

Abstract
Prophylactic pharmacological activation of astrocytic gene expression driven by the transcription factor Nrf2 boosts antioxidant defences and protects against neuronal loss in ischemia and other disease models. However, the role of Nrf2 in mediating endogenous neuroprotective responses is less clear. We recently showed that Nrf2 is activated by mild oxidative stress in both rodent and human astrocytes. Moreover, brief exposure to ischemic conditions was found to activate Nrf2 both in vivo and in vitro, and this was found to contribute to neuroprotective ischemic preconditioning. Here we show that transient ischemic conditions in vitro and in vivo cause an increase in the expression of Nrf2 target genes associated with the glutathione pathway, including those involved in glutathione biosynthesis and cystine uptake. Taken together, these studies indicate that astrocytic Nrf2 may represent an important mediator of endogenous neuroprotective preconditioning pathways.
1. Introduction
1.1. Nrf2 Is a Master Regulator of Antioxidant Gene Expression
Many acute and chronic neurological disorders are associated with oxidative stress, caused by an imbalance in the production and detoxification of reactive oxygen species (ROS). Nuclear factor-erythroid 2-related factor 2 (Nrf2), a member of the cap“n”collar transcription factor family, is a master regulator of antioxidant defense genes and drug-metabolizing enzymes [1, 2]. The binding of Nrf2 to a cisacting DNA promoter sequence, called the antioxidant response element (ARE), allows transactivation of a group of cytoprotective genes [1, 2]. Under normal conditions Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1), and through a two-site interaction, the transcription factor is ubiquitinated by Cul3/Rbx1 and targeted for degradation [3]. However, under oxidative stress conditions the two-site interaction between Nrf2 and Keap1 is disrupted, allowing Nrf2 to evade Keap1-mediated ubiquitination and accumulate in the nucleus where it activates genes with an ARE sequence within their promoters, leading to an induction of antioxidant machinery [2]. Upregulation of the ARE-gene battery has a significant impact on the ability of the cell to withstand and survive sustained oxidative insults. Prophylactic Nrf2 activation by small molecules is protective against a host of oxidative insults in vitro, including free radical donors and oxygen glucose deprivation (OGD), as well as toxic levels of glutamate or N-methyl-D-aspartate (NMDA, [4–6]). Nrf2 activation in vivo is similarly protective, reducing neurotoxin or stroke-induced injury [5, 7, 8].
1.2. Nrf2-Mediated Neuroprotection in the CNS
Nrf2 has an integral role in mediating antioxidant responses within the CNS. Nrf2-dependent gene expression in astrocytes can protect neurons against a variety of trauma and disease-causing agents [9–14]. In these studies, however, Nrf2-dependent gene expression was achieved artificially, either via astrocyte-specific overexpression of Nrf2, or through treatment with well-characterized small molecule activators of the pathway. The latter is in essence a form of chemoprevention, whereby prophylactic administration of small molecule Nrf2 activators confers significant neuroprotection [4, 5, 7, 15]. Indeed, fibroblasts are also rendered resistant to many electrophiles, peroxides, and redox-cycling agents, in an Nrf2-dependent fashion, by pretreatment or “priming,” with nontoxic doses of chemopreventive agents such as sulforaphane [16]. Prophylactic administration of Nrf2 activators can be considered an artificial pharmacological form of preconditioning, whereby exogenous activation of the antioxidant response renders the cell better able to defend itself from a subsequent insult. However, little was known as to what endogenous signals might trigger an Nrf2-dependent, physiologically relevant, endogenous, neuroprotective response.
2. Results and Discussion
2.1. Ischemia and Oxidative Stress Activate Nrf2 in Astrocytes
In nonneural cells, oxidative stress is known to activate Nrf2 through inhibition of Keap1-dependent Nrf2 degradation. This is effected at least in part via the modification of key cysteine residues in Keap1 and may result in the formation of disulfide bonds [17] and possibly conformational changes in the adaptor protein [2]. To investigate whether oxidative stress activates Nrf2 in neural cells, we chose two experimental models which recreate this in vitro, OGD and H2O2 application [18]. Exposure times or concentrations were selected, which bridged both lethal and sublethal doses in order to confirm oxidative-stress-dependent Nrf2 activation in physiologically relevant, viable conditions. Peroxide is a ROS with clearly identified roles in contributing to oxidative stress, while OGD and subsequent reoxygenation trigger a complex cascade of detrimental events including excitoxicity, cytosolic and mitochondrial ROS generation, and inflammatory responses (for review see [19]). Anoxia causes an accumulation of reducing equivalents within the electron transport chain, which upon reperfusion triggers a burst of ROS production [20]. Oxidative stress and ROS production also arise from impaired glutamate signaling due to excessive transmitter release and a reversal of glutamate uptake transporters. As a result excessive NMDAR activity triggers superoxide production via NADPH oxidase activation and xanthine oxidase [21].
We found that both OGD and mild oxidative stress (subtoxic H2O2) applied to mixed neuronal/astrocytic cultures [18] induced expression of Nrf2 target genes [18], including the classical target gene, hemeoxygenase (Hmox1) and the more recently identified target gene, sulfiredoxin (Srxn1). Given that the Srxn1 and Hmox1 genes can respond to factors other than Nrf2 [22], whether Nrf2 was the specific transcription factor mediating the gene induction following ischemia or H2O2 application remained unclear. To determine this, the same protocols were employed on Nrf2 −/− cultures revealing no significant gene induction after OGD or after H2O2 application in the absence of Nrf2, highlighting the central role of endogenous Nrf2 in mediating this gene induction [18]. Of note, we found that the locus of Nrf2 activation within the mixed cultures is centred on the astrocytes. Induction of Nrf2 target genes by OGD or oxidative stress was not observed in pure neuronal cultures devoid of astrocytes, while activation in pure astrocytic cultures was found to be highly robust [18]. Immunofluorescent analysis in mixed cultures of Hmox1 induction by oxidative stress also revealed induction that was specific to GFAP-positive astrocytes [18].
2.2. Astrocytic Nrf2 Contributes to Neuroprotective Ischemic Preconditioning
We next assessed whether the increase in Nrf2-regulated gene expression translated into enhanced protection, by quantifying neuronal viability following OGD in Nrf2 −/− and wild-type mixed cultures. Following OGD no difference in survival was observed between the Nrf2 wild-type and −/− neurons [18], which was perhaps surprising given the known protective capacity of Nrf2, although easily explained by the fact that Nrf2 target gene induction by this toxic insult was arising too late to confer protection. We next hypothesized that a sublethal insult might be sufficient to activate Nrf2, potentially contributing to neuroprotective ischemic preconditioning [23]. We established a preconditioning protocol, where a nontoxic 1.5 h exposure to OGD conferred significant neuroprotection against a toxic 3 h OGD insult applied 24 h later [18]. To determine whether the initial 1.5 h OGD insult led to recruitment of the Nrf2 pathway, Nrf2-dependent gene expression was assessed following the preconditioning stimulus. The 1.5 h OGD preconditioning stimulus led to a significant upregulation in both Srxn1 and Hmox1 gene expression, demonstrating activation of the Nrf2 pathway [18]. This suggested a possible contribution of astrocytic Nrf2 activation in the neuroprotection incurred by our preconditioning protocol.
To determine the role of Nrf2 in the protective effect of ischemic preconditioning, Nrf2 wild-type and −/− mixed cultures were exposed to the preconditioning protocol. While a preconditioning-induced increase in viability was apparent in both Nrf2 wild-type and −/− neurons, the magnitude of the protective effect of preconditioning was significantly diminished in the absence of Nrf2 [18]. Preconditioning reduced the total amount of OGD-induced death by around 60% in wild-type neurons, but by under 30% in Nrf2-deficient neurons [18]. Thus, Nrf2 activation is responsible for a significant portion of the protective effect of ischemic preconditioning in vitro. We next determined whether the Nrf2 pathway might also be implicated in in vivo preconditioning. To achieve this, Nrf2 target gene expression was assessed in mice subjected to a 15-minute occlusion of the middle cerebral artery (MCA), a stimulus known to trigger preconditioning and confer significant protection from subsequent ischemic episodes in vivo [24]. Cortical extracts from the ipsilateral hemisphere revealed a significant upregulation of both Srxn1 and Hmox1 following transient occlusion [18], demonstrating that an ischemic preconditioning episode in vivo activates the Nrf2 pathway.
2.3. Activation of Nrf2-Regulated Glutathione Pathway Genes by Preconditioning Stimuli
In addition to Srxn1 and Hmox1, Nrf2 also regulates the expression of enzymes involved in glutathione synthesis and utilisation, a system which contributes substantially to cells' antioxidant capacity. Indeed, activation of Nrf2 in astrocytes by overexpression of small molecules is thought to promote neuroprotection due to the coordinated upregulation of glutathione synthesis and release, leading to increased availability of glutathione precursors for nearby neurons to use to enhance their own glutathione pool [25].
To determine whether glutathione pathway genes are induced following preconditioning, gene expression of representative Nrf2-controlled glutathione pathway members were quantified by quantitative real-time PCR (qPCR) 4 or 8 h following exposure to the 1.5-hour OGD preconditioning stimulus. Expression of glutamate-cysteine ligase (Gclc) and glutamate/cystine antiporter (xCT) was assessed; Gclc encodes the catalytic subunit of the rate-limiting enzyme of glutathione synthesis, and xCT encodes a cystine/glutamate transporter and represents the major mechanism for obtaining cysteine for glutathione synthesis. Exposure to the ischemic preconditioning stimuli conferred a significant increase in Gclc gene expression (Figure 1(a), *P < .05, unpaired t-test, n = 3) and a near significant increase in xCT expression 4 h following OGD (Figure 1(b), P = .06, unpaired t-test, n = 5). To assess whether recruitment of the glutathione pathway might also occur in vivo, adult mice were subjected to a 15-minute occlusion of the MCA, which as mentioned above is a stimulus known to trigger in vivo preconditioning. Four hours later, mice were sacrificed and cortical tissue was harvested from the ipsilateral and contralateral hemispheres for RNA isolation and qPCR. Transient occlusion of the MCA triggered a significant upregulation in the mRNA expression of both Gclc and xCT in the ipsilateral cortex (see Figures 2(a) and 2(b), **P < .01, ***P < .001, Student's t-test, n = 6), as well as in the expression of another Nrf2 target gene, Glutamate-cysteine ligase regulatory subunit (Gclm) (Figure 2(c), ***P < .001, Student's t-test, n = 6), suggesting a potential contribution of these gene products to the resultant neuroprotection acquired by ischemic preconditioning in vivo. Thus, a variety of Nrf2 target genes are induced by transient ischemia in vivo.
Figure 1.
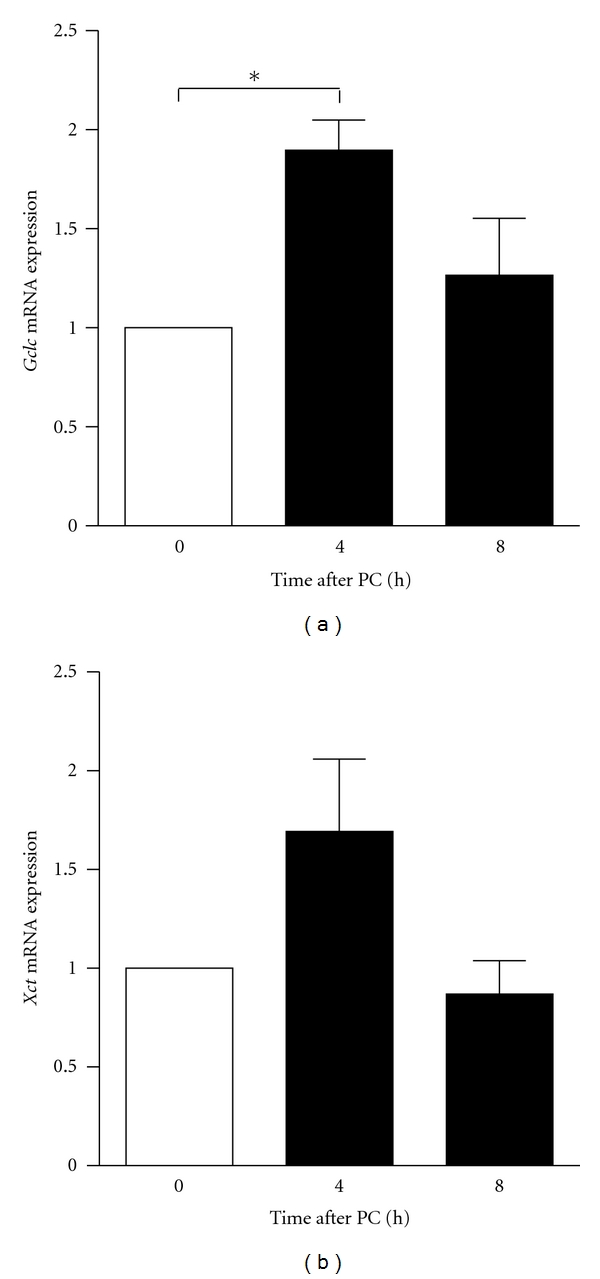
In vitro preconditioning activates Gclc expression. neurons were exposed to the 1.5 h OGD preconditioning stimulus and the expression of Nrf2-target genes. Gclc and xCT were determined by qPCR. Expression of both xCT and Gclc was significantly upregulated by the preconditioning stimulus, highlighting a potential contribution of their gene products in the neuroprotection acquired by preconditioning (bars represent mean ± SEM, *P < .05, unpaired t-test, n = 3 for Gclc, n = 5 for xCT).
Figure 2.
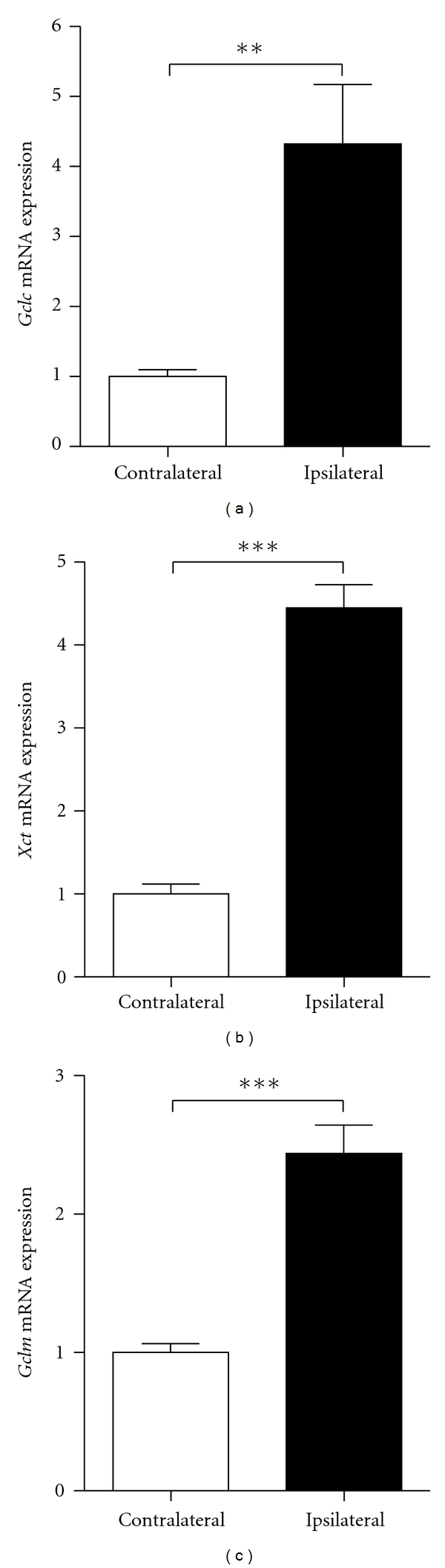
Upregulation of glutathione pathway components following in vivo ischemic preconditioning. Gene expression of glutathione pathway components, Gclc (a), xCT (b), and Gclm (c), was quantified by qPCR in mice subjected to a transient 15-minute occlusion of the middle cerebral artery, a stimulus known to trigger neuroprotective preconditioning in vivo. Transient ischemia significantly increased the cortical expression of Gclc, xCT, and Gclm in the ipsilateral hemisphere, as compared to the contralateral hemisphere, suggesting a specific recruitment of the ARE/Nrf2/glutathione pathway in the protective effects of preconditioning in vivo (bars represent mean ± SEM,**P < .01, ***P < .001, Student's t-test, n = 6).
2.4. Nrf2-Dependent and Nrf2-Independent Mechanisms of Astrocyte-Mediated Neuroprotection
Our finding that astrocytic Nrf2 plays a role in preconditioning is in line with studies identifying ROS production as a necessary event in the establishment of preconditioning [26] and the finding that superoxide radicals or H2O2 is capable of activating the preconditioning response [27]. Moreover, our finding that the gene expression of key markers of the glutathione pathway is increased following in vitro and in vivo preconditioning supports a neuroprotective role for the glutathione pathway in the Nrf2-dependent component of ischemic preconditioning. Indeed, numerous studies have identified glutathione as being involved in preconditioning [28, 29], and chemopreventive protection is associated with a marked upregulation of glutathione biosynthesis [16], as is Nrf2 activation in the brain [7, 15].
In contrast to our own findings, a recent study identified an Nrf2-independent neuroprotective effect following subtoxic H2O2 generation in astrocytes [30]. The authors utilized an elegant system whereby the expression of a H2O2-producing enzyme in astrocytes led to a specific and quantifiable level of H2O2 production following application of D-alanine. This in turn led to a protective response in astrocytes that rendered neurons resistant to an oxidative insult. However, this response was not dependent on astrocytic Nrf2 activation and potentially involved tyrosine phosphatase inhibition [30]. While the potential reasons behind the disparities between the two studies are discussed elsewhere [18], it is feasible that both Nrf2-dependent and Nrf2-independent mechanisms can contribute to adaptive neuroprotective responses by astrocytes to mild oxidative insults. Moreover, the importance of individual pathways may depend on developmental stage or the severity or nature of the oxidative insult. Notwithstanding these issues, it is clear that astrocytes are important mediators of adaptive neuroprotective responses to subtoxic insults.
2.5. Concluding Remarks
Our identification of astrocytic Nrf2, a mediator of endogenous ischemic preconditioning, underlines the importance of astrocytes in shaping neuronal vulnerability to insults. Moreover it emphasizes the potential value of astrocytic Nrf2 as a therapeutic target in a variety of disorders associated with oxidative stress [31]. Since Nrf2 controls multiple components of both the glutathione system and the thioredoxin-peroxiredoxin system [2, 31, 32], it has the capacity to mount a coordinated antioxidant response to oxidative insults in the brain. While studies have been focussed on rodent systems, it will be important to determine whether human astrocytes are capable of mediating a neuroprotective response to Nrf2-activating stimuli, something that human stem-cell-based approaches are now capable of answering.
3. Materials and Methods
3.1. Neuronal Cultures
Cortical mouse mixed cultures of neurons and astrocytes were prepared as described [33] from E17.5 CD1 mice with neurobasal growth medium supplemented with B27 (Invitrogen, Carlsbad, Calif, USA). These cultures involve approximately 90% NeuN-positive neurons and 10% GFAP-positive astrocytes [34]. Experiments were carried out on cultured neurons following a period of 8–10 days during which cortical neurons develop a network of processes, express functional NMDA-type and AMPA/kainate-type glutamate receptors, and form synaptic contacts. Prior to the start of experiments, neurons were subjected to trophic deprivation by transferring them from growth medium to TMo for two hours, a medium containing 10% MEM (Invitrogen) and 90% salt-glucose-glycine (SGG) medium (SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 0.5 mM sodium pyruvate, 0.1% phenol red; osmolarity 325 mosm/l, [35]).
3.2. Oxygen Glucose Deprivation
OGD was performed on DIV = 9 cultured mouse neurons. Cells were transferred from TMo, washed once in a glucose-free, balanced salt solution (SGG with mannitol substituted for glucose, SGG-Mann): 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM mannitol, 0.5 mM sodium pyruvate, 0.1% phenol red; osmolarity 325 mosm/l solution, which had previously been degassed by flushing the solution with 95% N2-5% Co2 for 30 min Cells were placed in degassed glucose-free SGG-Mann and put in a modular incubator chamber, which was flushed with 95% N2-5% Co2 for 4 min at a flow rate of 20 L/min, according to manufacturer's instructions (Billups-Rothenburgh, Del Mar, Calif, USA), in order to fully expel any remaining oxygen within the chamber. The chambered cells were then left in OGD at 37°C for 3 h, before being returned to normoxic conditions and glucose-containing media (TMo). No OGD control cells were placed in SGG and maintained in normoxic conditions for 3 h before also being returned to TMo. All cells were left in TMo until RNA isolation for the time point indicated. Anaerobic conditions within the modular incubator chamber were confirmed with Dry Anaerobic Indicator Strips (Fisher Scientific, Loughborough, UK).
3.3. RNA Isolation, RT-PCR, and qPCR
RNA was isolated using the Stratagene Absolutely RNA Miniprep kit as directed by the manufacturer, including the optional DNAse treatment (Stratagene, Amsterdam, Netherlands). For qPCR, cDNA was synthesized from 1–3 μg RNA using the Stratascript QPCR cDNA Synthesis kit (Stratagene) according to the manufacturer's instructions and as described previously [6, 34]. Briefly, the required amount of RNA (up to 3 μg) was diluted in RNase-free water (up to 7 μL final volume) and mixed on ice with 2x cDNA Synthesis master mix (10 μL), random primers: oligo-dT primers 3:1 (total 2 μL–200 ng), and either 1 μL RT/RNase block enzyme mixture (for RT reactions) or 1 μL water (for no-RT control reactions). Reaction mixtures were mixed, spun down, and incubated for 2 min at 25°C, 40 min at 42°C, and 5 min at 95°C. cDNA was stored at −20°C. Dilutions of this cDNA were subsequently used for real-time PCR (cDNA equivalent to 6 ng of initial RNA per 15 μL qPCR reaction for all genes). qPCR was performed in an Mx3000P qPCR System (Stratagene) using Brilliant SYBR Green qPCR Master Mix (Stratagene) according to the manufacturer's instructions. Briefly, the required amount of template was mixed on ice with 2x Brilliant SYBR Green Master Mix, forward and reverse primers at 200 nM each final concentration, 30 nM final concentration ROX passive reference dye, and water to the required reaction volume. Technical replicates as well as no-template and no-RT negative controls were included, and at least 3 biological replicates were studied in each case. The sequence of the utilized primers is as follows (all at 200 nM final): xCT-F: 5′-ATACTCCAGAACACGGGCAG-3′, xCT-R: 5′-AGTTCCACCCAGACTCGAAC-3′, Gclc-F: 5′-CCAACCATCCGACCCTCTG-3′, Gclc-R: 5′-TGTTCTGGCAGTGTGAATCC-3′, Gclm-F: 5′-GCACAGCGAGGAGCTTC-3′, Gclm-R: 5′-GAGCATGCCATGTCAACTG-3′, GAPDH-F: 5′-GGGTGTGAACCACGAGAAAT-3′, GAPDH-R: 5′-CCTTCCACAATGCCAAAGTT-3′. The qPCR cycling programme was 10 min at 95°C; 40 cycles of 30 sec at 95°C, 40 sec at 60°C with detection of fluorescence, and 30 sec at 72°C; 1 cycle (for dissociation curve) of 1 min at 95°C and 30 sec at 55°C with a ramp up to 30 sec at 95°C (ramp rate: 0.2°C/sec) with continuous detection of fluorescence on the 55–95°C ramp. Data were analysed using the MxPro qPCR analysis software (Stratagene), and the expression of the gene of interest was normalized to GAPDH, a commonly used control.
3.4. In Vivo Focal Cerebral Ischemia
All experiments were carried out in adult male C57Bl/6J mice (Charles River, UK) under an appropriate Home Office Licence and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986). Transient focal ischemia (15 min) was induced by intraluminal filament occlusion of the right middle cerebral artery (MCA). Animals were anaesthetized and maintained with isoflurane (2%) in a mixture of 30% O2 and 70% N2O by face mask. Focal cerebral ischemia was induced by the occlusion of the right MCA with an 8–0 nylon monofilament (Ethicon, Kirkton, Scotland) coated with a mixture of silicone resin (Xantoprene, Bayer Dental, Osaka, Japan) and hardener (Elastomer Activator, Bayer Dental). Briefly, the right common carotid (CCA), external carotid (ECA), and internal carotid (ICA) arteries and their branches were exposed through a midline cervical incision. A 6–0 silk suture was tied around the CCA proximal to the bifurcation of the ECA and ICA and then a second suture tied around the ECA distal to the superior thyroid artery (STA). The STA and occipital artery (OA) were closed by electrocoagulation. The silicone-coated monofilament (diameter 220 μm) was introduced into the CCA via a small incision and advanced 10 mm distal to the carotid bifurcation so as to occlude the MCA. Following the occlusion, mice were then recovered from anaesthesia briefly and placed in an incubator (30°C) before being reanesthetized in order to remove the monofilament to allow reperfusion. Wounds were sutured closed and anaesthesia discontinued. After reperfusion of 3.75 h (n = 6), mice were reanesthetized briefly with 5% isoflurane and decapitated. The brains were rapidly removed, and the regions supplied by the MCA (striatum and cortex) were dissected within the ipsilateral and contralateral MCAO territory, frozen in liquid nitrogen, and kept at −80°C for subsequent RNA analysis. Frozen tissue samples (ipsi- and contralateral) were weighed and homogenised in a 1 ml glass Dounce homogeniser, and RNA was isolated as described above using the Stratagene Absolutely RNA mini prep kit (Stratagene). Sham-operated animals were treated as above except that no occlusion of the MCA was performed.
Acknowledgments
This work was funded by the Wellcome Trust, a Royal Society University Research Fellowship (GH), Medical Research Scotland, Tenovus Scotland, and the BBSRC. Karen F. S. Bell is the recipient of a CIHR postdoctoral fellowship. Jill. H. Fowler is in receipt of a Alzheimer's Society Fellowship. The authors declare no conflict of interests. This Extra View article is linked to the following manuscript: Bell KFS, Mubarak B, Fowler J, Baxter PS, Gupta K, Tsujita T Chowdhry S, Horsburgh K, Hayes JD and Hardingham GE (2011) Mild oxidative stress activates Nrf2 in astrocytes which contributes to neuroprotective ischemic preconditioning. Proc. Natl. Acad. Sci. USA 108, E1-2.
References
- 1.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metabolism Reviews. 2006;38(4):769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 2.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual Review of Pharmacology and Toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 3.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate Cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. The Journal of Biological Chemistry. 2006;281(34):24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 4.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. Journal of Neuroscience. 2004;24(5):1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satoh T, Okamoto SI, Cui J, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proceedings of the National Academy of Sciences. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soriano FX, Léveillé F, Papadia S, et al. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. Journal of Neurochemistry. 2008;107(2):533–543. doi: 10.1111/j.1471-4159.2008.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satoh T, Kosaka K, Itoh K, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. Journal of Neurochemistry. 2008;104(4):1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shih AY, Imbeault S, Barakauskas V, et al. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. The Journal of Biological Chemistry. 2005;280(24):22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 9.Shih AY, Johnson DA, Wong G, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. Journal of Neuroscience. 2003;23(8):3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. Journal of Neuroscience. 2004;24(5):1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. Journal of Neuroscience. 2008;28(50):13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen PC, Vargas MR, Pani AK, et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Reviews in Molecular Medicine. 2009;11, article e17 doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calkins MJ, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicological Sciences. 2010;115(2):557–568. doi: 10.1093/toxsci/kfq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. Journal of Neuroscience. 2005;25(44):10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins LG, Kelleher MO, Eggleston IM, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicology and Applied Pharmacology. 2009;237(3):267–280. doi: 10.1016/j.taap.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of Biological Chemistry. 2010;285(11):8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell KF, Al-Mubarak B, Fowler JH, et al. Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):E1–E2. doi: 10.1073/pnas.1015229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertz L. Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology. 2008;55(3):289–309. doi: 10.1016/j.neuropharm.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clinical Neurology and Neurosurgery. 2009;111(6):483–495. doi: 10.1016/j.clineuro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiology of Disease. 2011;42(3):341–348. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soriano FX, Baxter P, Murray LM, Sporn MB, Gillingwater TH, Hardingham GE. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Molecules and Cells. 2009;27(3):279–282. doi: 10.1007/s10059-009-0050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. The Lancet Neurology. 2009;8(4):398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stenzel-Poore MP, Stevens SL, Xiong Z, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. The Lancet. 2003;362(9389):1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- 25.Shih AY, Johnson DA, Wong G, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. Journal of Neuroscience. 2003;23(8):3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tauskela JS, Brunette E, Monette R, Comas T, Morley P. Preconditioning of cortical neurons by oxygen-glucose deprivation: tolerance induction through abbreviated neurotoxic signaling. American Journal of Physiology. 2003;285(4):C899–C911. doi: 10.1152/ajpcell.00110.2003. [DOI] [PubMed] [Google Scholar]
- 27.Ohtsuki T, Matsumoto M, Kuwabara K, et al. Influence of oxidative stress on induced tolerance to ischemia in gerbil hippocampal neurons. Brain Research. 1992;599(2):246–252. doi: 10.1016/0006-8993(92)90398-s. [DOI] [PubMed] [Google Scholar]
- 28.Arthur PG, Lim SCC, Meloni BP, Munns SE, Chan A, Knuckey NW. The protective effect of hypoxic preconditioning on cortical neuronal cultures is associated with increases in the activity of several antioxidant enzymes. Brain Research. 2004;1017(1-2):146–154. doi: 10.1016/j.brainres.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 29.Choi YS, Cho KO, Kim EJ, Sung KIW, Seong YK. Ischemic preconditioning in the rat hippocampus increases antioxidant activities but does not affect the level of hydroxyl radicals during subsequent severe ischemia. Experimental and Molecular Medicine. 2007;39(4):556–563. doi: 10.1038/emm.2007.61. [DOI] [PubMed] [Google Scholar]
- 30.Haskew-Layton RE, Payappilly JB, Smirnova NA, et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(40):17385–17390. doi: 10.1073/pnas.1003996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calkins MJ, Johnson DA, Townsend JA, et al. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxidants and Redox Signaling. 2009;11(3):497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell KFS, Hardingham GE. CNS peroxiredoxins and their regulation in health and disease. Antioxidants and Redox Signaling. 2011;14(8):1467–1477. doi: 10.1089/ars.2010.3567. [DOI] [PubMed] [Google Scholar]
- 33.Léveillé F, Papadia S, Fricker M, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. Journal of Neuroscience. 2010;30(7):2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papadia S, Soriano FX, Léveillé F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nature Neuroscience. 2008;11(4):476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. Journal of Neuroscience. 2005;25(17):4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]