

Abstract
The Lactococcus lactis group II intron Ll.ltrB is similar to mobile yeast mtDNA group II introns, which encode reverse transcriptase, RNA maturase, and DNA endonuclease activities for site-specific DNA insertion. Here, we show that the Lactococcal intron can be expressed and spliced efficiently in Escherichia coli. The intron-encoded protein LtrA has reverse transcriptase and RNA maturase activities, with the latter activity shown both in vivo and in vitro, a first for any group II intron-encoded protein. As for the yeast mtDNA introns, the DNA endonuclease activity of the Lactococcal intron is associated with RNP particles containing both the intron-encoded protein and the excised intron RNA. Also, the intron RNA cleaves the sense-strand of the recipient DNA by a reverse splicing reaction, whereas the intron-encoded protein cleaves the antisense strand. The Lactococcal intron endonuclease can be obtained in large quantities by coexpression of the LtrA protein with the intron RNA in E. coli or reconstituted in vitro by incubating the expressed LtrA protein with in vitro-synthesized intron RNA. Furthermore, the specificity of the endonuclease and reverse splicing reactions can be changed predictably by modifying the RNA component. Expression in E. coli facilitates the use of group II introns for the targeting of specific foreign sequences to a desired site in DNA.
Keywords: Key Words: Gene therapy, intron mobility, Lactococcus lactis, retrotransposition, ribozyme
Group II introns are mobile genetic elements with an extraordinary catalytic repertoire (Curcio and Belfort 1996; Pyle 1996). Their catalytic RNAs promote RNA splicing and intron mobility reactions through their ability to act on both RNA and DNA substrates. Furthermore, they encode multifunctional proteins, which again act on both RNA and DNA to promote splicing and mobility. In addition to this unprecedented functional versatility, the splicing and mobility reactions of group II introns are of interest from an evolutionary perspective, because group II introns have been suggested to be progenitors of both spliceosomal introns (Sharp 1991; Michel and Ferat 1995) and non-LTR-retrotransposons (Zimmerly et al. 1995b).
Group II introns are characterized by a conserved secondary structure consisting of six double helical domains (Saldanha et al. 1993; Michel and Ferat 1995). Domains I and V are minimally required for catalytic activity, whereas domain VI contains the branch point nucleotide. Mobile group II introns encode open reading frames (ORFs), which are located primarily in the loop of domain IV, but in some cases extend upsteam to be contiguous with the 5′ exon (Saldanha et al. 1993; Michel and Ferat 1995). Previous studies of group II intron mobility have focused on the yeast group II introns aI1 and aI2, which characteristically move to cognate intronless alleles (homing) (Lazowska et al. 1994; Moran et al. 1995). The proteins encoded by aI1 and aI2 have been shown to have three activities: reverse transcriptase (RT) for intron duplication, maturase to promote splicing, and DNA endonuclease for site-specific cleavage of recipient alleles (Carignani et al. 1983; Kennell et al. 1993; Moran et al. 1994; Zimmerly et al. 1995a,b). The maturase activity, which has thus far only been shown genetically, presumably reflects that the protein binds specifically to the intron RNA and promotes or stabilizes formation of the catalytically active RNA structure (Lambowitz and Perlman 1990; Saldanha et al. 1993).
Remarkably, the studies with aI1 and aI2 showed that the intron-encoded DNA endonuclease activity, which initiates mobility, is associated with ribonucleoprotein (RNP) particles that contain both the intron-encoded protein and the excised intron RNA (Zimmerly et al. 1995a,b; Yang et al. 1996). Moreover, both the RNA and protein components function catalytically in DNA cleavage, with the intron RNA cleaving the sense strand of the recipient DNA by reverse splicing at the intron insertion site, and the intron-encoded protein cleaving the antisense strand at position +10 of the 3′ exon. After DNA cleavage, intron homing occurs by a target DNA-primed reverse transcription mechanism in which the 3′ end of the cleaved antisense strand is used as a primer for reverse transcription of either unspliced precursor RNA or the intron RNA that had reverse spliced into the sense strand of the recipient DNA (Zimmerly et al. 1995b; Eskes et al. 1997). The RT-mediated mobility of group II introns has been referred to as retrohoming (Curcio and Belfort 1996).
Further studies of the aI2 endonuclease showed that both the RNA and protein components contribute to recognition of the DNA target site (Guo et al. 1997). The DNA target site for sense-strand cleavage and reverse splicing extends from position −21 in the 5′ exon to +1 in the 3′ exon, with additional sequences between +1 and +10 required for antisense-strand cleavage (positions refer to distance from the splice junction). A 13-nucleotide region of the DNA target site extending from position −12 to +1 is recognized primarily by base-pairing with the intron RNA. This region contains short sequence elements IBS1 and IBS2 (intron binding sites 1 and 2) and δ′, which base pair with complementary sequences EBS1 and EBS2 (exon binding sites 1 and 2) and δ, located in different regions of domain I of the intron RNA. These base-pairing interactions had been shown previously to play a role in the RNA-catalyzed splicing of group II introns from precursor RNAs and in the reverse splicing of group II introns into RNA substrates (Saldanha et al. 1993; Michel and Ferat 1995). The regions of the DNA target site flanking the IBS seqences are presumably recognized by the intron-encoded protein. In the case of aI2, the protein primarily recognizes a subset of key nucleotide residues in the distal 5′ exon region of the DNA target site for reverse splicing and appears to promote DNA unwinding enabling base pairing of the intron RNA (Guo et al. 1997). Because recognition of the DNA target site involves base-pairing, it is possible to change the specificity of the endonuclease simply by modifying the RNA component (Eskes et al. 1997; Guo et al. 1997). The ability to design group II introns to target desired DNA sequences for site-specific cleavage and insertion could have widespread future applications in genetic engineering and gene therapy.
To fully exploit this approach as well as to further study the behavior of group II introns, it was desirable to express large amounts of the group II intron-encoded protein and RNP particles. Expression of active aI1 and aI2 proteins has proven difficult, however, in part because of differences in the genetic code and codon usage in yeast mitochondria. Recently, a number of group II introns have been found in several species of bacteria (Ferat and Michel 1993; Ferat et al. 1994; Knoop and Brennicke 1994; Mills et al. 1996; Mullany et al. 1996; Shearman et al. 1996). These introns utilize the standard genetic code and provide a seemingly desirable alternative for expression and mechanistic studies. In most cases, however, splicing or mobility of the intron have not been reported to occur in vivo. One exception is the Ll.ltrB intron, which was found in the putative relaxase gene (ltrB) of conjugative element pRS01 in Lactococcus lactis ML3 (Fig. 1A; Mills et al. 1996). The same intron (IntL) was found independently in the putative relaxase gene of the conjugative element inserted in the chromosome of L. lactis 712 (Shearman et al. 1996). The intron is spliced in Lactococcus and gene disruption experiments suggest that the intron-encoded protein, denoted LtrA, is needed for splicing.
Figure 1.
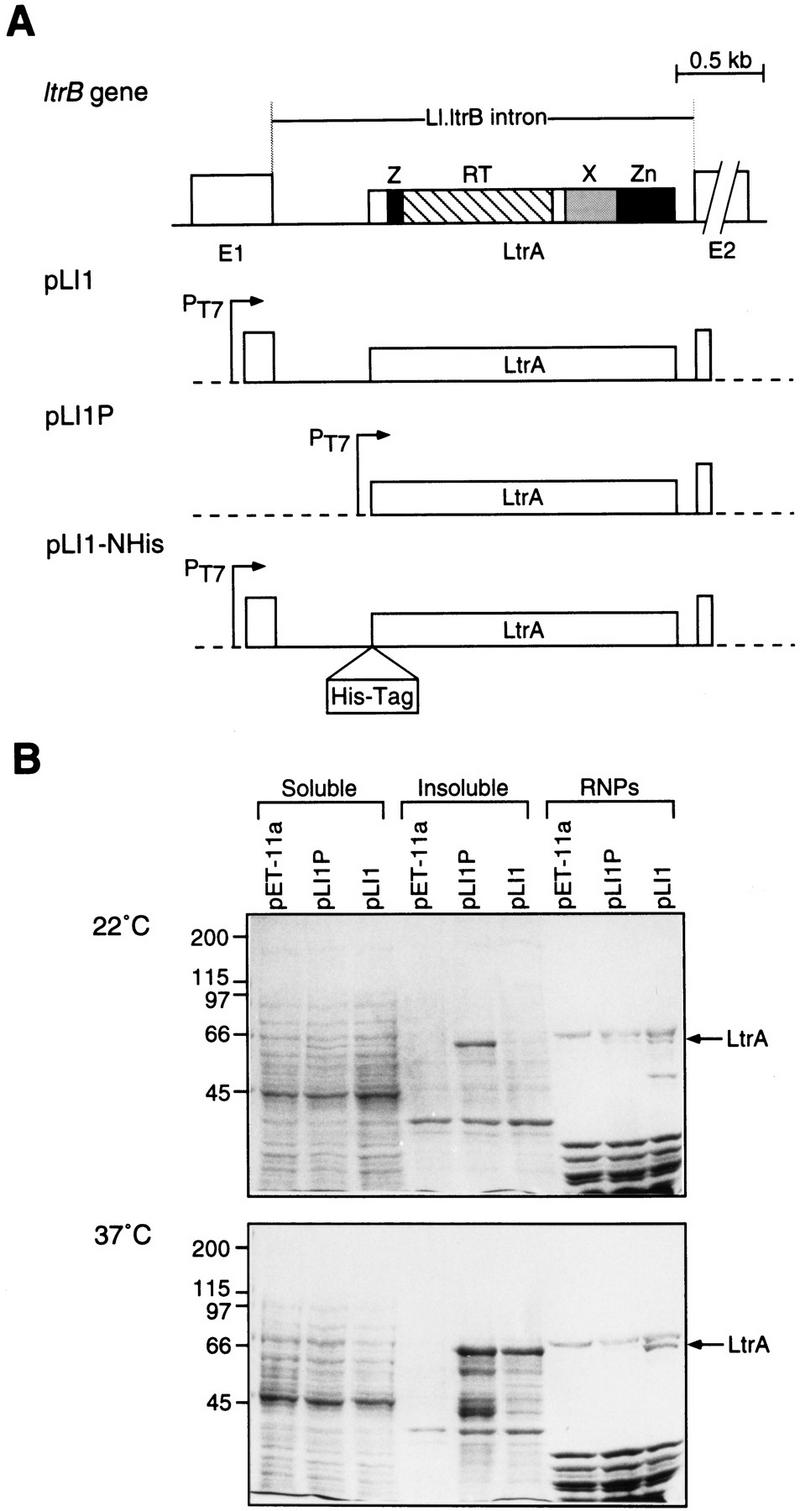
Expression of the LtrA protein in E. coli. (A) Diagram of the Ll.ltrB intron and constructs pLI1, pLI1P, and pLI1–NHis used for expression of the LtrA protein in E. coli. The protein domains shared by other group II intron ORFs are indicated (Mohr et al. 1993). Exons (E1 and E2) are open boxes, the intron is a line, and the LtrA ORF is a box within the intron. Vector sequences are indicated by broken lines. (B) SDS–PAGE of expressed proteins. Constructs were expressed in E. coli BL21(DE3) grown at 22°C or 37°C, and proteins in the soluble, insoluble, and RNP fractions (0.24 OD260 units) were analyzed by SDS–PAGE (Laemmli 1970) with a 3% stacking gel and an 8% resolving gel. Shown is a Coomassie blue-stained gel. Numbers at left indicate molecular mass markers (kD).
Here, we show that the intron-containing segment of the Lactococcus ltrB gene can be readily expressed in Escherichia coli, that the RNA precursor can be spliced efficiently, and that large quantities of active intron-encoded LtrA protein can be produced. The expressed protein forms functional RNP particles with the intron RNA and is shown to have RT, maturase, and endonuclease activities. Our results show the generality of the reaction mechanisms used by the yeast mtDNA group II introns. We also show that the system is amenable to the targeting of foreign sequences to a specific site in DNA.
Results
Expression of the LtrA protein in E. coli
In initial experiments, we tested two constructs for expression of the LtrA protein in E. coli. The first construct, pLI1, contains the Ll.ltrB intron and flanking exons cloned behind the phage T7 promoter in the expression vector pET-11a (Fig. 1A). This construct could potentially express both the intron RNA and the LtrA ORF, the latter with its own Shine–Dalgarno-like sequence for translation. The second construct, pLI1P, is a derivative of pLI1, which places the intron ORF immediately downstream of the phage T7 promoter and Shine–Dalgarno sequence of the vector. SDS–polyacrylamide gels stained with Coomassie blue showed that both constructs produced a protein that ran just below the 66-kD marker and was not produced by cells containing the vector pET-11a (Fig. 1B). This protein is approximately the size expected for expression of the entire ORF (70 kD), and we confirmed by sequencing that the protein produced by pLI1 has the expected amino terminus (not shown). The small discrepancy in apparent molecular mass likely reflects anomalous migration of the basic LtrA protein relative to size markers.
Figure 1B compares the recovery of LtrA protein in soluble and insoluble fractions and in RNP particle preparations, which presumably contain LtrA protein bound to endogenous RNAs. LtrA protein from pLI1, which coexpresses the intron and flanking exons, was ∼30% soluble in cells grown at 22°C and became more insoluble when expressed at 37°C. The protein was readily detectable in RNP particle preparations at either temperature, but the yield of LtrA protein in RNP particles was two- to threefold higher at 37°C. Notably, the LtrA protein from pLI1P, which lacks exon 1 and the 5′ segment of the intron, was largely insoluble at either 22°C or 37°C (70% and >95% insoluble, respectively), and its recovery in RNP particles was reduced compared with the pLI1 construct. Thus, coexpression of the intron-containing RNA in pLI1 appears to improve the solubility of the LtrA protein and increase its recovery in RNP particles.
Optimization of LtrA synthesis was carried out with pLI1–NHis, a derivative of pLI1 in which His6 and Xpress (DLYDDDDK) epitope tags were linked to the amino terminus of the LtrA ORF (Fig. 1A). In cells grown at 22°C, the yield of the tagged protein was 0.4–2 mg/liter of culture (2%–5% of the total protein), with ∼30% being soluble and 40%–90% of the soluble protein recovered in RNP particles (0.3–3 μg of LtrA protein/OD260) (not shown). Based on the above experiments, pLI1 and its derivatives were used under the 22°C conditions to obtain large amounts of soluble protein, and under the 37°C conditions to obtain large quantities of active RNP particles.
Assay of RT activity by use of exogenous substrates
The RT activity of the expressed LtrA protein in the RNP particle preparations was first assayed with the artificial template–primer substrate poly(rA)–oligo(dT)18 in the presence of RNase A to degrade the endogenous template RNA. Figure 2A shows that RNP particles from cells expressing pLI1 had high RT activity, whereas no activity was detected with RNP particles from cells harboring the vector pET-11a. RNP particles from cells expressing the pLI1–NHis-tagged protein also had RT activity proportionate to the amount of LtrA protein in the preparations (not shown). As expected, the RT activity of the LtrA protein was abolished by a frameshift mutation after amino acid 124 of the LtrA ORF (pLI1–FS) or by point mutations in the conserved YADD motif of the RT domain (pLI1–DD−). The YADD mutation did not substantially affect the recovery of LtrA protein in RNP particles (Fig. 2D). Notably, LtrA protein expressed from pLI1P, which lacks the 5′ segment of the intron, was detectable in RNP particles but had only low RT activity (10% of pLI1), which was less than expected for the amount of protein. Additional experiments showed that the RT activity of the LtrA protein is unstable under some conditions in the absence of the intron RNA (R. Saldanha and A.M. Lambowitz, unpubl.). These results suggest that binding to unspliced precursor and/or the intron RNA helps stabilize the LtrA protein in the active conformation, consistent with the findings above that coexpression of the full-length intron in pLI1 increases the solubility and RNP particle association of the LtrA protein.
Figure 2.

RT activity in RNP particle preparations from E. coli. (A) RT assays with poly(rA)–oligo(dT)18. RNP particles were isolated from cells grown at 37°C, and RT activity was assayed, as described in Materials and Methods. For pLI1P, the data shown are for cells grown at 22°C, because no RT activity was detected in RNP preparations from cells grown at 37°C. (B) Endogenous reverse transcription reactions in the RNP particles. Reactions were carried out in the presence or absence of 20 pmoles of 3′ exon, 5′ exon, or ampR primers. (C) Control for accessibility of primers to endogenous RNA templates. Endogenous reactions with pLI1 RNP particles were carried out with different DNA primers in the presence (shaded bars) or absence (open bars) of M-MLV RT. (D) SDS–PAGE of RNP particle proteins. SDS–PAGE was carried out as in Fig. 1, with 0.5 OD260 units of RNP particles. Numbers at left indicate molecular mass markers (kD). Error bars in the RT assays indicate standard deviations for three determinations.
RT activity with endogenous RNA templates in RNP particles
To identify the RNA species to which the LtrA protein is bound, RNP particle preparations were also assayed for RT activity with endogenous RNA templates, either alone or in the presence of DNA oligonucleotide primers complementary to different regions of the plasmid transcripts. Figure 2B shows that RNP particles from cells expressing pLI1 had high RT activity with endogenous RNAs in the presence of a primer complementary to the 3′ exon of the ltrB gene, whereas little or no RT activity was observed in the absence of this primer or with primers complementary to the 5′ exon or the ampR gene of the vector. Controls for annealing and accessibility showed that all three primers could be utilized by Moloney murine leukemia virus (M-MLV) RT added to the RNP particle preparations (Fig. 2C). Similarly high levels of LtrA RT activity were observed with 3′ exon primers having 3′ ends at different positions: E2 + 15 (Fig. 2B), E2 + 10, or E2 + 20 (not shown). No endogenous RT activity was detected with any primer in RNP particles from cells containing the vector or expressing the mutant FS or DD− proteins, and only minimal activity (<1% pLI1) was detected in cells containing pLI1P, which expresses the LtrA protein in the absence of intact intron RNA (Fig. 2B). We confirmed that the endogenous RT activity of pLI1 RNP particles in the presence of the 3′ exon primer was sensitive to pretreatment with RNase A and insensitive to actinomycin D (61% of control values at 20 μg/ml), as expected for an RT with an RNA template.
The finding that endogenous RT activity in the pLI1 RNPs is detected only with the 3′ exon primers suggests that the LtrA protein binds to unspliced precursor RNA near the 3′ end of the intron. In accord with this location, Southern hybridizations showed that 32P-labeled cDNAs synthesized from endogenous RNAs in the presence of the 3′ exon primer hybridized predominantly to the Lactococcus intron (not shown). The finding that the RT is associated with unspliced precursor RNA is expected if the LtrA protein functions in RNA splicing. Furthermore, the finding that the LtrA protein is positioned to initiate reverse transcription in the 3′ exon is similar to findings for the yeast aI2 intron (Kennell et al. 1993; Zimmerly et al. 1995b; S. Zimmerly and A.M. Lambowitz, unpubl.).
The Ll.ltrB intron is spliced in Escherichia coli and requires the LtrA protein
To determine if the intron in the construct pLI1 is spliced in Escherichia coli, Northern hybridization analysis was performed (Fig. 3). The precursor RNA was detected with a 3′ splice site probe (probe A), and the ligated exons with a splice junction probe (probe B). An in vitro (IV) spliced precursor RNA was included for comparison (lanes 7). Precursor processing and exon ligation in vivo began within 0.5 hr after the induction of synthesis of the Lactococcal RNA and persisted for 4 hr. The disappearance of precursor at later times likely reflects both splicing and RNA turnover, as evidenced by the RNA fragments detected with probe A and by the extensive degradation of the intron observed in blots hybridized with intron-specific probes (not shown).
Figure 3.

RNA splicing in E. coli. Northern hybridizations were performed with RNA extracted from E. coli 0 to 4 hr after induction of transcription from plasmid pLI1 (lanes 1–6). Oligonucleotide probe A hybridizes to the 3′ splice site of the precursor RNA, whereas probe B hybridizes to the splice junction of the ligated exons. (Lanes 7) RNA transcribed from pLI2–ΔORF and self-spliced in vitro (IV). The hybridization to self-spliced RNA shows the specificity of the probes and provides RNA size markers (precursor, 1.25 kb, ligated exons 0.35 kb). (Lanes 8) DNA size markers, with sizes (kb) indicated to the right. In the schematic below, exons are □, the intron is a shaded box, and vector sequences are represented by a line. Probes are shown as black bars.
To circumvent problems associated with rapid RNA turnover in E. coli, the accuracy of splicing and its dependence on the LtrA protein were assessed by primer extension assay. The precision of 5′ splice-site cleavage was determined by performing cDNA synthesis with an intron primer (primer 1; Fig. 4A,B). A cDNA corresponding precisely to the 5′ end of the intron, as determined from parallel DNA sequencing lanes (not shown), was observed within 0.5 hr of induction of pLI1 transcription, along with a doublet corresponding to 5′ ends of unspliced precursor RNAs. Ligated exon RNAs were detected as a corresponding cDNA doublet with an exon 2 primer with the same time course of appearance as the excised intron (Fig. 4A,B). The longer 5′ exon end corresponds to the predicted transcription start site at the T7 promoter of pLI1, whereas the shorter 5′ end coincides with the junction of vector and Lactococcal sequences and may reflect the creation of a new transcription start or processing site.
Figure 4.


RNA splicing is accurate in E. coli. (A) Primer extension analysis. RNAs extracted from E. coli at the indicated times after induction of transcription from plasmids pLI1 (lanes 1–6) or pLI2–ΔORF (lanes 7–9) were reverse-transcribed with intron-specific primer 1 or exon 2-specific primer 2. The cDNA bands, of lengths indicated in nucleotides to the right of each panel, correspond to precursors (Pre) and intron lariat for primer 1, and ligated exons (E1E2) for primer 2. (B) Schematic of RNAs, cDNAs, and PCR products. RNAs are depicted as in Fig. 3, cDNA extension products are shown as broken lines, PCR products as wavy lines, and primers as black bars.
The dependence of splicing on the LtrA protein was assessed by use of the construct pLI2–ΔORF, which contains an intact catalytic RNA, but has a deletion corresponding to amino acids 40–572 of the LtrA coding sequence in the loop of domain IV. In this case, the RNA preparations still showed the precursor doublet, but the intron and ligated exon bands were completely absent, suggesting that the expressed LtrA protein is required for splicing of the Lactococcal intron in E. coli (Fig. 4A,B). Additional experiments showed that splicing was also inhibited in cells expressing the frameshift mutant pLI1–FS or LtrA proteins with point mutations in the putative maturase domain (domain X; not shown). Sequencing of the ligated exon product via RT–PCR with primers P3 and P4 (see Fig. 4B) confirmed that splicing had occurred correctly (data not shown).
Protein-dependent splicing of the Ll.ltrB intron in vitro
In vitro splicing by the expressed LtrA protein was investigated by use of 32P-labeled precursor RNA synthesized from the construct pLI2–ΔORF, which lacks most of the intron ORF (Fig. 5). This transcript self-spliced in high salt (HS) reaction medium containing 500 mm NH4Cl and 50 mm MgCl2 to yield ligated exons and intron lariat (lanes 1–3). Under low salt (LS) conditions (100 mm NaCl and 10 mm MgCl2), the transcript did not self-splice (lanes 4–6), presumably reflecting the inability of the intron to fold into the catalytically active structure under these conditions, but splicing could be induced by the addition of micrococcal nuclease (MNase)-digested RNP particles containing LtrA protein expressed from pLI1 (lanes 7–9). In contrast, no splicing was detected under these conditions with RNP particles from cells containing the vector pET–11a (lanes 10–12), or from cells expressing the frameshift mutant (pLI1–FS; lanes 13–15), or two other LtrA mutant proteins having missense mutations in the putative maturase domain (domain X; not shown). We confirmed by RT–PCR and sequencing that exon ligation with the pLI1 protein had occurred correctly (not shown). For unknown reasons, the ligated exon band appeared as a doublet in the self-splicing reaction, but was more homogeneous in the protein-assisted reaction (cf. lanes 2 and 3 with lanes 8 and 9). In some lanes, incubation with the RNP particles resulted in increased nonspecific degradation of the 32P-labeled RNA, presumably as a result of E. coli nucleases. The protein-dependent splicing reaction was not stimulated by addition of ATP (not shown). In kinetic experiments, the initial rates of self-splicing and protein-dependent splicing were 3.2 ± 0.6 × 10−4/sec and 2.9 ± 0.8 × 10−3/sec, respectively, comparable to rates of self-splicing of well-studied yeast mtDNA group II introns (6.8 × 10−3 to 6.3 × 10−4/sec; Daniels et al. 1996; Costa et al. 1997). These results constitute the first biochemical demonstration of the maturase activity of a group II intron-encoded protein.
Figure 5.
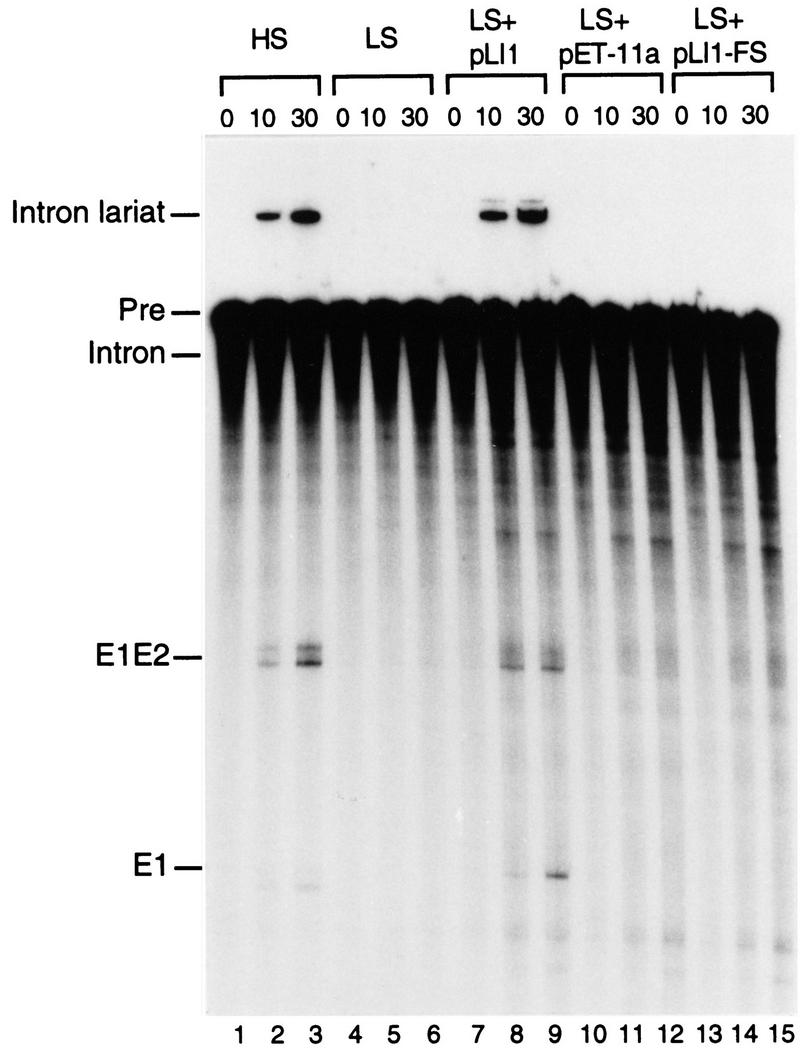
Protein-dependent in vitro splicing of the Ll.ltrB intron. 32P-Labeled in vitro transcript pLI2–ΔORF/EcoRI was incubated for 0, 10, or 30 min under high salt (HS) (lanes 1–3) or low salt (LS) (lanes 4–15) conditions (see Materials and Methods) in the presence or absence of MNase-digested RNP particles (0.1 OD260 unit) from E. coli expression strains grown at 37°C. The products were analyzed in a denaturing 4% polyacrylamide gel, which was dried and autoradiographed. (Lanes 1–6) Precursor RNA incubated in the absence of RNP particles; (lanes 7–15) precursor RNA incubated under LS conditions with MNase-digested RNP particles from E. coli expressing pLI1 (lanes 7–9), pET-11a (lanes 10–12), or pLI1–FS (lanes 13–15). (Pre) Unspliced precursor RNA; (E1E2) ligated exons; (E1) exon 1.
Characterization of DNA endonuclease activity
To assay DNA endonuclease activity of the Ll.ltrB intron, RNP particle preparations from cells expressing the LtrA protein were incubated with small (129 bp) labeled DNA substrates that contain the intron insertion site at the junction of exons 1 and 2 of the ltrB gene. In Figure 6, A and B, the DNA substrates were generated by PCR with 5′ end-labeled primers to separately label each of the two DNA strands, and the products were analyzed in denaturing polyacrylamide gels to detect the 5′ fragment of each strand. The RNP particles from cells expressing pLI1 cleaved each strand of the DNA substrate at a single major site (Fig. 6, lanes 1), whereas these cleavages were not detected with RNP particles from cells expressing the truncated FS protein (lanes 7) or containing the vector alone (lanes 6). Alignment with the sequencing ladder showed that the sense strand was cleaved precisely at the intron insertion site, and the antisense-strand was cleaved at position +9 of exon 2 (Fig. 6A,B,E). The sense strand cleavage is at the same position as for the yeast aI1 and aI2 introns, whereas the antisense-strand cleavage site is one nucleotide closer to the intron insertion site (cf. Zimmerly et al. 1995b; Yang et al. 1996). In the reaction with the 5′ sense-strand-labeled substrate, the pLI1 RNP particles gave an additional high molecular weight band that migrated above the substrate and likely results from complete reverse splicing of the intron RNA into the sense strand of the DNA substrate (Fig. 6A, lane 1; see schematic Fig. 6D). This band disappeared after RNase digestion with the concommitant appearance of a band one nucleotide longer than the major sense-strand cleavage product, presumably caused by incomplete removal of ribonucleotides from the fully reverse spliced product (lane 2). Approximately 5% of the DNA substrate was cleaved by the pLI1 RNP particles in these experiments.
Figure 6.

Assay of DNA endonuclease activity with E1E2 DNA substrates. (A,B) Endonuclease assays with 5′ sense and 5′ antisensestrand-labeled substrates, respectively. RNP particles (0.025 OD260 unit) from the indicated strains grown at 37°C were incubated with the labeled DNA substrates, and the cleavage products were analyzed in a denaturing 6% polyacrylamide gel, alongside sequencing ladders obtained from pLHS with the same 5′-end-labeled primer used to generate the substrate. (Lanes 1) pLI1 RNP particles; (lanes 2) cleavage products from the reaction of lane 1 incubated with RNase A; (lanes 3) DNA substrate incubated in the absence of RNP particles; (lanes 4,5) pLI1 RNP particles pretreated with RNase A or proteinase K; (lanes 6–8) RNP particles from cells containing the vector pET-11a, pLI1–FS, and pLI1P, respectively. The diffuse bands above the major sense strand cleavage product were not detected with internally labeled DNA substrate (see below) and are presumably an artifact of the 5′-end-labeled substrate. (C) Endonuclease assays with internally labeled DNA substrate. (Lane 1) pLI1 RNP particles; (lane 2) cleavage products from the reaction of lane 1 incubated with RNase A, as above. (D) Schematic showing the products expected for partial and complete reverse splicing of the Ll.ltrB intron into the DNA substrate. The dashed line depicts the cleaved region of the intron RNA (see Fig. 7). (E) Sequence of the Ll.ltrB target site showing the location of the cleavage sites (arrows) on the sense (top) and antisense (bottom) strands.
As expected for an RNP complex containing the intron-encoded protein and the intron RNA, the endonuclease activity of the Lactococcal intron was inhibited by pretreatment with either RNase or protease (Fig. 6A,B, lanes 4,5), and RNP particles from cells containing pLI1P, which express LtrA protein in the absence of intact intron RNA, had no detectable endonuclease activity (lane 8). In other experiments, the endonuclease activity was inhibited both by a mutation that deletes the Zn domain of the intron-encoded protein (ΔZn; Δ amino acids 491–599) and by a mutation (ΔD5) that deletes domain V of the intron RNA but continues to synthesize active intron-encoded RT protein (H. Ma, M. Matsuura, and A.M. Lambowitz, unpubl.), mirroring results for the yeast mtDNA introns (Zimmerly et al. 1995a,b). Furthermore, a smaller carboxy-terminal truncation (ΔConZn; Δ amino acids 543–599) that deletes just the conserved Zn2+-finger-like and endonuclease motifs continued to carry out the RNA-catalyzed sense-strand cleavage but showed no detectable antisense-strand cleavage, which is presumably catalyzed by this region of the LtrA protein (H. Ma, M. Matsuura, and A.M. Lambowitz, unpubl.).
Endonuclease assays with internally labeled DNA substrates to detect all of the DNA fragments provided further evidence that the sense-strand cleavage occurs by reverse splicing. The 73-nucleotide product, corresponding to the 3′ exon fragment of the sense-strand, was detected in polyacrylamide gels only after RNase digestion, suggesting attachment to a large RNA as expected for either partial or complete reverse splicing (Fig. 6C,D). Furthermore, with the internally labeled substrate, high molecular weight, RNase-sensitive bands corresponding to both partially and fully reverse spliced products could be detected migrating above the substrate band in the polyacrylamide gels (see below for explanation of bands).
Sense-strand cleavage occurs by reverse splicing
To further characterize the putative reverse spliced products, the RNP particle preparations were incubated with small 32P-labeled DNA substrates, and the products were analyzed in an agarose gel. As shown in Figure 7A, high molecular weight (>0.5 kb) products were detected with RNP particles from cells expressing pLI1 (lane 2), but not with RNP particles from cells expressing the vector pET–11a (lane 9), pLI1-FS, which has the frameshift mutation, or pLI1P, which produces LtrA protein but not intact intron RNA (lanes 10,11). The major products of ∼1 kb are shorter than expected, presumably caused by degradation of the intron RNA, as observed in the Northern hybridizations (not shown). Small amounts of larger products, including some of the size expected for full-length intron RNA (2.5 kb), were also detected in darker exposures. The identity of a smaller band that migrates just above the DNA substrate is not known. The reverse splicing reaction was inhibited by pretreating the RNP particles with RNase A or protease (Fig. 7, lanes 7 and 8), and the products were sensitive to RNase, alkali, S1 nuclease, and DNase I, as expected (lanes 3–6). The residual light band with DNase I presumably reflects short 32P-labeled DNA fragments that remain attached to the intron RNA.
Figure 7.

Reverse splicing of the Ll.ltrB intron into the E1E2 DNA substrate. (A) Reverse splicing reactions with internally labeled DNA substrate. RNP particles (0.025 OD260 unit) from cells grown at 37°C were incubated with the 129-bp 32P-labeled E1E2 DNA substrate (150,000 cpm, ∼125 fmoles). The products were denatured with glyoxal and analyzed in a 1% agarose gel. (Lane 1) DNA substrate incubated in the absence of RNP particles; (lane 2) DNA substrate incubated with RNP particles from cells expressing pLI1; (lanes 3–6) reverse spliced products of pLI1 RNP particles treated with RNase A, alkali, S1 nuclease, or DNase I; (lanes 7,8) pLI1 RNP particles pretreated with RNase A or proteinase K prior to reverse splicing (see Materials and Methods); (lanes 9–11) DNA substrate incubated with RNP particles from cells containing pET-11a, pLI1–FS, and pLI1P, respectively. (B) Reverse splicing reaction with E1E2 DNA substrates labeled separately at each of the four termini. pLI1 RNP particles were incubated with 5′- or 3′-end-labeled DNA substrates (150,000 cpm; ∼250 fmoles of 5′-end-labeled substrates and ∼200 fmoles of 3′-end-labeled substrates), and the products were analyzed as above. Numbers to the left indicate DNA size markers (kb).
Assays with DNA substrates labeled to similar specific activity at each of four termini showed that the reverse spliced products were detected only with DNA substrates labeled at the 5′ or 3′ ends of the sense strand (Fig. 7B). Both major products that were detected with internally labeled DNA substrate (lane 1) were also detected with the 3′ sense-strand-labeled substrate (lane 3), indicating that they contain intron RNA linked to 3′ exon DNA. The additional minor product detected with the 5′ sense-strand-labeled DNA presumably corresponds to the fully reverse spliced product in which the intron RNA is also attached to the 5′ exon DNA. Cloning and sequencing of the intron–exon junctions of the putative fully reverse spliced product by RT–PCR confirmed that the intron RNA had accurately reverse spliced into the DNA target site (not shown).
As noted above, the predominant reverse spliced products are shorter than expected because of degradation of the intron RNA, which likely occurred in E. coli. Estimates of the sizes of RNA fragments attached to the 5′- and 3′-exon DNAs in polyacrylamide gels (Fig. 6C) suggests that the fragments result from cleavages in the loop of domain IV at approximately at positions 850 and 2360, respectively. The cleavages at these locations apparently leave the catalytic core of the intron intact to catalyze reverse splicing.
Changing endonuclease specificity by modifying EBS1
In the cases of aI1 and aI2, the specificity of the intron endonuclease could be changed by modifying the intron’s EBS sequences, which base-pair with the IBS sequences in the DNA target site (Eskes et al. 1997; Guo et al. 1997). To test whether this is also the case for the Ll.ltrB intron, we made a modified intron expression construct pLI1–EBS1-6C, which has a G → C change at EBS1 position −6 (i.e., the intron nucleotide that base-pairs with E1–6 of IBS1). The putative IBS1/EBS1 interactions for the wild-type and modified intron are shown in Figure 8D (see Mills et al. 1996 for identification of the Ll.ltrB intron’s EBS and IBS sequences). The construct expressing the intron with the modified EBS1 sequence also contained a complementary change (E1–6G) in IBS1, enabling the modified intron to splice efficiently in E. coli. RNP particles from cells expressing the modified intron had wild-type levels of RT activity (not shown).
Figure 8.

Modifiable specificity of the Ll.ltrB endonuclease and reverse splicing activity. (A,B) Endonuclease assays with DNA substrates labeled at the 5′ ends of the sense and antisense strands, respectively. RNP particles from cells grown at 37°C expressing pLI1 (WT; lanes 1,2) or pLI1-EBS1-6C (lanes 3,4) were incubated with either the wild-type E1E2 DNA substrate (lanes 1,3) or a modified DNA substrate having a C → G change at E1–6 (E1–6G; lanes 2,4). The products were analyzed in a denaturing 6% polyacrylamide gel, which was dried and autoradiographed. The diffuse bands above the major sense strand cleavage product are presumably an artifact of the 5′-labeled substrate, as in Fig. 6. (C) Reverse splicing assays. The RNP particles were incubated as above with internally labeled DNA substrates, and the products were denatured with glyoxal and analyzed in a 1% agarose gel. Numbers to the left indicate DNA size markers (kb). (D) EBS1/IBS1 interactions for wild-type and modified forms of the LI.ltrB intron and DNA target sites. Watson–Crick and G-T pairings are indicated by short vertical bars and a dot, respectively.
Figure 8 shows endonuclease (A,B) and reverse splicing (C) assays comparing the target-site specificity of RNP particles containing the wild-type (EBS1-6G) and modified (EBS1-6C) introns. In both assays, the wild-type intron was active with the wild-type DNA target site, but not with a modified DNA target site that has a C → G change resulting in mispairing at E1–6 (lanes 1,2). In contrast, the modified intron no longer cleaved the wild-type target site but did cleave the modified target site that has the complementary change, C → G, at E1–6 (lanes 3,4). Thus, a single nucleotide change in IBS1 of the target DNA was sufficient to almost totally block DNA endonuclease and reverse splicing activity, but these activities could be restored by introducing the compensatory change in the intron’s EBS1 sequence.
Reconstitution of endonuclease activity with in vitro-synthesized intron RNA
To overcome the difficulty of preparing RNP particles with intact intron RNA from E. coli, we reconstituted RNP particles with LtrA protein and in vitro-synthesized intron RNA. The RNA component for reconstitution was obtained by self-splicing the in vitro transcript pLI2–ΔORF/BamHI, which contains the 1-kb intron-derivative with a deletion in the intron ORF. The in vitro transcript was incubated under high salt conditions, in which the intron splices efficiently in the absence of the LtrA protein (see above), and reconstitution was performed by incubating the spliced products with LtrA protein released from pLI1 RNP particles by MNase digestion.
Figure 9A shows that the reconstituted RNP particles reverse spliced efficiently into 32P-labeled DNA substrate containing the Lactococcal intron target site to yield products of ∼1 kb, the size expected for insertion of the intact ΔORF intron RNA (lane 4). Reverse-spliced products were not detected with either the in vitro spliced RNA or the RNP protein fraction alone (lanes 2 and 3). The MNase-digested RNP particle fraction by itself gave a smaller band that results from reverse splicing of incompletely digested endogenous intron RNA. As expected, reverse splicing by the reconstituted RNP particles was inhibited by pretreatment with either RNase or protease (lanes 5 and 6), indicating that both the RNA and protein components are required. Additional experiments with 5′ and 3′ end-labeled DNA substrates showed that the reconstituted RNP particles produced predominantly partially reverse spliced product with only a small proportion (<1%) of fully reverse spliced product, as found above for native RNP particles (not shown). Both of the two major bands observed with internally labeled substrate (Fig. 9A) presumably correspond to partially reverse spliced products, possibly containing different conformers of intron lariat RNA, as appears to be the case for the yeast mtDNA introns (Zimmerly et al. 1995a). Again, accurate intron–exon sequences were confirmed by RT–PCR (not shown).
Figure 9.

Reconstitution of Ll.ltrB intron endonuclease activity. (A) Reconstitution and sensitivity to pretreatment with RNase and protease. The endonuclease was reconstituted with spliced pLI2–ΔORF /BamHI RNA and MNase-digested RNP particles and used for reverse splicing assays with internally labeled E1E2 DNA substrate (∼30 fmoles). (Lane 1) DNA substrate incubated without RNP particles; (lane 2) DNA substrate incubated with in vitro-spliced pLI2–ΔORF RNA alone; (lane 3) DNA substrate incubated with MNase-digested RNP particles alone; (lane 4) DNA substrate incubated with endonuclease reconstituted by mixing MNase-digested RNP particles with in vitro spliced pLI2–ΔORF RNA; (lanes 5,6) reconstituted endonuclease preincubated with RNase or proteinase K prior to the reverse splicing reaction (see Methods). Reverse-spliced products were denatured with glyoxal and analyzed in a 1% agarose gel. Numbers to the left indicate size markers. (B) Reconstitution with full-length and modified intron RNAs. Reverse splicing into the E1E2 DNA substrate (∼60 fmoles) was carried out with endonucleases reconstituted with the following in vitro spliced RNAs: (Lane 1) pLI2–ΔORF; (lane 2) pLI1; (lane 3) pLI2–ΔORFkanR. The products were analyzed as above.
Figure 9B shows that reconstitution could also be carried out with the full-length intron RNA or with a modified form of the ΔORF intron that contains a kanR gene inserted in the loop of domain IV in place of the LtrA ORF. The domain IV loop region was presumed to be a desirable site for the insertion of additional genetic information, because this region normally contains the ORF and is not part of the catalytic core (Jarrell et al. 1988). The modified intron reverse spliced into the DNA target site, and although the efficiency of the reaction was lower than with the shorter pLI2–ΔORF intron, a product of the correct size was obtained (Fig. 9B). Further, cloning and sequencing via RT–PCR confirmed that the insertion had occurred at the correct position (not shown). These findings show that a group II intron can accommodate new genetic information and introduce it at a defined site in double-stranded DNA.
Discussion
Our results show that the L. lactis Ll.ltrB group II intron and its encoded protein (LtrA) can be expressed efficiently in E. coli. The LtrA protein, like the yeast aI1 and aI2 proteins, has RT, maturase, and DNA endonuclease activities. Further, as found for the mobile yeast group II introns, the Lactococcal intron endonuclease, which site-specifically cleaves DNA at the intron insertion site, is an RNP complex containing the intron-encoded protein and the intron RNA, and the intron RNA cleaves the sense-strand of the recipient DNA by a reverse splicing reaction, whereas the intron-encoded protein cleaves the antisense strand. A minor difference is that antisense-strand cleavage by the Lactococcal endonuclease occurs after position +9, compared with +10 for the yeast introns. Our results establish the generality of the reaction mechanisms used by the yeast mtDNA introns. The finding that a bacterial group II intron has the same spectrum of activities as the yeast introns predicts that the Lactococcus intron will be mobile in Lactococcus and possibly in E. coli. The yield of active LtrA protein (>0.1–0.5 mg/liter) is suitable for a wide variety of structural and mechanistic studies.
The active LtrA protein is recovered in RNP particle preparations, in which it appears to be associated both with unspliced precursor RNA, as expected for its role in RNA splicing, and with the excised intron RNA, in which it forms an integral component of the DNA endonuclease. The finding that RT activity with endogenous RNA templates is greatly stimulated by addition of DNA oligonucleotide primers complementary to the 3′ exon, but not by primers complementary to the 5′ exon or the ampR gene of the vector, suggests that the LtrA protein may be bound specifically near the 3′ end of the unspliced intron. This behavior is similar to that of the yeast aI2 RT, which also initiates reverse transcription just downstream of the intron in the 3′ exon of unspliced precursor RNA (Kennell et al. 1993; S. Zimmerly and A.M. Lambowitz, unpubl.).
Significantly, the correct folding of the LtrA protein appears to be facilitated by binding to the unspliced precursor or intron RNA. In the construct pLI1P, which is missing exon 1 and the 5′ segment of the intron, the LtrA protein was expressed, but had decreased, unstable RT activity and was less soluble than the protein expressed from pLI1, which contains the full-length intron and flanking exons. An analogous requirement for coexpression of an RNA-binding site was found previously for the hepatitis B virus RT (Wang et al. 1994). Because the LtrA protein remains active in RNA splicing when released from the endogenous RNA by MNase-digestion (see below), it appears that the RNA serves in part as a chaperone, which promotes folding of the LtrA protein into an active conformation, but is not required after folding has been achieved. We note, however, that some proportion of the LtrA protein appears to fold correctly, even in the absence of intact intron RNA. Coexpression of the intron RNA may be key for expression of fully active group II intron-encoded and related RT proteins.
The RNP particle preparations provide a source of active LtrA protein for biochemical analysis. We show here that the LtrA protein released from RNP particles by MNase-digestion promotes splicing of the Ll.ltrB intron under low salt conditions, the first biochemical demonstration of the maturase activity of a group II intron-encoded protein. This activity does not require exogenous ATP and presumably reflects that binding of the protein to the intron RNA promotes or stabilizes the formation of the catalytically active RNA structure. The ability of the LtrA protein to promote splicing is readily detected in E. coli and should be amenable to genetic analysis. The protein-dependent in vitro and in vivo splicing systems should permit detailed analysis of the maturase activity of a group II intron-encoded protein.
The site-specific endonuclease activity can be obtained either by coexpression of the LtrA protein and intron RNA or by reconstitution of LtrA protein from RNP particle preparations with in vitro-synthesized intron RNA. Interestingly, the RNP particles synthesized in E. coli remain active in reverse splicing into the DNA target site despite degradation of the intron RNA, which, judged by the sizes of RNA fragments, appears to be cleaved in the loop of domain IV. These cleavages apparently leave 5′ and 3′ RNA halves that remain associated to form an active catalytic core. The situation is analogous to trans-spliced group II introns that are split in domain IV and reconstitute the catalytic core from separate 5′ and 3′ halves (Jarrell et al. 1988; Bonen 1993; Michel and Ferat 1995). The specific cleavages in domain IV of the Ll.ltrB intron presumably reflect its accessibility to E. coli nucleases, whereas the catalytic core may be protected either by RNA structure or bound protein.
The reconstitution approach with Ll.ltrB intron RNA synthesized in vitro yields endonuclease with intact intron RNA. Furthermore, the reconstitution experiments show that it is possible to insert additional genetic information, a 1.3-kb kanR cassette, in domain IV, while retaining functionality of the intron RNA. Although the length of additional sequences that can be inserted in domain IV is not known, it could, in principle, be quite large in view of the finding that the trans-spliced group II introns reconstitute the catalytic core from 5′ and 3′ RNA halves synthesized at distant sites in the genome (Bonen 1993; Michel and Ferat 1995).
Group II introns are potentially useful for targeting insertion of foreign sequences at selected sites. In the case of the yeast group II introns, both the RNA and protein components contribute to the recognition of the DNA target site, and the target specificity can be modified by changing the EBS2, EBS1, or δ sequences in the intron RNA (Guo et al. 1997). We show here that a similar specificity change can be effected by modifying the EBS1 sequence in the Lactococcal intron. Further, we show that the endonuclease reconstituted with an intron RNA containing an inserted kanR genetic marker undergoes reverse splicing, resulting in the insertion of the kanR marker into the DNA target site. These methods based on the E. coli genetic system therefore hold great promise for the development of group II introns that target specific foreign sequences to a desired site in DNA.
Materials and methods
E. coli strains
E. coli strains used for the expression of the Ll.ltrB intron were BL21(DE3)lon–ompT (Studier and Moffatt 1986) and BLR(DE3)lon–ompT–recA::Tn10(TcR) (Novagen, Madison, WI). Both strains contain the DE3 prophage for the synthesis of phage T7 RNA polymerase. The strains were grown in LB or SOB medium (Sambrook et al. 1989). Antibiotics were added: 100 μg/ml of ampicillin; 25 μg/ml of chloramphenicol; 25 μg/ml of tetracycline.
Oligonucleotides
Oligonucleotides used in this study were 5′exon, 5′-GTTATGGATGTGTTCACGATCGACGTGGG; 3′exon, 5′-CGGAATTCCCAGTATAAAGATTCGTAGAA; amp, 5′-AGTTGCCTGACTCCCCGTCG; ex2, 5′-TTGGGATCCTCATAA GCTTTGCCGC; expr, 5′-AAAACCTCCATATGAAACCAACAATG; KS, 5′-TCGAGGTCGACGGTATC; LtrB-35, 5′-CGGGATCCTTGCAACCCACGTCGATCGTG; N-LtrA, 5′-CAAA GGATCCGATGAAACCAACAATGGCAA; NdeILTR5, 5′AGTGGCTTCCATATGCTTGGTCATCACCTCATC; Nde ILTR3, 5′-GGTAGAACCATATGAAATTCCTCCTCCCTAA TCAATTTT; primer 1, 5′-CCGTGCTCTGTTCCCGTATC AGC; primer 2, 5′-CTTTAGGAATGACTTTCCAGTC; primer 3, 5′-AAGCTTAGAGAAAAATAATGCGGTGC; primer 4, 5′-CTTTAGGAATGACTTTCCAGTC; probe A, 5′-AAAAATGATATGGTGAAGTAGGGA; probe B, 5′-AAAAATGATA TGGTTATGGATGTG; SK, 5′-CGCTCTAGAACTAGTGG ATC; Ssch2, 5′-TTGCAACCCACGTCGATCGTGAACACAT CGATAACCATATCATTTTTAATTCTACGAATCTTTATACTGG.
Recombinant plasmids
Plasmids in the pLI1 series are derivatives of pET-11a (Studier et al. 1990). pLI1 contains the Ll.ltrB intron and flanking exons cloned downstream of the phage T7 promoter in pET-11a. The plasmid was constructed by blunt-end ligation of the 2.8-kb HindIII fragment of pLE12 (Mills et al. 1996) into the XbaI site of pET-11a (Novagen), after filling in the 5′ overhangs with Klenow polymerase (Life Technologies, Gaithersburg, MD).
pLI1P contains the LtrA ORF cloned directly downstream of the phage T7 promoter in pET-11a. To construct this plasmid, the LtrA ORF of pLE12 was amplified by PCR with the primers expr and ex2. The PCR product was digested with NdeI and BamHI and cloned between the corresponding sites of pET-11a. To ensure that no unintended changes were introduced via PCR, the 2.4-kb SspI fragment, which includes most of the LtrA ORF, was replaced with that from pLI1, and the remaining 147-nucleotide region of the ORF between the ATG and SspI site was sequenced.
pLI1–NHis has His6 and Xpress (DLYDDDDK) epitope tags at the amino terminus of the LtrA ORF. To construct this plasmid, the ORF and 3′ exon of pLE12 were amplified by PCR with the primers N-LtrA and ex2. The PCR product was digested with BamHI, gel-purified, and cloned into the BamHI site of pRSET B (Invitrogen, San Diego, CA) to generate the intermediate plasmid pInt-N. In the second step, a region of pLE12 extending from the 5′ exon to the initiation codon of the LtrA ORF was amplified by PCR with the primers NdeILTR5 and NdeILTR3. The PCR product was cleaved with NdeI, gel purified, and cloned in the NdeI site of pInt-N. Transformants were screened to identify a clone containing the insert in the correct orientation. To ensure that no unintended changes were introduced by PCR, the 2.4-kb SspI fragment was replaced with that of pLI1, and the remaining region was sequenced as described above for pLI1P.
pLI1–FS is a derivative of pLI1, which has a frameshift after codon 124 of the LtrA ORF, introduced by linearizing pLI1 at the AgeI site in the LtrA ORF and religating after filling in the 4-nucleotide 5′ overhangs with Klenow DNA polymerase (Life Technologies). pLI1–DD− and pLI1–EBS1-6C are derivatives of pLI1 that were constructed by PCR mutagenesis. pLI1–DD− has the conserved YADD sequence in the RT domain changed to YAAA, and pLI1–EBS1-6C has a single nucleotide change in EBS1 (G284C) and a complementary change in IBS1 (E1–6G) to permit splicing. In each case, the region generated by PCR was sequenced completely to insure that no adventitious mutations had been introduced.
Plasmids in the pLI2 series are derivatives of pBSIIKS+ (Stratagene, La Jolla, CA). pLI2 contains the 2.8-kb Ll.ltrB HindIII fragment cloned into the HindIII site of pBSIIKS+ (Stratagene) in transcriptional alignment with the phage T3 and lac promoters.
pLI2–ΔORF, which has a large deletion in the intron ORF, was derived from pLI2 by inverse PCR, such that amino acids 40–572 of LtrA were replaced with the amino acids TR, and an MluI site was created at the deletion joint.
pLI2–ΔORFkanR was constructed by blunt-end ligation of the 1.3-kb SalI fragment containing the kanR gene from pUC4K (Pharmacia, Piscataway, NJ) into the MluI site of pLI2–ΔORF, after filling in both the 5′ overhangs with Klenow polymerase (Life Technologies).
pLHS, which was used to synthesize DNA target sites for endonuclease and reverse splicing assays, contains a 70-nucleotide sequence of ligated ltrB exons 1 and 2, extending from 35 nucleotides upstream to 35 nucleotides downstream of the intron insertion site, cloned between BamHI and EcoRI sites of pBSKS+ (Stratagene). The insert was generated from pLtrBH (D. Mills and G.M. Dunny, unpubl.) by PCR with the primers LtrB–35 and 3′ exon. pLHS–IBS1-6G, which has a C → G change at E1–6, was constructed similarly to pLHS, except that the DNA template was the 70-mer Ssch2.
Expression of LtrA protein
On the basis of optimization experiments with pLI1–NHis, the following protocol was adopted for expression of the LtrA protein in E. coli. E. coli strains BL21(DE3) or BLR(DE3) were transformed with the expression plasmid, and single colonies were selected on LB plates containing the appropriate antibiotics. For starter cultures, the single colonies were inoculated into 2 ml of LB medium containing antibiotics, and the cultures were shaken at 300 rpm at 37°C overnight. One milliliter of the overnight culture was then inoculated into 100 ml of LB medium containing antibiotics in a 500-ml Ehrlenmeyer flask and grown at 37°C in a rotary shaker (300 rpm) for 2–4 hr, until OD595 was 0.4–0.8. For induction, IPTG was added to a final concentration of 1 mm, and the incubation was continued for 3 hr. After induction, cells were collected by centrifugation in a Beckman JA-14 rotor (2200g for 5 min at 4°C), and washed with 25 ml of ice-cold 150 mm NaCl. The washed cell pellet was resuspended in 4 ml of ice-cold buffer A [50 mm Tris-HCl at pH 7.5, 1 mm EDTA, 1 mm DTT, 10% (vol/vol) glycerol], and lysozyme (Sigma, St. Louis, MO) was added to a final concentration of 2 mg/ml. The cells were then lysed by three cycles of freeze–thawing between −70°C and 37°C, followed by addition of 4 volumes of HKCTD (500 mm KCl, 50 mm CaCl2, 25 mm Tris-HCl at pH 7.5, 5 mm DTT) and sonication for 5 sec or until the mixture was no longer viscous.
Preparation of RNP particles
To prepare RNP particles, 5 ml of the E. coli lysate, which had been centrifuged in a Beckman JA-20 rotor (14,000g for 15 min at 4°C) to remove insoluble material, was layered over 5 ml of 1.85 m sucrose containing HKCTD and centrifuged in a Beckman Ti50 rotor (50,000g for 17 hr at 4°C). The resulting RNP pellet was washed with 1 ml of ice-cold distilled water and then dissolved in 25 μl of ice-cold 10 mm Tris-HCl at pH 8.0, and 1 mm DTT. After removal of insoluble material in a microcentrifuge (15,000g for 5 min at 4°C), the RNPs were stored frozen at −70°C. The yield of RNP particles was 25–50 OD260 units per 100 ml of culture, with 1 OD260 unit of RNPs containing 0.3–3 μg of LtrA protein. For in vitro splicing or reconstitution experiments, to minimize nuclease activity, the RNPs were further purified by an additional centrifugation through a 1.85 m sucrose cushion, as described above.
RT assays
RT activity with poly(rA)–oligo(dT)18 was assayed in 10 μl of reaction medium containing 10 mm KCl, 10 mm MgCl2, 50 mm Tris-HCl at pH 7.5, 5 mm DTT, 0.025 OD260 units of RNP particles, 1 μg of poly(rA)–oligo(dT)18, 10 μCi of [α-32P]dTTP (3000 Ci/mmole; DuPont-New England Nuclear), and 1 μg of RNase A (Sigma), as described for the yeast aI2 intron (Moran et al. 1995). The products were spotted on a DE81 filter (Whatman, Fairfield, NJ), which was processed and counted for Cerenkov radioactivity. In contrast to the yeast intron, where RNase A is necessary to release the RT from endogenous RNA templates (Moran et al. 1995; S. Zimmerly and A.M. Lambowitz, unpubl.), the RNP particle preparations containing the LtrA protein had essentially the same RT activity in the presence or absence of RNase A (not shown). The RT activity with the artificial template was optimal at 0–25 mm KCl and 5–10 mm Mg2+ and was strongly inhibited at higher salt concentrations.
Endogenous reverse transcription reactions were carried out in 10 μl of reaction medium containing 10 mm KCl, 10 mm MgCl2, 50 mm Tris-HCl at pH 7.5, 5 mm DTT, 0.025 OD260 units of RNP particles, 200 μM each of dATP, dCTP, and dGTP, and 10 μCi of [α-32P]dTTP (3000 Ci/mmole; DuPont-New England Nuclear). In some cases, a synthetic DNA oligonucleotide primer (20 pmoles) complementary to endogenous RNA was added to the reactions (see Fig. 2). Reactions were initiated by addition of the RNP particles, incubated for 10 min at 37°C, and assayed for incorporation of [32P]-dTTP into high molecular weight material by spotting on a DE81 filter, as described above for exogenous substrate assays. RT activity with endogenous RNA templates was optimal at 0–50 mm KCl and 5–20 mm MgCl2.
Analysis of in vivo splicing products
Total cellular RNA was isolated from E. coli BL21(DE3) containing the indicated plasmids. Cells were grown in TBYE (1.0% tryptone, 0.5% NaCl, 0.1% yeast extract) with ampicillin to an OD650 of 0.2 and induced with 1 mm IPTG. RNA was extracted by the lysozyme freeze–thaw method, followed by phenol extraction and ethanol precipitation (Belfort et al. 1990).
For Northern hybridizations, total cellular RNA (15 μg) was separated in a 1.5% agarose/2.2 m formaldehyde gel in 10 mm sodium phosphate (pH 6.5) running buffer and blotted to a Hybond N membrane (Amersham, Arlington Heights, IL), as described (Sambrook et al. 1989). The RNA was cross-linked to the membrane with a UV Stratalinker 2400 (Stratagene) under auto-cross-link conditions. Blots were hybridized in Rapid-hyb buffer (Amersham) for 2 hr at 45°C with 5′ 32P-labeled DNA oligonucleotide probes.
For primer extension analysis, each reaction contained 5 μg of cellular RNA in 62.5 mm Tris-HCl at pH 8.0, 75 mm NaCl, 12.5 mm DTT with 0.2 pmole of 5′ 32P-labeled primers. Samples were incubated at 60°C for 3 min and placed on ice. Buffer containing 50 mm Tris-HCl at pH 8.0, 60 mm NaCl, 10 mm DTT, 6 mm MgOAc, 0.47 mm each dNTP, and 3 units of AMV RT (Life Technologies) was added, and the samples were incubated at 48°C for 30 min. The reaction was stopped by adding formamide–dye mix (Belfort et al. 1990). Samples were heated to 95°C for 3 min before loading on a 10% polyacrylamide (19:1)/7 m urea gel. DNA sequencing ladders generated with primers 1 and 2 were run in parallel lanes.
In vitro splicing
Protein-assisted splicing reactions were carried out with MNase-digested RNP particles purified through two ultracentrifugation cycles (see above). After decanting the supernatant and washing the tube walls with ice-cold distilled water, the final RNP pellet was dissolved in 25–100 μl of 10 mm Tris-HCl at pH 8.0, 1 mm DTT to a concentration of 0.1 to 0.2 OD260 units/μl. For MNase digestion, an equal volume of 40 mm Tris-HCl at pH 7.5, 10 mm CaCl2, 20 mm DTT was added. Digestion with MNase (15 units; Pharmacia) was carried out for 10 min at 22°C and then terminated by addition of 10 mm EGTA.
Substrates for in vitro splicing were 32P-labeled in vitro transcripts synthesized with phage T3 RNA polymerase (Stratagene) from pLI2–ΔORF linearized with EcoRI. In vitro transcription was in 50 μl of reaction medium (Caprara et al. 1996) containing 1 μg of template DNA, 0.5 mm each of ATP, GTP, CTP, 0.4 mm UTP, and 5 μCi [α-32P]-UTP (3000 Ci/mmole) for 2 hr at 37°C. The DNA template was digested with DNase I (Pharmacia; 10 units for 20 min at 37°C), and the precursor RNA was purified in a denaturing 4% polyacrylamide gel.
For splicing reactions, the 32P-labeled precursor RNA (16 nm; ∼100,000 cpm) was dissolved in 10 μl of low salt reaction medium containing 100 mm NaCl, 10 mm MgCl2, 40 mm Tris-HCl at pH 7.5, 10 mm DTT, and 2 units of RNasin (Amersham), then incubated at 65°C for 1 min and slowly cooled to 37°C in a waterbath. After addition of 1 μg of E. coli tRNA (Sigma) carrier as protection against nonspecific nucleases, splicing reactions were initiated by addition of the MNase-treated RNP particles (0.1 OD260 units), incubated at 37°C for different times (see Fig. 5), and terminated by addition of EDTA to a final concentration of 15 mm. The products were incubated with proteinase K (Sigma; 1 μg for 5 min at 25°C), extracted with phenol–chloroform–isoamyl alcohol (25:24:1), ethanol precipitated, and analyzed in a denaturing 4% polyacrylamide gel. Self-splicing reactions were carried out similarly in high salt (HS) reaction medium containing 500 mm NH4Cl, 50 mm MgCl2, 50 mm Tris-HCl at pH 7.5, 10 mm DTT, 2 units of RNasin, and 1 μg of E. coli tRNA (Sigma) carrier, and terminated by addition of EDTA to a final concentration of 75 mm.
Preparation of DNA substrates for reverse splicing and endonuclease reactions
For most experiments, 129-bp internally or 5′ end-labeled DNA substrates containing the junction of exons 1 and 2 of the ltrB gene were synthesized from pLHS by PCR with primers KS and SK, by use of procedures described previously (Zimmerly et al. 1995b; Yang et al. 1996). The DNA substrates were purified in a 6% nondenaturing polyacrylamide gel.
For the experiment of Figure 7B, DNA substrates labeled at the 5′ end of the sense or antisense strands were prepared from pLHS linearized at the NotI or XhoI sites, respectively. The digested plasmids were dephoshorylated with calf intestine alkaline phosphatase (Boehringer Mannheim, Indianapolis, IN), 5′-end-labeled with phage T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ-32P]ATP (3000 Ci/mmole; DuPont-New England Nuclear) (Sambrook et al. 1989), and redigested with KpnI or SacI for the sense and antisense strand substrates, respectively. DNA substrates labeled at the 3′ end of the sense or antisense strands were prepared by digesting pLHS with KpnI or SacI, respectively. The digested plasmids were 3′-end-labeled with terminal deoxynucleotidyl transferase (TdT) (Life Technologies) and [α-32P]dGTP (3000 Ci/mmole; Dupont-New England Nuclear) (Collins and Hunsaker 1985), and then digested with NotI or XhoI for the sense and antisense strand DNA substrates, respectively. The 5′- and 3′-end-labeled DNA substrates (NotI–KpnI; 144 bp, XhoI–SacI; 145 bp) were purified in a 2% agarose gel.
DNA endonuclease and reverse splicing assays
DNA endonuclease and reverse splicing assays were essentially as described for the yeast aI1 and aI2 introns (Zimmerly et al. 1995a,b; Yang et al. 1996), except that the reaction medium (10 mm KCl, 10 mm MgCl2, 50 mm Tris-HCl at pH 7.5, and 5 mm DTT) was optimized for the Ll.ltrB intron. Unless specified otherwise, 150,000 cpm of the DNA substrate (∼125 fmoles of 5′-end-labeled substrate or ∼30 fmoles of internally-labeled substrate) were used per reaction. RNP particles were tested for sensitivity to pretreatment with RNase A or proteinase K, and reaction products were tested for sensitivity to nucleases, as described (Zimmerly et al. 1995a,b). Alkali treatment of reaction products was carried out in 50 μl of 0.2 n NaOH and 2 mm EDTA for 5 min at 95°C.
Reconstitution of the Lactococcus intron endonuclease with LtrA protein and in vitro-synthesized intron RNA
The RNA component for reconstitution was generated by in vitro transcription of pLI2–ΔORF, pLI2–ΔORFkan, or pLI1. The plasmids were linearized with BamHI and transcribed with phage T3 or T7 RNA polymerases (Caprara et al. 1996). To generate the intron RNA for reconstitution, the in vitro transcript (30–50 μg) was self-spliced for 60 min at 37°C in 100 μl of 1 m NH4Cl, 100 mm MgCl2, 50 mm Tris-HCl at pH 7.5. Prior to reconstitution, the RNA was heated to 85–90°C for 2 min, then stored on ice. The protein fraction for reconstitution was prepared by MNase digestion of purified RNP particles containing LtrA protein expressed from pLI1. The RNP particles (1.0 OD260) were resuspended in 40 μl of 10 mm Tris-HCl at pH 7.5, 10 mm MgCl2, 2.5 mm CaCl2, 5 mm DTT and incubated with MNase (36 units; Pharmacia) for 10 min at 22°C, after which the MNase was inactivated by addition of EGTA to 7.5 mm. For reconstitution and reverse splicing, 0.05 OD260 units of the MNase-treated RNP particles was mixed with 1 μg of the spliced RNA in 20 μl of the standard reverse splicing reaction medium (see above) and 30 or 60 fmoles of 32P-labeled DNA substrate. In the experiment of Figure 9B, the mixture was preincubated on ice for 10 min before initiating the reverse splicing reaction by raising the temperature to 37°C. The reconstitution mixtures were tested for sensitivity to pretreatment with 2 μg of RNase A (Sigma) plus 2.5 units of RNase T1 (Boehringer Mannheim) or proteinase K (2 μg; Sigma) for 5 min at 22°C prior to adding the DNA substrate.
Acknowledgments
We thank Michelle Simons for technical assistance, Benoit Cousineau and Michael Cusick for maturase mutants, and Richard Lease for comments on the manuscript. This work was supported by National Institutes of Health grants GM37949 to A.M.L., GM39422 and GM44844 to M.B., and a grant from the Minnesota–South Dakota Dairy Research Center to G.M.D. H.W. is the recipient of an Erwin Schroedinger Postdoctoral Fellowship from the Austrian Science Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lambowitz.1@osu.edu; FAX (614) 688-3555.
References
- Belfort M, Ehrenman K, Chandry PS. Genetic and molecular analysis of RNA splicing in Escherichia coli. Methods Enzymol. 1990;181:521–539. doi: 10.1016/0076-6879(90)81149-o. [DOI] [PubMed] [Google Scholar]
- Bonen L. Trans-splicing of pre-mRNA in plants, animals, and protists. FASEB J. 1993;7:40–46. doi: 10.1096/fasebj.7.1.8422973. [DOI] [PubMed] [Google Scholar]
- Caprara MG, Mohr G, Lambowitz AM. A tyrosyl-tRNA synthetase protein induces tertiary folding of the group I intron catalytic core. J Mol Biol. 1996;257:512–531. doi: 10.1006/jmbi.1996.0182. [DOI] [PubMed] [Google Scholar]
- Carignani G, Groudinsky O, Frezza D, Schiavon E, Bergantino E, Slonimski P. An mRNA maturase is encoded by the first intron of the mitochondrial gene for the subunit I of cytochrome oxidase in S. cerevisiae. Cell. 1983;35:733–742. doi: 10.1016/0092-8674(83)90106-x. [DOI] [PubMed] [Google Scholar]
- Collins ML, Hunsaker WR. Improved hybridization assays employing tailed oligonucleotide probes: A direct comparison with 5′-end-labeled oligonucleotide probes and nick-translated plasmid probes. Anal Biochem. 1985;151:211–224. doi: 10.1016/0003-2697(85)90168-x. [DOI] [PubMed] [Google Scholar]
- Costa M, Dème E, Jacquier A, Michel F. Multiple tertiary interactions involving domain II of group II self-splicing introns. J Mol Biol. 1997;267:520–536. doi: 10.1006/jmbi.1996.0882. [DOI] [PubMed] [Google Scholar]
- Curcio MJ, Belfort M. Retrohoming: cDNA-mediated mobility of group II introns requires a catalytic RNA. Cell. 1996;84:9–12. doi: 10.1016/s0092-8674(00)80987-3. [DOI] [PubMed] [Google Scholar]
- Daniels D, Michels WJ, Pyle AM. Two competing pathways for self-splicing by group II introns: A quantitative analysis of in vitro reaction rates and products. J Mol Biol. 1996;256:31–49. doi: 10.1006/jmbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- Eskes R, Yang J, Lambowitz AM, Perlman PS. Mobility of yeast mitochondrial group II introns: Engineering a new site specificity and retrohoming via full reverse splicing. Cell. 1997;88:865–874. doi: 10.1016/s0092-8674(00)81932-7. [DOI] [PubMed] [Google Scholar]
- Ferat J-L, Michel F. Group II self-splicing introns in bacteria. Nature. 1993;364:358–361. doi: 10.1038/364358a0. [DOI] [PubMed] [Google Scholar]
- Ferat J-L, Le Gouar M, Michel F. Multiple group II self-splicing introns in mobile DNA from Escherichia coli. CR Acad Sci Paris. 1994;317:141–148. [PubMed] [Google Scholar]
- Guo, H., S. Zimmerly, P.S. Perlman, and A.M. Lambowitz. 1997. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA, EMBO J. (in press). [DOI] [PMC free article] [PubMed]
- Jarrell AK, Dietrich RC, Perlman PS. Group II intron domain 5 facilitates a trans-splicing reaction. Mol Cell Biol. 1988;8:2361–2366. doi: 10.1128/mcb.8.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennell JC, Moran JV, Perlman PS, Butow RA, Lambowitz AM. Reverse transcriptase activity associated with maturase-encoding group II introns in yeast mitochondria. Cell. 1993;73:133–146. doi: 10.1016/0092-8674(93)90166-n. [DOI] [PubMed] [Google Scholar]
- Knoop V, Brennicke A. Evidence for a group II intron in Escherichia coli inserted into a highly conserved reading frame associated with mobile DNA sequences. Nucleic Acids Res. 1994;22:1167–1171. doi: 10.1093/nar/22.7.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Perlman PS. Involvement of aminoacyl-tRNA synthetases and other proteins in group I and group II intron splicing. Trends Biochem Sci. 1990;15:440–444. doi: 10.1016/0968-0004(90)90283-h. [DOI] [PubMed] [Google Scholar]
- Lazowska J, Meunier B, Macadre C. Homing of a group II intron in yeast mitochondrial DNA is accompanied by unidirectional co-conversion of upstream-located markers. EMBO J. 1994;13:4963–4972. doi: 10.1002/j.1460-2075.1994.tb06823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Ferat J-L. Structure and activities of group II introns. Annu Rev Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]
- Mills D, McKay LL, Dunny GM. Splicing of a group II intron involved in the conjugative transfer of pRS01 in Lactococci. J Bacteriol. 1996;178:3531–3538. doi: 10.1128/jb.178.12.3531-3538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr G, Perlman PS, Lambowitz AM. Evolutionary relationships among group II intron-encoded proteins and identification of a conserved domain that may be related to maturase function. Nucleic Acids Res. 1993;21:4991–4997. doi: 10.1093/nar/21.22.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Mecklenburg KL, Sass P, Belcher SM, Mahnke D, Lewin A, Perlman PS. Splicing defective mutants of the COXI gene of yeast mitochondrial DNA: Initial definition of the maturase domain of the group II intron aI2. Nucleic Acids Res. 1994;22:2057–2064. doi: 10.1093/nar/22.11.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Zimmerly S, Eskes R, Kennell JC, Lambowitz AM, Butow RA, Perlman PS. Mobile group II introns of yeast mitochondrial DNA are novel site-specific retroelement. Mol Cell Biol. 1995;15:2828–2838. doi: 10.1128/mcb.15.5.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullany P, Pallen M, Wilks M, Stephen JR, Tabaqchali S. A group II intron in a conjugative transposon from the gram-positive bacterium, Clostridium difficile. Gene. 1996;174:145–150. doi: 10.1016/0378-1119(96)00511-2. [DOI] [PubMed] [Google Scholar]
- Pyle AM. Inside an intron invasion. Nature. 1996;381:280–281. doi: 10.1038/381280a0. [DOI] [PubMed] [Google Scholar]
- Saldanha RJ, Mohr G, Belfort M, Lambowitz AM. Group I and group II introns. FASEB J. 1993;7:15–24. doi: 10.1096/fasebj.7.1.8422962. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sharp PA. Five easy pieces. Science. 1991;254:663. doi: 10.1126/science.1948046. [DOI] [PubMed] [Google Scholar]
- Shearman C, Godon J, Gasson M. Splicing of a group II intron in a functional transfer gene of Lactococcus lactis. Mol Microbiol. 1996;21:45–53. doi: 10.1046/j.1365-2958.1996.00610.x. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Wang GH, Zoulim F, Leber EH, Kitson J, Seeger C. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J Virol. 1994;68:8437–8442. doi: 10.1128/jvi.68.12.8437-8442.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zimmerly S, Perlman PS, Lambowitz AM. Efficient integration of an intron RNA into double-stranded DNA by reverse splicing. Nature. 1996;381:332–335. doi: 10.1038/381332a0. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Eskes R, Yang J, Perlman PS, Lambowitz AM. A group II intron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell. 1995a;83:529–583. doi: 10.1016/0092-8674(95)90092-6. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995b;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]