

Abstract
Introduction
Over the past ten years, ankyrin polypeptides have emerged as critical players in cardiac excitation-contraction coupling. Once thought to solely play only a structural role, loss-of-function variants in genes encoding ankyrin polypeptides have highlighted how this protein mediates the proper subcellular localization of the various electrical components of the excitation-contraction coupling machinery. A large body of evidence has revealed how the disruption of this localization is the primary cause of various cardiomyopathies, ranging from long QT syndrome 4, to sinus node disease, to more common forms of arrhythmias.
Areas Covered
This review details the varied roles that ankyrin polypeptides play in excitation-contraction coupling in the heart and the development of ankyrin-specific cardiomyopathies. It will further discuss how ankyrin polypeptides may be involved in structural and electrical remodeling of the heart, post-myocardial infarct. Attention is given to how ankyrin interactions with membrane bound ion channels may regulate these channels’ response to stimuli. Special attention is given to exciting new data, which may offer the potential for unique therapies, for not only combating heart disease, but which also holds promise for wider applications to various disease states.
Expert Opinion
The ankyrin family of adapter proteins is emerging as an intimate player in cardiac excitation-contraction coupling. Until recently, these proteins have gone largely unappreciated for their importance in proper cardiac function. New insights into how these proteins function within the heart are offering potentially new avenues for therapies against cardiomyopathy.
1. Introduction
The function of all excitable tissues and organs relies upon the proper, coordinated electrical response to a stimulus. This coordination depends on the spatial and temporal organization of individual electrical components within single cells—i.e. a neuron, cardiomyocyte, or pancreatic beta cell. Through the evolution of membrane ion channels, transporters, receptors, signaling proteins, and specialized cytoskeletal elements, the excitable cell has developed robust molecular machinery that exquisitely integrates these component parts into a dynamic electrical system.
The excitation-contraction (EC) coupling machinery of the heart is an excellent example of how vertebrates have evolved both critical structural and molecular components to synchronize and constantly tune their physiological response to a wide variety of stimuli. Briefly, upon generation of an action potential (AP) in the sinoatrial node (SAN), the heart’s natural pacemaker, a highly coordinated cascade of electrical events is initiated. This cascade ultimately leads to the rhythmic contraction of the heart and proper perfusion of blood throughout the cardiovascular system [1;2].
As the AP wavefront propagates through the heart it depolarizes the membrane potential (Em) of the ventricular cardiomyocyte by opening voltage-gated sodium channels (Nav) embedded in the sacrolemma (SL) membrane. This depolarization activates voltage-gated calcium channels (Cav). Ca2+ entry though Cav channels triggers further Ca2+ release from internal sarcoplasmic reticulum (SR) Ca2+ stores. The combined Ca2+ influx into the cytosol raises free cytosolic [Ca2+] ([Ca]i) allowing Ca2+ to activate myofilaments, thus causing contraction. For relaxation to occur, Ca2+ must be rapidly and efficiently removed from the cytosol. The two largest contributors to Ca2+ removal are the SR Ca ATPase (SERCA) and the Na/Ca exchanger (NCX). SERCA extrudes Ca2+ out of the cytosol and sequesters it into the SR for release on the next beat [3], while the (NCX) mediates Ca2+ efflux out of the cell and into the extracellular space. As Nav and CaV channels inactivate, the myocyte begins to repolarize, primarily via activation of a diverse family of voltage-gated potassium channels (Kv). Open Kv channels mediate outward polarizing currents. This resets the membrane potential and returns the myocyte back to resting conditions, and the sequence is repeated upon the next AP. Even in this simplified example of EC coupling, the sequence and magnitude of each event are critical to proper cardiac function (See Figure 1). Disruption of any of these events may have devastating consequences at the level of the cell, organ, and/or organism [4-16].
Figure 1).
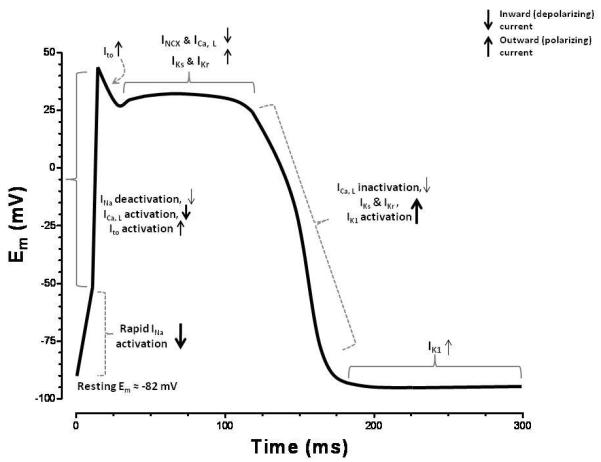
Diagram detailing the AP wavefront of a single contraction in a cardiac myocyte and the relative contributions of the pertinent ionic currents. The rapid upstroke in the Em is largely facilitated by the opening of the voltage gated Na-channels. Secondary to this, voltage-gated Ca channels also assist in depolarization. As the membrane further depolarizes, voltage gated K-channels open allowing an outward polarizing current to pass. The balance of these depolarizing and polarizing currents is represented in the AP plateau. As the depolarizing channels slowly begin to deactivate, the K-channels start to dominate and quickly repolarize the cell to its resting Em. The relative strength of each current is represented by the thickness of the arrows at each variable stage of the AP.
The importance of Na+, Ca2+, and K+ ion homeostasis in membrane excitability is well established (in the case of Ca2+, for more 125 years [17]). With the advancement of voltage-clamp and patch-clamp techniques, the role of single ion channels and transporters could be quantitatively studied. This enabled investigators to isolate the role each played in the varied electrical currents present in all excitable cells [18;19]. However, for all that was known regarding the contribution of individual channels and transporters, little was known regarding the importance of their specific subcellular localization. In 1987 Rios and Brum highlighted the significance of proper cellular organization by demonstrating that proper Ca2+ release in the skeletal muscle requires a direct coupling of the SL Ca channel with the SR-bound Ca release channel, ryanodine receptor (RyR) [20], suggesting a high degree of protein compartmentalization. Disruption of this complex effectively destroys skeletal muscle contractility. Subsequent studies would show similar importance of subcellular localization of the electrical components and regulatory molecules of the EC coupling machinery in the heart [21-26].
Despite the obvious importance of proper subcellular localization, a critical, unanswered question remained: how does a myocyte properly traffic, target and retain each of these constituent parts to the appropriate subcellular domain? Over the past 15 years this question has become an area of expanding research. This emerging field is yielding interesting and surprising answers while unmasking unique human disorders and potentials for new therapies in the fight against many excitable cell diseases.
This review focuses on the role of the ankyrin family of adapter proteins in the coordination of the EC coupling machinery in the heart. Particular attention will be paid to disease states very recently linked to primary ankyrin dysfunction, such as ankyrin-B syndrome and SAN dysfunction, as well as current therapies in use. Additionally, this review will discuss recent findings demonstrating ankyrin-based regulation of the ion channel response to unique stimuli. Finally, we will review possible pathways for ankyrin targeting and trafficking within the heart and how this is regulated following myocardial infarction.
2. Ankyrin Genes and Structure
Ankyrin polypeptides have emerged as an unexpected and intimate player in EC coupling. For decades, ankyrin polypeptides were thought to solely play a structural role, based on initial studies of ankyrin-R (AnkR) in erythrocytes [27;28]. However, in the 1980’s ankyrin expression was demonstrated in the brain, implicating a more general role in many tissues [29]. Indeed, ankyrins are now known to be present, and play unique roles, in tissues as varied as cardiac myocardium, photoreceptors, neurons, epithelial cells, and pancreatic beta cells. [30-33].
Much is known regarding the structure and genetic organization of ankyrin polypeptides [34;35]. Specificity of cellular and tissue expression is achieved through the alternative splicing of three ankyrin genes: ANK1 (encoding ankyrin-R), ANK2 (ankyrin-B), and ANK3 (ankyrin-G). Although it is known to be expressed in neurons and striated muscle [34;36;37], AnkR is largely restricted to erythrocytes. Ankyrin-B (AnkB) and ankyrin-G (AnkG) are ubiquitously expressed and are the primary subtypes in the heart. All ankyrin subtypes share a high degree of homology [38]. Prototypically, ankyrin polypeptides are composed of four functional domains: the membrane-binding domain (MBD), the spectrin-binding domain (SBD), the death domain, and the regulatory C-terminal domain (CTD) [39]. Each of these domains plays a specific role in the function of the protein.
The MBD is a sequence of 24 ANK repeats comprising a suprahelical structure. The ANK repeat is a common motif within over one hundred vertebrate proteins and mediates protein-protein interactions. In fact the ANK repeat is found in a host of critical cardiac and non-cardiac proteins including ANKRD1 (CARP), transient receptor potential cation (TRPC) channels, and SH3 and multiple ankyrin (SHANK) repeat domains proteins. The MBD structure mediates the association of ankyrins with nearly all respective binding partners. The MBD binds to cell adhesion molecules thus conferring structural stability of the cell and tissue [34]. Critically, this domain also binds to integral membrane proteins including the NCX, the ATP-sensitive potassium channel (KATP), the Na/K ATPase (NKA), the voltage gated sodium channel (Nav1.5), the inositol 1,4,5-trisphosphate receptor (IP3R) and the anion exchanger in many cell types, the cardiomyocyte included [35;40-43]. More importantly, the MBD acts as a multivalent binding domain. Thus, it plays a significant role in the subcellular coordination of functionally coupled proteins and their regulatory substrates to microdomains [26].
As its name implies, the spectrin-binding domain binds the cytoskeletal protein β-spectrin and confers structural continuity between ankyrin-associated proteins and the cytoskeleton [44;45]. As with the MBD, the role of the SBD extends beyond structural integrity. In the heart, this domain also binds to the multifunctional protein phosphatase PP2A, a key regulator of EC coupling via its B56α subunit [46;47].
Collectively, the death domain and CTD make up the ankyrin regulatory domain. This domain provides two unique functions. First, it regulates how integral membrane proteins (such as Na channels or the NCX) bind to the MBD, while also regulating ankyrin/spectrin interactions. Second, this domain is involved in the subcellular targeting of ankyrins to specific cellular domains [48-52]. The importance of the CTD for ankyrin function is highlighted by the fact that nearly all ankyrin-B human arrhythmia loss-of-function mutations are found within this domain [53;54].
The significance of the ability of ankyrin polypeptides to coordinate the localization of functionally coupled proteins should be noted. Michaely et al. determined the crystal structure of the 24 ANK repeats of the MBD [55]. They determined that the axial length of this suprahelical segment was 132 Å, with a radius of only 45 Å. It can quickly be appreciated that the binding of functionally related proteins (such as NKA and NCX linked by [Na]i) to such a small area would immediately attenuate any limitations introduced by diffusion. Even if one assumes 1:1 binding of the channels in the complex and then those being diametrically opposed, they would be no more than ~9 nm apart. This idea further extends to PP2A, CaMKII and KATP localization within ankyrin-based complexes. Not only would functionally related proteins be spatially linked to a single microdomain, so would their regulatory substrates.
3.0 Ankyrin-B Syndrome
In 1995, linkage between a mutation on chromosome 4q25-27 and a rare form of long QT syndrome (LQTS) was described [56]. It would later be shown that this mutation leads to an A-to-G mis-sense mutation at base 4274 in the ANK2 gene, resulting in a glutamic acid to glycine substitution at residue 1425 (E1425G). This specific variant results in a loss-of-function of AnkB [57;58]. Notably, this was the first example of LQTS not associated with an ion channel mutation, confirming that ankyrin polypeptides play primary roles in cardiovascular disease states [59].
The LQTS phenotype was initially characterized in a large French family with a history of unexplained sudden death [58]. Determined to be a unique form of LQTS (called LQTS 4), the disease is described by the common LQT-related phenotypes of prolonged rate corrected QT interval, syncope and risk of sudden death. However, beyond these typical LQTS signatures, LQTS 4 patients exhibited sinus node bradycardia and arrhythmia, delayed conduction or conduction block, catecholaminergic polymorphic ventricular tachycardia (CPVT), and idiopathic ventricular fibrillation. ECGs revealed high incidence of atrial fibrillation (AF), junctional escape rhythm, and a notched biphasic T wave [58]. Since the latter are atypical of LQTS, the combined cardiac phenotypes were later renamed “ankyrin-B syndrome” [53]. Since the initial discovery of the E1425G mutation, multiple other AnkB mutations (nine total) in additional families have been characterized; each demonstrating the same general phenotype, with varying degrees of penetrance and severity in the human population [54;57;58;60]. ANK2 loss-of-function variant carriers are generally treated with pacemakers and/or beta-blockers [58].
3.1 Ankyrin-B Syndrome and the EC Coupling Machinery
The cellular phenotypes of ankyrin-B syndrome can ultimately be traced to the disruption of the EC coupling machinery. Since in-depth studies on human myocytes are impractical, a mouse model of AnkB deficiency was developed. The mouse is heterogeneous for a null mutation in murine Ank2 (AnkB+/−), and recapitulates many of the human phenotypes, including: a significant decrease in AnkB activity, sinus node bradycardia, and CPVT. Importantly, attempts to rescue the phenotype with expression of an exogenous GFP-tagged WT AnkB construct were successful, while exogenous expression of GFP-tagged AnkB containing the human E1425G mutation was not [54;58].
While the NCX has been a known binding partner of AnkB, the critical importance of this association was only recently elucidated [26;61]. Along with NCX, AnkB binds to the Na/K ATPase. This complex is localized to the Z-line and coupled to the T-tubular domain. Together, these proteins are critical for Na and Ca homeostasis in cardiac EC coupling. Primarily, the NCX uses the energy of the Na+ gradient to extrude Ca2+ out of the cytosol during contraction. In turn, the Na/K ATPase actively pumps Na+ out of and K+ into the myocyte, thus establishing the useful chemical gradient. Both the Na/K ATPase and the NCX are down-regulated in AnkB+/− hearts. This shifts the balance of fluxes in favor of Na accumulation [62;63]. Much like the effects of cardiac glycosides on contraction, this results in increased SR Ca2+ load ([Ca]SRT), and increased contractility [64]. Critically, the loss of ventricular NCX and NKA is coupled with no change in L-type Ca2+ current (ICa,L), the primary mode of Ca2+ entry into the myocyte. Since decreased NCX results in a decreased ability to extrude Ca2+, on a beat-to-beat basis more Ca2+ would enter the myocyte than would leave. Ca2+ would quickly accumulate in the SR via SERCA-mediated uptake, leading to Ca2+ overload at a new steady-state (Figure 2).
Figure 2).
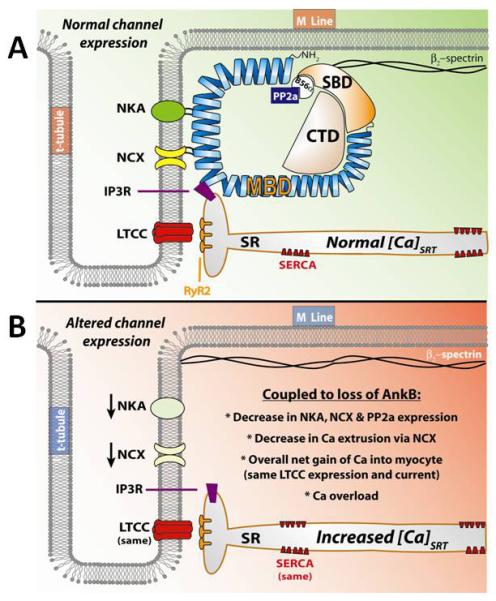
Ankyrin-B expression in the cardiac ventricular myocyte. A) In a healthy myocyte, ankyrin-B is directly involved in the proper trafficking and localization of the Na/K ATPase (NKA), the Na/Ca exchanger (NCX), the IP3 receptor (IP3R), and the serine/threonine protein phosphatase PP2A. The correct expression and localization of these proteins assure the proper balance of Ca fluxes. B) In myocytes isolated from AnkB +/− hearts AnkB expression is severely altered. This results in a loss of both NKA and NCX from the membrane, and a loss of PP2A localization. Critically, no difference is L-type Ca channel (LTCC) or Ca pump (SERCA) is observed. This shifts the balance of Ca fluxes to increased influx, until a new steady-state is reached in which the sarcoplasmic reticulum (SR) Ca stores become overloaded.
The result is a destabilization of Ca homeostasis leading to increases in spontaneous SR Ca2+ release, spontaneous Ca wave formation, and propensity for arrhythmogenic delayed or early after depolarizations [12;64-67]. This disruption of the EC coupling machinery reasonably explains why patients with ankyrin-B syndrome suffer from non-reentrant arrhythmias and CPVT, while partially explaining the high rate of AF (see section 3.3 below). While the actual mechanism for the often observed prolongation of the QT interval remains unknown, models point to the Na+ accumulation arising from the loss of NCX and Na/K ATPase resulting in slowed or blocked conduction [62].
AnkB also binds the IP3R in a ternary complex with the NCX and Na/K ATPase [26;68]. While no singular function of the IP3R in EC coupling is known, an interesting speculation is that it could in part be acting as a “pressure valve” against Ca2+ overload. A consistent finding in mammalian myocytes is a persistent diastolic SR Ca2+ leak which is not mediated through the RyR [69-71]. Since the relationship of Ca2+ release to [Ca]SRT is highly non-linear and unstable at very high [Ca]SRT [72], it has been proposed that under conditions of chronic SR Ca2+ overload an increased leak could perform the task of unloading the SR of Ca2+, stabilizing Ca2+ release. The IP3R has been demonstrated to mobilize SR Ca2+ stores [73], and it could be ideally situated to alleviate Ca2+ overload. As Ca2+ overload persists, the IP3R could open in response, allowing Ca2+ efflux out of the SR into the cytosol. The preferential localization of the IP3R near the NCX in the AnkB complex could quickly mediate Ca2+ extrusion and attenuate Ca2+ overload. A test of this hypothesis would be to express an IP3R mutant unable to bind AnkB and test whether non-RyR mediated Ca2+ leak is mitigated.
3.2 Ankyrin-B Targets PP2A
In cardiac EC coupling, PP2A is a key regulator of many of the constituent processes [74;75]. This serine/threonine phosphatase regulates channels as diverse as the RyR, the L-type Ca channel, the IP3R and the Na/K ATPase. Recently, AnkB was identified as a binding partner for the PP2A regulatory subunit B56α [47]. In cardiomyocytes, AnkB mediates the localization of PP2A to the Z-lines and M-lines where localization requires the presence of Rho-GEF, obscurin [49]. This localization nicely situates the phosphatase within the same microdomains with potential target substrates. While the PP2A-specific targets within this AnkB complex have yet to be determined, these data extend the prospect that AnkB may play unknown roles in the actual regulation of EC coupling by localizing the phosphatase near its potential substrates.
Extending this possibility, work from Terentyev et al. demonstrated that disruption of B56α signaling in cardiac myocytes leads to increased spontaneous Ca2+ release from the RyR and arrhythmogenesis [76]. This was shown to be CaMKII-dependent. This is consistent with data from studies by Curran et al. which demonstrated that increases in CaMKII-dependent diastolic SR Ca2+ leak resulted in a higher incidence of arrhythmogenic spontaneous Ca2+ waves in both failing and healthy hearts [65]. Taken together, these data open the possibility that AnkB could mediate RyR regulation in the heart via CaMKII where an AnkB association is known [50]. Disrupting PP2A activity in this microdomain may have the effect of chronically increasing phosphorylation levels of the RyR, thereby destabilizing Ca release and making the myocyte more prone to spontaneous Ca release.
3.3 Ankyrin-B Deficiency and Sinus Node Disease
Similar to individuals harboring AnkB mutations, AnkB+/− mice display decreased AnkB expression, decreased heart rate, high incidence of AF, highly-penetrant sinus node disease (SND) and risk of sudden death. Recent investigations by Le Scouarnec et al. into the mechanism behind the sinus node abnormalities present in AnkB+/− mice yielded similar results to ventricular myocytes [57]. Similarly, isolated AnkB+/− mouse SAN cells show a 30-40% decrease in both NCX and Na/K ATPase expression, along with a 40% loss of IP3R, with a concomitant 50% decrease in INCX. The atrial voltage gated L-type Ca channel, Cav1.3, expression was the same compared to WT SAN cells, but ICa,L was decreased by 50%, with no observed changes in ICa,T expression or function. Notably, this differs from ventricular studies where ICa,L current was unchanged. However, the ventricle almost exclusively expresses the Cav1.2 subtype, stressing the specificity of AnkB-mediated protein trafficking and targeting.
Regulation of Ca homeostasis has been demonstrated to be a vital component of SAN pacemaker activity. Deemed the “calcium clock”, SR and SL Ca2+ fluxes can largely determine SAN firing rate [77]. This calcium clock requires the presence of a passive SR Ca2+ leak. This leak slowly depolarizes the membrane, likely via electrogenic NCX activity, bringing the SL to threshold for AP generation. As in ventricular myocytes, this SR Ca2+ leak would require maintaining the [Ca]SRT at sufficiently high levels [78]. Disruption of either the underlying Ca2+ loading or unloading mechanisms (i.e. ICa,L or NCX) would greatly alter the ability of the SAN to function properly. This dysregulation was evidenced by the extreme rate variability observed in AnkB+/− SAN cells. Overall resting firing rate was depressed in AnkB+/− compared to WT, and the cells are prone to sudden bursts of hyperactivity. This high variability was exacerbated by the beta-adrenergic agonist isoproterenol.
This work was followed up by an elegant study by Glukhov et al using whole atrial preparations isolated from AnkB+/− mice [5]. They showed the same large rate variability seen in SAN cells. Intriguingly, there were two competing loci of automaticity in this preparation. At varying times the SAN was outcompeted by the atrioventricular junction (AVJ), typically the secondary pacemaker of the heart. Presuming that similar rate variability present in the SAN cells is also present in AVJ cells, this would suffice as a plausible and simple mechanism for the junctional escape rhythm observed in humans with AnkB mutations.
3.4 Ankyrin B Regulates KATP Channel Expression and ATP-sensitivity
The ATP-sensitive potassium channel is involved in a cardioprotective response to ischemia/reperfusion injury [79]. During ischemia, ATP production decreases and the KATP channel opens. This results in an increased outward hyperpolarizing current and an abbreviated APD, thereby conserving energy. Recently, the KATP channel was found to directly associate with AnkB in cardiomyocytes through the α-subunit Kir6.2 [40;80]. In isolated AnkB+/− myocytes, KATP channel expression was downregulated and IK,ATP was decreased under conditions of metabolic distress. While the KATP channel has a limited role in EC coupling in a healthy myocyte, this downregulation could limit the available repolarization reserve during ischemia; thus impairing the proper cardioprotective response.
Furthermore, the KATP channel has been shown to be dysregulated in pancreatic beta cells isolated from AnkB+/− mice [80-82]. In pancreatic tissue, KATP channel activity is intimately involved in insulin secretion. In fact, a human mutation in the AnkB binding motif of Kir6.2 (E322K) has been recently linked to permanent neonatal diabetes mellitus [83]. This again highlights the importance of ankyrin-mediated protein trafficking and retention as being critical in a number of excitable tissues.
One of the most exciting findings in ankyrin research in the last 10 years came out of work by Kline, Kurata, and Li. Specifically, they demonstrated a new role for ankyrin-B in ion channel regulation. This work clearly implicates AnkB binding in the regulation of KATP channel ATP sensitivity [40;80]. In AnkB+/− mice, the K50 for ATP of the channel is significantly higher. This right shift in sensitivity was accompanied by an increased open probability of the channel in the absence of ATP. As there is an overall loss of IK,ATP observed in these mice, this increase in open probability may be a compensatory mechanism. The data demonstrates for the first time that ankyrin polypeptides not only act as targeting proteins, but are involved in regulating the activity of their substrates. This is a critical finding. Future therapies using based on ankyrin interactions may very well be able exploit this to alter channel function.
4.0 Ankyrin G: Brugada Syndrome, Nav1.5 Regulation, and the βIV-spectrin Complex
AnkG is highly expressed in cardiac tissue and is largely localized to the intercalated disc where it associates with the Nav1.5 channel [16;42]. In vertebrate cardiac physiology, the proper expression, localization and regulation of the Nav1.5 are all critical for efficient cardiac function. Mutations in the Nav1.5 gene (SCN5A) are linked to inherited arrhythmias, Brugada syndrome and LQTS 3 [84]. The vast majority of these mutations cause alterations in the biophysical properties of the channel. However, a particular mutation in SCN5A (E1053K) results in the disruption of the NaV1.5 AnkG binding motif. This alters Nav1.5 targeting and localization at both the intercalated disc and T-tubule domain; causing a unique form of Brugada syndrome, sick sinus syndrome and conduction defects [42;85].
Intriguingly, Nav1.5 E1053K mutant channels also show altered biophysical properties. Specifically, a shift in the activation potential to more negative potentials, faster inactivation, and a slower time to recovery were all observed in the mutant channel [42]. Taken together with the work by Kline and Li this is compelling evidence that ankyrin polypeptides not only mediate the proper localization of their various binding partners, but could simultaneously play a role in regulating their activity [40].
βIV-spectrin is an actin-associated polypeptide that binds AnkG and has been shown to be important for normal localization of AnkG and Nav channels in neurons [86]. Recently, Hund and colleagues identified an important role for a βIV-spectrin/AnkG based complex in regulating Nav1.5 and cell excitability at the cardiomyocyte intercalated disc [7]. Specifically, the authors showed that βIV-spectrin is expressed at the cardiomyocyte intercalated disc region with AnkG, Nav1.5 and the multifunctional Ca2+/calmodulin-dependent protein kinase II (CaMKII), which is known to regulate Nav1.5 activity (Figure 3) [87]. Interestingly, βIV-spectrin harbors a C-terminal amino acid sequence with high homology to validated CaMKII binding motifs in the β2a subunit of Cav1.2 and the NR2B subunit of the NMDA receptor as well as to the CaMKII autoregulatory domain itself. Hund et al. showed that βIV-spectrin binds directly to CaMKII via this C-terminal motif and forms a quaternary complex with AnkG, Nav1.5 and CaMKII in vivo. Furthermore, they found that spectrin-based regulation of Nav1.5 occurred through direct phosphorylation of the channel by CaMKII at S571 in the DI-DII linker domain.
Figure 3).
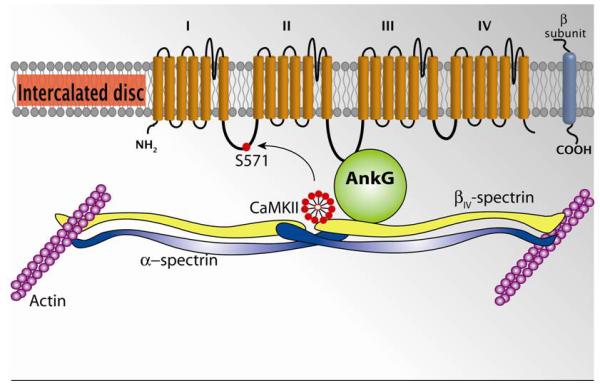
Ankyrin-G expression at the intercalated disc in the cardiac ventricular myocyte. AnkG binds to DII-DIII loop of the alpha-subunit of the Nav1.5 channel. This interaction is critical for proper localization of the Nav1.5 channel to the intercalated disc. AnkG binds to the cytoskeletal protein βIV-spectrin. In turn, βIV-spectrin binds to CaMKII near its carboxy terminus. The localization of CaMKII to this microdomain in the intercalated assures normal NaV1.5 activity, as lack of CaMKII-dependent phosphorylation at S571 in the DI-II loop alters the activity of the channel.
Using a spontaneous mouse model that expresses mutant βIV-spectrin lacking the CaMKII binding domain (qv3J), the authors demonstrated that the βIV-spectrin/CaMKII interaction was necessary for normal Nav1.5 regulation and cell excitability in heart. CaMKII-dependent phosphorylation results in an increased persistent late INa in WT myocytes not present in those from qv3J mice. This effect was greatly exaggerated in myocytes stimulated with the β-agonist isoproterenol. Decreased channel availability at nearly all potentials, and a significant depolarizing shift, were also observed in the S571 phosphorylated channel.
Furthermore, the WT mice showed a significantly higher propensity to develop EADs and arrhythmia versus the qv3J mice. The authors attributed this effect to the lack of the late persistent INa in these mice. Similar to the AnkB+/− effect on [Na+]i (section 3.1 above), a persistent late INa would lead to Na accumulation and increased SR Ca2+ loading. The resultant increase in [Ca]SRT would destabilize RyR regulation leading to spontaneous Ca2+ release. The resultant increase in INCX could depolarize the membrane, causing the observed EADs. Notably recent work using a mouse model overexpressing CaMKII demonstrated a strikingly similar phenotype, including the increase in late persistent INa [88]. This work again stresses how ankyrin polypeptides bring together various electrical components of EC coupling and their regulatory enzymes.
5.0 Regulation of Ankyrin-B Expression
Over the last decade, much of the work regarding ankyrin polypeptides has focused on their role in the subcellular localization and trafficking of integral membrane proteins. However, recent investigations are detailing how ankyrin polypeptides themselves are localized, trafficked and differentially expressed in the myocyte; particularly, ankyrin expression changes in response to myocardial infarct (MI), a period when active electrically remodeling of the heart is known to occur.
The border zone (BZ) area of an infarct in the heart is observed as an initiation site of non-reentrant arrhythmias [89]. This arrhythmogenic circuit is associated with the redistribution of ion channels and transporters in the SL membrane, many of which are binding partners of ankyrin polypeptides [90]. For this reason, Hund et al. hypothesized that the BZ would be an active site of electrical remodeling involving ankyrin-B. After isolating myocytes from within the BZ and comparing ankyrin expression to remote (unaffected by MI) areas of the heart, striking differences were apparent. At 5 days post-MI AnkB expression was significantly down-regulated in the BZ, while AnkB mRNA was increased in BZ tissue with no differences in remote myocardial tissue. By 14 days post-MI AnkB expression and mRNA levels in the BZ had returned to normal [91].
The loss of AnkB in the BZ was accompanied by a simultaneous decrease in Na/K ATPase expression and changes in its localization. This was persistent at 5 and 14 days post-MI, and returned to normal expression levels after 2 months. Interestingly, NCX and IP3R expression were not altered; however, gross redistribution away from their normal T-tubular junction localization was observed. This difference may represent divergent AnkB-dependent expression patterns for these functionally related proteins and warrants further investigation. Furthermore, this remodeling seems to be AnkB-specific, as no changes in the major cytoskeletal binding partner, β2-spectrin, were detected. For much the same reasons detailed above (section 3.1), this AnkB-dependent remodeling could lead to SR Ca2+ overload and increased propensity for spontaneous, arrhythmogenic SR Ca2+ release.
An additional role for AnkB mediating PP2A expression was also observed. Previous data has shown that transgenic expression of a mutant B56α unable to bind PP2A results in increased PP2A expression [92]. Further, PP2A overexpression has been shown to induce dilated cardiomyopathy [93]. In myocytes isolated from the BZ, PP2A expression was greatly increased during the same time period containing the downregulation of AnkB, an aforementioned binding partner of B56α (see section 3.2). This leads to the seductive possibility that AnkB downregulation may lead to structural as well as electrical remodeling of the heart and is a topic of further investigation.
Yet, for all that is now known and being investigated regarding the role of ankyrin polypeptides in the electrical stability of a cardiomyocyte, practically nothing is known about how ankyrin polypeptides themselves are trafficked and properly localized. In fact, the field completely lacks the knowledge of the identity of any critical, mediating membrane trafficking proteins. So, how do ankyrin polypeptides get to where they need to be in the cardiomyocyte?
Very recent data has emerged which begins to answer this question. Gudmundsson et al. presented exciting data in support of the hypothesis that a unique family of Eps15 homology domain-containing (EHD) proteins are required for proper ankyrin trafficking [94]. EHD proteins regulate anterograde and retrograde endosomal trafficking, membrane protein recycling and lipid homeostasis in many cell types [95;96]. This work identifies four members of EHD gene products (EHD1-4) expressed in the heart, and found a particular importance of EHD3 involvement with proper AnkB and NCX trafficking.
This work demonstrated that EHD1-4 all associate with AnkB in the heart. These EHDs are coexpressed in the perinuclear region of the myocyte, suggesting that they may be involved in the intracellular chaperoning of AnkB. Loss of AnkB (AnkB+/− and AnkB−/−) resulted in an increased expression of all EHDs, perhaps a compensatory mechanism in an effort to increase AnkB localization. This is the first known ankyrin-associated protein that is not negatively regulated by ankyrin expression. Interestingly, the interaction of EHDs with AnkB was mediated through the coiled-coil region of the EHD rather than the Eps15 homology domain itself. Classically, EHD proteins are were thought to interact with their targets at this highly conserved Eps15 homology domain. This opens the possibility that EHDs in general may act in a more diverse way than is currently appreciated.
Gene silencing experiments revealed that EHD3 is responsible for AnkB expression and localization. Similarly, EHD3 deficient cells displayed gross abnormalities in the localization of NCX, however expression remained the same. The expressed NCX protein was confined to the perinuclear region. As expected in AnkB−/− cells, both NCX expression and INCX were down-regulated. While overexpression of EHD3 in WT mice was able to increase INCX, no rescue was observed in AnkB−/− cells overexpressing EHDs. This is the first evidence that EHD3 is required for the proper localization of NCX. It is notable that this EHD3/AnkB system exhibits specificity. No changes in the expression or function of Cav1.2 (known to localize and express independently of AnkB) were observed when EHD3 was silenced. This is the first, direct evidence of a functional role of EHD3 in cardiomyocyte membrane trafficking and proper expression of AnkB.
Perhaps the most exciting finding was the differential regulation of EHD3 post-MI. In line with the time course observed for AnkB expression post-MI, EHD3 expression was significantly increased in BZ myocytes as early as 48 hour post-MI. EHD3-4 expression remained increased at 5 days post-MI, the same time period observed to have loss of AnkB expression. No changes were observed in EHD1 or EHD2. No changes in expression of EHD1-4 were observed in myocytes isolated from tissue remote to the MI. This is compelling evidence that EHD3 and EHD4 are involved in the electrical (and perhaps structural) remodeling of the heart post-MI, particularly with the restoration of AnkB expression and localization. An immediate question to be answered is whether the same might be observed in other states of cardiac remodeling—such as athletic hypertrophy, chronic hypertension or congestive heart failure.
6.0 Expert Opinion
First characterized in erythrocytes, ankyrin polypeptides were thought to play a structural role by linking integral membrane proteins with the underlying spectrin-based cytoskeleton. Over the last 25 years, ankyrin polypeptides have been demonstrated to be expressed in tissues as diverse as the eye, pancreas, neuron, and skeletal and cardiac muscle. They are near ubiquitously expressed and always serve a critical structural function. Emerging evidence over the past 15 years has highlighted the critical importance of ankyrin-mediated expression and localization of integral membrane proteins in excitable and non-excitable tissues. Mutations affecting the ability of these proteins to properly bind to ankyrin lead to diseases like Brugada syndrome, neonatal diabetes and hemolytic anemia. These diseases highlight the importance of proper subcellular localization of electrical components within a cell.
We are now aware of several mutations in ankyrin genes that are the primary cause of many vertebrate phenotypes. Mutations in ANK2 cause decreased expression and abnormal localization of the AnkB protein. These single point mutations can have devastating effects on the electrical stability of excitable cardiac tissues within the carrier [54]. A single mutation in AnkB has been demonstrated to lead to what is now known as “ankyrin-B syndrome.” This disease is characterized by the presence of type 4 long QT syndrome (unique to the AnkB mutation), sinus node sickness, arrhythmia, CPVT, and potenially sudden death. Notably, ankyrin-B is expressed in a number of cell types beyond heart and mice lacking ankyrins display a number of non-cardiac phenotypes including metabolic dysfunction, neurodegenerative disorders [97], early senescence, possible stem cell dysfunction, and kyphosis [82]. This wide diversity of pathologies emphasizes the critical importance of ankyrin polypeptides in an equally wide diversity of cell and tissue types. While knowledge of how ankyrins mediate cardiac and neuronal electrical signaling is expanding, little work in other tissues types has been demonstrated.
With this expanding knowledge comes the possibility for new therapies based on manipulation of ankyrin levels and/or activity. A potentially new and exciting avenue of approach was highlighted in the work of Kline and Kurata [40;80]. They were able to demonstrate that the KATP channel’s response to ATP is actually regulated by binding of AnkB. This opens up the possibility of being able to exploit the property of this protein-protein interaction and pharmacologically alter channel function. Work to further characterize the site of interaction on both proteins is a point of interest. Then, in theory, a small peptide could be synthesized which mimics this binding interaction, thus restoring proper channel function independent of AnkB. These strategies might produce therapeutic results for cardiac arrhythmia, but also have ramifications on other disease states such as hypo- or hyperinsulinemia, or enhancing cardioprotection during ischemia/reperfusion. Speculatively, these protein-protein regulatory sequences need not be limited to the KATP channel. Similar sites could be mapped on other ion channels and exploited for an increasing number of disease states.
The toughest challenge facing the development of therapeutic approaches to ankyrin-based diseases is that ankyrins are complex and multifunctional proteins. This means that a single mutation in the ANKx gene leads to the disruption of multiple cellular proteins at once. Further complicating this issue, ankyrins are widely expressed in nearly all tissue types. No subtype specificity for any tissue is known, i.e. heart expresses all three ankyrin subtypes as do neurons, etc.
This idea of manipulating ion channel and exchanger trafficking immediately calls to mind the data of Gudmundsson et al [94]. Two immediate questions to be answered are: 1) are EHDs involved in cardiac electrical and structural cardiac remodeling during CHF, and 2) can EHD proteins be manipulated pharmacologically? If so, can this be used to attenuate the remodeling? Vice versa, can EHDs be used to hasten the recovery from myocardial infarct? The discovery of EHDs in cardiac remodeling could prove vastly important in the years to come. While much work remains to be done exploring these proteins, the potential promise for therapies is striking.
6.0.1 Future Directions
While much has been learned regarding the function of cardiac ankyrin polypeptides over the past 10 years, many questions remain. Of note, the pathways for AnkB loss/degradation post-MI have yet to be explored. While this idea has not been directly addressed, evidence from other studies in heart and neurons suggests that the Ca2+ activated protease, calpain, may play a significant role [98;99]. Investigations into the possible regulatory role of ankyrin polypeptides by calpain post-MI may offer further therapeutic targets. Cardiac-specific inhibition of calpain could, in theory, attenuate electrical and/or structural remodeling associated with AnkB loss.
Additionally, there is little knowledge regarding the role of AnkR in the heart. While it is known to be expressed and serve as a structural component in cardiac muscle, little else is known [36]. Intriguing data have linked a splice variant of ANK1 to SR expression in striated muscle [100]. This opens the possibility that AnkR could localize to the SR junctional microdomain in cardiomyocytes. This domain is an important component of EC coupling in the heart. Understanding a role for AnkR within it would further our understanding of this critical domain.
The province of the ankyrin family of adapter proteins has expanded over the last three decades from purely a structural protein to a primary player in the electrical stability of excitable tissues. With each incremental step forward, our knowledge of this unique family of proteins is offering unpredicted, exciting and novel avenues for therapies. As this field of research expands, the next 10 years should prove equally exciting.
Article Highlights.
Single point mutations in ankyrin-B result in the complex phenotype called “Ankyrin-B Syndrome.”
Ankyrin polypeptides are critical for the trafficking and localization of ion channels to their proper microdomains in the heart.
Ankyrin polypeptides and their accessory proteins may be involved in cardiac remodeling after myocardial infarct.
Pharmacologic manipulation of these accessory proteins may hold the potential for new therapies against maladaptive remodeling associated with heart failure.
Ankyrin polypeptides can regulate ion channel activity and response to stimuli.
The ability of ankyrins to regulate ion channel activity could potentially be exploited as new avenue for therapies.
List of Abbreviations
- Å
angstrom
- AF
atrial fibrillation
- Ank-B
ankyrin B
- Ank-G
ankyrin G
- Ank-R
ankyrin R
- AP
action potential
- APD
action potential duration
- AVJ
atrioventricular junction
- BZ
border zone
- [Ca]i
cytosolic free Ca2+ concentration
- CPVT
catelcholaminergic polymorphic ventricular tachycardia
- [Ca]SRT
total sarcoplasmic reticulum Ca content
- CTD
C-terminal domain
- CaMKII
calcium-calmodulin-dependent protein kinase type II
- Cav
voltage-gated calcium channel
- EC
excitation-contraction
- Em
membrane potential
- EHD
Eps15 homology domain
- GFP
Green fluorescent protein
- ICa,L
L-type calcium channel-mediated current
- IK
potassium channel-mediated current
- INCX
Na/Ca exchanger-mediated current
- INa
Sodium channel-mediated current
- IP3R
inositol 1,4,5-trisphosphate receptor
- K50
Substrate concentration causing half-maximal inhibition
- KATP
ATP sensitive potassium channel
- Kv
voltage-gated potassium channel
- LQTS
Long QT syndrome
- LTCC
L-type calcium channel
- MBD
membrane binding domain
- MI
myocardial infarct
- [Na]i
cytosolic free Na+ concentration
- Nav
voltage-gated sodium channel
- NCX
Na/Ca exchanger
- NKA
Na/K ATPase
- nm
nanometer
- PP2A
protein phosphatase type 2A
- RyR
ryanodine receptor
- SAN
sinoatrial node
- SBD
spectrin binding domain
- SERCA
sarco- endoplasmic Ca2+ ATPase
- SL
sarcolemma
- SND
sinus node disease
- SR
sarcoplasmic reticulum
Footnotes
Declaration of interest The authors declare no conflict of interest.
PJ Mohler acknowledges funding from the National Institutes of Health: HLO8 4583 and HLO8 3422.
Reference List
- 1.Bardou AL, Auger PM, Birkui PJ, et al. Modeling of cardiac electrophysiological mechanisms: from action potential genesis to its propagation in myocardium. Crit Rev Biomed Eng. 1996;24:141–221. doi: 10.1615/critrevbiomedeng.v24.i2-3.20. [DOI] [PubMed] [Google Scholar]
- 2.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ.Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 3.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J.Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 5.Glukhov AV, Fedorov VV, Anderson ME, et al. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am J Physiol Heart Circ Physiol. 2010;299:H482–H491. doi: 10.1152/ajpheart.00756.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hund TJ, Mohler PJ. Ankyrin-based targeting pathway regulates human sinoatrial node automaticity. Channels (Austin.) 2008;2:404–406. doi: 10.4161/chan.2.6.7220. [DOI] [PubMed] [Google Scholar]
- 7.Hund TJ, Koval OM, Li J, et al. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J.Clin.Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kline CF, Hund TJ, Mohler PJ. Ankyrin regulates KATP channel membrane trafficking and gating in excitable cells. Channels (Austin.) 2010;4:55–57. doi: 10.4161/chan.4.1.10362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koval OM, Guan X, Wu Y, et al. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl.Acad.Sci U.S.A. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Kline CF, Hund TJ, et al. Ankyrin-B regulates Kir6.2 membrane expression and function in heart. J.Biol.Chem. 2010;285:28723–28730. doi: 10.1074/jbc.M110.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pogwizd SM, Qi M, Yuan W, et al. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ.Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 12.Pogwizd SM, Bers DM. Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann.N.Y.Acad.Sci. 2002;976:454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- 13.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 14.Szlufcik K, Missiaen L, Parys JB, et al. Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol.Cell. 2006;98:1–14. doi: 10.1042/BC20050031. [DOI] [PubMed] [Google Scholar]
- 15.Wehrens XH, Marks AR. Sudden unexplained death caused by cardiac ryanodine receptor (RyR2) mutations. Mayo Clin.Proc. 2004;79:1367–1371. doi: 10.4065/79.11.1367. [DOI] [PubMed] [Google Scholar]
- 16.Lowe JS, Palygin O, Bhasin N, et al. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180:173–186. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ringer S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J.Physiol. 1883;4:29–42. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodgkin AL, Huxley AF. Movement of sodium and potassium ions during nervous activity. Cold Spring Harb.Symp.Quant.Biol. 1952;17:43–52. doi: 10.1101/sqb.1952.017.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Neher E, Sakmann B, Steinbach JH. The extracellular patch clamp: a method for resolving currents through individual open channels in biological membranes. Pflugers Arch. 1978;375:219–228. doi: 10.1007/BF00584247. [DOI] [PubMed] [Google Scholar]
- 20.Rios E, Brum G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 1987;325:717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- 21.McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol.Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Verdonck F, Mubagwa K, Sipido KR. [Na(+)] in the subsarcolemmal ‘fuzzy’ space and modulation of [Ca(2+)](i) and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Petrecca K, Atanasiu R, Grinstein S, et al. Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am.J.Physiol. 1999;276:H709–H717. doi: 10.1152/ajpheart.1999.276.2.H709. [DOI] [PubMed] [Google Scholar]
- 24.Gao T, Yatani A, Dell’Acqua ML, et al. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 25.Wibo M, Bravo G, Godfraind T. Postnatal maturation of excitation-contraction coupling in rat ventricle in relation to the subcellular localization and surface density of 1,4-dihydropyridine and ryanodine receptors. Circ.Res. 1991;68:662–673. doi: 10.1161/01.res.68.3.662. [DOI] [PubMed] [Google Scholar]
- * 26.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS.Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. This study details how ankyrin-B is critical for the proper localization of integral membrane ion proteins and ion channels.
- 27.Bennett V. Purification of an active proteolytic fragment of the membrane attachment site for human erythrocyte spectrin. J.Biol.Chem. 1978;253:2292–2299. [PubMed] [Google Scholar]
- 28.Bennett V, Stenbuck PJ. Association between ankyrin and the cytoplasmic domain of band 3 isolated from the human erythrocyte membrane. J.Biol.Chem. 1980;255:6424–6432. [PubMed] [Google Scholar]
- 29.Bennett V, Davis J. Spectrin and ankyrin in brain. Cell Motil. 1983;3:623–633. doi: 10.1002/cm.970030527. [DOI] [PubMed] [Google Scholar]
- 30.Ayalon G, Davis JQ, Scotland PB, et al. An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell. 2008;135:1189–1200. doi: 10.1016/j.cell.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Kizhatil K, Baker SA, Arshavsky VY, et al. Ankyrin-G promotes cyclic nucleotide-gated channel transport to rod photoreceptor sensory cilia. Science. 2009;323:1614–1617. doi: 10.1126/science.1169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li ZP, Burke EP, Frank JS, et al. The cardiac Na+-Ca2+ exchanger binds to the cytoskeletal protein ankyrin. J.Biol.Chem. 1993;268:11489–11491. [PubMed] [Google Scholar]
- 33.Kizhatil K, Sandhu NK, Peachey NS, et al. Ankyrin-B is required for coordinated expression of beta-2-spectrin, the Na/K-ATPase and the Na/Ca exchanger in the inner segment of rod photoreceptors. Exp.Eye Res. 2009;88:57–64. doi: 10.1016/j.exer.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 34.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 35.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. J.Mol.Cell Cardiol. 2009;47:203–209. doi: 10.1016/j.yjmcc.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett PM, Baines AJ, Lecomte MC, et al. Not just a plasma membrane protein: in cardiac muscle cells alpha-II spectrin also shows a close association with myofibrils. J.Muscle Res.Cell Motil. 2004;25:119–126. doi: 10.1023/b:jure.0000035892.77399.51. [DOI] [PubMed] [Google Scholar]
- 37.Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, et al. Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol.Biol.Cell. 2003;14:1138–1148. doi: 10.1091/mbc.E02-07-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. J.Mol.Cell Cardiol. 2009;47:203–209. doi: 10.1016/j.yjmcc.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohler PJ, Gramolini AO, Bennett V. Ankyrins. J.Cell Sci. 2002;115:1565–1566. doi: 10.1242/jcs.115.8.1565. [DOI] [PubMed] [Google Scholar]
- * 40.Li J, Kline CF, Hund TJ, et al. Ankyrin-B regulates Kir6.2 membrane expression and function in heart. J.Biol.Chem. 2010;285:28723–28730. doi: 10.1074/jbc.M110.147868. This study describes how ankyrin interactions with ion channels can regulate the channel’s activity and response to stimuli.
- 41.Lowe JS, Palygin O, Bhasin N, et al. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J.Cell Biol. 2008;180:173–186. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * 42.Mohler PJ, Rivolta I, Napolitano C, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc.Natl.Acad.Sci.U.S.A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. This study demonstrates how a single mutation in the ankyrin binding domain of the Na channel leads to Brugada syndrome.
- 43.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS.Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett V, Stenbuck PJ. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature. 1979;280:468–473. doi: 10.1038/280468a0. [DOI] [PubMed] [Google Scholar]
- 45.Bennett V, Stenbuck PJ. Identification and partial purification of ankyrin, the high affinity membrane attachment site for human erythrocyte spectrin. J.Biol.Chem. 1979;254:2533–2541. [PubMed] [Google Scholar]
- 46.Cunha SR, Mohler PJ. Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J Biol Chem. 2008;283:31968–31980. doi: 10.1074/jbc.M806050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhasin N, Cunha SR, Mudannayake M, et al. Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am.J.Physiol Heart Circ.Physiol. 2007;293:H109–H119. doi: 10.1152/ajpheart.00059.2007. [DOI] [PubMed] [Google Scholar]
- 48.Abdi KM, Mohler PJ, Davis JQ, et al. Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J.Biol.Chem. 2006;281:5741–5749. doi: 10.1074/jbc.M506697200. [DOI] [PubMed] [Google Scholar]
- 49.Cunha SR, Mohler PJ. Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J.Biol.Chem. 2008;283:31968–31980. doi: 10.1074/jbc.M806050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohler PJ, Gramolini AO, Bennett V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (-/-) neonatal cardiomyocytes. J.Biol.Chem. 2002;277:10599–10607. doi: 10.1074/jbc.M110958200. [DOI] [PubMed] [Google Scholar]
- 51.Mohler PJ, Hoffman JA, Davis JQ, et al. Isoform specificity among ankyrins. An amphipathic alpha-helix in the divergent regulatory domain of ankyrin-b interacts with the molecular co-chaperone Hdj1/Hsp40. J.Biol.Chem. 2004;279:25798–25804. doi: 10.1074/jbc.M401296200. [DOI] [PubMed] [Google Scholar]
- 52.Davis LH, Davis JQ, Bennett V. Ankyrin regulation: an alternatively spliced segment of the regulatory domain functions as an intramolecular modulator. J.Biol.Chem. 1992;267:18966–18972. [PubMed] [Google Scholar]
- 53.Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc.Natl.Acad.Sci.U.S.A. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohler PJ, Le SS, Denjoy I, et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 2007;115:432–441. doi: 10.1161/CIRCULATIONAHA.106.656512. [DOI] [PubMed] [Google Scholar]
- 55.Michaely P, Tomchick DR, Machius M, et al. Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 2002;21:6387–6396. doi: 10.1093/emboj/cdf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ** 56.Schott JJ, Charpentier F, Peltier S, et al. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am.J.Hum.Genet. 1995;57:1114–1122. This is the first paper which describes a unique LQTS syndrome associated with a mutation in the ANK2 gene.
- 57.Le Scouarnec S, Bhasin N, Vieyres C, et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl.Acad.Sci U.S.A. 2008;105:15617–15622. doi: 10.1073/pnas.0805500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ** 58.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. Seminal paper, first paper to describe LQTS 4 being caused by an Ank-B specific mutation. First description of an LQTS which was not caused by a mutation in a gene encoding for an ion channel.
- 59.Eber SW, Gonzalez JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat.Genet. 1996;13:214–218. doi: 10.1038/ng0696-214. [DOI] [PubMed] [Google Scholar]
- 60.Sedlacek K, Stark K, Cunha SR, et al. Common genetic variants in ANK2 modulate QT interval: results from the KORA study. Circ.Cardiovasc.Genet. 2008;1:93–99. doi: 10.1161/CIRCGENETICS.108.792192. [DOI] [PubMed] [Google Scholar]
- 61.Li ZP, Burke EP, Frank JS, et al. The cardiac Na+-Ca2+ exchanger binds to the cytoskeletal protein ankyrin. J.Biol.Chem. 1993;268:11489–11491. [PubMed] [Google Scholar]
- 62.Wolf RM, Mitchell CC, Christensen MD, et al. Defining new insight into atypical arrhythmia: a computational model of ankyrin-B syndrome. Am.J.Physiol Heart Circ.Physiol. 2010;299:H1505–H1514. doi: 10.1152/ajpheart.00503.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu.Rev.Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 64.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc.Natl.Acad.Sci.U.S.A. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Curran J, Brown KH, Santiago DJ, et al. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008 doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ.Res. 2005;96:535–542. doi: 10.1161/01.RES.0000159387.00749.3c. [DOI] [PubMed] [Google Scholar]
- 68.Mohler PJ, Davis JQ, Davis LH, et al. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J.Biol.Chem. 2004;279:12980–12987. doi: 10.1074/jbc.M313979200. [DOI] [PubMed] [Google Scholar]
- 69.Curran J, Hinton MJ, Rios E, et al. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ.Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 70.Szlufcik K, Missiaen L, Parys JB, et al. Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol.Cell. 2006;98:1–14. doi: 10.1042/BC20050031. [DOI] [PubMed] [Google Scholar]
- 71.Zima AV, Bovo E, Bers DM, et al. Ca2+ Spark Dependent and Independent Sarcoplasmic Reticulum Ca2+ Leak in Normal and Failing Rabbit Ventricular Myocytes. J.Physiol. 2010 doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shannon TR, Ginsburg KS, Bers DM. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys.J. 2000;78:334–343. doi: 10.1016/S0006-3495(00)76596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mackenzie L, Roderick HL, Proven A, et al. Inositol 1,4,5-trisphosphate receptors in the heart. Biol.Res. 2004;37:553–557. doi: 10.4067/s0716-97602004000400008. [DOI] [PubMed] [Google Scholar]
- 74.Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J.Biol.Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- 75.Klein G, Schroder F, Vogler D, et al. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation. role of phosphatase 2A. Cardiovasc.Res. 2003;59:37–45. doi: 10.1016/s0008-6363(03)00357-2. [DOI] [PubMed] [Google Scholar]
- 76.Terentyev D, Belevych AE, Terentyeva R, et al. miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ.Res. 2009;104:514–521. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ. 2007;16:335–348. doi: 10.1016/j.hlc.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Y, Gao Z, Chen B, et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc.Natl.Acad.Sci.U.S.A. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suzuki M, Li RA, Miki T, et al. Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ.Res. 2001;88:570–577. doi: 10.1161/01.res.88.6.570. [DOI] [PubMed] [Google Scholar]
- 80.Kline CF, Kurata HT, Hund TJ, et al. Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl.Acad.Sci U.S.A. 2009;106:16669–16674. doi: 10.1073/pnas.0907138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Healy JA, Nilsson KR, Hohmeier HE, et al. Cholinergic augmentation of insulin release requires ankyrin-B. Sci.Signal. 2010;3:ra19. doi: 10.1126/scisignal.2000771. [DOI] [PubMed] [Google Scholar]
- 82.Mohler PJ, Healy JA, Xue H, et al. Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence. PLoS.One. 2007;2:e1051. doi: 10.1371/journal.pone.0001051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vaxillaire M, Populaire C, Busiah K, et al. Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes. 2004;53:2719–2722. doi: 10.2337/diabetes.53.10.2719. [DOI] [PubMed] [Google Scholar]
- 84.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 85.Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–1347. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 86.Yang Y, Lacas-Gervais S, Morest DK, et al. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J.Neurosci. 2004;24:7230–7240. doi: 10.1523/JNEUROSCI.2125-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wagner S, Dybkova N, Rasenack EC, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J.Clin.Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sossalla S, Maurer U, Schotola H, et al. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na (+) current. Basic Res.Cardiol. 2010 doi: 10.1007/s00395-010-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pinto JM, Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovasc.Res. 1999;42:284–297. doi: 10.1016/s0008-6363(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 90.Nattel S, Maguy A, Le BS, et al. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- *91.Hund TJ, Wright PJ, Dun W, et al. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc.Res. 2009;81:742–749. doi: 10.1093/cvr/cvn348. This study describes how ankyrin pathways may be involved in cardiac remodeling post myocardial infarction.
- 92.Ruediger R, Brewis N, Ohst K, et al. Increasing the ratio of PP2A core enzyme to holoenzyme inhibits Tat-stimulated HIV-1 transcription and virus production. Virology. 1997;238:432–443. doi: 10.1006/viro.1997.8873. [DOI] [PubMed] [Google Scholar]
- 93.Brewis N, Ohst K, Fields K, et al. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am.J.Physiol Heart Circ.Physiol. 2000;279:H1307–H1318. doi: 10.1152/ajpheart.2000.279.3.H1307. [DOI] [PubMed] [Google Scholar]
- *94.Gudmundsson H, Hund TJ, Wright PJ, et al. EH domain proteins regulate cardiac membrane protein targeting. Circ.Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. This study is the first to describe EHD proteins being involved in membrane trafficking in the heart, and how they may be involved in remodeling post-myocardial infarct.
- 95.Miliaras NB, Wendland B. EH proteins: multivalent regulators of endocytosis (and other pathways) Cell Biochem.Biophys. 2004;41:295–318. doi: 10.1385/CBB:41:2:295. [DOI] [PubMed] [Google Scholar]
- 96.Daumke O, Lundmark R, Vallis Y, et al. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- 97.Bennett V, Lambert S. Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltage-gated sodium channels. J.Neurocytol. 1999;28:303–318. doi: 10.1023/a:1007005528505. [DOI] [PubMed] [Google Scholar]
- 98.Harada K, Fukuda S, Kunimoto M, et al. Distribution of ankyrin isoforms and their proteolysis after ischemia and reperfusion in rat brain. J.Neurochem. 1997;69:371–376. doi: 10.1046/j.1471-4159.1997.69010371.x. [DOI] [PubMed] [Google Scholar]
- 99.Inserte J, Garcia-Dorado D, Hernando V, et al. Calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ.Res. 2005;97:465–473. doi: 10.1161/01.RES.0000181170.87738.f3. [DOI] [PubMed] [Google Scholar]
- 100.Porter NC, Resneck WG, O’Neill A, et al. Association of small ankyrin 1 with the sarcoplasmic reticulum. Mol.Membr.Biol. 2005;22:421–432. doi: 10.1080/09687860500244262. [DOI] [PubMed] [Google Scholar]