK cell activation and induced neutrophil apoptosis. Nevertheless, the exacerbated hepatic injury in DC depleted mice challenged with APAP was independent of NK cells and neutrophils or numerous immune modulatory cytokines and chemokines.
Conclusions
Taken together, these data indicate that liver DC protect against APAP toxicity while their depletion is associated with exacerbated hepatotoxicity.
Keywords: liver, immunity, cellular depletion, neutrophils, necrosis
Introduction
Acetaminophen (APAP) is a widely used over the counter analgesic and antipyretic agent. Although usually considered safe at therapeutic doses, at higher doses, APAP causes acute liver failure, characterized by centrilobular hepatic necrosis. APAP-induced hepatoxicity is the leading cause of acute liver failure, accounting for nearly 50% of all cases (1–4). The spectrum of APAP related liver failure ranges from individuals taking an intentional overdose (42%) to accidental overdose (49%) (3, 4). In the United States, APAP overdose is a major burden to the health care system. It is responsible for over 56,000 emergency room visits, 2,600 hospitalizations, and over 450 deaths from acute liver failure annually (1, 3).
APAP is a dose-dependent hepatotoxin. When taken at therapeutic doses, over 90% of APAP is metabolized by gluconylation and sulphation and its metabolites are rapidly excreted in the urine. Of the remaining APAP, approximately 2% is excreted intact in urine, and 5–9% is metabolized by the cytochrome P450 system to N-acetyl-p-benzo-quinoneimine (NAPQ1), a highly reactive metabolite (1, 5, 6). At therapeutic doses of APAP, hepatic glutathione (GSH), a major intracellular antioxidant, induces the formation of a safely excretable APAP-protein adduct. However, at toxic doses of APAP, GSH becomes overwhelmed and severely depleted in both the cytoplasm and mitochondria (1, 7). Once GSH is depleted, NAPQ1 is able to exert its harmful effects by forming covalent bonds with cellular proteins. Covalent bonding to mitochondrial proteins causes mitochondrial dysfunction by inhibition of the Ca2+-Mg2+-ATPase, resulting in accumulation of cytosolic calcium. This disturbance leads to a decrease in ATP synthesis, disruption of cellular membrane, and eventually necrotic cell death (1, 7–9).
While toxic metabolites of APAP account for the primary hepatic insult, the liver’s innate immune system has also been shown to play a major role in APAP-induced liver injury in what is akin to a “two-hit” mechanism. That is, although GSH depletion and the resulting toxic metabolites are prerequisites for APAP hepatotoxicity, there is evidence that the severity of liver injury may depend on subsequent downstream participation of inflammatory mediators (1, 10–17). NK and NKT cell activation have been purported to be a crucial component in the progression of APAP-induced hepatotoxicity (12). Hepatic NK and NKT cells are a major source of IFN-γ, which has been shown to mediate hepatocyte apoptosis, leukocyte infiltration, as well as cytokine and chemokine production in APAP-induced liver injury (11). However, more recent evidence suggests that NK cells are less critical to APAP toxicity (15). Kupffer cells have also been shown to contribute to APAP-mediated hepatotoxicity. Michael et al (16) showed that mice treated with gadolinium chloride, a deactivator of macrophages, had dramatically decreased APAP-induced liver injury. Kupffer cells are thought to exacerbate liver injury by increasing the synthesis of oxygen free radicals (16). However, there is also evidence to the contrary. Ju et al (13) found that following depletion of Kupffer cells APAP-induced liver injury was exacerbated. The mechanism was purported to be related to decreased expression of several hepato-regulatory cytokines, including Interleukin-10 (IL-10), which functions to limit inducible nitric oxide synthase expression and peroxynitrite-induced liver injury (13). The role of neutrophils in APAP-induced hepatotoxicity is also controversial. Liu et al (18) demonstrated that depletion of neutrophils using RB6-8C5 protected mice against APAP-induced liver injury, as evidenced by reduced serum ALT levels, decreased centrilobular necrosis, and improved survival. In contrast, Lawson et al (14) demonstrated that neutrophils contribute to the removal of necrotic debris rather than directly affecting the pathogenesis of APAP-induced injury. Thus, it appears that secondary induction of the hepatic innate immune system likely plays a part in APAP-induced liver injury, however, the precise role of its components remains incompletely understood.
Dendritic cells (DC) are the principal antigen presenting cells in lymphoid organs and in the periphery, including the liver, and initiate both innate and adaptive immune responses (19, 20). We and others have shown that liver DC are characterized by their immaturity and more commonly mediate tolerance rather than immunogenicity (21–24). DC are primarily responsible for hepatic, oral, and portal venous tolerance (22, 23) and have been purported to play a role in sundry other aspects of hepatic tolerance including the acceptance of liver allografts with minimal immune suppression and the frequent deposition of hepatic tumor metastases (24). However, while in their steady-state liver, DC have tolerogenic properties, there is emerging evidence for a reversal of their immune-phenotype after hepatic insult. We have recently reported that in chronic liver fibrosis, DC mature in vivo, become highly immunogenic, and govern intra-hepatic inflammation via production of TNF-α thereby modulating activation of NK cells and liver T cells (25). Our preliminary investigations also showed that DC become pro-inflammatory after APAP challenge. Based on these data, we postulated that in acute liver injury induced by APAP, DC play a central role in exacerbating secondary hepatic injury. Our results confirm an important role for DC in APAP, but suggest that rather than worsening liver insult DC are protective.
Methods
Animals and In Vivo Models
Male C57BL/6 (H-2Kb), CD11c-DTR, OT-I (B6.Cg-RAG2tm1Fwa-TgN), and OT-II (B6.Cg-RAG2tm1Alt-TgN) mice (4–8 week old) were purchased from Taconic Farms (Germantown, NY) and then bred in-house. Mice were housed in a pathogen-free environment. To induce acute hepatic injury, mice were treated intraperitoneal (i.p.) with 500 μg/g APAP diluted in PBS. In selected experiments, mice were treated with for 10 days with Flt3L (10μg; Celldex, Fall River, MA) before APAP challenge. To effect DC depletion, CD11c.DTR mice were treated with a single i.p. dose of diphtheria toxin (4ng/g; Sigma-Aldrich, Saint Louis, MO). In vivo plasmacytoid DC depletion was accomplished using 120G8 (200ug; Imgenex, San Diego, CA) (26). In selected experiments, the novel immune-modulator VAG539 (30mg/kg, Novartis, Basel, Switzerland) was used to partially inactivate DC (27). To deplete Gr1+ cells, RB6-8C5 was employed (150μg/day; Monoclonal Antibody Core Facility, Sloan-Kettering Institute, New York, NY). For in vivo NK cell depletions, 100 μl of a 1:5 dilution of anti-asialo GM1 (Wako Chemical, Richmond, VA) was injected i.p. 3 days prior to APAP treatment. In selected experiments, antibodies directed against IFN-α (1μg, F18, Sigma), TNF-α (200μg, AB-410, R&D, Minneapolis, MN), IL-6 (200μg, AB-406, R&D), or MCP-1 (50 μg, AB-479, R&D) were administered in vivo before APAP challenge. Changes in serum liver enzymes, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), were determined using the Olympus AU400 Chemistry Analyzer (Center Valley, PA). In survival experiments, animals were euthanized when they were moribund and death was imminent. Animal procedures were approved by the New York University School of Medicine Animal Care and Use Committee.
Cellular Isolation
Liver DC were isolated as previously described (25). Briefly, immediate post-mortem laparotomy was performed and the portal vein was cannulated and infused with 1% Collagenase IV (Sigma-Aldrich). Hepatectomy was then performed and livers mechanically minced before incubation with Collagenase IV at 37° for 10 minutes. Low speed (30g) centrifugation was performed to exclude the pelleted hepatocytes followed by high speed (300g) centrifugation to isolate the hepatic non-parenchymal cells (NPC). The NPC were then further enriched over a 40% Optiprep (Sigma-Aldrich, Saint Louis, MO) density gradient. To purify the DC population, NPC were incubated with 1 μg of anti-FcγRIII/II (2.4G2, Fc block; Monoclonal Antibody Core Facility, Sloan-Kettering Institute, New York, NY) per 106 cells, labeled with fluorescently conjugated anti-CD11c and anti-MHC II (both BD Biosciences, Franklin lakes, NJ) and FACS sorted using a MoFlo cell sorter (Beckman Coulter, Fullerton, CA). Splenocytes were prepared by mechanical disruption and specific cellular subgroups were purified by FACS.
T cell proliferation assays
For in vitro T cell proliferation assays, peptide-pulsed DC (3×104) were added to CD8+OT-I TCR-transgenic T cells (1×105) specific for Ova257–264, or CD4+OT-II TCR-transgenic T cells specific for Ova323–339 in 96-well plates for 48–72 hours before pulsing with 3H-Thymidine as described (25). DC were loaded with the relevant Ova peptide (10 μg/ml; AnaSpec, San Jose, CA) for 90 minutes before co-culture with respective T cells. In selected experiments, VAF347 (5mM, Novartis), a low-molecular-weight compound which binds thearyl hydrocarbon receptor, was used to prevent DC induction of CD4+ T cells (28).
Western Blotting and qPCR
Western blotting was performed as we have described (29). Livers were minced in PBS with Protease Inhibitor cocktail (Roche, Pleasanton, CA) and homogenized. The whole organ lysate was spun at 500 x g, and the post-nuclear supernatant was obtained. After determination of total protein by the Lowry assay, 10 % polyacrylamide gels were equiloaded with samples, electrophoresed at 90 V, electrotransferred to PVDF membranes and probed with primary mouse monoclonal antibodies for HMGB-1 or glutathione (Abcam, Cambridge, UK). Secondary goat anti-mouse HRP was used (1: 4,000). Blots were developed by ECL (Thermo Scientific, Asheville, NC). Total GSH from whole liver homogenates was measured using the Glutathione Assay Kit (Cayman Chemicals, Ann Arbor, MI) according to the manufacturer’s protocol. For PCR assays, RNA was isolated from pancreas using a Qiagen RNEasy isolation kit (Qiagen, Germantown, MD). qPCR was performed using a standardized pre-configured PCR array (SA Biosciences, Frederick, MD) on the Stratagene MX3000P (Promega, Madison, Wisconsin) according to the respective manufacturers’ protocols.
See Additional Supplemental Methods
Results
DC depletion exacerbates APAP-mediated hepatic injury
To evaluate a possible role for DC in either exacerbating or protecting against APAP toxicity, we employed CD11c.DTR mice in which transient DC depletion can be effected (Supplemental Figure 1). Mice were depleted of DC or mock depleted and then challenged with APAP. At 12 hours, mice were sacrificed and the extent of liver injury determined by histopathology. Mice treated with APAP and depleted of DC (APAP-DC) had markedly more extensive centrilobular necrosis compared with controls (Figure 1a, b). Consistent with these findings, serum liver enzymes were highly elevated in APAP-DC mice (Figure 1c). Similarly, NPC production of inflammatory mediators after APAP challenge, including MCP-1 and IL-6, were higher in mice depleted of DC (Figure 1d). DC depletion alone, in absence of APAP challenge, had no effect (Figure 1a-d). Similarly, effects of APAP were similar in both CD11c.DTR and WT mice, in absence of DC depletion (not shown). Notably, APAP metabolism appeared unchanged in APAP-DC mice compared with APAP treatment alone based on tissue glutathione assay and glutathione adduct formation (Supplemental Figure 2a, b). To determine if there was higher systemic toxicity in APAP-challenged mice after DC depletion, we measured serum levels of inflammatory mediators. We found that serum MCP-1, IL-6 and TNF- α, were elevated in APAP-DC mice (Figure 1e, f). However, as expected, organ damage was limited to the liver as the lungs, kidneys, pancreas, and intestine were histologically normal (Supplemental Figure 3).
Figure 1. DC depletion exacerbates APAP toxicity.

(a) Liver from saline-treated, DC depleted, APAP, and APAP-DC mice were assessed by H&E staining of paraffin embedded sections and (b) the percentage of necrotic hepatocyte area determined by counting 10 high-powered fields per mouse. (c) Serum liver enzymes were measured as were (d) NPC production of MCP-1 and IL-6 in cell culture supernatant and (e) serum levels of MCP-1, IL-6, and (f) TNF-α. Experiments were performed in triplicate using 3–5 mice per group (*p<0.05; **p<0.01; ***p<0.001).
To determine whether DC depletion resulted in higher APAP-mediated mortality, mice were treated with APAP and depleted of DC or mock depleted and then observed for up to 2 weeks. Remarkably, approximately half of APAP-DC mice died within 48 hours of challenge while death was rare in control animals (Figure 2). There was no further mortality observed in APAP-DC mice after 48 hours from the time of APAP challenge.
Figure 2. DC depletion exacerbates APAP-related mortality.
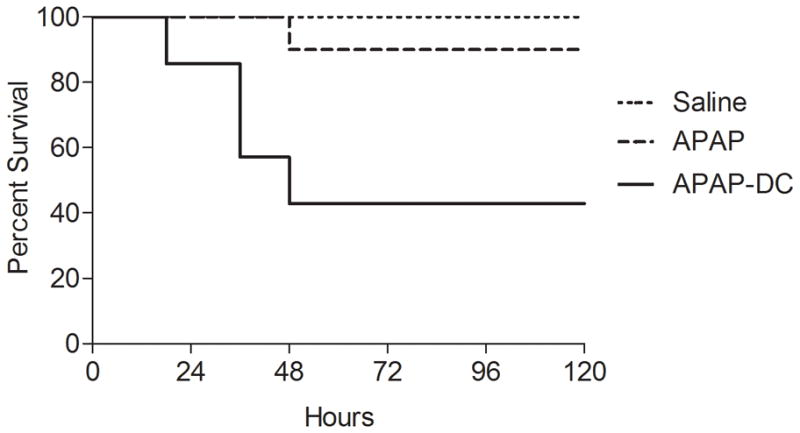
Mice were treated with saline, DC depletion alone, APAP, or APAP-DC and survival measured according to the Kaplan-Meier method (n=7–10 mice per group; p<0.05 for comparison between APAP-DC and other groups).
We next tested whether depletion of specific DC subsets were responsible for the observed higher toxicity associated with APAP-challenge. Mice were depleted exclusively of plasmacytoid DC using 120G8 before APAP challenge. However, plasmacytoid DC depletion did not exacerbate APAP liver toxicity (Supplemental Figure 4). Similarly, we tested whether activation of the aryl hydrocarbon receptor on DC, which inhibits liver DC maturation in vivo and mitigates their ability to induce adaptive Th2 responses (27, 30) (Supplemental Figure 5a, 5b), would also lead in exacerbated injury when administered to APAP challenged mice. However, VAG539 did not modulate APAP toxicity (Supplemental Figure 5c–e).
DC immune-phenotype is altered after APAP challenge
Since the absence of DC results in exacerbated APAP-mediated injury, we interrogated the immune-phenotype of DC in animals challenged with APAP. Both the absolute number and fraction of liver DC among hepatic leukocytes did not change after APAP challenge (Figure 3a, b); however, DC underwent an increase in maturation and alteration in subset composition in acute APAP hepatotoxicity (Figure 3c). In particular, DC harvested from APAP injured liver had elevated expression of MHC II and CD86, exhibited a lower B220+ plasmacytoid fraction, and underwent a “myeloid shift” expressing higher CD11b and lower CD8 (Figure 3c). Conversely, spleen DC phenotype was unchanged after APAP challenge (Figure 3c). Liver DC also increased their expression of TLR2, TLR4, TLR7, and TLR9 after APAP challenge (Supplemental Figure 6a). However, there was no measureable increase in selected byproducts of sterile inflammation in APAP-DC liver compared with APAP alone (Supplemental Figure 6b, c). In addition to an altered surface phenotype, the immunogenicity of DC harvested from the APAP injured liver was altered as DC from APAP liver produced higher IL-6, MCP-1, and TNF-α (Figure 3d) compared with liver DC from saline-treated mice. Furthermore, consistent with their increased TLR expression, liver DC had an exaggerated cytokine response to TLR ligation after APAP toxicity (Figure 3e). Spleen DC did not produce altered levels of cytokines after APAP challenge (not shown). Despite changes in hepatic DC surface phenotype and cytokine production after APAP challenge, their capacity to stimulate antigen-restricted CD4+ and CD8+ T cells was not enhanced (Figure 3f).
Figure 3. DC immune-phenotype is altered after APAP challenge.

(a) The total number and (b) fraction of CD11c+ liver NPC per mouse was determined by flow cytometry. (c) Liver or spleen DC expression of surface markers in APAP or saline treated mice were measured. Median fluorescence indices are listed below histograms. (d) Liver DC production of IL-6, TNF-α, MCP-1, and IL-10 in cell culture supernatant was measured. (e) Liver DC production from IL-6 was also measured after incubation with ligands for TLR9 (CpG ODN 1826), TLR3 (Poly I:C), and TLR4 (LPS). (f) DC induction of antigen-restricted T cell proliferation was measured using appropriate peptide pulsed DC to stimulate CD4+OT-II and CD8+OT-I T cells. All in vitro experiments performed in triplicate and repeated at least three times (*p<0.05; **p<0.01).
DC expansion ameliorates the hepato-toxic effects of APAP
Since DC depletion exacerbates APAP-mediated hepatotoxicity, we postulated that expansion of DC populations would mitigate liver injury. To test this, we employed Flt3L which we have previously shown expands DC populations in vivo, more than 10-fold (31, 32). Mice were treated with Flt3L for 10 days before APAP challenge followed by sacrifice at 12 hours. Notably, the maturation level of liver DC in Flt3L treated mice was similar to controls except for a greater fraction of B220+ plasmacytoid DC in the Flt3L treated group (Supplemental Figure 7a, b). However, after APAP treatment, the fraction of plamsamcytoid DC were similar in both the Flt3L + APAP and in the APAP only group (Supplemental Figure 7b). Mice treated with Flt3L were partially protected from liver injury as indicated by reduced serum liver enzymes (Figure 4a) and histologic measurement of necrosis (Figure 4b, c). Notably, adoptive transfer of DC to APAP-DC mice did not protect mice from exacerbated toxicity (not shown). However, based on our investigations tracking adoptively transferred DC, it is likely that DC transfer is insufficient for protection as adoptively transferred DC do not populate the liver at early or late time points after administration (Supplemental Figure 8).
Figure 4. Flt3L treatment protects from APAP injury.

Mice were treated with saline, APAP, Flt3L, or APAP+Flt3L. (a) serum liver enzymes as well as (b, c) histological evidence of liver damage were determined (n=4–5 mice/group; *p<0.05; **p<0.01).
DC depletion alters the composition of hepatic leukocytes after APAP challenge
To determine whether secondary alterations come into effect upon DC depletion which may be, in part, responsible for the exacerbated toxicity in APAP-DC mice, we examined the changes in hepatic leukocyte composition after DC depletion. There was a shift in composition of NPC, including a marked increase in the number of neutrophils in APAP-DC liver compared with APAP treatment alone (Figure 5).
Figure 5. Hepatic NPC composition is altered after APAP challenge.

The cellular composition of hepatic leukocytes was determined by flow cytometry using an average of 5 mice per group.
Since neutrophils expand in APAP-DC challenged mice, we postulated that DC induce neutrophil apoptosis after APAP injury and, conversely, DC depletion would result in increased neutrophil viability. To test this, we measured the fraction of Gr1+CD11b+Annexin V+ cells in the normal liver, APAP challenged liver, and in the liver of APAP-DC mice. Consistent with our hypothesis, we found that APAP treatment resulted in increased neutrophil apoptosis. Conversely, the fraction of apoptotic neutrophils was sharply decreased upon DC depletion in the context of APAP injury (Supplemental Figure 9a). DC depletion alone in absence of APAP administration had no effect on the neutrophil apoptotic fraction (not shown).
Since NK1.1+ cells have also been implicated in the mechanism of APAP-mediated hepatotoxicity (12), we postulated that DC may prevent the activation of NK cells after APAP injury. To test this, hepatic NK1.1+ cells were purified, co-cultured with DC, and simultaneously stimulated with PMA + Ionomycin. Notably, DC from normal liver further enhanced NK1.1+ cell production of IFN-γ. Conversely, APAP liver DC prevented NK1.1+ cellular activation (Supplemental Figure 9b). Similarly, NK cells treated with PMA + Ionomycin and simultaneously co-cultured with DC from control livers were potently cytolytic. Conversely, APAP liver DC did not stimulate NK-mediated cytolysis of Yac-1 targets (Supplemental Figure 9c). Taken together, out data suggests that in acute APAP hepatotoxicity, liver DC inhibit neutrophil viability and NK cell activation.
DC depletion exacerbates APAP-mediated hepatic injury independently of neutrophils, NK cells, or inflammatory mediators
Both neutrophils and NK cells have been implicated in the pathogenesis of APAP (12, 14, 18). Furthermore, since APAP-DC treated animals experience an expansion of neutrophils and our data shows that DC affect neutrophil viability and NK activation status, we postulated that the exacerbated centrilobular necrosis associated with DC depletion in APAP challenged animals was secondary to an expanded neutrophil population or activated NK cells, rather than directly related to the absence of DC. To test this, we simultaneously depleted either NK cells or neutrophils in conjunction with DC before APAP administration. However, neither depletion of NK cells or neutrophils, along with DC, mitigated the exacerbated hepatotoxicity associated with DC depletion (Figure 6) suggesting that the protective effects of DC is not simply secondary to expansion of other leukocyte populations. Similarly, since DC depletion results in elevated serum levels of TNF-α, IL-6, and MCP-1 after APAP administration, we tested whether blockade of these cytokines in vivo would prevent the exacerbated liver injury. However, none of these cytokine blockades protected APAP-DC animals (Supplemental Figure 10). Similarly, IFN-α blockade (33) failed to protect APAP-DC animals (Supplemental Figure 10)
Figure 6. Neutrophil or NK cell depletion do not protect APAP-DC mice.

(a, b) CD11c-DTR mice were depleted of DC and simultaneously depleted of either neutrophils or NK cells before challenge with APAP.
Discussion
There is evidence to suggest that APAP-induced liver toxicity is the result of a “two-hit” mechanism. The first hit being depletion of Glutathione which in turn allows the toxic metabolite NAPQ1 to exert harmful effects by forming covalent bonds with cellular proteins. The second hit being the downstream activation of cells of the innate immune system. Since DC have a central function in liver immunity and inflammation, we postulated a critical role for DC in APAP-mediated toxicity.
Previously, we showed that DC expand 5-fold and undergo a transformation in function from a tolerogenic to an immunogenic role in chronic liver fibrosis (25). We reported that DC contribute to the pro-inflammatory cascade in liver fibrosis via production of TNF-α and subsequent T cell activation as well as induction of innate immune responses (25). Similar to liver fibrosis, in APAP toxicity, DC are highly pro-inflammatory producing elevated levels of IL-6, TNF-α, and MCP-1 (Figure 3d, e). However, in contrast to chronic liver disease, in acute liver injury as a result of APAP overdose, DC populations remained stable in number. Furthermore, whereas chronic liver injury resulted in the transformation of DC from weak purveyors of tolerance to potent immunogenicity, in the current context, DC did not gain enhanced capacity to stimulate CD4+ or CD8+ T cells (Figure 3f) or NK cells (Supplemental Figure 9b, c). The trigger in the hepatic microenvironment that thrusts DC in certain inflammatory contexts towards immunogenicity is uncertain but may be the key to understanding hepatic tolerance.
Furthermore, whereas DC appear to contribute to the pathologic environment in chronic liver disease, in the current context, DC are protective. This is evidenced by reduced liver enzymes and histologic measurement of necrosis in APAP treated mice co-treated with Flt3L which expands DC populations 10-fold. Furthermore, mice depleted of DC had significantly more extensive centro-lobular necrosis (Figure 1a, b) and increased mortality (Figure 2) when compared to mock depleted mice. In addition, APAP-DC mice produced markedly higher serum liver enzyme levels (Figure 1c), and inflammatory mediators MCP-1, IL-6, and TNF-α (Figure 1e, f) compared with APAP challenge in absence of DC depletion. These findings suggest that DC have a novel protective role in APAP hepatotoxicity. Again, the contextual switch governing DC behavior in diverse states of liver injury remains uncertain.
The role of Kupffer cells in APAP-induced liver toxicity is controversial (13, 16, 18) but may perhaps be similar to the role of DC. Ju et al (13) showed an increased susceptibility to APAP-induced liver injury when effectively depleting mice of Kupffer cells with gadolinium chloride. The mechanism of the Kupffer hepato-protective effect is thought to be related to decreased expression of IL-10, an anti-inflammatory cytokine, in Kupffer cell depleted mice. In our current study, DC did not produce detectible IL-10 after APAP treatment and their depletion did not affect serum or liver IL-10 levels (Figure 3d). Interestingly, Bamboat et al (34) demonstrated that in liver ischemia/reperfusion injury, liver DC were responsible for IL-10 production via TLR9 activation. DC-mediated IL-10 production was shown to suppress monocyte inflammatory function and reduce hepatic injury (34). However, since DC do not produce detectible IL-10 after APAP challenge, it appears that the mechanism responsible DC protective role in APAP-induced hepatotoxicity is distinct from that of ischemia/reperfusion liver injury. Our mechanistic investigations further show that the exacerbated liver toxicity associated with DC depletion is independent of associated elevations in neutrophils or inflammatory monocytes as well as independent of systemic elevations in TNF-α, MCP-1, and IL-6 and unrelated to IFN-α (Supplemental Figure 10).
Our study implicates DC in APAP-induced liver injury as an innate protector which can be compared to the role of DC in sepsis. Sepsis is characterized by an intense hyper inflammatory response designed to eliminate an underlying infectious source (35). However, there is growing evidence that an initial pro-inflammatory cascade is thought to be followed by an activation of a compensatory anti-inflammatory response syndrome which leads to immune suppression and subsequent poorer clinical outcomes (35, 36). DC loss appears to play a significant role in the pathogenesis of sepsis (37). By using cecal ligation and puncture (CLP) surgery as a model for sepsis in rodents, recent investigations have shown that decline in DC counts occur in conjunction with immune dysfunction, suggesting that DC may even have a protective role against the development of immunosuppression in sepsis (37). The mechanism of DC loss appears to be related to extensive apoptosis of DC during sepsis. This was demonstrated in a study in which mouse spleens showed a significant increase in Caspase 3 mediated apoptosis in follicular dendritic cells 36–48 hours after CLP insult compared to controls (37, 38). Furthermore, Scumpia et al (39) showed that DC deficient mice had increased mortality after CLP. Similarly, in our study, DC depletion exacerbated APAP-mediated hepatic injury and lead to a significant increase in mortality.
Sterile inflammation is the activation of the innate immune system and subsequent creation of an inflammatory response in the absence of an infectious source. Inciting stimuli include mechanical trauma, ischemia, and organ specific toxins such as APAP induced liver (40,41). Endogenous signals of tissue injury known as damage associated molecular patterns (DAMPs) have been shown to activate antigen presenting cells such as DCs. DAMPs can induce DC maturation by ligating their TLRs. However, our mechanistic studies indicate that whereas DC express elevated levels of TLRs (Supplemental Figure 6a) and produce exaggerated responses to TLR ligation (Figure 3e), there was no difference between APAP and APAP-DC liver in regards to the levels of measurable DAMPs including HMGB1 (Supplemental Figure 6b), heat shock proteins (Supplemental Figure 6c), and S100A9 (not shown). Exact understanding of the regulatory role of DC and its interplay with sterile inflammation could be an important step in the development of immune-directed therapy in APAP-induced liver injury and may have implications to other disease processes regulated by immunity and inflammation. Furthermore, the translational potential of this study to the protective role of DC in acute APAP toxicity in humans requires further exact investigation using human specimens.
Supplementary Material
Acknowledgments
Grant Support: This work was supported in-part by grants from a Liver Scholar Award from the American Liver Foundation (GM), a Society of University Surgeons Junior Faculty Grant (GM), and National Institute of Health Award DK085278 (GM) and CA155649 (GM).
List of abbreviations
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- APAP
Acetaminophen
- DC
Dendritic cells
- APAP-DC
Acetaminophen-treated with depletion of dendritic cells
- Flt3L
FMS-like tyrosine kinase 3 ligand
- GSH
Glutathione
- NAPQ1
N-acetyl-p-benzo-quinoneimine
- NPC
Non-parenchymal cells
- i.p
intraperitoneal
- DAMP
Damage associated molecular patterns
Contributor Information
Michael K. Connolly, Email: mkcmkc444@hotmail.com.
Diego Ayo, Email: diego.ayo@nyumc.org.
Ashim Malhotra, Email: ashim.malhotra@nyumc.org.
Michael Hackman, Email: michael.hackman@nyumc.org.
Andrea S. Bedrosian, Email: andrea.bedrosian@nyumc.org.
Junaid Ibrahim, Email: junaid.ibrahim@nyumc.org.
Napoleon E. Cieza-Rubio, Email: napoleon.rubio@nyumc.org.
Andrew H. Nguyen, Email: andrew.nguyen@nyumc.org.
Justin R. Henning, Email: justin.henning@nyumc.org.
Monica Dorvil-Castro, Email: drdorvil@gmail.com.
H. Leon Pachter, Email: leon.pachter@nyumc.org.
George Miller, Email: george.miller@nyumc.org.
Reference List
- 1.Chun LJ, Tong MJ, Busuttil RW, Hiatt JR. Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol. 2009 Apr;43(4):342–349. doi: 10.1097/MCG.0b013e31818a3854. [DOI] [PubMed] [Google Scholar]
- 2.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;(196):369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004 Jul;40(1):6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- 4.Lee WM. The case for limiting acetaminophen-related deaths: smaller doses and unbundling the opioid-acetaminophen compounds. Clin Pharmacol Ther. 2010 Sep;88(3):289–292. doi: 10.1038/clpt.2010.164. [DOI] [PubMed] [Google Scholar]
- 5.Han D, Shinohara M, Ybanez MD, Saberi B, Kaplowitz N. Signal transduction pathways involved in drug-induced liver injury. Handb Exp Pharmacol. 2010;(196):267–310. doi: 10.1007/978-3-642-00663-0_10. [DOI] [PubMed] [Google Scholar]
- 6.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003 Dec;31(12):1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 7.Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007 Aug;11(3):525–48. vi. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H, Bait ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006 Jan;89(1):31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 9.Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003 Jul 31;349(5):474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 10.Gardner CR, Laskin JD, Dambach DM, Sacco M, Durham SK, Bruno MK, et al. Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor-alpha and interleukin-10. Toxicol Appl Pharmacol. 2002 Oct 1;184(1):27–36. [PubMed] [Google Scholar]
- 11.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002 Aug;16(10):1227–1236. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 12.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004 Dec;127(6):1760–74. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 13.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002 Dec;15(12):1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- 14.Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci. 2000 Apr;54(2):509–516. doi: 10.1093/toxsci/54.2.509. [DOI] [PubMed] [Google Scholar]
- 15.Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology. 2008 Sep;48(3):889–897. doi: 10.1002/hep.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. 1999 Jul;30(1):186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- 17.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006 Jun;43(6):1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 18.Liu ZX, Kaplowitz N. Role of innate immunity in acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2006 Aug;2(4):493–503. doi: 10.1517/17425255.2.4.493. [DOI] [PubMed] [Google Scholar]
- 19.Plitas G, Burt BM, Stableford JA, Nguyen HM, Welles AP, DeMatteo RP. Dendritic cells are required for effective cross-presentation in the murine liver. Hepatology. 2008 Apr;47(4):1343–1351. doi: 10.1002/hep.22167. [DOI] [PubMed] [Google Scholar]
- 20.Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity. 2008 Sep 19;29(3):319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Pillarisetty VG, Shah AB, Miller G, Bleier JI, Dematteo RP. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J Immunol. 2004 Jan 15;172(2):1009–1017. doi: 10.4049/jimmunol.172.2.1009. [DOI] [PubMed] [Google Scholar]
- 22.Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. 2008 Sep 19;29(3):464–475. doi: 10.1016/j.immuni.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood. 2008 Oct 15;112(8):3175–3185. doi: 10.1182/blood-2008-05-159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crispe IN, Giannandrea M, Klein I, John B, Sampson B, Wuensch S. Cellular and molecular mechanisms of liver tolerance. Immunol Rev. 2006 Oct;213:101–118. doi: 10.1111/j.1600-065X.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 25.Connolly MK, Bedrosian AS, Malhotra A, Henning JR, Ibrahim J, Vera V, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J Immunol. 2010 Aug 15;185(4):2200–2208. doi: 10.4049/jimmunol.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asselin PC, Brizard G, Pin JJ, et al. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 27.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, Roncarolo MG. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell–mediated effects on regulatory T cells. Blood. 2008 Aug;112(4):1214–1222. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 28.Ettmayer P, Mayer P, Kalthoff F, Neruda W, Harrer N, Hartmann G, Epstein MM, Brinkmann V, Heusser C, Woisetschlager M. A Novel Low Molecular Weight Inhibitor of Dendritic Cells and B Cells Blocks Allergic Inflammation. Am J Respir Crit Care Med. 2005 December;173:599–606. doi: 10.1164/rccm.200503-468OC. [DOI] [PubMed] [Google Scholar]
- 29.Xu Y, Malhotra A, Ren, Schlame M. The enzymatic function of tafazzin. The enzymatic function of tafazzin. J Biol Chem. 2006 Dec 22;281(51):39217–24. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 30.Matsuda H, Suda T, Hashizume H, Yokomura K, Asada K, Suzuki K, Chida K, Nakamura H. Alteration of Balance between Myeloid Dendritic Cells and Plasmacytoid Dendritic Cells in Peripheral Blood of Patients with Asthma. Am J Respir Crit Care Med. 2002 July 10;166:1050–1054. doi: 10.1164/rccm.2110066. [DOI] [PubMed] [Google Scholar]
- 31.Kingham TP, Chaudhry UI, Plitas G, Katz SC, Raab J, Dematteo RP. Murine liver plasmacytoid dendritic cells become potent immunostimulatory cells after Flt-3 ligand expansion. Hepatology. 2007 Feb;45(2):445–454. doi: 10.1002/hep.21457. [DOI] [PubMed] [Google Scholar]
- 32.Miller G, Pillarisetty VG, Shah AB, Lahrs S, Dematteo RP. Murine Flt3 ligand expands distinct dendritic cells with both tolerogenic and immunogenic properties. J Immunol. 2003 Apr 1;170(7):3554–3564. doi: 10.4049/jimmunol.170.7.3554. [DOI] [PubMed] [Google Scholar]
- 33.Krug A, Rothenfusser S, Hornung V, Jahrsdörfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN- α/β in plasmacytoid dendritic cells. Eur J Immunol. 2001 Apr 17;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 34.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, Dematteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010 Feb;120(2):559–569. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001 Aug;16(2):83–86. doi: 10.1097/00024382-200116020-00001. [DOI] [PubMed] [Google Scholar]
- 36.Efron P, Moldawer LL. Sepsis and the dendritic cell. Shock. 2003 Nov;20(5):386–401. doi: 10.1097/01.SHK.0000092698.10326.6f. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Venet F, Chung-Shiang C, Lomas-Neira J, Ayala A. Changes in dendritic cell function in the immune response to sepsis. Expert OpinBiolTher. 2007 July;7(7):929–938. doi: 10.1517/14712598.7.7.929. [DOI] [PubMed] [Google Scholar]
- 38.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. 2003 Dec 15;171(2):909–914. doi: 10.4049/jimmunol.171.2.909. [DOI] [PubMed] [Google Scholar]
- 39.Scumpia PO, McAuliffe PF, O’Malley KA, Ungaro R, Uchida T, Matsumoto T, Remick DG, Clare-Salzler MJ, Moldawer LL, Efron PA. CD 11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol. 2005 Nov;175(5):3282–3286. doi: 10.4049/jimmunol.175.5.3282. [DOI] [PubMed] [Google Scholar]
- 40.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010 Mar;28:321–42. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang D, La Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends in Immunology. 2009 Aug;30(11):531–537. doi: 10.1016/j.it.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.