

Abstract
The familial melanoma gene (INK4a/MTS1/CDKN2) encodes potent tumor suppressor activity. Although mice null for the ink4a homolog develop a cancer-prone condition, a pathogenetic link to melanoma susceptibility has yet to be established. Here we report that mice with melanocyte-specific expression of activated H-rasG12V on an ink4a-deficient background develop spontaneous cutaneous melanomas after a short latency and with high penetrance. Consistent loss of the wild-type ink4a allele was observed in tumors arising in ink4a heterozygous transgenic mice. No homozygous deletion of the neighboring ink4b gene was detected. Moreover, as in human melanomas, the p53 gene remained in a wild-type configuration with no observed mutation or allelic loss. These results show that loss of ink4a and activation of Ras can cooperate to accelerate the development of melanoma and provide the first in vivo experimental evidence for a causal relationship between ink4a deficiency and the pathogenesis of melanoma. In addition, this mouse model affords a system in which to identify and analyze pathways involved in tumor progression against the backdrop of genetic alterations encountered in human melanomas.
Keywords: Ink4a, Ink4b, Ras, p53, melanoma, transgenic
Efforts to elucidate the genetic program governing the genesis and progression of melanoma have been fueled by the rapid rise in its incidence (Rigel et al. 1996), its high metastatic propensity and its poor clinical response to existing therapeutic modalities (Herlyn 1993). Whereas several epigenetic factors, such as exposure to UV irradiation, seem to contribute to the development of this disease, predisposition to melanoma appears to have a strong genetic component, distinct from determinants of skin type and melanin composition (Herlyn 1993 and references therein). In the search for genetic loci responsible for melanoma susceptibility, several potential chromosomal hot spots have been uncovered, including frequent loss of 6q and 10q and nonrandom karyotypic alterations of chromosome 1 (for review, see Herlyn 1993). By far, the most compelling etiological link to melanoma has mapped to 9p21. An exhaustive series of cytogenetic, linkage, and molecular analyses have documented a high incidence of 9p21 germ-line and somatic mutations in familial and sporadic melanomas (see below), arguing strongly for the presence of a melanoma susceptibility gene within this locus.
In mouse and man, the genomic organization of the 9p21 locus is quite complex. This region contains the closely linked INK4a and INK4b genes that encode the highly related G1 cyclin-dependent kinase inhibitors, p16Ink4a and p15Ink4b, respectively (Serrano et al. 1993; Hannon and Beach 1994; Kamb et al. 1994; Quelle et al. 1995a). In addition to p16Ink4a, the mouse and human INK4a genes, through alternative first exon usage and reading frames, also encode a novel growth inhibitor protein, known as p19ARF (alternative reading frame) (Quelle et al. 1995b). Thus, the 9p21 locus has the capacity to encode at least three potent growth inhibitors, all within a relatively short genomic distance. This genomic organization and the common occurrence of large homozygous 9p21 codeletions (Jen et al. 1994; Kamb et al. 1994; Orlow et al. 1995; Flores et al. 1996) hampered initial efforts to evaluate the role of each gene product in melanoma suppression and to assign definitively a melanoma susceptibility gene. A partial resolution of this issue has come from the more recent genetic and biological data pointing to ink4a, as opposed to ink4b, as the key tumor suppressor gene. Most noteworthy are the observations of germ-line mutations that exclusively targeted the INK4a gene in susceptible individuals (Hussussian et al. 1994; Gruis et al. 1995) and of the cancer-prone phenotype resulting from deletion of ink4a (p16Ink4a and p19ARF) in the mouse genome (Serrano et al. 1996). In contrast, ink4b-deficient mice do not develop cancer with high frequency (E. Latres, C. Cordon-Cardo, and M. Barbacid, unpubl.). The striking difference in phenotypes between the ink4a and ink4b knockout mice is quite remarkable given the high degree of functional and biochemical similarity between p16Ink4a and p15Ink4b. This raised the possibility that the second ink4a gene product may contribute to the anti-neoplastic activity of this gene (see below). Although this view is conceptually attractive, germ-line mutations exclusively targeting the p16Ink4a reading frame have been identified in a few melanoma-prone individuals (FitzGerald et al. 1996). Although this observation confirms the importance of p16Ink4a, it does not disprove a potential contribution by p19ARF to ink4a tumor suppressor activity in some settings.
In addition to loss of tumor suppressor loci (e.g., 9p21), the stepwise phenotypic progression from normal melanocyte to metastatic malignant melanoma is thought to require the activation of various oncogenes, particularly those encoding components of the receptor tyrosine kinase (RTK)–Ras–mitogen-associated protein kinase (MAPK) pathway. For example, transgenic studies showed that overexpression of activated RTKs in melanocytes resulted in the generation of melanoma in mice (Iwamoto et al. 1991; Takayama et al. 1997). An etiological role for activation of the Xmrk receptor [another RTK with epidermal growth factor (EGF) receptor homology] in melanoma formation has been firmly established in a fish melanoma model (Wittbrodt et al. 1992). In addition, the human EGF receptor gene has been mapped to 7p11–13 (Davies et al. 1980), a chromosomal region frequently rearranged in melanomas (for review, see Herlyn 1993), and EGF receptor overexpression has been shown in a human melanoma cell line (Huang et al. 1996). Although activating ras mutations themselves do not occur with high frequency in human melanomas (see below), these genetic clues underscore the importance of the Ras signaling pathway in the development of melanoma.
Historically, activating ras mutations have been among the most common genetic lesions in human cancers. That activating ras mutations are indeed oncogenic in vivo has been well documented by tumor phenotypes resulting from its cell-type-specific expression in transgenic mouse models [e.g., breast carcinoma (Tremblay et al. 1989; Mangues et al. 1990)]. Although N- and H-ras gene mutations have been observed in melanomas, a causal role for activated Ras in melanocyte transformation has yet to be definitively established in vivo. A previous transgenic mouse study in which activated H-ras was overexpressed in melanocytes yielded a hyperproliferative phenotype without evidence of transformation (Powell et al. 1995). On the other hand, stable transfection of activated H-ras in cultured mouse (Wilson et al. 1989; Ramon y Cajal et al. 1991) and human (Albino et al. 1992) melanocytes was shown to induce phenotypic characteristics of fully transformed melanoma cells (such as growth inhibition by protein kinase C activator, anchorage independence, tumorigenicity in nude mice, and chromosomal instability). A number of groups have examined the mutational status of ras family genes in primary melanoma tissues and melanoma cell lines; however, conflicting results have served to fuel the debate surrounding the pathogenetic relevance of ras mutations. For instance, Albino et al. (1989) reported that the frequency of activating ras mutations (predominantly N-ras) was significantly higher in cultured melanoma cell lines (24%) than in noncultured melanoma cell lines (5%–6%), raising the possibility that ras mutations may be a consequence of the inherent genomic instability of transformed cells further accentuated by adaptation to culture. In contrast, van’t Veer et al. (1989) have observed a high frequency of N-ras mutations in primary melanomas approaching 20%. Moreover, several other groups (Ball et al. 1994; Jafari et al. 1995; Wagner et al. 1995) have investigated noncultured melanoma samples at different stages and found a higher frequency of ras mutations (N-ras more than H-ras) in metastatic and recurrent tumors, suggesting a role for Ras activation in disease progression rather than initiation. These data correlate well with expression studies showing increased Ras immunoreactivity in late stage melanomas (Yasuda et al. 1989). In addition, activating N-ras and H-ras mutations have been documented in metastatic melanoma cell lines through use of the NIH-3T3 transformation assay (Albino et al. 1984). Taken together, the consensus view would favor a role for N- and H-ras mutations in promoting progression toward a more advanced stage of melanoma, although no formal genetic proof exists to support this hypothesis.
A curious genetic feature of human melanoma is the low incidence of p53 mutations. Both single-strand conformation polymorphism (SSCP) and direct sequence analysis have revealed an absence of point mutation (exons 5–8) or allelic loss of p53 gene in >50 surgical specimens of primary (superficial spreading or nodular) and metastatic melanomas (Lubbe et al. 1994; Papp et al. 1996). This finding is striking in light of the fact that loss of p53 function is observed in >55% of human cancers overall (Hollstein et al. 1991). The basis for the low frequency of p53 mutation in melanoma is not understood, but could relate to overlapping tumor suppressor functions of INK4a and p53 genes, thereby reducing the biological requirement for p53 elimination in the INK4a deficient state (see Discussion).
In this study, we assess in vivo the roles of ink4a and Ras in the development of melanoma. We show that melanocyte-specific expression of mutant H-Ras in transgenic mice that are null for ink4a leads to the spontaneous development of multiple cutaneous melanomas with high penetrance. This faithful mouse model of human melanoma enabled us to address a number of key issues including whether ink4a deletions consistently eliminate both p19ARF and p16Ink4a coding sequences, whether loss of ink4b is an obligate causal event, and whether the lack of p53 mutations remains an evolutionarily conserved feature of this cancer type.
Results
Tyr–ras transgenic mice
In previous studies, melanocyte-specific transgene expression has been achieved with the aid of the mouse tyrosinase gene promoter (5.5 kb of 5′-flanking sequences), albeit with variable expressivity possibly resulting from position effects (Ganss et al. 1994 and references therein). More recent efforts have identified a strong melanocyte-specific enhancer far upstream of the tyrosinase promoter region that confers enhanced and copy number-related expression of a tyrosinase transgene in melanocytes (Ganss et al. 1994). Here, both the proximal promoter and upstream enhancer element were used to direct melanocyte-specific expression of activated H-Ras (G12V) in transgenic mice.
An unusually low frequency of founder production (3 founders from a total of 420 pronuclear injections and 74 surviving offspring) raised suspicion of biological selection against high level tyr–ras transgene expression. This explanation is supported by the compromised clinical presentation of one founder mouse (TR59) that appeared runted, displayed hyperpigmentation of paws and pinnae, and suffered from an unstable gait (Fig. 1A). The unstable gait may relate to disturbances in the development of the stria vascularis of the inner ear, a process known to be dependent on normal melanocyte function (Steel and Barkway 1989) or may reflect growth of a melanocytic hamartoma in the cochlea space (Powell et al. 1995). Although early postnatal death of TR59 precluded the establishment of transgenic offspring and a morphological analysis of the inner ear, antemortem skin biopsies revealed a marked increase in melanization of suprabasal keratinocytes without obvious melanocytic proliferation (data not shown). The two remaining tyr–ras transgenic founders (TR39 and TR60) possessed low transgene copy number and H-rasG12V expression level (see below), appeared grossly normal, and transmitted the transgene to their offspring. In the case of TR60, all transgenic offspring were male, indicating transgene integration onto the Y chromosome. TR60 developed left exophthalmos early in life (8 weeks); on enucleation, a proliferating melanocytic mass was detected arising from the pigmented retinal epithelium (data not shown). TR39 and TR60 founders were crossed with mice harboring a targeted deletion of ink4a exons 2 and 3 that contribute to the open reading frame (ORF) of both p16Ink4a and p19ARF. The mice analyzed in this study were of a mixed genetic background that consisted primarily of C57BL/6 (∼65%), CBA (∼25%), and 129Sv (∼10%).
Figure 1.
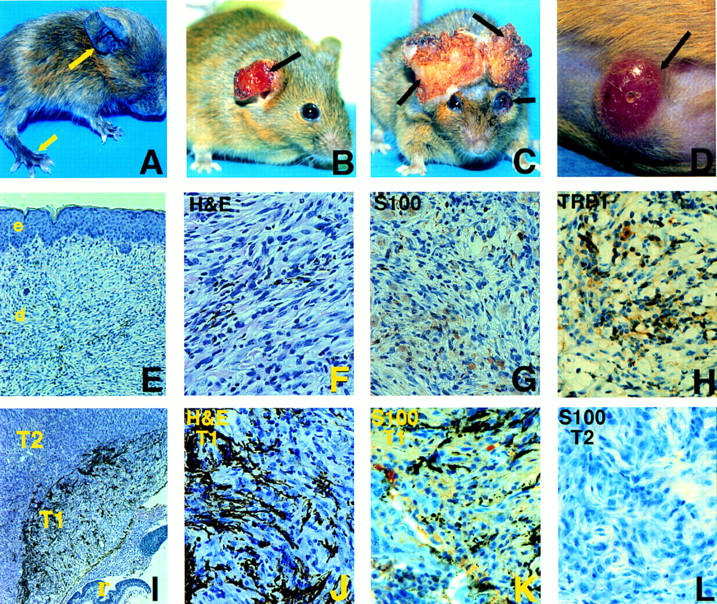
Tyr–Ras transgenics developed multiple primary cutaneous and ocular melanomas. (A) Photomicrograph depicting founder TR59; note focal hyperpigmentation evident on paws and pinnae (arrows). (B) Photomicrograph of eroded cutaneous tumor (arrow) developing on pinna, early stage, line TR60. (C) Photomicrograph of late stage cutaneous tumors of pinnae with destruction of local structure. Also note hemorrhagic exophthalmus (arrow) representing an ocular melanoma, line TR39. (D) Photomicrograph of an early lesion on the abdomen, line TR60. Note the well-circumscribed pink dermal nodule with superficial telangiectasia (arrow). (E–H) Photomicrographs of histological sections of the cutaneous tumor in B. (E) Hematoxylin and eosin (H&E) 100×. Dermal proliferation of spindle-shape cells with varying degrees of pigmentation. (e) epidermis; (d) dermis. (F) H&E, 400×. Note the prominent cellular atypia at higher magnification. (G) S100 immunoperoxidase reaction, 400×. Nuclear as well as cytoplasmic immunoreactivity is present. (H) TRP1 immunoperoxidase reaction, 400×. Strong cytoplasmic immunoreactivity is detected. (I–L) Photomicrographs of histologic sections of the ocular tumor in C. (I) H&E, 50×. At this power, note the remnant of the sensory retina (r) destroyed by adjacent proliferating spindle cell mass. Distinct transition from melanotic (T1) to amelanotic (T2) tumors clearly visible at this power. (J) H&E, 400×. At higher power, heavy melanization present in T1 region of I. (K) S100 immunoperoxidase, 400×. Positive S100 immunoreactivity detected in T1 region of I. (L) S100 immunoperoxidase, 400×. S100 immunoreactivity is lost as tumor progresses beyond the transition zone to the amelanotic portion, T2 region of I.
Characterization of tyr–ras tumors and transgene expression
TR39- and TR60-derived tyr–ras transgenic mice placed on all three ink4a genotypes developed cutaneous or ocular tumors spontaneously. They presented with either amelanotic dermal nodules with marked telangiectasia that eventually ulcerated or exophthalmos resulting from enlarging retro-orbital mass. The most common anatomical site of tumor formation was the pinna of the ears (30%), followed by the torso (23%), tail (20%), eye (20%), perineum (3%), and periorbital area (3%) (Fig. 1). Although the incidence and latency of Ras-induced tumors differed depending on the presence or absence of ink4a mutation (see below), the clinical behavior and histological characteristics of established tumors were similar regardless of baseline ink4a status. The tumors were locally invasive but without evidence of macrometastasis at autopsy in all tumor-bearing mice. Micrometastases were not detected on full histological survey of four advanced tumor-bearing mice (data not shown).
The cutaneous tumors originated in the dermis with no apparent epidermal involvement, except for moderate epidermal hyperplasia in early lesions (Fig. 1E). As the tumor expanded, epidermal atrophy and ulceration ensued (data not shown). Histologically, all cutaneous tumors were composed predominantly of spindle cells with prominent epithelioid features and contained varying degrees of melanization within the specimens (Fig. 1E,F). Cellular atypia was frequently present, as evidenced by nuclear pleomorphism and hyperchromasia. The melanocytic origin of these tumors was established by their positive immunoreactivity for S100 as well as for tyrosinase-related protein (TRP) 1, a melanocyte-specific marker (Thomson et al. 1988) (Fig. 1G, S100, and H, TRP1). The ocular melanomas appeared to emerge from the pigmented retinal epithelium (Fig. 1I). In early stages of tumorigenesis, these ocular neoplasms consisted of proliferating spindle and epithelioid cells that were heavily pigmented and exhibited strong S100 immunoreactivity (Fig. 1J, H&E, and K, S100). These lesions subsequently underwent a distinct morphological transition characterized by loss of pigmentation and S100 immunoreactivity (Fig. 1I, cf. I with K and H for pigmentation and S100 stain). Interestingly, this transition was present in tumors arising in the ink4a+/− and ink4a−/− mice, indicating that the underlying genetic event occurred at loci other than those of ink4a or ras.
Total cellular RNAs derived from both nontransgenic and TR39 and TR60 adult eyes and skin, TR39 and TR60 primary tumors, and tyr–ras tumor cell lines were assayed by Northern blot analysis for specific hybridization to a human ras probe. This tyr–ras transcript was not detected in nontransgenic or normal transgenic eye and skin (Fig. 2A, lanes 1,2; skin not shown). In contrast, a strong hybridization signal was obtained in all tumor RNA samples (for representative samples, see lanes 3–5) as well as their derivative cell lines (data not shown), with levels comparable to those detected in myc/ras-transformed fibroblasts (Fig. 2, lane 6). Because the lack of detectable H-rasG12V transcripts in nontumor transgenic skins is likely caused by both the low level of transgene expression and the low number of melanocytes relative to other cell types, tyr–ras expression was also examined on the protein level by immunohistochemistry. As shown in Figure 2B, transgenic (a), but not nontransgenic (b), samples exhibited scattered immunoreactivity to anti-H-Ras-specific antisera in the base of hair follicles and in dermal dendritic cells, representing melanocytes from which the cutaneous tumors likely originate. Strong Ras immunoreactivity was evident in primary tumor samples (Fig. 2B, c) as well as in early passage tumor cell cultures (e). In these cultures, the dendritic nature of the melanocytes is better appreciated and further highlighted with anti-Ras immunostaining. No signal was obtained when anti-Ras antibody was omitted from the incubations (d and f).
Figure 2.
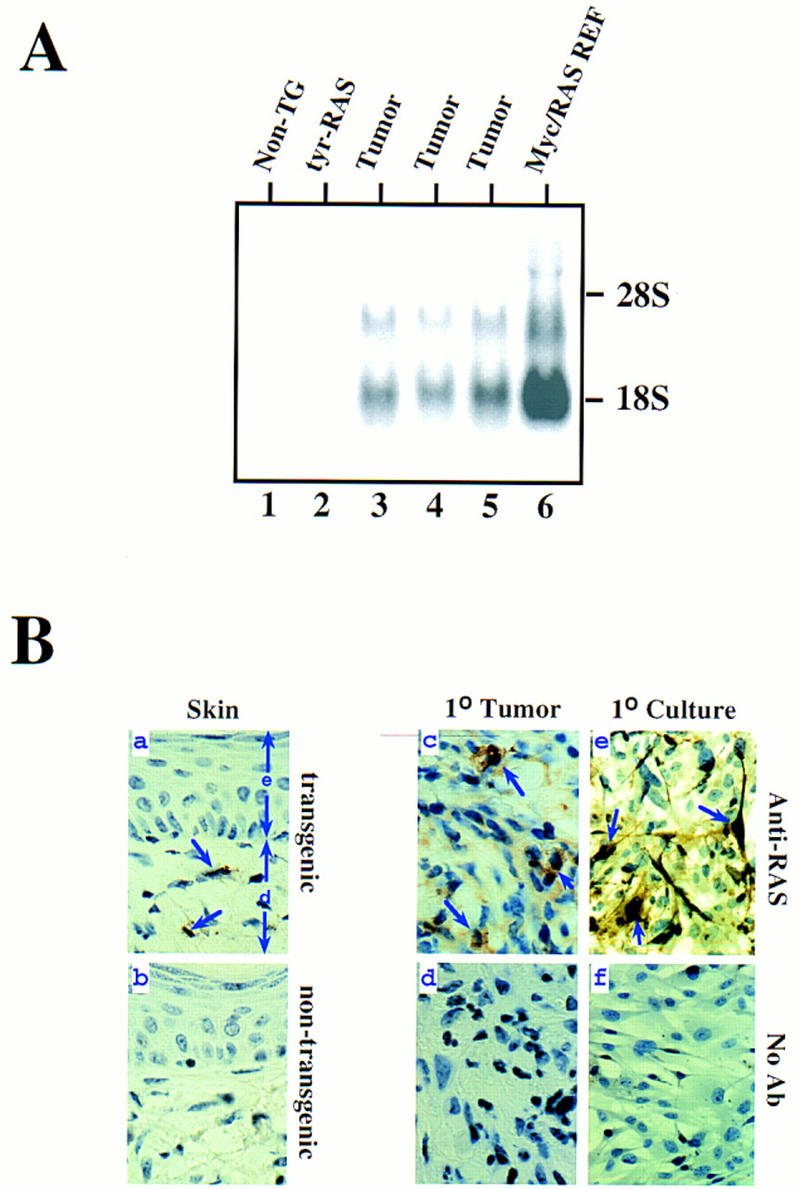
Cutaneous melanoma expressed high level of the transgene, H-rasG12V. (A) Northern blot analysis of nontumor and tumor tissue assaying activated H-rasG12V transgene expression. (Lane 1) Normal nontransgenic eye RNA; (lane 2) normal transgenic eye RNA; (lanes 3–5) RNA isolated from cutaneous tumors arising from transgenic line TR39 (lane 3) and TR60 (lanes 4,5); (lane 6) Myc/RAS-transformed rat embryo fibroblasts. (B) Photomicrographs of anti-Ras immunohistochemistry, 400×. (a) Normal skin from line TR60. (e) epidermis; (d) dermis. (b) Normal skin from a nontransgenic mouse. (c–d) Cutaneous melanoma sections, line TR60. (e–f) derivative cell line of c grown in tissue culture chamber slides.
Consistent loss of ink4a, but not ink4b, in tyr–ras tumors
The frequent mutation/deletion of the ink4a/ink4b genes in human melanoma (Hayward 1996 and references therein) prompted an assessment of the status of their mouse homologs in the tyr–ras melanomas. Southern blot analysis of DNA derived from a tumor arising in TR60 founder mouse revealed diminished hybridization signal to both ink4a and ink4b probes compared with the signal obtained with normal tail DNA from the same mouse (e.g., Fig. 3B, cf. lane 2 and lane 1). This hybridization pattern was consistent with a large homozygous codeletion. To further assess for loss of heterozygosity (LOH) of ink4a and ink4b in these melanomas, Ras-induced melanomas from both transgenic lines on ink4a heterozygous background were analyzed by Southern blot and multiplex PCR assays.
Figure 3.




LOH of ink4a in ras-induced malignant melanoma. (A) Targeting strategy of ink4a locus. Restriction map of the wild-type and knockout ink4a alleles and probes used. (B) Southern blot analysis of tumor DNAs extracted from a panel of Ras-induced cutaneous melanomas and their respective normal DNAs extracted from tails. (Top) Hybridization to the 3′-flanking probe (probe A), which distinguishes the wild-type and knockout alleles. Note consistent reduction in hybridization signal from the remaining wild-type allele in tumor samples examined when compared with that in the corresponding tail DNA samples. The strength of the residual wild-type signals correlates well with the amount of normal tissue composition within the corresponding tumor. More importantly, the derivative cell line of tumor 4a (lane 9) reveals a complete loss of wild-type signal by Southern blot analysis (data not shown). The signal ratios of wild-type to knockout alleles, calculated as described (see Materials and Methods), are as follows: (Lanes 3,4) 1.0/0.76; (lanes 5–7) 1.0/0.63/0.43; (lanes 8–10) 1.0/0.40/0.83. (Middle) Hybridization to an internal probe (probe B) that corresponds to the deleted region in the knockout allele. (Bottom) Hybridization to exon 2 probe of ink4b (probe C). (C) Multiplex PCR analysis. (Top) Generation of standard curve; (left) autoradiograph of ink4a exon 2 signal; (right) linear regression performed on exon 2 signals, as determined by PhosphoImager quantitation, plotted against amount of wild-type DNA. (Bottom) Amplification of exon 2 signal and determination of reduction to homozygosity in tumor samples. (D) Southern blot analysis of normal and tumor DNAs from melanomas arising in ink4a−/− background. Loading is normalized against hybridization to control, an irrelevant single copy gene probe (see Materials and Methods). ink4b hybridization signal intensities, calculated as described (see Materials and Methods), are as followes: (Lanes 1,2) 1.0/1.2; (lanes 3,4) 1.0/1.0; (lanes 5,6) 1.0/1.1; (lanes 7,8) 1.0/0.87; (lanes 9,10) 1.0/0.84; (lanes 11,12) 1.0/1.2; (lanes 13,14) 1.0/1.0.
In the Southern blot analysis, the restriction fragment length polymorphism of the knockout allele permits a clear view of the integrity of the remaining wild-type ink4a allele following tumor development. In such analyses, the knockout allele not only serves as a loading control with which to monitor possible reduction to homozygosity for ink4a, but also enables one to address whether homozygous loss of ink4b is a required or incidental event. Stated differently, because the knockout allele is functionally inert with respect to ink4a only, one would anticipate that its neighboring ink4b gene would not be deleted unless loss of ink4b function was also an essential pathogenetic event. As shown in Figure 3B, with the exception of tumor 4b (see PCR below), Southern blot analysis of DNA derived from dissected tyr–ras ink4a+/− tumors showed a marked reduction in hybridization intensity of the wild-type allele compared with the knockout allele when assayed with a 3′-flanking probe (probe A, lanes 1–9). When the same blot was hybridized to an ink4a exon 2/3 probe that recognizes the wild-type allele only, an equivalent decrease in signal intensity in tumor-derived DNAs was observed (probe B, lanes 1–9). These analyses suggested that reduction to homozygosity at ink4a is a consistent event coincident with the development of Ras-induced melanoma in vivo. In contrast, reduction in hybridization signal for ink4b was not observed in all of the tail/tumor DNA samples (probe C, lanes 1–9); when present, the decrease was more modest than that observed for the ink4a gene.
Semiquantitative multiplex PCR assays allowed us to examine with greater precision tumor-associated deletions of ink4a exons 2 and 3 (p16Ink4a/p19ARF) and ink4b (p15Ink4b) exon 2 sequences. Analysis of ink4a revealed a reduction to homozygosity in all six tumors examined for exon 2 (Fig. 3C) and in five of six examined for exon 3 (data not shown). These multiplex PCR results matched those obtained by Southern blot analysis for tumors 3a, 3b, and 4a. For tumor 4b, which did not show LOH by Southern blot analysis (Fig. 3B, lane 10), the multiplex PCR assay revealed a decreased amplification signal of the ink4a allele consistent with a reduction to homozygosity (see Fig. 3C). Two ocular tumors (tumors 1b and 2b) were analyzed by multiplex PCR alone because of their small size and low DNA yield. Tumor 2a sustained a barely detectable exon 2/3 deletion by Southern blot analysis (Fig. 3B, lane 4) and was excluded from the multiplex PCR analysis as a noninformative case, because this specimen contained a proportionally greater amount of normal epidermal and dermal components intermingled with tumor cells than did the other specimens. In such a sample, the amplification of normal DNA precludes the detection of a deletion by this semi-quantitative PCR method.
By multiplex PCR, a modest decrease in the amplification signal for ink4b was observed in tumors 2b, 4a, and 4b (data not shown). As these tumors also sustained a deletion of ink4a, the more modest reduction in ink4b hybridization signals may be caused by the presence of a large deletion of the wild-type allele and the retention of ink4b sequences on the ink4a knockout allele. Sequence analyses of exons 1 and 2 of the remaining ink4b alleles from these three tumors revealed only wild-type sequences, providing evidence for a functionally intact ink4b in these tumors.
Lack of ink4b deletion in tyr–ras tumors arising in mice homozygous null for ink4a
One interpretation of the codeletion of ink4a and ink4b in tumors analyzed above, as well as in published studies of cell lines and clinical tumor samples (Jen et al. 1994; Kamb et al. 1994; Orlow et al. 1995; Flores et al. 1996), may be that the loss of ink4b is a result of ink4a deletional events that randomly extend to surrounding sequences rather than the result of a biological scenario favoring codeletion of both genes. To provide support for this innocent bystander concept, the integrity of the ink4b gene was ascertained in tyr–ras tumors arising in mice homozygous null for ink4a, where tumor-associated deletion of ink4a would not be genetically required. In seven melanomas examined, no loss in ink4b signal was detected compared with that obtained in non-tumor tail DNAs (Fig. 3D, top) or with an internal loading control (Fig. 3D, bottom; see legend for quantitation ratios). Together, these multilevel analyses strongly suggest that ink4a is the principal target for tumor-associated chromosomal loss and that the occasional involvement of ink4b likely reflects its status as a bystander in deletions extending beyond the ink4a gene. These results do not exclude a tumor suppressor role for p15Ink4b under some circumstances, however, and a final resolution of this issue will require an analysis of tumor incidence in tyr–ras mice harboring ink4b deletions.
ink4a deficiency cooperates with activated RAS to induce melanoma in vivo
In previous studies, we showed that homozygous null ink4a mice spontaneously develop highly aggressive malignancies with tumor formation first apparent at 4–5 months and with an average latency of 7–8 months for 50% tumor incidence (Serrano et al. 1996). Fibrosarcomas and B cell lymphomas were the predominant tumor types and melanomas were not observed (L. Chin and R.A. DePinho, unpubl.). Notwithstanding these species differences, the above LOH studies strongly implicated the loss of ink4a function as an important pathogenetic event in Ras-induced melanomas. Thus, a more direct assessment for the role of ink4a was performed by comparing the incidence of melanoma development in the TR60 line in the presence or absence of ink4a for a period of 6.5 months (Fig. 4; +/+, n = 41; +/−, n = 50; −/−, n = 35).
Figure 4.

ink4a deficiency accelerates development of Ras-induced malignant melanomas. Time course of incidence and latency of tumor development in ink4a−/− transgenic (n = 28), ink4a+/− transgenic (n = 50), and ink4a+/+ transgenic (n = 41) animals. In the ink4a−/− transgenic cohort, a total of 21 tumors developed in 17 animals, 16 of which were melanomas. Fifty percent of these animals developed tumors by 5.5 months. In the ink4a+/− transgenic cohort, four primary melanomas developed in two mice. In the ink4a+/+ transgenic cohort, one primary melanoma developed in one mouse. Mice used in this study were derived from intercrosses between ink4a knockout and tyr–ras transgenic mice. The genetic composition of the knockout consists of 80% C57BL/6J, 18.7% 129/Sv, and 1.25% SJL/J. The composition of the transgenic consists of 50% C57BL/6J and 50% CBA/J. The final genetic composition of mice utilized in this study is complex and consists of 65% C57BL/6J, 25% CBA/J, 9.4% 129/Sv, and 0.6% SJL/J.
During this period of observation, only 1 of the 41 tyr–ras ink4a+/+ mice developed a primary melanoma at an ocular site. Among the 50 tyr–ras ink4a+/− mice, 2 animals developed 4 primary melanomas, all at cutaneous sites. In the tyr–ras ink4a−/− cohort, seven mice died suddenly of unknown causes without obvious tumors on autopsy, similar to our previous observations (Serrano et al. 1996). Among the remaining 28 mice, a total of 21 tumors developed in 17 mice; 16 of those were melanomas arising at both cutaneous and ocular sites. Of these 17 tumor mice, 12 developed melanomas only, 2 developed fibrosarcomas only, and 3 developed both melanomas and fibrosarcomas. Fifty percent of mice succumbed to tumors by 5.5 months. In summary, tyr–ras mice are particularly susceptible to the development of melanomas in the absence of ink4a gene function.
Lack of p53 gene mutation/deletion in ink4a-deficient tyr–ras tumors
Deletion or mutation of the p53 gene is common in many different tumor types but is strikingly rare in human melanoma as presented above. To determine whether a similar genetic profile exists in our animal model, the integrity of the p53 gene and level of p53 protein were examined in the tyr–ras tumors and derivative cell lines. First, tumor and control DNAs, when assayed for hybridization to a mouse p53 cDNA probe spanning the ORF, showed no gross deletions or rearrangments by Southern blot analysis (Fig. 5A). Second, Western blot analysis and immunohistochemistry on tumor tissues were performed to measure the steady-state amount of p53 protein. Typically, p53 levels are very low in normal cells. In tumors bearing dominant-negative mutants of p53, stabilization of the mutant protein leads to a dramatic accumulation of the protein in the nucleus (Levine 1997). In all tyr–ras melanomas and their derived cell lines, low, or undetectable levels of p53 protein were detected by Western blotting and immunohistochemistry, respectively (Fig. 5B, Western data not shown). In addition, no immunoreactivity was observed with the use of anti-p53 antibodies that are capable of recognizing p53 in its mutant conformation (data not shown). Finally, DNAs extracted from seven independently derived melanomas were amplified by PCR and gel-purified products were subjected to automated bidirectional nucleotide sequence analysis. These analyses focused on exons 4–8 for six tumors and exons 5 and 7 for one small tumor sample with a limited amount of DNA. Exons 4–8 were selected for examination because these exons encode mutational hot spots comprising >90% of the p53 mutations catalogued in human cancers (Levine 1997). As anticipated from the immunohistochemical and Western blot data above, all exons examined in these seven tumor samples were found to be free of mutations. Thus, similar to the human condition, the wild-type status of the p53 gene is maintained in mouse melanomas.
Figure 5.
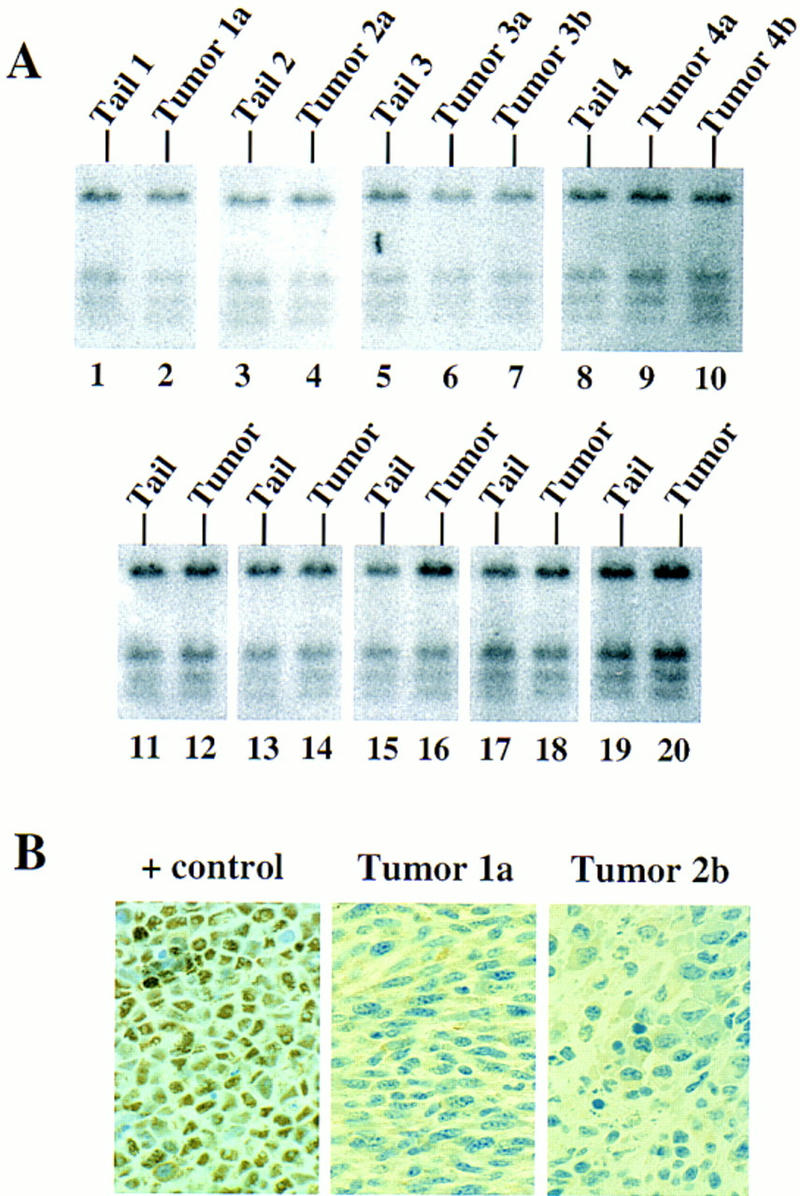
Status of p53 gene in Ras-induced malignant melanomas. (A) Southern blot analysis of tumors arising in ink4a+/− background (lanes 1–10) and ink4a−/− background (lanes 11–20). (B) Anti-p53 immunoperoxidase reaction, 1000×. (+ control) SVT2 cell line. (Tumor 1a) Cutaneous melanoma. (Tumor 2b) Ocular melanoma. Note negative immunoreactivity to anti-p53 antibody Ab-7, which recognizes both mutant and wild-type products.
Discussion
The ink4a gene and melanoma suppression in vivo
We have previously reported that mice carrying a targeted deletion of the ink4a gene that eliminates both p16Ink4a and p19ARF are viable and spontaneously develop a cancer-prone condition at an early age. Moreover, ink4a-deficient primary fibroblasts exhibited increased proliferative potential, high rate of spontaneous immortalization, and efficient transformation by H-Ras alone. These findings clearly established a role for ink4a as a potent and important tumor suppressor gene (Serrano et al. 1996). Unlike humans with germ-line ink4a mutations, however, melanomas were not observed in either spontaneous or carcinogen-induced tumors arising in mice harboring one or two copies of the null ink4a allele (L. Chin and R.A. DePinho, unpubl.). Species-specific differences in tumor spectrum have been reported for other tumor suppressor genes as well, most notably retinoblastoma (Rb) (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992), and the mechanistic basis for these differences is not understood at present. The absence of melanoma in the ink4a-deficient mouse could relate to a number of factors, including the presence or absence of important genetic modifiers and/or differences in the tissue microenvironment (e.g., most mouse melanocytes reside in the follicular epithelium rather than the epidermis). As previous studies have shown that mice can develop melanoma (Bradl et al. 1991; Iwamoto et al. 1991; Takayama et al. 1997), we reasoned that the genetic context (i.e., susceptibility modifiers or need for cooperating oncogene mutations) was a critical parameter influencing the tumor spectrum in the ink4a-deficient mice.
This line of reasoning provided the conceptual basis for the experimental approach used in this study. The selection of H-rasG12V as the cooperating oncogene in vivo was based on (1) the capacity of ink4a-deficient fibroblasts to be transformed efficiently by activated H-Ras alone in vitro (Serrano et al. 1996), in contrast to normal fibroblasts that require an additional immortalizing event [e.g., myc overexpression (Land et al. 1983), loss of ink4a or p53 function (Serrano et al. 1997)], and (2) the presence of H-rasG12V mutations in some human melanomas (see introductory section). Mutant H-rasG12V expression directed to the melanocytes of mice leads to the spontaneous development of multiple primary cutaneous melanomas with the highest penetrance observed in the ink4a−/− cohort. In accord with these findings, detailed molecular analyses of the Ras-induced melanomas arising in ink4a+/− mice revealed consistent deletion of the remaining wild-type ink4a allele. In contrast, a very different mutational profile was obtained for the ink4b gene. Although some deletions of ink4b did take place in ink4a+/− tumors, the loss of ink4b sequences was always accompanied by the reduction to homozygosity of the ink4a gene. Most importantly, in the tumors arising in the ink4a−/− mice, there was a complete absence of ink4b deletion/mutation. Based on these data, we conclude that loss of ink4a in cooperation with activated H-Ras represent powerful initiating factors in the induction of melanoma and that codeletion of ink4b is not an essential genetic event in the development of these tumors. This study, therefore, provides the most direct in vivo experimental evidence to date that ink4a is a melanoma suppressor gene. It does not exclude the possibility, however, that the ink4b gene may play a role in melanoma susceptibility and/or progression under some circumstances. In this regard, an examination of whether ink4b deficiency can cooperate with the tyr–ras transgene to generate melanomas in vivo will be highly informative.
The distinctively strong connection between tumorigenesis and mutation of the ink4a gene is unique among all of the cyclin-dependent kinase inhibitors, such as p21Cip1, p27Kip1, p57Kip2, and other Ink4 family proteins (Kamb 1995; Elledge et al. 1996). Specifically, mice lacking p21Cip1 do not exhibit an increased rate of spontaneous tumor formation (Deng et al. 1995), and although p27Kip1-deficient mice can develop intermediate lobe pituitary hyperplasia or adenoma, these neoplasms rarely progress to malignant pituitary tumors (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996). Similarly, in human cancers, the frequent alteration of INK4a contrasts sharply with an overall lower rate of INK4b mutation/deletion (Hirama and Keoffler 1995; Stone et al. 1995), infrequent mutation in p21Cip1 and p27Kip1 (Bathia et al. 1995; Gao et al. 1995; Vidal et al. 1995; Spirin et al. 1996; Lancombe et al. 1997), and lack of reported genetic lesions for p57Kip2 (Orlow et al. 1996). Such biological correlates would not have been anticipated in light of the highly similar biochemical profiles and cell culture activities of these inhibitors. These observations raise the possibility that the prominent role of the ink4a gene in tumor suppression may relate to its remarkable capacity to encode a second, structurally unrelated, growth-inhibitory product, p19ARF (Quelle et al. 1995b). A potential anti-oncogenic role for p19ARF is supported by its capacity to inhibit cell cycle progression (Quelle et al. 1995b, 1997) and to block cellular transformation by Myc/Ras or E1a/Ras (N. Liegeois and R.A. DePinho, in prep.).
The theoretical requirement for elimination of both p16Ink4a and p19ARF during tumorigenesis can also explain the high incidence of ink4a gene mutations that dually affected both reading frames (Newcomb et al. 1995; Brenner et al. 1996; FitzGerald et al. 1996; Hangaishi et al. 1996; Heinzel et al. 1996; Kinoshita et al. 1996) and the frequent occurrence of 9p21 homozygous deletions (Jen et al. 1994; Kamb et al. 1994; Orlow et al. 1995; Flores et al. 1996; Quesnel et al. 1996). This concept is consistent with the existence of cancer cell lines that have sustained a p19ARF-specific exon 1β deletion while remaining wild type for p16Ink4a but have deleted the related cyclin-dependent kinase inhibitor gene ink4b instead (Glendening et al. 1995; Flores et al. 1996). Homozygous deletions are common for the ink4a locus but represent an unusual loss-of-function mechanism for a tumor suppressor that classically presents with inactivating point mutations on the remaining wild-type allele after sustaining deletion of the other allele (Cordon-Cardo 1995; Levine 1997). Based on the above observations, it is tempting to speculate that the genetic mechanisms leading to the development of these cancers require disruption of two functionally distinct tumor suppressors, achieved through the elimination of a cyclin-dependent kinase inhibitor (e.g., p16Ink4a or p15Ink4b) and p19ARF. It is important to note, however, that several ink4a mutations have been described that target only p16Ink4a and spare p19ARF sequences (FitzGerald et al. 1996). In such tumors, it is possible that another genetic component positioned along a putative p19ARF tumor suppressor pathway (see below) has been targeted.
Role for the RAS pathway in malignant melanoma
Although activating ras mutations have an established role in the genesis of many different cancers, the results reported here provide strong support for its oncogenic role in malignant melanoma as well. Our findings stand in contrast to previous studies suggesting a role for Ras activation in more advanced stages of melanoma (progression rather than initiation), particularly in promoting a metastatic phenotype (see introductory section). Here, the development of multiple, locally invasive primary melanomas without evidence of metastatic spread clearly indicates that additional genetic events beyond Ras activation and ink4a deficiency are required for progression to metastatic disease.
The initiator role served by Ras is undoubtedly a necessary molecular step in our model. The prolonged tumor latency in the absence of ink4a deficiency, however, suggests that it is not, by itself, a potent inducer of melanoma. The weak oncogenic activity of mutant H-ras in melanocytes in vivo was also evident in another transgenic study in which melanocyte-expression of mutant H-ras resulted in melanocytic hyperplasia and no melanomas (Powell et al. 1995). The lack of melanomas in this previous study contrasts with that reported here and could relate to our use of the far upstream tyrosinase enhancer element (Ganss et al. 1994), our utilization of germ-line ink4a mutations, and variability in genetic background. With regard to the last parameter, an increase in the C57BL/6 composition appears to prolong the tumor latency when the tyr–ras transgene is placed on the ink4a+/+ genotype (L. Chin, J. Pomerantz, and R.A. DePinho, unpubl.). On another level, the modest oncogenic actions of Ras in melanocyte transformation may be in accord with recent studies showing that overexpression of activated H-ras in primary fibroblasts induces a G1 arrest and premature cellular senescence, and that H-Ras-induced mitogenesis or oncogenesis requires an accompanying immortalizing event such as ink4a or p53 deficiency (Serrano et al. 1997). This requirement of antecedent or concomitant immortalization events to elicit Ras-induced transformation of cultured cells matches well with the synergistic actions of Ras activation and ink4a deficiency in melanocyte transformation observed in this study.
Absence of p53 mutations in melanoma
Although p53 mutations represent the most common genetic abnormality in human cancers (Hollstein et al. 1991; Harris and Hollstein 1993), human melanomas and those generated in the tyr–ras model are remarkably free of p53 mutations and deletions. A possible explanation for this phenomenon is that some other component of the p53 pathway renders melanomas functionally deficient for p53, for example, mdm-2 gene amplification (Gelsleichter et al. 1995; Poremba et al. 1995). Alternatively, it is possible that some degree of functional overlap in tumor suppressor activity exists between p53 and ink4a. If such scenarios are indeed the case, then the very high frequency of ink4a deletion could obviate the need for p53 elimination in such tumors. Along these lines, both p53 and ink4a encode potent growth and tumor suppressive activities and their loss of function correlates with cellular immortalization and transformation by activated Ras (Serrano et al. 1997). Although a direct mechanistic link between p53 and ink4a pathways has yet to be established, it is intriguing that high levels of p19ARF have been observed in p53-deficient cells (Quelle et al. 1995b), leaving open the possibility of a regulatory feedback loop. Moreover, whereas p19ARF can block transformation by Myc/Ras or E1a/Ras, it has no effect on the capacity of SV40 Large T-antigen to cooperate with Ras to transform primary cells (N. Liegeois and R.A. DePinho, in prep.). This result gains significance in light of the ability of SV40 Large T antigen to render cells functionally deficient for p53 (Van Dyke 1994 and references therein). Although the actions of p19ARF have yet to be positioned along a known tumor suppressor pathway, it is tempting to speculate that a functional link between p19ARF and p53 could account for the reciprocal relationship of mutations in these genes in human (N. Liegeois and R.A. DePinho, in prep.) and mouse (this study) melanomas; that is, ink4a-deficient (p16Ink4a + p19ARF) cancers rarely exhibit p53 mutant products.
A new mouse model for malignant melanoma
The tyr–ras mouse bears several potentially useful attributes that distinguish it from existing mouse models of melanoma. First and foremost, the tyr–ras model is built on the use of endogenous proto-oncogenes (ras) and tumor suppressor genes (ink4a) strongly implicated in the genesis of human melanoma. As such, it may provide a more appropriate genetic context in which to identify new cooperating genes/pathways required for disease progression or resistance. Second, the lack of metastatic disease in the tyr–ras model could be exploited to evaluate candidate metastatic genes through transgenesis. Finally, tyr–ras induces nonmetastatic, locally invasive cutaneous melanomas, making it a potential system in which to assess therapeutic modalities for early disease processes.
Materials and methods
Production of transgenic mice
The tyr–ras transgene consists of a 1.8-kb SalI/NotI genomic fragment isolated from pRIPI–cH–Ras (kind gift of Shimon Efrat, Albert Einstein College of Medicine, Bronx, NY) encoding the mutant human H-Ras (G12V) placed under the control of the previously described tyrosinase promoter element (5.5 kb of 5′-flanking sequences) and the newly identified distal enhancer element (3.6 kb located 12 kb upstream of the promoter region) (Ganss et al. 1994) and followed by the SV40 splice and polyadenylation sequences (Gorman et al. 1982). Gel-purified transgenic inserts were introduced into the germ line of C57Bl/6 × CBA (B6/CBA) F1 mice (JAX) by pronuclear microinjection, and genomic DNA was prepared from tail tips as described previously (Hogan et al. 1994) and assayed for the presence of the transgene by Southern blotting or DNA slot blot with transgene-specific probes (Sambrook et al. 1989).
RNA and DNA isolation, Southern and Northern blot analyses, and multiplex PCR and PCR–SSCP assays of microdissected tumor samples
Total RNA was prepared by the LiCl method (Auffray and Rougeon 1980). RNAs were checked to be evenly loaded and intact by ethidium bromide staining. Genomic DNA was extracted from either fresh-frozen tumor samples (only for Southern blots) or from deparaffinized tissue sections by use of the Qiamp Tissue Kit (Qiagen). For the LOH studies, Southern blot analysis of PstI-digested tail and tumor DNA samples from the same animal was performed as described previously (Serrano et al. 1996) with an ink4a 3′-flanking genomic probe and an ink4a exon 2/3 cDNA probe defined previously (Serrano et al. 1996) and graphically illustrated in Figure 3A. To determine the status of the ink4b gene in these same tumors, an ink4b exon 2 AccI-digested 470-bp fragment (Fig. 3A) was used for hybridization to the same nitrocellulose filter probed previously with ink4a. To examine the integrity of the p53 gene, PstI-digested, Southern blotted DNAs from normal or tumor tissues were assayed for hybridization to the mouse p53 cDNA spanning the entire ORF (Tan et al. 1986). Quantitation of signals was performed with PhosphorQuant software (Bas 1000-Mac, Bio-Imaging System Fujix, Fuji). To determine LOH in tumors arising in ink4a heterozygous animals, the signal intensity of the knockout alleles was used as a loading control, and ratio of wild-type/knockout bands from tail DNA was taken as 1.0. For ink4b hybridization in tumors arising in ink4a homozygous animals, the polycystic kidney disease gene (pdk2) exon 1 probe, 192-bp EcoRI fragment, was used as a single copy loading control (Mochizuki et al. 1996). Signal intensity of the corresponding tail DNA was taken as 1.0.
DNA amplification was performed by use of two sets of primers. One set of primers corresponded to specific ink4a or ink4b exon sequences (ink4a-2, F, GTGATGATGATGGGCAACGTTC; R, GGGCGTGCTTGAGCTGAAGC. ink4a-3, F, AGGGCCCTGGAACTTCGCGGC; R, GCTAGACACGCTAGCATCGC. ink4b-2, F, AGGTCATGATGATGGGCAGC; R, ATACCTCGCAATGTCACGG). The other set of primers was directed to murine GAPDH. Three primer pairs were used to generate fragments of 452 bp (F, ACCACAGTCCATGCCATCAC; R, TCCACCACCCTGTTGCTGTA) (Clontech), 313 bp (F, CAGTCCATGCCATCACTGC; R, ACCTGGTCCTCAGTGTAGCC), or 180 bp (F, CAGAAGACTGTGGATGGCC; R, ATCCACGACGGACACATTGG); these primers were used as internal controls in coamplification with the ink4a or ink4b primers. PCR products were mixed with 1 μl of a nondenaturing loading buffer and 5 μl of each sample was loaded in a 6% nondenaturing acrylamide gel. After drying, the gels were exposed to an X-ray film and then to a phosphorimage plate for 30 min. The image plate was analyzed by a PhosphorImager as described above. The intensity of the ink4a and ink4b, and control bands was determined and expressed as a ratio of target/control.
To validate the quantitative nature of the multiplex PCR assays, varying mixtures of tail DNAs extracted from either ink4a+/+ or ink4b+/+ and ink4a−/− and ink4b−/− mice were used to generate standard curves for each PCR assay. ink4a−/− DNA contains 0% exon 2/3 content, whereas ink4b−/− DNA contains 0% exon 2 content. Mixtures of vayring amounts of ink4a+/+ or ink4b+/+ and ink4a−/− and ink4b−/− DNAs represent the range of exon 2/3 or exon 2 contents, respectively. The signal intensity of PCR products from tail DNA of +/+ mice alone represents the positive control and was normalized to a value of 100%. The validity of these curves was further assessed by assaying tail DNA extracted from ink4a+/− or ink4b+/− mice. Samples presenting <20% of the control signal were considered completely deleted for the ink4a DNA fragment (reduction to homozygosity); those presenting <40% of control were considered to have LOH for ink4b.
For PCR, 10–15 ng of DNA was amplified in the presence of 1–3.75 mm MgCl2, 80–160 μm dNTP mix, 5% DMSO, 1 μl of 10× buffer, 5 units of Taq polymerase (Promega), and 4.5–8 pmole of each primer in a final volume of 10 μl. For the radiolabeled reactions, 1 μCi of [α-33P]dCTP (NEN) was added into each reaction tube. Samples were incubated at 95°C for 3 min. This was followed immediately by primer-specific cycling, parameters as follows: ink4a–E2 and GAPDH (452 bp) (95°C–61°C–72°C) × 30 cycles; ink4a–E3 and GAPDH (180 bp) (95°C–64°C–72°C) × 3 cycles (95°C–59°C–72°C) × 25 cycles; ink4b–E2 and GAPDH (313 bp) (95°C–60°C–72°C) × 3 cycles (95°C–57°C–72°C) × 23 cycles. All products were extended at 72°C for 5 min after cycling.
Exons 4–8 of p53 were analyzed by direct automated fluorescent sequencing of gel purified (Qiagen) PCR products with a Perkin Elmer/Applied Biosystems Model 373 DNA Stretch Sequence. Primer pairs spanned intron-exon boundaries in all cases except exon 6 in which the last 6 nucleotides at the 3′ end were not amplified. Primer sequences used were as follows. p53–Exon 4, 4F, CCATCCACAGCCATCACCTC; 4R, CCACTCACCGTGCACATAAC; p53–Exon 5, 5F, TCTCTTCCAGTACTCTCCTCCC; 5R, TTACCATCACCATCGGAGC; p53–Exon 6, 6F, TCTTAGGCCTGGCTCCTCC; 6R, TGGCTCATAAGGTACCACCAC; p53–Exon 7, 7F, GCCGCCTCTGAGTATACCAC; 7R, CCTTCCTACCTGGAGTCTTCC; p53–Exon 8, 8F, TCCCGGATAGTGGGAACCTTC; 8R, CCTGCGTACCTCTCTTTGCG. PCR amplification conditions for p53 exons 5–7 were 95°C × 3 min followed by 95°C × 30 sec, 60°C × 30 sec, and 72°C × 1 min for 30 cycles. The conditions for exons 4 and 8 were 95°C × 3 min followed by 95°C × 30 sec, 64°C × 30 sec, and 72°C × 1 min for 3 cycles, then 95°C × 30 sec, 60°C × 30 sec, and 72°C × 1 min for 25 cycles. In both cases, the PCR reactions were extended at 72°C for 5 min after cycling.
Histological analysis and immunohistochemistry
Samples were fixed in 10% buffered formalin and processed through paraffin embedding by standard procedures as described previously (Serrano et al. 1996). For p53 studies, a pellet of cells from line SVT2 (American Type Culture Collection, Rockville, MD) was fixed and processed as above to serve as a positive control. For immunohistochemistry, 3 μm paraffin-embedded sections were rehydrated, rinsed in PBS, and blocked in 3% BSA in PBS at room temperature for 20 min. Affinity-purified polyclonal antisera or monoclonal antibodies against H-Ras, protein S100, TRP-1 (clone TA99, kind gift of A. Houghton, Memorial Sloan-Kettering Cancer Center, New York, NY), and p53 (pAb240 and Ab-7, Oncogene Science) were diluted in 3% BSA/PBS, incubated on tissue sections overnight at 4°C, and washed in PBS. Secondary biotinylated antibodies included goat anti-rabbit (1:1000 dilution), rabbit anti-sheep (1:1000 dilution), and horse anti-mouse (1:500 dilution) (Vector Laboratories, Burlingame, CA), and were incubated for 1 hr at room temperature. Avidin–biotin peroxidase complexes were then incubated for 30 min (Vector Laboratories, 1:25 dilution). Diaminobenzidine was used as the final chromogen and hematoxylin was used as the nuclear counterstain.
Tumor-derived cell lines were seeded in chamber slides (Lab-Tek) at a density of 20,000–50,000 cells per well and fixed with methanol/acetone (1:1) at −20°C for 10 min followed by immunohistochemical protocol as above.
Acknowledgments
We thank Nicole Schreiber-Agus, Alan Houghton, and Glenn Merlino for critical reading of the manuscripts and Alice Tam and Ken Olive for technical assistance. J.P. is a recipient of a Howard Hughes Medical Institute Medical Student Research Training Fellowship and the Oncogene Obiwon Award. D.P. is supported in part by the Charles A. Dana Foundation and grant T32 CA-09512-12. C.C.C. is supported by grants from the National Institutes of Health (CA47538, CA47179, and CA-DK97650). R.A.D. is supported by grants from the National Institutes of Health (R01HD28317, R01EY09300, and R01EY 11267) and is a recipient of the Irma T. Hirschl Career Scientist Award. Support from the Cancer Core grant P30CA13330 is also acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL depinho@aecom.yu.edu; FAX (718) 430-8972.
References
- Albino AP, Le Strange R, Oliff AI, Furth ME, Old LJ. Transforming ras genes from human melanoma: A manifestation of tumour heterogeneity? Nature. 1984;308:69–72. doi: 10.1038/308069a0. [DOI] [PubMed] [Google Scholar]
- Albino AP, Nanus DM, Mentle IR, Cordon-Cardo C, McNutt NS, Bressler J, Andreeff M. Analysis of ras oncogenes in malignant melanoma and precursor lesions: Correlation of point mutations with differentiation phenotype. Oncogene. 1989;4:1363–1374. [PubMed] [Google Scholar]
- Albino AP, Sozzi G, Nanus DM, Jhanwar SC, Houghton AN. Malignant transformation of human melanocytes: Induction of a complete melanoma phenotype and genotype. Oncogene. 1992;7:2315–2321. [PubMed] [Google Scholar]
- Auffray C, Rougeon F. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP. Ras mutations in human melanoma: A marker of malignant progression. J Invest Dermatol. 1994;102:285–290. doi: 10.1111/1523-1747.ep12371783. [DOI] [PubMed] [Google Scholar]
- Bathia K, Fan S, Spangler G, Wintraub M, O’Connor PM, Judde J-G, Magrath IA. A mutant p21 cyclin-dependent kinase inhibitor isolated from a Burkitt’s lymphoma. Cancer Res. 1995;55:1431–1435. [PubMed] [Google Scholar]
- Bradl M, Klein-Szanto A, Porter S, Mintz B. Malignant melanoma in transgenic mice. Proc Natl Acad Sci. 1991;88:164–168. doi: 10.1073/pnas.88.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner A, Paladugu A, Wang H, Olopade OI, Dreyling MF, Aldaz CM. Preferential loss of expression of p16INK4a rather than p19ARF in breast cancer. Clin Cancer Res. 1996;2:1993–1998. [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C. Mutations of cell cycle regulators. Biological and clinical implications for human neoplasia. Am J Pathol. 1995;147:545–560. [PMC free article] [PubMed] [Google Scholar]
- Davies RL, Grosse VA, Kucherlapati R, Bothwell M. Genetic analysis of epidermal growth factor action: Assignment of human epidermal growth factor receptor gene to chromosome 7. Proc Natl Acad Sci. 1980;77:4188–4192. doi: 10.1073/pnas.77.7.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder PJ. Mice lacking p21Cip1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Winston J, Harper JW. A question of balance: The role of cyclin-kinase inhibitors in development and tumorigenesis. Trends Cell Biol. 1996;6:388–392. doi: 10.1016/0962-8924(96)10030-1. [DOI] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow C, Firpo E, Polyak K, Tsai L-H, Broudy V, Perlmutter RM, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- FitzGerald MG, Harkin DP, Silva-Arrieta S, MacDonald DJ, Lucchina LC, Unsal H, O’Neill E, Koh J, Finkelstein DM, Isselbacher KJ, et al. Prevalence of germ-line mutations in p16, p19ARF, and CDK4 in familial melanoma: Analysis of a clinic-based population. Proc Natl Acad Sci. 1996;93:8541–8545. doi: 10.1073/pnas.93.16.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JF, Walker GJ, Glendening JM, Haluska FG, Castresana JS, Rubio MP, Pastorfide GC, Boyer LA, Kao WH, Bulyk ML, et al. Loss of the p16INK4a and p15INK4b genes, as well as neighboring 9p21 markers, in sporadic melanoma. Cancer Res. 1996;56:5023–5032. [PubMed] [Google Scholar]
- Ganss R, Montoliu L, Monaghan AP, Schutz G. A cell-specific enhancer far upstream of the mouse tyrosinase gene confers high level and copy number-related expression in transgenic mice. EMBO J. 1994;13:3083–3093. doi: 10.1002/j.1460-2075.1994.tb06607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Chen YQ, Wu N, Grignon DJ, Sakr W, Porter AT, Honn KV. Somatic mutations of the WAF1/CIP1 gene in primary prostate cancer. Oncogene. 1995;11:1395–1398. [PubMed] [Google Scholar]
- Gelsleichter L, Gown AM, Zarbo RJ, Wang E, Coltrera MD. p53 and mdm-2 expression in malignant melanoma: An immunocytochemical study of expression of p53, mdm-2, and markers of cell proliferation in primary versus metastatic tumors. Mod Pathol. 1995;8:530–535. [PubMed] [Google Scholar]
- Glendening JM, Flores JF, Walker GJ, Stone S, Albino AP, Fountain JW. Homozygous loss of the p15INK4b gene (and not the p16INK4 gene) during tumor progression in a sporadic melanoma patient. Cancer Res. 1995;55:5531–5535. [PubMed] [Google Scholar]
- Gorman CM, Moffat LF, Howard BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruis NA, van der Velden PA, Sandkuijl LA, Prins DE, Weaver-Feldhaus J, Kamb A, Bergman W, Frants RR. Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nature Genet. 1995;10:351–353. doi: 10.1038/ng0795-351. [DOI] [PubMed] [Google Scholar]
- Hangaishi A, Ogawa S, Imamura N, Miyawaki S, Miura Y, Uike N, Shimazaki C, Emi N, Takeyama K, Hirosawa S, et al. Inactivation of multiple tumor-suppressor genes involved in negative regulation of the cell cycle, MTS1/p16INK4A/CDKN2, MTS2/p15INK4b, p53, and Rb genes in primary lymphoid malignancies. Blood. 1996;87:4949–4958. [PubMed] [Google Scholar]
- Hannon GJ, Beach D. p15Ink4b is a potential effector of cell cycle arrest mediated by TGF-β. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med. 1993;329:1318–1327. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- Hayward NK. The current situation with regard to human melanoma and genetic inferences. Curr Opin Oncol. 1996;8:136–142. doi: 10.1097/00001622-199603000-00011. [DOI] [PubMed] [Google Scholar]
- Heinzel PA, Balaram P, Bernard HU. Mutations and polymorphisms in the p53, p21 and p16 genes in oral carcinomas of Indian betel quid chewers. Intl J Cancer. 1996;68:420–423. doi: 10.1002/(SICI)1097-0215(19961115)68:4<420::AID-IJC3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Herlyn M. Molecular and cellular biology for melanoma. Austin, TX: R.G. Landes; 1993. [Google Scholar]
- Hirama T, Keoffler HP. Role of cyclin-dependent kinase inhibitors in the development of cancer. Blood. 1995;86:841–854. [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Huang TS, Rauth S, Das Gupta TK. Overexpression of EGF receptor is associated with spontaneous metastases of a human melanoma cell line in nude mice. Anticancer Res. 1996;16:3557–3563. [PubMed] [Google Scholar]
- Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Jr, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nature Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Takahashi M, Ito M, Hamatani K, Ohbayashi M, Wajjwalku W, Isobe K, Nakashima I. Aberrant melanogenesis and melanocytic tumour development in transgenic mice that carry a metallothionein/ret fusion gene. EMBO J. 1991;10:3167–3175. doi: 10.1002/j.1460-2075.1991.tb04878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jafari M, Papp T, Kirchner S, Diener U, Henschler D, Burg G, Schiffmann D. Analysis of ras mutations in human melanocytic lesions: Activation of the ras gene seems to be associated with the nodular type of human malignant melanoma. J Cancer Res & Clin Oncol. 1995;121:23–30. doi: 10.1007/BF01202725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen J, Harper JW, Bigner SH, Bigner DD, Papadopoulos N, Markowitz S, Willson JKV, Kinzler KW, Vogelstein B. Deletion of p16 and p15 genes in brain tumors. Cancer Res. 1994;54:6353–6358. [PubMed] [Google Scholar]
- Kamb A. Cell-cycle regulators and cancer. Trends Genet. 1995;11:136–140. doi: 10.1016/s0168-9525(00)89027-7. [DOI] [PubMed] [Google Scholar]
- Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day III RS, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- Kinoshita I, Dosaka-Akita H, Mishina T, Akie K, Nishi M, Hiroumi H, Hommura F, Kawakami Y. Altered p16INK4 and retinoblastoma protein status in non-small cell lung cancer: Potential synergistic effect with altered p53 protein on proliferative activity. Cancer Res. 1996;56:5557–5562. [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova KO, Soares VC, Hofmann ES, Ono M, Khanam D, Hayday A, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27KIP1. Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Lancombe L, Orlow I, Silver D, Gerald W, Fair WR, Reuter WE, Cordon-Cardo C. Analysis of p21WAF1/CIP in primary bladder tumors. Oncol Res. 1997;8:409–414. [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lubbe J, Reichel M, Burg G, Kleihues P. Absence of p53 gene mutations in cutaneous melanoma. J Invest Dermatol. 1994;102:819–821. doi: 10.1111/1523-1747.ep12381544. [DOI] [PubMed] [Google Scholar]
- Mangues R, Seidman I, Pellicer A, Gordon JW. Tumorigenesis and male sterility in transgenic mice expressing a MMTV/N-ras oncogene. Oncogene. 1990;5:1491–1497. [PubMed] [Google Scholar]
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Lon DF. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal displasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Newcomb EW, Rao LS, Giknavorian SS, Lee SY. Alterations of multiple tumor suppressor genes (p53 (17p13), p16INK4 (9p21), and DBM (13q14)) in B-cell chronic lymphocytic leukemia. Mol Carcin. 1995;14:141–146. doi: 10.1002/mc.2940140302. [DOI] [PubMed] [Google Scholar]
- Orlow I, Lacombe L, Hannon GJ, Serrano M, Dalbagni G, Pellicer I, Reuter VE, Zhang Z-F, Beach D, Cordon-Cardo C. Deletion of the p16 and p15 genes in human bladder tumors. J Natl Cancer Inst. 1995;87:1524–1529. doi: 10.1093/jnci/87.20.1524. [DOI] [PubMed] [Google Scholar]
- Orlow I, Iavarone A, Crider-Miller SJ, Bonilla F, Latres E, Lee MH, Gerald WL, Massague J, Weissman BE, Cordon-Cardo C. Cyclin-dependent kinase inhibitor p57KIP2 in soft tissue sarcomas and Wilms’ tumors. Cancer Res. 1996;56:1219–1221. [PubMed] [Google Scholar]
- Papp T, Jafari M, Schiffmann D. Lack of p53 mutations and loss of heterozygosity in non-cultured human melanocytic lesions. J Cancer Res & Clin Oncol. 1996;122:541–548. doi: 10.1007/BF01213550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poremba C, Yandell DW, Metze D, Kamanabrou D, Bocker W, Dockhorn-Dworniczak B. Immunohistochemical detection of p53 in melanomas with rare p53 gene mutations is associated with mdm-2 overexpression. Oncol Res. 1995;7:331–339. [PubMed] [Google Scholar]
- Powell MB, Hyman P, Bell OD, Balmain A, Brown K, Alberts D, Bowden GT. Hyperpigmentation and melanocytic hyperplasia in transgenic mice expressing the human T24 Ha-ras gene regulated by a mouse tyrosinase promoter. Mol Carcin. 1995;12:82–90. doi: 10.1002/mc.2940120205. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter H, Walker C, Beach D, Sherr CJ, Serrano M. Cloning and characterization of murine p16INK4a and p15INK4b genes. Oncogene. 1995a;11:635–645. [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995b;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Cheng M, Ashmun RA, Sherr CJ. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci. 1997;94:669–673. doi: 10.1073/pnas.94.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel B, Preudhomme C, Fenaux P. p16ink4a gene and hematological malignancies. Leukem & Lymphom. 1996;22:11–24. doi: 10.3109/10428199609051724. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S, Suster S, Halaban R, Filvaroff E, Dotto GP. Induction of different morphologic features of malignant melanoma and pigmented lesions after transformation of murine melanocytes with bFGF-cDNA and H-ras, myc, neu, and E1a oncogenes. Am J Pathol. 1991;138:349–358. [PMC free article] [PubMed] [Google Scholar]
- Rigel DS, Friedman RJ, Kopf AW. Lifetime risk for development of skin cancer in the U.S. population: Current estimate is now 1 in 5. J Am Acad Dermatol. 1996;35:1012–1013. doi: 10.1016/s0190-9622(96)90139-5. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/cdk4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Willer WM, Koeffler HP. p27/Kip1 mutation found in breast cancer. Cancer Res. 1996;56:2400–2404. [PubMed] [Google Scholar]
- Steel KP, Barkway C. Another role for melanocytes: Their importance for normal stria vascularis development in the mammalian inner ear. Development. 1989;107:453–463. doi: 10.1242/dev.107.3.453. [DOI] [PubMed] [Google Scholar]
- Stone S, Jiang P, Dayananth P, Tavtigian SV, Katcher H, Parry D, Peters G, Kamb A. Complex structure and regulation of the p16(MTS1) locus. Cancer Res. 1995;55:2988–2994. [PubMed] [Google Scholar]
- Takayama H, LaRochelle WJ, Sharp R, Otsuka T, Kriebel P, Anver M, Aaronson SA, Merlino G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci. 1997;94:701–706. doi: 10.1073/pnas.94.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TH, Wallis J, Levine AJ. Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen-p53 protein complex. J Virol. 1986;59:574–583. doi: 10.1128/jvi.59.3.574-583.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson TM, Real FX, Murakami S, Cordon-Cardo C, Old LJ, Houghton AN. Differentiation antigens of melanocytes and melanoma: Analysis of melanosome and cell surface markers of human pigmented cells with monoclonal antibodies. J Invest Dermatol. 1988;90:459–466. doi: 10.1111/1523-1747.ep12460906. [DOI] [PubMed] [Google Scholar]
- Tremblay PJ, Pothier F, Hoang T, Tremblay G, Brownstein S, Liszauer A, Jolicoeur P. Transgenic mice carrying the mouse mammary tumor virus ras fusion gene: Distinct effects in various tissues. Mol Cell Biol. 1989;9:854–859. doi: 10.1128/mcb.9.2.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ’t Veer LJ, Burgering BM, Versteeg R, Boot AJ, Ruiter DJ, Osanto S, Schrier PI, Bos JL. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol. 1989;9:3114–3116. doi: 10.1128/mcb.9.7.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke TA. Analysis of viral-host protein interactions and tumorigenesis in transgenic mice. Semin Cancer Biol. 1994;5:47–60. [PubMed] [Google Scholar]
- Vidal M, Loganzo F, Jr, de Oliveira AR, Hayward NK, Albino AP. Mutations and defective expression of the WAF1 p21 tumour-suppressor gene in malignant melanomas. Melanoma Res. 1995;5:243–250. doi: 10.1097/00008390-199508000-00006. [DOI] [PubMed] [Google Scholar]
- Wagner SN, Ockenfels HM, Wagner C, Hofler H, Goos M. Ras gene mutations: A rare event in nonmetastatic primary malignant melanoma. J Invest Dermatol. 1995;104:868–871. doi: 10.1111/1523-1747.ep12607039. [DOI] [PubMed] [Google Scholar]
- Wilson RE, Dooley TP, Hart IR. Induction of tumorigenicity and lack of in vitro growth requirement for 12-O-tetradecanoylphorbol-13-acetate by transfection of murine melanocytes with v-Ha-ras. Cancer Res. 1989;49:711–716. [PubMed] [Google Scholar]
- Wittbrodt J, Lammers R, Malitschek B, Ullrich A, Schartl M. The Xmrk receptor tyrosine kinase is activated in Xiphophorus malignant melanoma. EMBO J. 1992;11:4239–4246. doi: 10.1002/j.1460-2075.1992.tb05518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Kobayashi H, Ohkawara A, Kuzumaki N. Differential expression of ras oncogene products among the types of human melanomas and melanocytic nevi. J Invest Dermatol. 1989;93:54–59. doi: 10.1111/1523-1747.ep12277350. [DOI] [PubMed] [Google Scholar]