

Abstract
The cyclic interactions that occur between the subunits of the yeast mitochondrial RNA polymerase can serve as a simple model for the more complex enzymes in prokaryotes and the eukaryotic nucleus. We have used two-hybrid and fusion protein constructs to analyze the requirements for interaction between the single subunit core polymerase (Rpo41p), and the σ-like promoter specificity factor (Mtf1p). We were unable to define any protein truncations that retained the ability to interact, indicating that multiple regions encompassing the entire length of the proteins are involved in interactions. We found that 9 of 15 nonfunctional (petite) point mutations in Mtf1p isolated in a plasmid shuffle strategy had lost the ability to interact. Some of the noninteracting mutations are temperature-sensitive petite (ts petite); this phenotype correlates with a precipitous drop in mitochondrial transcript abundance when cells are shifted to the nonpermissive temperature. One temperature-sensitive mutant demonstrated a striking pH dependence for core binding in vitro, consistent with the physical properties of the amino acid substitution. The noninteracting mutations fall into three widely spaced clusters of amino acids. Two of the clusters are in regions with amino acid sequence similarity to conserved regions 2 and 3 of σ factors and related proteins; these regions have been implicated in core binding by both prokaryotic and eukaryotic σ-like factors. By modeling the location of the mutations using the partial structure of Escherichia coli σ70, we find that two of the clusters are potentially juxtaposed in the three-dimensional structure. Our results demonstrate that interactions between σ-like specificity factors and core RNA polymerases require multiple regions from both components of the holoenzymes.
Keywords: RNA polymerase, σ factor, transcription initiation, mitochondrial transcription, MTF1, RPO41, two-hybrid
The complex RNA polymerases of eukaryotes and prokaryotes require auxiliary factors specific for the initiation phase of transcription. These factors associate with the core polymerases to form holoenzymes competent for promoter recognition, selective DNA binding, and opening of the double-stranded DNA at the start site of transcription. Shortly after initiation, the factors are released as the RNA polymerase makes the transition into its elongating form. The factors associated with eukaryotic nuclear RNA polymerase II (Pol II) before initiation, and released shortly after transcription is initiated include TFIIB, TFIID, TFIIE, TFIIF, and TFIIH (Conaway and Conaway 1993; Zawel and Reinberg 1995). In bacterial cells, members of the family of σ factors carry out most of the functions of the many eukaryotic nuclear factors (for review, see Helmann 1994). The interaction of a σ factor with the core polymerase alters the conformation of both the polymerase and the σ factor to expose amino acids critical for promoter recognition, and to allow the loading of the polymerase onto the DNA (Dombroski et al. 1993; Polyakov et al. 1995). Although much has been learned about how σ factors and other sequence-specific binding factors interact with DNA, relatively little is known about the interactions of these factors with the subunits of core polymerases and how these interactions influence conformational changes in both components of the holoenzyme. In this work, we have used the yeast mitochondrial RNA polymerase (mt RNAP) as a simple model to examine the interaction between a promoter specificity factor and a core polymerase.
The core mt RNAP is a single polypeptide encoded by the nuclear gene RPO41. Rpo41p shares nine regions of amino acid sequence similarity with the single subunit RNA polymerases of the T7 and T3 bacteriophage (Masters et al. 1987; Jang and Jaehning 1994). These regions include the amino acids known to be required for structure and function of the catalytic domain of the phage polymerases (Delarue et al. 1990; Sousa et al. 1993). However, unlike the phage polymerases that function independently, Rpo41p requires a specificity factor encoded by the nuclear MTF1 gene. Mtf1p has only limited amino acid similarity to σ factors (Jang and Jaehning 1991), but functions in many ways like σ in that it is required for promoter recognition and initiation of transcription. Although Mtf1p does not bind to its simple nine-base promoter (consensus ATATAAGTA; Osinga et al. 1982) on its own, it interacts with the core polymerase in solution to create a holoenzyme capable of promoter recognition (Mangus et al. 1994). Mtf1p is released after a short transcript has been synthesized and is available for interaction with a new core subunit, also reminiscent of σ factors (Mangus et al. 1994). The mitochondrial RNA polymerase therefore undergoes the same cycle of interactions as do the more complicated prokaryotic and eukaryotic nuclear enzymes, but requires only two polypeptides rather than the four to more than 30 used in the more complex systems.
In this work we have investigated the requirements for interaction between Mtf1p and Rpo41p. A comprehensive deletion analysis of both proteins failed to identify a simple interaction region, indicating that several regions of both proteins may be involved in the protein–protein interactions. We have demonstrated that this is the case for Mtf1p by identifying three regions required for interactions with Rpo41p. Two of these regions are similar to regions of σ-like factors shown to have a role in interactions with the prokaryotic and eukaryotic core polymerases. This analysis establishes the yeast mt RNAP as a useful model for the analysis of protein–protein interactions during the transcription cycle, and demonstrates that core polymerase/accessory factor interactions involve complex-binding surfaces on both components of the holoenzyme.
Results
The interaction between Mtf1p and Rpo41p can be detected in two-hybrid constructs
The analysis of interactions between σ-like factors and core polymerases is complicated because of the number of polypeptides in most core RNA polymerases. With the two-component mitochondrial RNA polymerase it is possible to use the powerful technique of two-hybrid analysis (Bartel et al. 1993; Phizicky and Fields 1995) to determine regions and/or specific amino acids of the Rpo41p core and the Mtf1p σ-like specificity factor necessary for protein–protein interactions. Although two-hybrid analyses of proteins that normally interact in the cytoplasm have been successful in the nuclear environment required for the technique, it was critical that we establish that the relatively weak interactions between the mtRNAP subunits (Mangus et al. 1994) could be detected in two-hybrid constructs. Initially, full-length polymerase subunits were tested for interaction in this assay. Mtf1p was fused to the LexA DNA-binding domain, whereas Rpo41p was fused to the Vp16 transcriptional activator in vectors described by Hollenberg et al. (1995; Materials and Methods). β-Galactosidase activity was undetectable with the fusion constructs on their own (except for a weak positive signal with the LexA:Mtf1p construct), or in combination with the unfused vector constructs. High levels of β-galactosidase were only observed when the LexA:Mtf1p construct was present with the VP16:Rpo41p construct (see below).
Deletions of Rpo41p and Mtf1p fail to identify a discrete interaction region
Two-hybrid analyses have been used to delineate small regions of proteins necessary and sufficient for protein–protein interactions (Bartel et al. 1993; Phizicky and Fields 1995). Therefore, we asked if deletion constructs could be used to define the region of interaction between the two proteins. As shown in Figure 1, several deletion constructs were made for both Mtf1p (Fig. 1A) and Rpo41p (Fig. 1B) in two-hybrid vector backbones. The six Mtf1p deletions included three carboxy-terminal deletions, one amino-terminal deletion, and two internal deletions. The two internal deletions removed amino acids conserved with σ regions 2.1 and 2.2. All constructs were expressed in yeast cells and produced proteins of the predicted sizes (Fig. 2A; data not shown). Furthermore, the deletion constructs were expressed at levels that were equivalent to the full-length product (Fig. 2A). However, none of the Mtf1p deletion constructs showed any interaction, as measured by β-galactosidase activity, with the core polymerase in this assay (data not shown). These results suggest that it is not a single domain but multiple regions of the folded structure of Mtf1p whose contact with Rpo41p is required to produce a stable interaction.
Figure 1.

Two-hybrid deletion constructs. (A) Mtf1p deletions were fused to the LexA DNA-binding region and tested for interaction with a full-length Rpo41p two-hybrid construct . (B) Rpo41p deletions were fused to the Vp16 activator region and tested for interaction with a full-length LexA:Mtf1p construct. Strains, vectors, and protocols are described in Materials and Methods. Numbers refer to amino acid positions retained in the constructs. Shaded boxes in the full-length maps of MTF1 and RPO41 refer to regions conserved between σ factors and Mtf1p (Jang and Jaehning 1991) or between Rpo41p and T7 RNA polymerase (Masters et al. 1987; Jaehning 1993).
Figure 2.

Confirmation of the expression of the two-hybrid constructs. Two-hybrid fusion proteins were detected in extracts of whole-cell yeast grown at 30°C. Rpo41p fusion proteins were visualized using an anti-Rpo41p antibody, whereas Mtf1p fusion proteins were detected with an anti-LexA antibody (Materials and Methods). Equal amounts of yeast cell extracts were analyzed for each sample. (A) Mtf1p carboxy-terminal deletion constructs. (B) Rpo41p deletion constructs. (C) Mtf1p point mutations that are defective for interaction with Rpo41p in the two-hybrid system.
A similar analysis was conducted with Rpo41p. Rpo41p is larger than its phage relatives and contains an amino-terminal extension of ∼300 amino acids that has no similarity to the phage polymerases (Masters et al. 1987). Because the phage polymerases do not require a specificity factor, we have speculated that the amino-terminal extension may have a role in the interaction with the specificity factor (Jaehning 1993). To determine if this sequence alone is capable of interacting with Mtf1p, we fused the amino-terminal sequence (amino acids 1–311) of Rpo41p to the Vp16 activation region. The amino-terminal fragment showed no interaction with Mtf1p by this analysis (data not shown). However, this fusion construct was not detected in vivo by Western blot analysis. Four additional Rpo41p constructs (Fig. 1B) were tested for interaction with Mtf1p. Each of the constructs accumulated in yeast cells (Fig. 2B), but none was capable of interaction with Mtf1p (data not shown). Two constructs (Rpo41p1-597 and Rpo41p1-918) contain the amino-terminal extension, but are unable to mediate interaction with Mtf1p. The carboxy-terminal polymerase region, Rpo41p311–1351, is also incapable of interacting with Mtf1p. Mtf1p may therefore interact with amino acids in both the amino-terminal extension and the polymerase domain.
Isolation of point mutations in Mtf1p that confer a petite phenotype
Because deletions in Mtf1p and Rpo41p failed to identify discrete regions sufficient for interaction, we turned to analysis of point mutations to identify specific residues required for the interaction. Although MTF1 is not essential for yeast cell growth, it is required for the stable replication and transmission of the mitochondrial genome (Lisowsky and Michaelis 1988). Yeast strains lacking functional Mtf1p rapidly lose their full-length mitochondrial DNA, producing a petite phenotype that cannot be complemented by the subsequent expression of MTF1 (Jang and Jaehning 1991). Isolation of nonfunctional MTF1 mutations therefore requires the use of the plasmid shuffle technique where a plasmid bearing a wild-type copy of the gene is used to cover a chromosomal mutation (Sikorski and Boeke 1991). A yeast strain was created for this procedure as outlined in Figure 3.
Figure 3.

Plasmid shuffle isolation of PCR-generated mtf1 mutants. Plasmids, strains, and protocols are described in Materials and Methods. A haploid strain (yJH71) containing a single functional copy of MTF1 on a plasmid was transformed with plasmids bearing mutant mtf1 genes on a LEU2 selectable plasmid. The transformants were plated onto glucose medium containing 5-FOA to select for the loss of the URA3 plasmid bearing the wild-type copy of MTF1. The mutant alleles were then tested for Mtf1p function by plating the cells on a nonfermentable carbon source (YPG) . Petite and ts petite mutants were identified.
For mutagenesis, MTF1 was amplified using low fidelity PCR conditions to create random mutations throughout the gene (Materials and Methods). The mutant fragments were transformed into the recipient strain and then grown on 5-fluoro-orotic acid (5FOA) to select for cells that had lost the wild-type MTF1 plasmid with the URA3 marker. The mtf1 mutants were tested subsequently for mitochondrial function by growth on a nonfermentable carbon source. Strains that were unable to grow on glycerol plates (petite), or unable to grow on glycerol plates at 37°C temperature-sensitive petite (ts petite) were collected for further analysis.
From 10,000 primary transformants, 22 petite and ts petite mutants were identified and sequenced to determine the mutation responsible for the petite phenotype. Six of the mutants contained nonsense codons, whereas the other 17 mutants contained either single (6), double (9), or triple (2) missense mutations (Table 1). Missense mutations were identified between codons 40 and 247. To increase the pool of single missense mutations, we reconstructed many of the mutations as single point mutations using site-directed mutagenesis. Oligonucleotides based on the sequenced mutations were designed for mutagenesis of the wild-type sequence (Materials and Methods). The isolated single mutations were retested using the plasmid shuffle to determine the effect of the single mutations in vivo. Nine of the 10 reconstructed mutations were either petite or ts petite when present as single mutations (Table 1). Although not all of the identified mutations were reconstructed, we did isolate a substantial pool of defective Mtf1p mutations. In total, 15 petite or ts petite point mutations were identified.
Table 1.
Petite and ts petite mutations identified by PCR mutagenesis of MTF1
| Mutations
|
Phenotype
|
Mutations
|
Phenotype
|
|---|---|---|---|
| Nonsense | single amino acid | ||
| K35Stop | petite | Y42C | ts petite |
| Q60Stop | petite | L53H | ts petite |
| K71Stop | petite | I154T | ts petite |
| R189Stop | petite | K157E | ts petite |
| K203Stop | petite | S218R | ts petite |
| I221K | petite | ||
| Multiple amino acid | single amino acid | ||
| H44P/V135A | petite | isolated from multiple | |
| Y54F/E114V | petite | H44P | ts petite |
| R79H/M247T | petite | Y54F | petite |
| I59V/K71E/S81N | petite | R79H | wild type |
| C192F/E233D | ts petite | S81N | petite |
| Q219R/D225G | petite | E114V | ts petite |
| S190P/L228S | petite | V135A | ts petite |
| K40R/Q60R | ts petite | C192F | ts petite |
| K67E/Y108N | ts petite | Q219R | ts petite |
| Y68H/D104G/T134S | ts petite | D225G | petite |
| L82P/E141V | ts petite | L228S | petite |
Twenty-two petite or ts petite mtf1 mutants were identified by a plasmid shuffle technique (Materials and Methods). Mutants containing nonsense, missense, and multiple missense mutations were identified. Many of the multiple missense mutations were isolated so that single mutations (shown in bold) could be analyzed separately.
Transcription of all classes of mitochondrial genes is reduced in ts mtf1 mutants
It was possible that some of the defects in Mtf1p function could be attributable to failure to recognize one or more of the promoters in the mitochondrial genome. We therefore analyzed patterns of mitochondrial transcription at the permissive (30°C) and nonpermissive (36°C) temperatures for several ts petite mtf1 mutant strains (Fig. 4; data not shown). Strains of yeast bearing the wild-type or temperature-sensitive (L53H and I154T) alleles of MTF1 were grown in minimal glucose media at the permissive temperature and then shifted to the nonpermissive temperature. Samples of yeast were collected at intervals and total RNA was isolated, fractionated, and hybridized with oligonucleotide probes specific for mitochondrial 14S rRNA, mRNAs for the COB and OLI1 genes, and the tRNAs for Glu, Ser1, Trp, Thr(ACN), f-Met, and Phe, as well as the cytoplasmic 18S rRNA as a normalization control. These genes represent 7 of the 12 known mitochondrial transcription units and include several of the different promoter sequence variations (Dieckmann and Staples 1994). Some of the hybridization analyses are shown in Figure 4A and the quantitation of these data are presented in Figure 4B.
Figure 4.

Ts mtf1 mutants are defective in all classes of mitochondrial gene transcription. Wild-type (wt) and ts petite strains bearing mtf1 mutations L53H and I154T were grown in minimal glucose medium at 30°C and shifted to 37°C. Total RNA was harvested from cells immediately before and at the indicated time intervals after the shift to the nonpermissive temperature. (A) Blots of the isolated RNAs were hybridized with labeled 14S rRNA, COB, or tRNATHR(ACN) oligonucleotide probes to analyze the abundance of mitochondrial transcripts. (B) Hybridization to the blots was quantitated by PhosphorImager analysis to directly compare levels of transcripts. The blots were hybridized to an 18S rRNA oligonucleotide probe to normalize for loading and transfer. Strains, plasmids, and protocols are described in Materials and Methods.
Three conclusions can be drawn from the results in Figure 4. First, mitochondrial gene expression is reduced significantly in the ts mutants relative to wild-type expression, even at the permissive temperature (Fig. 4A). Because the strains bearing the mutant alleles of Mtf1p grow almost as well as wild type on glycerol medium, mitochondrial function can be maintained apparently with only 10%–20% of the wild-type level of mitochondrial transcripts. Second, there is a rapid decrease in mitochondrial RNA abundance in both the wild-type and mutant strains after the shift to the nonpermissive temperature, but the mutants do not recover and RNA levels decrease to almost undetectable levels (Fig. 4B).
Finally, the abundance of all mitochondrial transcripts that we analyzed was reduced to a similar extent (Fig. 4A,B; data not shown). These mtf1 mutations therefore affect the transcription of all mitochondrial genes and are not specifically defective for recognition of a subset of mitochondrial promoters. Under the conditions used in these assays, petites do not start to accumulate in the mutant strains until about 24 hr after the shift to the nonpermissive temperature (data not shown). During this period the mutants maintained levels of mitochondrial DNA similar to those found in the wild-type strain (data not shown). Therefore, the petite phenotype caused by the mtf1 mutations is caused by a defect in transcription of mitochondrial genes, not by a rapid loss of mitochondrial DNA. This defect could be caused by failure to interact with the core polymerase, inability to recognize or bind to the mitochondrial promoter, or loss-of-function in other steps in initiation. In the following sections we have tested these mutations for the first of these possible defects.
Some of the mtf1 mutations no longer interact with Rpo41p in the two-hybrid assay
We introduced each of the point mutations into the two-hybrid LexA fusion vector (Materials and Methods). Unlike the deletion mutations described above, most (10 of 15) of the mutants retained the ability to interact with Rpo41p, as determined by qualitative production of β-galactosidase in filter assays (data not shown). However, five of the 15 mutations (L53H, V135A, I154T, S218R, and D225G; see Fig. 5) were negative for β-galactosidase production, indicating that they had lost the ability to interact with Rpo41p. The inability of these mutants to interact was not attributable to lowered or abolished expression of the fusion constructs, as all of the noninteracting mutants are expressed at levels equivalent to wild-type (Fig. 2C). Because many of the petite MTF1 mutants were isolated as temperature-sensitive mutations, we repeated the filter assays after growing cells at 37°C (data not shown). Two additional mutations (Y42C and K157E) fail to interact at the nonpermissive temperature.
Figure 5.

Identification of point mutations in Mtf1p that fail to interact with Rpo41p. β-Galactosidase activity is shown for the LexA:Mtf1p construct alone and the LexA:Mtf1p construct with the VP16:Rpo41p construct. Interaction between the Mtf1p mutants and Rpo41p was determined for cells grown at 30°C (shaded bar). Activity for ts mutants Y42C and K157E is also shown for cells grown at 37°C (hatched bar) or 23°C (crosshatched bar), respectively, to demonstrate the temperature-sensitive nature of the interaction. β-Galactosidase activity is expressed in Miller units (Miller 1972).
When the qualitative assays were followed by quantitative measurement of β-galactosidase activity, we found that many of the petite mutations associate with Rpo41p at levels that are indistinguishable from the wild-type protein (Fig. 5). However, consistent with the filter assays the noninteracting mutants L53H, V135A, I154T, S218R, and D225G produce no β-galactosidase activity above background levels. We also identified two mutants that have intermediate levels of interaction. Mutant I221K generates <50% and mutant H44P produces <25% of the β-galactosidase activity of the wild-type construct. The two temperature-sensitive mutations Y42C and K157E produce β-galactosidase at levels that are equivalent to the wild-type protein at their permissive temperatures (30°C and 23°C, respectively), but are reduced severely in interaction at their nonpermissive temperatures (36°C and 30°C, respectively). In all, nine mtf1 mutations are fully, partially, or conditionally defective for interaction with the core polymerase.
Confirmation of the two-hybrid analyses with biochemical association assays
To confirm the two-hybrid data we used a biochemical affinity assay (Mangus et al. 1994) to demonstrate that Rpo41p and Mtf1p interact in solution in the absence of DNA. For this assay the mtf1 mutants were fused to glutathione S-transferase (GST) and mutant and wild-type Mtf1p–GST fusions were bound to a glutathione agarose column (Materials and Methods). A whole-cell yeast extract containing Rpo41p was loaded onto the fusion protein columns that were washed subsequently and step-eluted to release any Rpo41p that had bound to the Mtf1p fusions on the columns (Materials and Methods). Rpo41p that was bound to and eluted from the columns was detected by an anti-Rpo41p antibody (Mangus et al. 1994).
As shown in Figure 6, the column chromatography results confirm the two-hybrid observations. As controls, the wild-type fusion and two interacting mutants, S81N and E114V (Fig. 6; data not shown) were tested for interaction. Rpo41p bound efficiently to all three fusion constructs. Consistent with the two-hybrid results, Rpo41p showed little ability to bind to the noninteracting mutants V135A and S218R (Fig. 6). Noninteracting mutants L53H, I154T, and D225G also show no Rpo41p binding in this assay (data not shown). Mutants H44P (Fig. 6) and I221K (data not shown) show intermediate levels of interaction with Rpo41p in this assay, consistent with the intermediate interaction observed in the two-hybrid assay (note β-galactosidase units in parentheses in Fig. 6). Additionally, mutant K157E, which is temperature-sensitive for interaction in the two-hybrid system, exhibited intermediate binding in this assay (data not shown).
Figure 6.

Biochemical confirmation of the noninteracting Mtf1p mutations. GST fusion constructs of wild-type and the indicated mutant Mtf1ps were isolated by glutathione agarose chromatography. Whole-cell yeast extracts containing Rpo41p were loaded onto the GST–Mtf1p columns. The columns were washed to eliminate nonspecific binding, then step-eluted to release Rpo41p bound to the column. Rpo41p in the input, wash, and elution column fractions was detected by Western blot analysis using an anti-Rpo41p antibody. Protocols are described in Materials and Methods. The percentage of two-hybrid interaction relative to wild-type Mtf1p (from Fig. 5) is shown in parentheses for each mutant.
In contrast to the results with the other conditional and partially defective mutants, the Y42C fusion protein did not interact at all with Rpo41p under the conditions of this assay (Fig. 7, top). The buffers used in all of the binding assays shown in Figure 6 contain Tris and are buffered to a pH of 7.9 at room temperature, which is pH 8.3 at the 4°C temperature used for the binding assay. Although the pH of the yeast mitochondrion has not been reported, it seemed likely that the in vivo pH experienced by the Y42C mutation in the nucleus for the two-hybrid assay or in the mitochondrion for the complementation assay could be significantly lower than pH 8.3. In addition, the change from tyrosine to cysteine alters the residue from hydrophobic to a potentially charged amino acid; the pK of cysteine is in the range of 8–9 depending on context (Walsh 1979). We therefore repeated the binding assay at pH 7.3 to determine if partial deprotonation of the cysteine residue had reduced binding to Rpo41p at the higher pH. As shown in Figure 7 (bottom), at pH 7.3 the Y42C mutant interacts with Rpo41p at levels equivalent to wild type. Furthermore, when this mutant is isolated at the nonbinding pH (pH 8.3) it still retains the ability to interact with Rpo41p when it is shifted to pH 7.3 (data not shown). Binding of the wild type and other mutant Mtf1p constructs is indistinguishable at the two different pHs (data not shown). The simplest explanation of these data is that partial negative charge on the deprotonated cysteine residue directly reduces the affinity of the mutant Mtf1p for Rpo41p. The reversible nature of the interaction is further support of the fact that these point mutations are not unfolded or unstable proteins—they have simply lost the ability to bind to Rpo41p at levels detectable in our assays or sufficient for transcription in vivo.
Figure 7.

High pH inhibits the interaction of ts petite mutant Y42C with Rpo41p. The GST–Mtf1p fusion construct bearing mutation Y42C was isolated and tested for interaction with Rpo41p as outlined in the legend to Fig. 6. The columns and the whole-cell yeast extract containing Rpo41p were equilibrated with buffers of either pH 8.3 (as shown in Fig. 6) or pH 7.3. Protocols for the detection of Rpo41p in the eluted fractions are described in Materials and Methods.
Discussion
In this work, we have confirmed further the functional homology of the mitochondrial RNA polymerase specificity factor, Mtf1p, with the large family of prokaryotic and eukaryotic nuclear σ-like factors. Despite the limited amino acid sequence similarity between many of the members of this family, there are many shared functions (for review, see Helmann and Chamberlin 1988; Helmann 1994) including roles in suppression of nonspecific interactions with DNA, selective promoter sequence recognition, promoter melting, and, as described in this work, interactions with the core RNA polymerase. The mutational strategy (PCR mutagenesis and plasmid shuffle screening) used in our studies resulted in the identification of a large number of petite and ts petite mutations useful for delineating the functional regions of Mtf1p. As shown in Figure 8, these mutations span much of the length of the MTF1 gene. The mutations that affect interactions with Rpo41p are shown above the linear map of the gene; the nine mutations fall in three discrete clusters that we have designated A, B, and C. Below the map are the six petite mutations that retain the ability to interact with Rpo41p. In several cases, the two classes of mutants are closely apposed.
Figure 8.

Position of Mtf1p mutations that affect interactions with Rpo41p. Noninteracting Mtf1p mutations cluster in three regions designated A, B, and C. Regions with amino acid sequence similarity to σ factors are shaded (Jang and Jaehning 1991).
Additional mutations in Mtf1p have been identified using a site-directed mutagenesis approach (Shadel and Clayton 1995). Although only three of the 14 mutations created in that study resulted in a petite phenotype, the position of the nonfunctional alterations confirmed that the regions of similarity with σ factor were essential for Mtf1p function. Our collection of 15 point mutations serves to establish further the importance of these conserved regions. Ten of the 15 petite mutations lie in the conserved regions identified previously (Fig. 8). The five mutations not localized to the conserved regions lie in the areas between regions 2.1/2.2 and 2.3/2.4, and between regions 2 and 3 (Fig. 8). Although we did not identify point mutations in the region carboxy-terminal to conserved region 3, there are likely to be additional essential residues in this part of the protein based on the deletion analysis reported by Shadel and Clayton (1995). They found that an additional 30 or more amino acids carboxy-terminal of region 3 were required for full function of Mtf1p.
One of our point mutations (Y42C) is in the same position identified as important by Shadel and Clayton (1995); another (L53H) is immediately adjacent to a mutation identified in that study (D52A). The fact that the adjacent ts petite mutant Y54F still interacts with the core (Fig. 5) means that we cannot predict whether the D52A mutation is defective for interaction or another step in the transcription reaction. This region of the protein therefore appears to have two distinct functions closely interdigitated in the folded structure. It is interesting that both mutations at amino acid 42 (Y42C and Y42R) result in a ts phenotype in vivo (Table 1; Shadel and Clayton 1995). Because we have shown that the Y42C mutation affects core interactions, this is probably also the defect for the Y42R mutation. In an in vitro binding assay we can correct the defect of the Y42C mutation by lowering the pH to create the uncharged form of cysteine (Fig. 7). The fact that both mutations are ts in vivo may therefore reflect alterations in pH or ionic environment in the mitochondrion that occur at elevated temperature. Under these altered conditions, a charged residue (cysteine or arginine) at position 42 does not support interactions with the core.
We found that the temperature shift dramatically decreases mitochondrial transcription even when wild-type Mtf1p is present. When Mtf1p is replaced by a ts noninteracting mutation (I154T or L53H), abundance of all classes of transcripts drops rapidly at the elevated temperature and does not recover to levels that support mitochondrial function. The fact that the reduction was similar for all the promoters we examined is consistent with the idea that the subunits of the holoenzyme could no longer interact in the altered conditions of the mitochondrion at the nonpermissive temperature.
We have not confirmed directly that RNA synthesis shuts off at the nonpermissive temperature. However, our observation that mitochondrial DNA levels do not change during the several cell generations represented by the extended time course of the experiment indicates that the decrease in transcript abundance is not attributable to a loss of template. This observation also calls into question the hypothesized role of the mtRNAP in replication of mitochondrial DNA (Clayton 1991). If, as we have shown, the mutated enzyme is nonfunctional for the synthesis of all classes of transcripts, how can it still be active for primer synthesis? This view is consistent with the work of Fangman et al. (1990), who have shown that mitochondrial DNA can be replicated in the absence of functional Rpo41p.
Comparison to the core interaction regions of σ factors: cluster A mutations/conserved region 2
Previous studies of the core interaction regions of bacterial σ factors have demonstrated clearly that amino acids in conserved regions 2.1 and 2.2 are of critical importance (Shuler et al. 1995; Tintut and Gralla 1995; Joo et al. 1997). The amino acid sequences of these regions of Escherichia coli σ70 and σ32, and Bacillus subtilis σE are shown in Figure 9A aligned with Mtf1p. Also included are a region of the human RAP30 subunit of RNA Pol II factor TFIIF shown to be protected when in a complex with the bacterial core RNA polymerase (McCracken and Greenblatt 1991), and a short region of E. coli σ54 demonstrated to be important for core interactions (Tintut and Gralla 1995).
Figure 9.
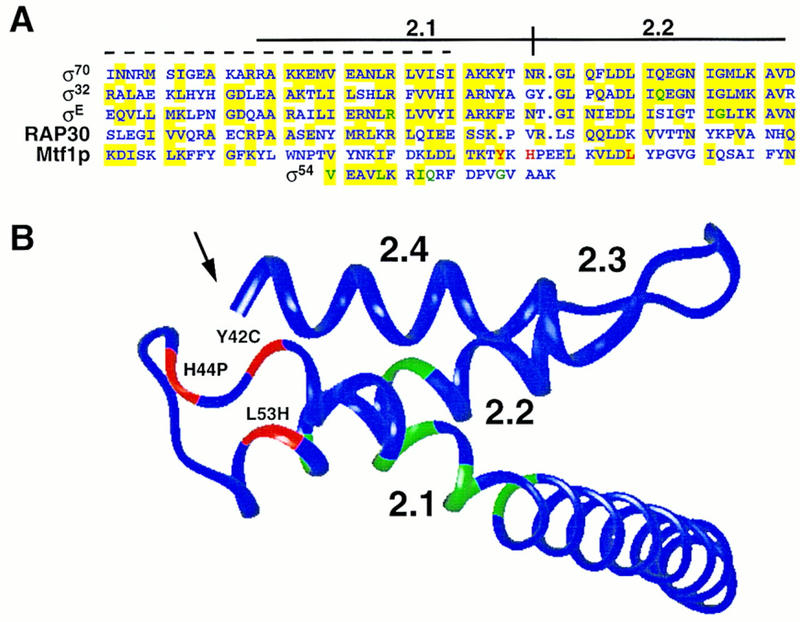
Alignment of the cluster A mutations with known core interaction regions of σ and σ-like factors, and location of this region on the structure of σ70. (A) Core-binding regions identified in σ70 [amino acids 361–417 (Lesley and Burgess 1989)], σ32 [amino acids 35–91 (Joo et al. 1997)], σE [amino acids 44–100 (Schuler et al. 1995)], Rap30 [amino acids 105–160 (McCracken and Greenblatt 1991)], Mtf1p [amino acids 9–66 (this work)] and σ54 [amino acids 175–193 (Tintut and Gralla 1995)] are shown. A 30-amino-acid peptide from σ70 with affinity for core polymerase (Lesley and Burgess 1989) is shown by a broken line above the σ70 sequence. The position of mutations that reduce interaction with core polymerase are shown in green for σE, σ32, and σ54, and in red for Mtf1p. Colored shading is used to indicate similarity between the amino acid sequences. (B) The locations of the noninteracting mutations shown in A are highlighted on the structure of region 2 of σ70 as determined by Malhotra et al. (1996) using InsightII (Biosym, San Diego, CA). The Mtf1p noninteracting cluster A mutations are shown in red. Positions required for interaction in σE, σ32, and σ54 are shown in green. Note that the carboxyl terminus of region 2.4 (marked with an arrow) is brought close to the position of the noninteracting mutations in regions 2.1 and 2.2 in the three-dimensional structure.
Although no point mutations of σ70 have been reported that abolish core interactions, Lesley and Burgess (1989) identified several deletions that decreased core binding. A deletion that encompassed region 2.1 had the most deleterious effect, and a synthetic peptide spanning this region (indicated by the broken line in Fig. 9A) was found to bind to core polymerase (Lesley and Burgess 1989). However, this peptide also bound with similar affinities to the holoenzyme form of E. coli RNA polymerase and to σ70 (Lesley and Burgess 1989). Severinova et al. (1996) have extended these observations using a tryptic fragment of σ70. They found that a fragment encompassing part of region 1 and most of conserved region 2 (amino acids 114–448) bound to the core polymerase. Binding by this fragment is specific, as full-length σ70 was able to compete with the fragment for core binding. However, the fragment binds core with an affinity 30-fold lower than full-length σ70, suggesting that other regions also make important contributions to core binding.
Recent work has focused on regions 2.1 and 2.2 of σ factors to identify individual residues critical for core interactions. Shuler et al. (1995) screened site-directed mutations in regions 2.1 and 2.2 of B. subtilis σE and identified two point mutations (highlighted in Fig. 9A) that interfered with core interactions. Tintut and Gralla (1995) noted that although σ54 and σ70 share very little amino acid sequence similarity, a short motif can be found in both proteins. Mutagenesis of this region of σ54 resulted in the identification of the residues highlighted in Figure 9A as critical for core interactions (Tintut and Gralla 1995). Joo et al. (1997) found that a mutation in region 2.2 in σ32 reduces affinity for the core polymerase. Note that the mutations identified in these screens (shown in green) flank the highlighted mutations in cluster A of Mtf1p (shown in red), and in one case, identify the same position in the alignment. Although some of the noninteracting mutations do overlap the original synthetic peptide of Lesley and Burgess (1989), it is clear from this comparison that core interaction requires many amino acids in regions 2.1 and 2.2 in addition to those included in the peptide. This conclusion is consistent with predictions made by Gribskov and Burgess (1986) and Helmann and Chamberlin (1988) based on the high level of amino acid sequence conservation in these regions of the bacterial σ factors.
The bacterial σ factors and the eukaryotic nuclear factor RAP30 (McCracken and Greenblatt 1991) all form complexes with the bacterial core polymerase. As shown in Figure 9A, the amino acid sequence of this region of the different proteins has not been highly conserved. It is possible, however, that structural elements of the interaction region are similar. We have used the recently reported structure of a portion of σ70 (Malhotra et al. 1996) to model the position of the cluster A noninteracting mutations from Mtf1p as well as the region 2.1 and 2.2 mutations of σ32, σE, and σ54. As shown in Figure 9B, the highlighted positions define a structural domain including the helical region of 2.1 and a turn or bend connecting this element to helical region 2.2.
It is of course difficult to predict the structure of these other σ-like factors accurately, especially as there are some single amino acid insertions and deletions in the region to be modeled (Fig. 9A). It is however of interest to note the location of the tyrosine residue at an accessible position near the beginning of the turn. We have shown that in Mtf1p, the ionization state of this residue in the Y42C mutation is critical for interaction. Most σ factors have a bulky hydrophobic residue in this position (Lonetto et al. 1992). The fact that σ54 and RAP30 do not share this residue (Fig. 9A) indicates that the determinants for binding are more complex than simple interactions between single amino acids.
Cluster B mutations
The reported structure of σ70 only extends to the carboxyl terminus of region 2 (Malhotra et al. 1996), so the additional noninteracting mutations in clusters B and C (Fig. 8) cannot be modeled relative to the cluster A mutations in region 2. However, as indicated by the arrow in Figure 9B, the folded structure does bring the carboxyl terminus of the region 2.4 helix into close juxtaposition with the region 2.1/2.2 interaction domain. This means that the noninteracting mutations that we identified in cluster B are probably very close to those in cluster A in the folded structure. Although there is no obvious amino acid sequence similarity between this region of Mtf1p and the σ factors (Jang and Jaehning 1991), common structures may exist. The amino acid differences may be important for the selective interactions with different types of core polymerases. Because none of the original deletion mutations tested by Lesley and Burgess (1989) selectively removed this part of the protein between regions 2 and 3, this question has not yet been addressed.
Cluster C mutations/conserved region 3
The third cluster of mutations identified in our screen lie in conserved region 3. All of these mutations (S218R, I221K, and D225G) are interposed very closely with petite mutations that retain the ability to interact with the core (Q219R and L228S). The spacing between these mutations indicates that they may define two important surfaces (potentially faces of a helical or sheet structure), one of which is critical for core interactions and the other required for another essential function. There is precedent for amino acids in region 3 also having a role in core interactions. Lesley and Burgess (1989) described one deletion that removed region 3 and reduced core interactions by a factor of 5 in vitro. In addition, Zhou et al. (1992) reported that a small deletion in σ32 reduced significantly the affinity for the core polymerase. Combined with the fact that we were unable to define deletions of either Rpo41p or Mtf1p that retained the ability to interact in vivo, all of these observations strongly support a model of a complex interaction surface created by distant regions in the amino acid sequence brought together in the folded structure. Because our mutagenesis was not exhaustive, it is possible that even more than the three regions that we have identified are important for interactions.
Interaction regions in the core polymerase
There is currently no information on the particular residues or regions of any core polymerase required for factor interactions. σ70 has been shown to make contacts with all of the subunits of the core [β, β′, and α (Coggins et al. 1977; McMahan and Burgess 1994; Greiner et al. 1996)]. It is probable that the eukaryotic nuclear σ-like factors also make contacts with more than one subunit of the core. The RAP30 and RAP74 subunits of TFIIF each contain core interaction regions in support of this idea (McCracken and Greenblatt 1991; Wang and Burton 1995). Although the analysis of interaction elements of the single subunit Rpo41p core should be simpler than for the multisubunit enzymes, our analysis of deletion mutations in Rpo41p (Fig. 2) support the idea that these interaction elements will encompass several widely spaced regions in the RPO41 gene. It will be interesting to ultimately determine if these elements share any amino acid sequence or structural similarity with elements in the multisubunit prokaryotic and eukaryotic core polymerases. These studies could help to elucidate the origins of this unusual RNA polymerase.
Although there are as yet no identified homologs of the yeast MTF1 gene, it appears that most eukaryotes do possess an Rpo41p-type mitochondrial core polymerase (Cermakian et al. 1996; Chen et al. 1996; Tiranti et al. 1997). The analysis of the interaction regions in the single polypeptide enzymes could therefore eventually explain the alterations that caused the phage-related proteins to lose the ability to recognize a promoter on their own and to substitute a required accessory factor. The conservation of structures and functions among the RNA polymerases of phage, bacteria, and the eukaryotic nucleus and mitochondrion will allow observations of the relatively simple mitochondrial RNA polymerase to guide further experiments with the multisubunit as well as the single polypeptide enzymes.
Materials and methods
Media and genetic methods
Standard media such as YP medium containing 2% of either glucose (YPD) or glycerol (YPG), synthetic complete medium (SC) lacking the appropriate amino acids, and sporulation medium were prepared as described by Guthrie and Fink (1991). 5-FOA medium was prepared by adding 5-FOA to synthetic medium at a concentration of 500 mg/liter (Sikorski and Boeke 1991). Yeast cells were transformed using the lithium acetate method (Ito et al. 1983). Mating, sporulation, and dissection were carried out by standard methods (Guthrie and Fink 1991).
Two-hybrid plasmid constructs and assays
MTF1 was cloned as a 1.0-kb EcoRI fragment from pJJ525 (Mangus et al. 1994) into the corresponding site of pBTM116 (Bartel et al. 1993) to create plasmid pJJ832. Insert orientation was confirmed by restriction enzyme analysis. For mutant mtf1 constructions, pJJ832 was digested with NsiI and religated to produce an internal deletion of 101 bp. The resulting plasmid was digested with either AflII and MscI or AflII and PstI. The fragments were treated with shrimp alkaline phosphatase. mtf1 mutations created by site-directed mutagenesis in pBluescript SK(+) were digested with AflII and PstI (vector sequence) and ligated to pJJ832. mtf1 mutants isolated as single point mutations (I221K, L53H, I154T, and S218R) were digested with AflII and MscI, and ligated to the corresponding digest of pJJ832. Resulting plasmids were tested by restriction analysis to confirm the replacement of the mtf1 deletion with the point mutation constructs. Mutants Y42C and K157E were first cloned into pBluescript SK(+), then the AflII to PstI fragments were cloned into pJJ832.
Two-hybrid deletion constructs
For carboxy-terminal deletions, plasmid pBTM116 was digested with EcoRI and SmaI. The vector was dephosphorylated with shrimp alkaline phosphatase (U.S. Biochemical), then separated by electrophoresis on an agarose gel and extracted from the gel using Qiaex beads (Qiagen). The vector was ligated to the following MTF1 fragments of pJJ525. For the Mtf1p1–224 construct, the EcoRI to BamHI fragment of MTF1 from pJJ525 was used as the insert. The EcoRI to BglII fragment of this same vector was used for the Mtf1p1–296 construct. And finally, the EcoRI to XmnI fragment was used to clone the Mtf1p1–315 construct. Inserts were prepared as follows. pJJ525 was digested with either BamHI, BglII, or XmnI. Blunt ends were then created by filling in the overhang with Klenow (New England Biolabs) followed by digestion with EcoRI. Fragments were separated by electrophoresis through 0.8% agarose gels and purified using Qiaex beads (Qiagen). For the amino-terminal deletion (Mtf1p52–341) a ScaI to PstI (partial ScaI digest) fragment from pJJ525 was inserted into the SmaI and PstI sites of pBTM116. Vector and insert were prepared as described above.
Rpo41p two-hybrid constructs were prepared using RPO41 fragments generated from the following PCR primers. Restriction sites in the primers are underscored: NOTI-5′, 5′-CAGGCC- TGCGCGGCCGCAGATGCTGAGACCGGCCTATAAATC-3′; NOTI-3′, 5′-GCGGATCCGGGCGGCCGCGAAAAAATATTGACTGTTTCTCAATAC-3′; 5′-318, 5′-GCCGCCGCGGATCCACGGTTCAACAGAAGTCTTG-3′; BAM-HI-5′, 5′-CGCGGGATCCTGATGCTGAGACCGGCCTATAAATCGC-3′; 3′-311, 5′-CGAAGAGGATCCAAGGAAGG-3′; 3′-597, 5′-GCGCGGATCCTAATACTCTGAGGC-3′.
The PCR fragments were cloned into TA cloning vectors (pGEMT, Promega; pCRII, Invitrogen) and then digested with the appropriate enzymes for cloning. Insert fragments as well as vector fragments were separated by electrophoresis and purified as described above. The Rpo41p1–1351 insert was prepared by the amplification of an RPO41 plasmid using the NOTI-5′ and NOTI-3′ primers. The insert was created by a NotI digest and cloned into the corresponding site of pVP16 (Hollenberg et al. 1995). The Rpo41p1–311 insert was prepared using the BAMHI-5′ and 3′-311 primers. The insert was created by a BamHI digestion and ligated into the BamHI site of pVP16. Primers 5′-318 and NOTI-3′ were used to amplify the Rpo41p318–1351 insert. The insert was cloned into the BamHI and NotI sites of pVP16 after digestion with the same enzymes. The Rpo41p1–918 insert was created by digesting the Rpo41p1–1351 clone with NotI and MscI. The fragment was ligated to pVP16 that had been digested with EcoRI, filled in with Klenow to make blunt ends, then digested with NotI. The Rpo41p318–597 insert was created with the 5′-318 and 3′-597 primers. The insert was cloned into the BamHI site of pVP16. The Rpo41p1–597 fragment was amplified with the BAMHI-5′ and 3′-597 primers. The insert was created by partial digestion with BamHI (there is a BamHI site in the Rpo41p sequence) and ligated to the corresponding site of pVP16. Two-hybrid constructs containing LexA and VP16 fusions were transformed into yeast strains AMR70 and L40, respectively (Hollenberg et al. 1995). To test the constructs for interactions, the haploid strains were mated and diploids were selected on Ura−, Trp−, Leu− medium. The strains were tested initially for β-galactosidase production by filter lift assays (Breeden and Nasmyth 1985), and for growth on plates lacking histidine and containing 5 mm 3-aminotriazole (Phizicky and Fields 1995). For quantitative assays, strains were grown to mid-log phase in selective medium and β-galactosidase activity was assayed in permeabilized cells (Miller 1972).
Plasmid and strain construction for the MTF1 plasmid shuffle
Plasmids pJH118, pJH121, and pJH124 were constructed by inserting a 1.5-kb EcoRI fragment containing the promoter and entire coding sequence of MTF1 (Mangus et al. 1994) into the EcoRI site of pUC18, pUC7, and pBLUESCRIPT SK(+). Plasmids pJH119 and pJH142 were made by cloning this EcoRI fragment into the URA3+ vector YCplac33, and LEU2+ vector YCplac111 (Gietz and Sugino 1988), respectively. To construct plasmid pJH133, a 3.8-kb BglII–BamHI fragment bearing a 3.8-kb hisG–URA3–hisG cassette was isolated from plasmid pNKY51 (Alani et al. 1987), and was inserted into the BglII site of plasmid pJH121, disrupting the MTF1-coding sequence.
The isogenic diploid yJH60 was made by crossing yJH58 (MATa his4Δ309 ura3-52 ino1-13 leu2-3,112) and yJH59 (MATα his4Δ309 ura3-52 ino1-13 leu2-3,112). The heterozygous MTF1/mtf1::hisG–URA3–hisG strain yJH61 was made from yJH60 by one step gene replacement (Rothstein 1983) of the 5.3-kb EcoRI fragment from pJH133. Strain yJH64 was made by selecting for loss of the URA3 gene from the heterozygous MTF1/mtf1 yJH61 on 5-FOA medium. To construct recipient strain yJH71, strain yJH64 was transformed with pJH119 containing a functional MTF1 gene and the resulting transformants were sporulated to generate mtf1:hisG (pJH119) haploid progeny whose genotype was tested on both YPG and 5-FOA media. All strain constructions were confirmed using a 1-kb EcoRI fragment of pJH117 (pJJ517) as an MTF1 probe.
PCR-based mutagenesis and isolation of mtf1 mutants
Mutagenic PCR was performed essentially as described previously (Leung et al. 1989) to generate in vitro random mutations of MTF1. Plasmid pJH118 containing a 1.5-kb EcoRI fragment of MTF1 was used as a DNA template. A 17-mer of universal sequencing primer and a 23-mer oligonucleotide, 5′-CACAGGAAACAGCTATGACCATG-3′, encompassing the reverse sequencing primer were used as primers. The PCR reactions contained 30 mm Tricine at pH 8.4, 10 ng of pJH118, 0.2 mm dNTPs, 7 mm MgCl2, 0.5 mm MnCl2, 20 pmoles of each primer, 5 mm β-mercaptoethanol, 0.01% gelatin, 1 unit of Taq polymerase (the gift of N. Pace, Indiana University). Reactions were done in 100 μl and preheated to 92°C for an initial denaturation step followed by 30 cycles. The PCR-amplified fragment was purified by electrophoresis on a 0.7% agarose gel and used for the introduction of mutations into yeast with the plasmid pJH142 gapped with BamHI. The gap–duplex recombinant mutagenesis was performed as described by Muhlrad et al. (1992). Yeast strain yJH71 was transformed to Leu+ with 500 ng of PCR-amplified fragment and 100 ng of gapped pJH142. Approximately 10,000 individual Leu+ transformants were plated on 5-FOA plates to select those that lost pJH119 containing the functional MTF1 gene. 5-FOA resistant Leu+ transformants were screened for loss of mitochondrial function caused by mtf1 mutations in plasmid pJH142 by plating them on both YPD and YPG media. The replica plates were subsequently incubated at 30°C and 37°C for 3–6 days. Strains that did not grow on YPG medium at 30°C were isolated as mtf1 mutants, and those that grew on YPG medium at 30°C but not 37°C were also isolated and retested for the conditional phenotype. Mutated plasmids were recovered, passaged through E. coli, and sequenced by the Sanger dideoxy method (Sanger et al. 1977). To ensure that the mtf1 mutant alleles were responsible for the observed phenotypes, the recovered plasmids were transformed into yJH71 followed by 5-FOA selection. When the recovered alleles contained multiple point mutations, oligonucleotide-directed mutagenesis reactions were performed to isolate the corresponding single point mutations. The mutagenic oligonucleotides were designed so that each alteration in coding sequence was accompanied by a silent mutation altering a restriction site: H44P, 5′-ATAAACCTCCAGAAGAATTGAAGGTACT-3′, delete ScaI; V135A, 5′-TTTCTAACAGCTGCTAATGT-3′, add PvuII; L228S, 5′-ATGGGACCCCATTTCATTTAG-3′, delete BamHI; S81N, 5′-GAAAAACGATCGAATCTCTACAA-3′, add PvuI; E114V, 5′-ATCTAATCGATGTAGAGCGA-3′, add ClaI; Y54F, 5′-GAAAGTGCTTGACCTCTTCCCTGG-3′, delete ScaI; D225G, 5′-AAGAATGGGGTCCCATTTT-3′, delete BamHI; C192F, 5′-CTAGATCTAAATTTTCAGTA-3′, add BglII; Q219R, 5′-GATAGTCGATGTATAGAAG-3′, delete NsiI; R79H, 5′-AGAAAAACACTCGAGTCTCT-3′, add XhoI; D2.1, 5′-CAATAAAATCTTTAAACATCCAGAAGAA-3′, delete 34–42, add DraI; D2.2, 5′-CCAGAAGAATTGAAGATCTTTTATAATAAA-3′, delete 50–62, add BglII.
Uracil-containing single-stranded DNA was produced from plasmid pJH124 in E. coli strain CJ236 [dut ung (Kunkel et al. 1987)]. Each mutagenic oligonucleotide was phosphorylated with T4 polynucleotide kinase and used separately in in vitro mutagenesis reactions. The resulting mutated DNAs were used to transform E. coli strain NM522 [dut ung (Kunkel et al. 1987)] to select for the mutated plasmids. Each mtf1 mutation was confirmed by sequencing and subcloned into the EcoRI site of YCplac111 to give individual constructs and retested in yJH71 as noted above.
RNA isolation and analysis
Yeast were grown to mid-log phase, collected by centrifugation and frozen as pellets in liquid nitrogen. Yeast total RNA was isolated and purified as described by Elder et al. (1983). Yeast total RNA (10–20 μg) was electrophoresed through agarose gels containing formaldehyde and blotted by capillary action to Zetaprobe nylon membranes (BioRad). Oligonucleotide probes specific for mitochondrial 14S, COB, and tRNAThr and cytoplasmic 18S RNAs (Ulery et al. 1994) were end-labeled with [γ-32P]ATP (Amersham) and hybridized at 40°C in 0.5 m sodium phosphate buffer (pH 7.5) and 7% SDS. An oligonucleotide specific for 18S rRNA was used to normalize for differences in RNA samples on the blot. Signal intensity was quantitated with a PhosphorImager (Molecular Dynamics).
Western blots
Yeast cell extracts were prepared by growing cells to an OD595 of 0.6. Cells (10 ml) were harvested and resuspended in 1 ml of ice-cold Z buffer (60 mm Na2HPO4, 40 mm NaHPO4, 10 mm KCl, 1 mm MgSO4, and 50 mm β-mercaptoethanol at pH 7.0). Cells were pelleted in a microfuge and resuspended in 200 μl of ice-cold Z buffer. Glass beads (300 μl) were added and the cells were vortexed for 5 min at 4°C. The extract was cleared by a 5-min centrifugation (maximum speed) and the supernatant was collected. Protein concentrations were determined by the Bradford method (Bradford 1976) using BSA as a standard. Total protein (50–100 μg) were separated by SDS-PAGE and blotted to Immobilon-P (Millipore). Rpo41p was detected by polyclonal antibodies as described previously (Mangus et al. 1994). Two-hybrid Mtf1p constructs were detected using anti-LexA antibody generously provided by Dr. Roger Brent (Harvard University, Cambridge, MA). Hybridization conditions were as described by Harlow and Lane (1988). Detection was by chemiluminescence using ECL kits from Amersham Corp.
GST–fusion plasmid constructs and GST–Mtf1p affinity chromatography
The internal AflII–BglII fragment from the mtf1 mutants was used to replace the wild-type sequence of MTF1 in pGEX-1 [plasmid pJJ526 (Mangus et al. 1994)]. pJJ526 was digested with NsiI and religated to produce a deletion of 101 bp. The resulting plasmid was digested with AflII and BglII followed by dephosphorylation of ends by shrimp alkaline phosphatase (U.S. Biochemical). The vector was isolated by gel electrophoresis and ligated to AflII–BglII fragments from the MTF1 mutants. The resulting plasmids were tested by restriction enzyme analysis to confirm insertion of the full-length mutant fragments. Interaction studies with GST–Mtf1p constructs were performed as described previously (Mangus et al. 1994) with the following adjustments. Proteins were eluted with a 5-column volume step of T(500) buffer. The pH of the T(50) and T(500) solutions was 7.9 at 25°C and 8.3 at 4°C. When chromatography was performed at pH 7.3, MOPS buffer was used instead of Tris-HCl in the solutions.
Acknowledgments
We thank Margaret Short and Anne Whalen for help with the two-hybrid constructs, Roger Brent for anti-LexA antibody, Mike Woontner for thoughtful discussions and comments on the manuscript, Tom Blumenthal for comments on the manuscript, Seth Darst for the σ70 coordinates, and Paul Hagerman and Elsi Vacano for help with computer modeling. This work was supported by grants from the National Institutes of Health (GM 36692 awarded to J.A.J., and P30 CA46934 to the University of Colorado Cancer Center DNA Sequencing Core Facility); S.-H.J. was supported by a grant from the Genetic Engineering Research Fund (1995) of the Korean Ministry of Education.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jaehning_j@defiance.UCHSC.edu; FAX (303) 315-3326.
References
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strain. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel PL, Chien C, Sternglanz R, Fields S. Using the two-hybrid system to detect protein-protein interactions. In: Hartley DA, editor. Cellular interactions in development: A practical approach. Oxford, UK: IRL Press; 1993. pp. 153–179. [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breeden L, Nasmyth K. Regulation of the yeast HO gene. Cold Spring Harb Symp Quant Biol. 1985;50:643–650. doi: 10.1101/sqb.1985.050.01.078. [DOI] [PubMed] [Google Scholar]
- Cermakian N, Ikeda TM, Cedergren R, Gray MW. Sequences homologous to yeast mitochondrial and bacteriophage T3 and T7 RNA polymerases are widespread throughout the eukaryotic lineage. Nucleic Acids Res. 1996;24:648–654. doi: 10.1093/nar/24.4.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Kubelik AR, Mohr S, Breitenberger CA. Cloning and characterization of the Neurospora crassa cyt-5 gene. J Biol Chem. 1996;271:6537–6544. [PubMed] [Google Scholar]
- Coggins JR, Lumsden J, Malcom AD. A study of the quaternary structure of E. coli RNA polymerase using bis(imido esters) Biochemistry. 1977;16:1111–1116. doi: 10.1021/bi00625a013. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. General initiation factors for RNA polymerase II. Annu Rev Biochem. 1993;62:161–190. doi: 10.1146/annurev.bi.62.070193.001113. [DOI] [PubMed] [Google Scholar]
- Clayton DA. Nuclear gadgets in mitochondrial DNA replication and transcription. Trends Biol Sci. 1991;16:107–111. doi: 10.1016/0968-0004(91)90043-u. [DOI] [PubMed] [Google Scholar]
- Delarue M, Poch O, Tordo N, Moras D, Argos P. An attempt to unify the structure of polymerases. Protein Eng. 1990;6:461–467. doi: 10.1093/protein/3.6.461. [DOI] [PubMed] [Google Scholar]
- Dieckmann CL, Staples RR. Regulation of mitochondrial gene expression in Saccharomyces cerevisiae. Int Rev Cytology. 1994;152:145–181. doi: 10.1016/s0074-7696(08)62556-5. [DOI] [PubMed] [Google Scholar]
- Dombroski A, Walter W, Gross C. Amino-terminal amino acids modulate σ-factor DNA-binding activity. Genes & Dev. 1993;7:2446–2455. doi: 10.1101/gad.7.12a.2446. [DOI] [PubMed] [Google Scholar]
- Elder RT, Loh EY, Davis RW. RNA from the yeast transposable element Ty1 has both ends in the direct repeats, a structure similar to retrovirus RNA. Proc Natl Acad Sci. 1983;80:2432–2436. doi: 10.1073/pnas.80.9.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fangman WL, Henly JW, Brewer BJ. RPO41-Independent maintenance of (rho−) mitochondrial DNA in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:10–15. doi: 10.1128/mcb.10.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Greiner DP, Hughes KA, Gunasekera AH, Meares CF. Binding of the sigma 70 protein to the core subunits of E. coli RNA polymerase, studied by iron-EDTA protein footprinting. Proc Natl Acad Sci. 1996;93:71–75. doi: 10.1073/pnas.93.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov M, Burgess RR. Sigma factors from E. coli, B. subtilis, phage SP01, and T4 are homologous proteins. Nucleic Acids Res. 1986;14:6745–6763. doi: 10.1093/nar/14.16.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:1–37. [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Helmann JD. Bacterial sigma factors. In: Conaway RC, Conaway JW, editors. Transcription: Mechanisms and regulation. New York, NY: Raven Press; 1994. pp. 1–17. [Google Scholar]
- Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H. Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol Cell Biol. 1995;15:3813–3822. doi: 10.1128/mcb.15.7.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkalai cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehning JA. Mitochondrial transcription: Is a pattern emerging? Mol Microbiol. 1993;8:1–4. doi: 10.1111/j.1365-2958.1993.tb01197.x. [DOI] [PubMed] [Google Scholar]
- Jang SH, Jaehning JA. The yeast mitochondrial RNA polymerase specificity factor, MTF1, is similar to bacterial sigma factors. J Biol Chem. 1991;266:22671–22677. [PubMed] [Google Scholar]
- ————— . Mechanisms of mitochondrial transcription. In: Conaway RC, Conaway JW, editors. Transcription: Mechanisms and regulation. New York, NY: Raven Press; 1994. pp. 171–184. [Google Scholar]
- Joo DM, Ng N, Calendar R. A σ32 mutant with a single amino acid change in the highly conserved region 2.2 exhibits reduced core RNA polymerase affinity. Proc Natl Acad Sci. 1997;94:4907–4912. doi: 10.1073/pnas.94.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Lesley SA, Burgess RR. Characterization of the Escherichia coli transcription factor sigma 70: Localization of a region involved in the interaction with core RNA polymerase. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique. 1989;1:11–15. [Google Scholar]
- Lisowsky T, Michaelis G. A nuclear gene essential for mitochondrial replication suppresses a defect of mitochondrial transcription in Saccharomyces cerevisiae. Mol & Gen Genet. 1988;214:218–223. doi: 10.1007/BF00337714. [DOI] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The sigma 70 family: Conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Severinova E, Darst SA. Crystal structure of a sigma 70 fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- Mangus DA, Jang S-H, Jaehning JA. Release of the yeast mitochondrial RNA polymerase specificity factor from transcription complexes. J Biol Chem. 1994;269:26568–26574. [PubMed] [Google Scholar]
- Masters BS, Stohl LL, Clayton DA. Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell. 1987;51:89–99. doi: 10.1016/0092-8674(87)90013-4. [DOI] [PubMed] [Google Scholar]
- McCracken S, Greenblatt J. Related RNA polymerase-binding regions in human Rap30/74 and Escherichia coli sigma 70. Science. 1991;253:900–902. doi: 10.1126/science.1652156. [DOI] [PubMed] [Google Scholar]
- McMahan SA, Burgess RR. Use of aryl azide cross-linkers to investigate protein-protein interactions: An optimization of important conditions as applied to E. coli RNA polymerase and localization of a sigma 70-alpha cross-link to the C-terminal region of alpha. Biochemistry. 1994;33:12092–12099. doi: 10.1021/bi00206a012. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Osinga KA, Haan MD, Christianson T, Tabak HF. A nonanucleotide sequence involved in promotion of ribosomal RNA synthesis and RNA priming of DNA replication in yeast mitochondria. Nucleic Acids Res. 1982;10:7993–8006. doi: 10.1093/nar/10.24.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions: Methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyakov A, Severinova E, Darst SA. Three-Dimensional structure of E. coli core RNA polymerase: Promoter binding and elongation conformations of the enzyme. Cell. 1995;83:365–373. doi: 10.1016/0092-8674(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Rothstein RJ. One step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;12:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinova E, Severinova K, Fenyo D, Marr M, Brody EN, Roberts JW, Chait BT, Darst SA. Domain organization of the Escherichia coli RNA polymerase sigma 70 subunit. J Mol Biol. 1996;263:637–647. doi: 10.1006/jmbi.1996.0604. [DOI] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. A Saccharomyces cerevisiae mitochondrial transcription factor sc-mtTFB, shares features with sigma factors but is functionally distinct. Mol Cell Biol. 1995;15:2101–2108. doi: 10.1128/mcb.15.4.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuler MF, Tatti KM, Wade KH, Moran Jr CP. A single amino acid substitution in sigma E affects its ability to bind core RNA polymerase. J Bacteriol. 1995;177:3687–3694. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Boeke JD. In vitro mutagenesis and plasmid shuffling: From cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- Sousa R, Chung Y, Rose J, Wang B. Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature. 1993;364:593–599. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- Tintut Y, Gralla JD. PCR mutagenesis identifies a polymerase-binding sequence of sigma 54 that includes a sigma 70 homology region. J Bacteriol. 1995;177:5818–5825. doi: 10.1128/jb.177.20.5818-5825.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Savoia A, Forti F, D’Apolito M-F, Centra M, Rocchi M, Zeviani M. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the expressed sequence tags database. Hum Mol Genet. 1997;6:615–625. doi: 10.1093/hmg/6.4.615. [DOI] [PubMed] [Google Scholar]
- Ulery TL, Jang S-H, Jaehning JA. Glucose repression of yeast mitochondrial transcription: Kinetics of derepression and role of nuclear genes. Mol Cell Biol. 1994;14:1160–1170. doi: 10.1128/mcb.14.2.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. Enzymatic reaction mechanisms. San Francisco, CA: W.H. Freeman and Co.; 1979. [Google Scholar]
- Wang BQ, Burton ZF. Functional domains of human RAP74 including a masked polymerase binding domain. J Biol Chem. 1995;270:27035–27044. doi: 10.1074/jbc.270.45.27035. [DOI] [PubMed] [Google Scholar]
- Zawel L, Kumar KP, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes & Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- Zhou YN, Walter WA, Gross CA. A mutant sigma 32 with a small deletion in conserved region 3 of sigma has reduced affinity for core RNA polymerase. J Bacteriol. 1992;174:5005–5012. doi: 10.1128/jb.174.15.5005-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]