

Abstract
Synaptic plasticity, or changes in synaptic strength, is thought to underlie learning and memory. Imaging studies, mainly in brain slices, have revealed that long-term synaptic plasticity of excitatory synapses in hippocampal neurons is coupled with structural plasticity of dendritic spines, which is thought to be essential for inducing and regulating functional plasticity. Using pharmacological and genetic manipulation, the signalling network underlying structural plasticity has been extensively studied. Furthermore, the recent advent of fluorescence resonance energy transfer (FRET) imaging techniques has provided a readout of the dynamics of signal transduction in dendritic spines undergoing structural plasticity. These studies reveal the signalling pathways relaying Ca2+ to the functional and structural plasticity of dendritic spines.
LINKED ARTICLES
This article is part of a themed section on Imaging. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2011.163.issue-8BJP has previously published an Imaging in Pharmacology themed section, edited by A Davenport and C Daly. To view this section visit http://dx.doi.org/10.1111/bph.2010.159.issue-4
Keywords: long-term potentiation, LTP, excitatory synapses, glutamate receptor, NMDA receptor, CaMKII, Ras, FLIM, fluorescence lifetime imaging microscopy
Introduction
Synaptic plasticity in the hippocampus is a prominent cellular model of learning and memory (Derkach et al., 2007). Information flows unidirectionally through the hippocampus, entering via the dentate gyrus (DG), before reaching CA3 and finally CA1 (this last synapse is called the Schaffer Collateral). In slices, specific patterns of stimulation to the Schaffer Collateral can induce long-lasting increases and decreases in synaptic strength, termed long-term potentiation (LTP) and depression (LTD) respectively.
The cell signalling underlying LTP at Schaffer Collateral synapses has been intensively studied, and a multitude of signalling molecules have been identified (Kennedy et al., 2005). Signalling for most forms of LTP starts with the flow of Ca2+ ions into postsynaptic sites through N-methyl-d-asparatic acid (NMDA)-type glutamate receptors (NMDAR) (Bliss and Collingridge, 1993). At resting membrane potential, NMDARs are blocked by Mg2+ at the channel pore, but the Mg2+ block can be released by postsynaptic depolarization. Thus, NMDARs act as a coincidence detector for presynaptic glutamate release and postsynaptic depolarization (Bliss and Collingridge, 1993). The Ca2+ elevation in spines activates numerous signalling proteins including protein kinase C (PKC), Ca2+/calmodulin-dependent kinase II (CaMKII) and small GTPase proteins such as Ras and Rho (Kennedy et al., 2005). These molecules lead to cellular processes important for LTP and LTD such as actin polymerization and depolymerization, membrane trafficking and exocytosis and endocytosis of glutamate receptors (Kennedy and Ehlers, 2006; Hotulainen and Hoogenraad, 2010). The end result of these processes is an increase in synaptic strength, which for Schaffer Collateral LTP is achieved by the insertion of 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid (AMPA)-type glutamate receptors (AMPAR) into the synapse (Derkach et al., 2007). It has also been reported that retrograde signalling from the post-synapse to the pre-synapse can occur, increasing the probability of presynaptic transmitter release (Lisman and Raghavachari, 2006; Enoki et al., 2009).
In the hippocampus, most excitatory postsynaptic terminals reside in dendritic spines, small (∼0.1 fL) mushroom-shaped structures emanating from dendrites. Many spines are connected to the dendrite via a narrow neck that acts as a diffusion barrier to compartmentalize signalling in spines (Svoboda et al., 1996; Holbro et al., 2009; Bloodgood and Sabatini, 2005). Spine volume is tightly coupled with function: larger spines have a wider postsynaptic density, more functional AMPARs, and likely produce larger excitatory postsynaptic potential (Harris et al., 1992; Matsuzaki et al., 2001; Kasai et al., 2010). The structure of spines is dynamically regulated in an activity-dependent manner (Kasai et al., 2010). LTP and LTD are associated with long-term enlargement (Matsuzaki et al., 2004; Okamoto et al., 2004; Park et al., 2006) and shrinkage (Zhou et al., 2004) of dendritic spines respectively. Also, the spine neck resistance has been found to be regulated in an activity-dependent manner (Bloodgood and Sabatini, 2005; Grunditz et al., 2008; Tanaka et al., 2008). In addition to modification of existing spines, new spine formation is associated with some forms of LTP (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999). These diverse forms of structural plasticity may be important for regulating spine function and synaptic plasticity.
Recent advances in two-photon imaging and photochemistry now enable one to image spine structural plasticity and the associated functional plasticity in brain slices (Matsuzaki et al., 2001; 2004;). Furthermore, signal transduction and molecular dynamics during spine structural plasticity have been imaged using two-photon fluorescence resonance energy transfer (FRET) techniques (Yasuda, 2006; Yasuda et al., 2006; Harvey et al., 2008; Lee et al., 2009). These new techniques have provided many insights into the mechanisms and roles of spine structural plasticity. Furthermore, by combining these imaging techniques with pharmacology, the signalling mechanisms underlying different steps of structural and functional plasticity of dendritic spines have been revealed.
Studying structural and functional plasticity of dendritic spines
To image structural plasticity of spines undergoing LTP or LTD, one must identify stimulated spines. This is not simple, because in a typical electrophysiology experiment, 10–100 synapses are activated among ∼10 000 spines. One can load a cell with Ca2+ indicator and find stimulated spines in response to synaptic stimulation (Mainen et al., 1999; Zhou et al., 2004; Enoki et al., 2009), but this is technically challenging. Alternatively, assuming that LTP is associated with spine enlargement, one could search for spines enlarged in response to synaptic stimulation (Kopec et al., 2006; Harvey and Svoboda, 2007; Yang et al., 2008).
Because imaging spines during electrophysiological LTP is difficult, multiple techniques to chemically induce LTP in many spines have been developed. In slices, chemical LTP (cLTP) can be induced by bath application of forskolin, rolipram and picrotoxin in zero Mg2+ (Otmakhov et al., 2004; Kopec et al., 2006). In this cocktail, forskolin (an activator of adenylyl cyclase) and rolipram (phosphodiestratase inhibitor) increase cAMP in CA3 neurons (and other neurons), causing burst activity in Schaffer Collateral synapses onto CA1. Picrotoxin increases the overall circuit activity by blocking inhibitory synapses, and removing Mg2+ unblocks NMDA receptors. This cLTP protocol produces spine enlargements as well as increases in excitatory postsynaptic current (EPSC) (Otmakhov et al., 2004; Kopec et al., 2006; 2007;). Another method for cLTP uses the potassium channel blocker tetraethylammonium (TEA), which depolarizes cells, increases circuit activity and produces NMDA receptor–independent LTP and spine structural plasticity (Aniksztejn and Ben-Ari, 1991; Hosokawa et al., 1995; Gu et al., 2010). In dissociated neurons, bath application of glycine (which enhances NMDA receptor response) and bicuculline (a GABAA receptor inhibitor) produces spine enlargements and increase in miniature EPSC (Lu et al., 2001; Park et al., 2006). Because cLTP protocols strongly stimulate most synapses, they probably trigger other cell mechanisms like homeostasis or cell death.
In contrast to cLTP, two-photon uncaging of caged glutamate allows one to stimulate a single targeted dendritic spine, thus eliminating the need to search for stimulated spines (Matsuzaki et al., 2001). Caged glutamate does not bind to glutamate receptors, but photostimulation removes the caging group, releasing glutamate and activating glutamate receptors. To stimulate a selected spine, one aims a two-photon laser (720 nm for MNI-L-caged glutamate) near the spine head and delivers a series of short pulses (ms), uncaging glutamate near the spine, and activating glutamate receptors in the spine. With this method, one can directly measure synaptic strength by measuring the uncaging-evoked EPSC (uEPSC) due to AMPAR activation. Typically, the laser intensity is adjusted so that uEPSCs under the basal condition are ∼10 pA, an amplitude similar to mini-EPSCs (Matsuzaki et al., 2001; 2004; Steiner et al., 2008; Tanaka et al., 2008; Lee et al., 2009). Because stimulating with uncaging alone does not depolarize spines enough to release the Mg2+ block of NMDAR, inducing LTP requires either pairing with postsynaptic depolarization or the removal of Mg2+ from extracellular solution to remove the Mg2+ block (Matsuzaki et al., 2004; Steiner et al., 2008; Tanaka et al., 2008; Lee et al., 2009). It should be noted that synaptic plasticity induced by two-photon glutamate uncaging may be different from that induced by presynaptic fibre stimulation, as Ca2+ increase in presynaptic sites maybe important for some forms of plasticity and may omit other neurotransmitters. Protocols for LTD induction using two-photon glutamate uncaging have not been found yet.
Pharmacology of spine structural plasticity and LTP
Long-lasting synaptic plasticity and associated spine structural plasticity share pharmacological properties, which we will compare for individual signalling molecules. For example, both LTP and associated spine enlargement are sensitive to inhibitors of CaMKII and Ras–extracellular signal-regulated kinase (ERK) signalling (Matsuzaki et al., 2004; Harvey and Svoboda, 2007; Lee et al., 2009; Patterson et al., 2010) (Figure 1).Actin polymerization (Matsuzaki et al., 2004; Okamoto et al., 2004) and exocytosis of endosomes (Park et al., 2004; 2006; Yang et al., 2008) are involved in both processes. Similarly, LTD and associated spine shrinkage are inhibited by activation of calcineurin (Zhou et al., 2004). However, the pathways for LTD and spine shrinkage seem to branch from there (Zhou et al., 2004), as protein phosphatase 1/2A blockers calyculin A and okadaic acid inhibit LTD but not spine shrinkage. Conversely, phosphorylation of cofilin is involved only in spine shrinkage, but not in LTD (Zhou et al., 2004).
Figure 1.
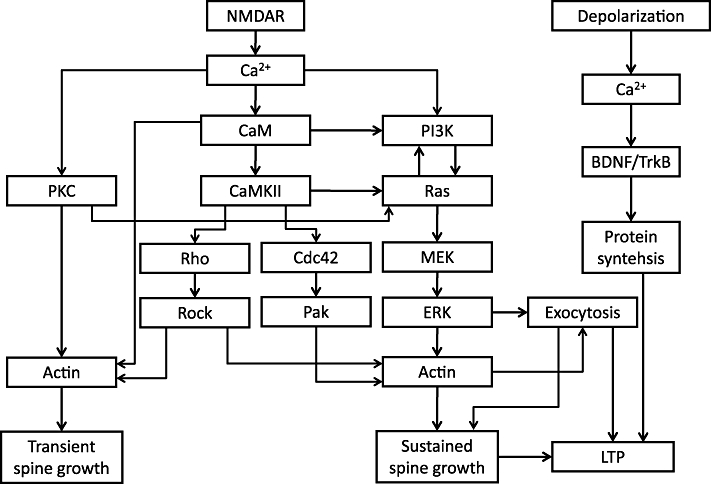
Hypothetical signalling pathways. The inputs are NMDAR activation and depolarization, while the outputs are transient and sustained spine growth and LTP. The signalling pathways in between have been studied for both LTP and for structural plasticity.
During LTP induced by 100 Hz tetanic electrical stimulation, or low-frequency two-photon glutamate uncaging in zero Mg2+, spines undergo enlargement in two distinct phases: first a transient phase which lasts 1–3 min and then a sustained plateau phase lasting more than one hour (Matsuzaki et al., 2004) (Figure 2). The amplitude of the sustained volume increase measured at 20–60 min is +50–100% (Harvey and Svoboda, 2007; Matsuzaki et al., 2004; Lee et al., 2009), while that of the transient phase (as defined by the peak volume change minus the sustained volume change) is +100–300% (Harvey and Svoboda, 2007; Matsuzaki et al., 2004; Lee et al., 2009) (Figure 2). The transient and sustained phases have different pharmacological properties.
Figure 2.
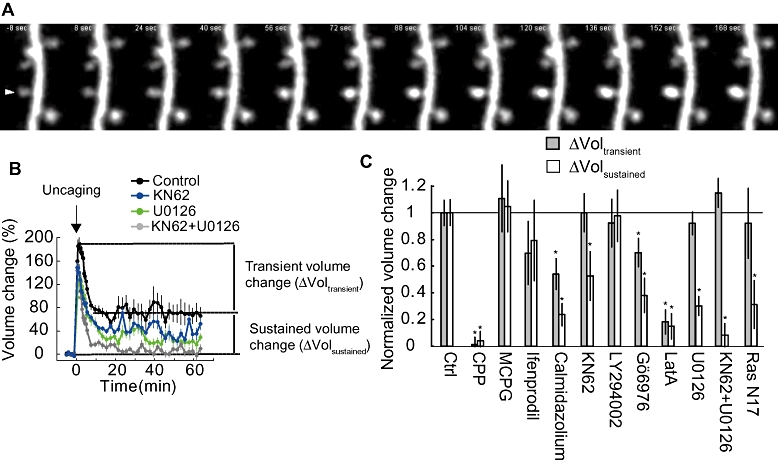
Pharmacological analysis of spine enlargement induced by 2-photon glutamate uncaging in zero Mg2+. A. Characteristic images of spine growth following 2-photon glutamate uncaging. Uncaging pulses were applied at 0.5 Hz for 1 min (30 pulses). B. Time course of spine structural plasticity. Incubation with either KN62 (10 µM) or U0126 (20 µM) partially blocked the sustained phase of structural plasticity. Incubation with the both KN62 and U0126 completely blocked sustained structural plasticity. C. Pharmacology of transient and sustained phases of structural plasticity. For signalling molecules each inhibitor affects, please refer to Table 1.Panels B and C are modified from Harvey et al. 2008.
Other stimulus protocols, like pairing uncaging with depolarization (either step or spikes) or electrical theta-burst stimulation, lead to a rapid increase in spine size with negligible decay (Harvey and Svoboda, 2007; Steiner et al., 2008; Tanaka et al., 2008; Yang et al., 2008; Lee et al., 2009). While these responses do not appear to have distinguishable transient and sustained phases, pharmacological manipulation reveals that these phases are nonetheless distinct (Tanaka et al., 2008; Yang et al., 2008). In the following sections, we review pharmacological analyses of spine structural plasticity associated with LTP under various conditions (Figure 1).
CaMKII
CaMKII is one of the most studied proteins involved in LTP and memory (Lisman et al., 2002). CaMKII subunits combine into a dodecamer wherein each subunit acts as a serine–threonine kinase (Rosenberg et al., 2005). When Ca2+ enters spines through NMDARs, Ca2+ binds to calmodulin, which in turn binds to CaMKII (Lisman et al., 2002). Active CaMKII subunits autophosphorylate the T286 site of adjacent subunits, thus allowing the enzyme to remain active even after Ca2+/CaM dissociation. The importance of T286 phosphorylation in LTP, learning and memory has been demonstrated by mutating this autophosphorylation site to alanine (T286A) in mice; these animals have impaired LTP and perform poorly in a Morris Water maze (Giese et al., 1998). CaMKII's kinase activity is also important, as mice with a kinase-dead mutation in CaMKIIα (K42R) have impaired LTP and memory (Yamagata et al., 2009). Furthermore, various pharmacological inhibitors for CaMKII have been developed with different mechanisms, and they all consistently inhibit the induction of LTP (Malinow et al., 1989; Ito et al., 1991; Hvalby et al., 1994; Otmakhov et al., 1997; Buard et al., 2010). When constitutively active CaMKII is injected or expressed in neurons, synaptic strength is potentiated, showing CaMKII activation is sufficient to induce LTP (Lledo et al., 1995; Hayashi et al., 2000).
The role of CaMKII in spine structural plasticity has also been extensively studied. KN62 and KN93, small molecule inhibitors of Ca2+-calmodulin kinases (CaMKs: they inhibit CaMKI, II, IV and CaMK kinase; Wayman et al., 2008) inhibit the sustained phase of structural plasticity induced by 2-photon glutamate uncaging, but not the transient phase (Matsuzaki et al., 2004; Harvey et al., 2008; Steiner et al., 2008; Lee et al., 2009). Some studies show almost complete inhibition of the sustained phase (Matsuzaki et al., 2004; Steiner et al., 2008), while others show only partial inhibition (∼50%) (Harvey et al., 2008; Lee et al., 2009) (Table 1). Overexpression of mutant CaMKIIα (T286A) or autocamtide 2 CaMKII inhibitory peptide (AIP2) inhibits the sustained phase of structural plasticity (Lee et al., 2009; Murakoshi et al., 2011). Also, mice with the kinase-dead mutation in CaMKIIα (K42R) exhibited deficits in sustained spine enlargement as well as in LTP (Yamagata et al., 2009).
Table 1.
The pharmacology and genetics of structural plasticity
| Target | Drug (concentration) or gene manipulation | Transient phase block | Sustained phase block |
|---|---|---|---|
| Two-photon glutamate uncaging in 0 Mg2+ | |||
| NMDAR | AP5 (50 µM)1 | + | + |
| CPP (10 µM)2 | + | + | |
| mGluR | MCPG (0.5–1 mM)1,2 | – | – |
| GluN2B | Ifenprodil (3 µM)2 | – | – |
| Calmodulin | W7 (20 µM)1 | + | + |
| Calmidazolium(30 µM)2 | Partial | + | |
| CaMKs | KN62 (4 µM)1 | – | + |
| KN62 (10 µM)2,3 | – | Partial | |
| KN93 (10 µM)4 | – | + | |
| CaMKII | CaMKII(T286A)3 | – | Partial |
| AIP25 | – | + | |
| MEK | U0126 (20 µM)2 | – | Partial |
| Ras | DN-Ras (S17N)2 | – | Partial |
| PKC | Gö6976 (1 µM)2 | Partial | Partial |
| Rho | C3 transferase5 | + | + |
| shRNA5 | Partial | – | |
| Rock | Glycyl-H1152 (2 µM)5 | + | Partial |
| Cdc42 | Wasp (210–321)5 | – | Partial |
| shRNA5 | – | + | |
| Pak | IAP3 (100 µM)5 | – | + |
| Actin | LatrunculinA (20nM)1 | – | Partial |
| LatrunculinA (100-200nM)1,2 | + | + | |
| Protein synthesis | Anisomycin (5–25 µM)6, 7 | – | – |
| Cyclohexiamide (300 µM)6, 7 | – | – | |
| 2-photon Glutamate uncaging paired with postsynaptic spiking | |||
| TrkB | K252a (200nM)6 | – | + |
| Anti-TrkB6 | – | + | |
| TrkB-Fc6 | – | + | |
| Protein synthesis | Anisomycin (5–25 µM)6 | – | + |
| Cyclohexiamide (300 µM)6 | – | + | |
| Electric stimulation (Theta burst or 100 Hz Tetanus) | |||
| Exocytosis | Botox8 | – | + |
| PKA | PKI9 | – | + |
| Protein synthesis | Anisomycin (20 µM)8 | – | + |
| Cyclohexiamide (60 µM)8 | – | + | |
| CaMKII | CaMKII (K42R knock-in)9 | – | + |
| Chemical LTP | |||
| CaMKI | STO-609 (10 µM)10 | NA | + |
| DN-CaMKI10 | NA | + | |
| Pak | DN-PAK10 | NA | + |
| Cofilin | Cofilin S3A11 | NA | + |
| Exocytosis | DN-Rab11 (S25N)12 | NA | + |
| DN-Rme1 (G429R)12 | NA | + | |
| AMPAR insertion | GluA1 C-tail13 | NA | – |
Drugs are listed with concentration in parentheses; mutants are listed in italics. +denotes blockade of structural plasticity by the manipulation; – denotes no block. Stimulus protocols are as follows: Mg2+ free uncaging means glutamate uncaging on spines in ACSF lacking Mg2+, but including TTX. Theta burst stands for theta burst protocol stimulation of Schaffer Collaterals (see (Yang et al., 2008) for details). Spike pairing means pairing glutamate uncaging with spikes delivered via whole-cell patch clamp. Transient block refers to structural plasticity immediately following stimulation, while sustained block refers to structural plasticity >20 min. after stimulation. Plus sign (+) indicates full inhibition (>∼80%), minus sign (–) indicates no inhibition (<∼20%) and ‘Partial’ indicates partial inhibition. Many experiments have been done using NMDAR antagonists; only two were listed here. NA: not applicable.
Ras/ERK
One of the many downstream pathways from CaMKII is the Ras–Raf–mitogen-activated protein kinase/ERK kinase (MEK)–ERK (Ras-Raf-MEK-ERK) signalling pathway (Figure 1). The Ras family of small GTPases is best known for its role in cancer (Schubbert et al., 2007). Small GTPases are activated by guanosine nucleotide exchange factors (GEFs) and inactivated by GTPase activation proteins (GAPs). Many GEFs and GAPs reside in or near the synapse and are activated during synaptic plasticity. For example, the GEF RasGRF1 is neuron specific and associates directly with the GluN2B subunit of NMDAR (Farnsworth et al., 1995; Krapivinsky et al., 2003). For GAPs, SynGAP associates with PSD-95, resides in the postsynaptic density (PSD) and is phosphorylated by CaMKII, which decreases its activity (Chen et al., 1998; Kim et al., 1998). Ras has multiple downstream effectors, including Raf–MEK–ERK (Thomas and Huganir, 2004) and phophotydilinositol-3 kinase (PI3K) (Qin et al., 2005). ERK signalling has been shown to be required for LTP in Schaffer Collateral synapses and some forms of memory by pharmacological inhibition of MEK (English and Sweatt, 1997; Atkins et al., 1998; Selcher et al., 1999; Selcher et al., 2003). Later, Ras was implicated in LTP: constitutively active and dominant negative Ras increased and decreased synaptic EPSCs respectively (Zhu et al., 2002). These results similarly occluded and precluded LTP.
For structural plasticity, uncaging on spines in the presence of the MEK inhibitor U0126 blocked sustained structural plasticity without effecting the transient phase, in a similar manner to KN62's block of late, but not early, structural plasticity (Figure 2) (Harvey et al., 2008; Patterson et al., 2010). Overexpression of dn-Ras (Ras 17N) also blocked sustained but not transient structural plasticity. These results suggest that the Ras–ERK pathway is important for spine structural plasticity as well as LTP. Finally, inhibitors of CaMKs (KN62) and ERK (U0126) show additive effects (Harvey et al., 2008): when either one of them is used, the sustained phase of structural plasticity is inhibited only partially (∼50%), while when added together, it completely inhibits structural plasticity (Figure 2C), suggesting that CaMK and ERK are in parallel pathways.
Rho GTPases
Rho GTPases, including Rac1, Cdc42 and RhoA, regulate actin organization (Hotulainen and Hoogenraad, 2010) and play important roles in regulating spine morphology (Luo, 2000; Tashiro and Yuste, 2004; Saneyoshi et al., 2010) and function (Wang et al., 2005; Asrar et al., 2009; Rex et al., 2009; Gu et al., 2010; McNair et al., 2010). Recently, the involvement of Rho GTPase proteins Rho and Cdc42 in glutamate uncaging-induced spine enlargement has been studied (Murakoshi et al., 2011). When Cdc42 signalling is inhibited by expressing shRNA against Cdc42 or the Cdc42 binding domain of Wasp [Wasp(210–321)], sustained spine growth is inhibited, while the transient phase remains intact. Furthermore, inhibition of Pak, one of Cdc42's downstream effectors, by IAP3 showed a similar phenotype, suggesting that the Cdc42–Pak pathway is important for maintenance of the sustained spine growth. Also, when Rho signalling is inhibited by expressing shRNA against Rho, the transient phase is preferentially inhibited. Stronger inhibition of Rho by C3 transferase, as well as pharmacological inhibition of downstream factor Rock (Glycyl-H1152), inhibited both transient and sustained phases of the spine growth, suggesting that the Rho–Rock pathway is important for both transient and sustained spine growth.
PI3K
PI3Ks are a class of phosphatidylinositol kinases that add a phosphate group to phosphatidylinositol (4,5)-triphosphate (PIP2), creating phosphatidylinositol (3,4,5)-triphosphate (PIP3) (Hawkins et al., 2006). Ras, besides activating the Raf–MEK–ERK pathway, can activate PI3K and the synthesis of PIP3 (Qin et al., 2005). PIP3 associates with many proteins that contain pleckstrin-homology (PH) domains specific to PIP3, including downstream Akt (Bjornsti and Houghton, 2004). While phosphoinositides are well known for their role in membrane trafficking and neurite growth, PI3K and PIP3's roles in LTP were more recently discovered. This was first shown when the application of the PI3K antagonist wortmannin blocked perforant path LTP in vivo in rats by a presynaptic mechanism (Kelly and Lynch, 2000). PI3K's role in Schaffer Collateral LTP was shown soon after, as both wortmannin and LY294002 were able to block LTP if applied during LTP induction (Sanna et al., 2002; Opazo et al., 2003; Qin et al., 2005). Antagonists applied during the maintenance phase were able to reduce LTP but only if applied at higher doses than necessary to block LTP induction (wortmannin: 200 nM vs. 5 µM; LY294002: 20 µM vs. 100 µM) (Sanna et al., 2002; Opazo et al., 2003). These higher doses of antagonists also caused a rundown in basal EPSC of the unstimulated pathway (Opazo et al., 2003; Karpova et al., 2006). Finally, transfecting neurons with PH domains of general receptor for phosphoinositides (GRP), which binds to and thus masks PIP3, blocks LTP (Arendt et al., 2009).
As for spine structural plasticity, the PI3K inhibitor LY294002 effects neither transient nor sustained structural plasticity (Table 1, Figure 3B) (Harvey et al., 2008). However, this could be due to relatively low dose (20 µM) used in the studies. A higher dose (100 µM) of LY294002 can inhibit the sustained phase of structural plasticity (Patterson and Yasuda, unpubl. data).
Figure 3.
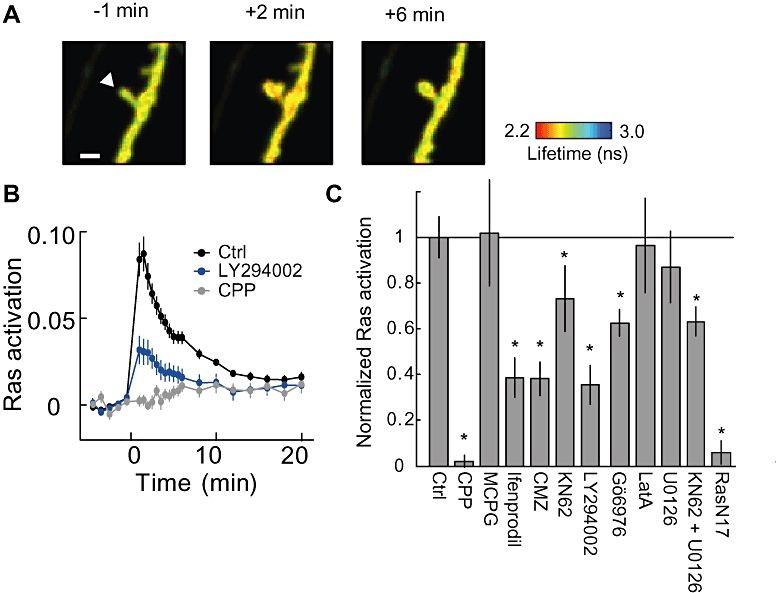
Pharmacology of spine Ras activation. A. Representative fluorescence lifetime images of Ras activation. B. Time course of average Ras activation in the spine. Ras activation was blocked by CPP (NMDAR inhibitor) and LY294002 (PI3K inhibitor). C. Pharmacology of Ras activation. For signalling molecules each inhibitor affects, please refer to Table 1. All panels adapted from (Harvey et al., 2008).
PKC
PKC was one of the first kinases to be implicated in LTP when it was found that intracellular injection of PKC into a CA1 neuron increased its EPSP and lowered its firing threshold (Hu et al., 1987). A series of papers then followed using a variety of non-specific PKC antagonists including K-252b, mellitin, PMB and H-7, which showed that these antagonists could block the induction of LTP in the Schaffer Collateral as well as the perforant path (Lovinger et al., 1987; Reymann et al., 1988; Malinow et al., 1989). The PKC specificity of these drugs were confirmed using the peptide inhibitor PKC(19–31) (Malinow et al., 1989; Wang and Feng, 1992; Wang and Kelly, 1995). PKC inhibitors could effectively reduce LTP when applied up to 3 h later (Lovinger et al., 1987; Wang and Feng, 1992). Several isoforms have been implicated in the induction and maintenance of LTP (Abeliovich et al., 1993). One isoform in particular, the atypical, constitutively active isoform PKMζ has been found to be important specifically for the maintenance of LTP (Ling et al., 2002) as well as some forms of memory (Shema et al., 2007).
In contrast to the intense study of PKC in LTP, much less is known about PKC's role in spine structural plasticity. Application of Gö6976, the inhibitor of Ca2+-dependent PKCα and β, impaired both transient and sustained phases of structural plasticity partially (Harvey et al., 2008).
Other kinases
CaMKI is activated by CaMK kinase and Ca2+/calmodulin (Wayman et al., 2008). It has been shown to be required for ERK activation and LTP by using specific inhibitor STO-609 (Schmitt et al., 2005). Also, during chemical LTP, CaMKI signals to Pak, leading to spine enlargement (Fortin et al., 2010).
Actin
In addition to the second messengers above, a variety of cellular processes have been investigated pharmacologically, foremost among them actin polymerization. Dendritic spines contain high concentrations of actin, of which 80–90% are filamentous (F-actin) (Star et al., 2002). The regulation of the actin cytoskeleton is important for spine morphology: actin polymerization and depolymerization are associated with spine enlargement and shrinkage during LTP and LTD respectively (Fukazawa et al., 2003; Okamoto et al., 2004). Furthermore, spine enlargement during LTP is inhibited by the inhibitor of action polymerization latrunculin A (LatA) in a dosage-dependent manner: at low doses (20 nM), it inhibits only the sustained phase (Matsuzaki et al., 2004), while at higher doses (100–200 nM), LatA inhibits both the transient and sustained phases of structural plasticity (Matsuzaki et al., 2004; Harvey et al., 2008); at an extremely high dose (10 µM), LatA causes spine shrinkage (Honkura et al., 2008; Murakoshi et al., 2008). These are consistent with the finding that multiple forms of F-actin assembly exist in dendritic spines (Honkura et al., 2008).
Importantly, pharmacological inhibition of actin polymerization with latrunculin A/B or cytochalasin D inhibits LTP (Kim and Lisman, 1999; Fukazawa et al., 2003) as well as spine structural plasticity (Matsuzaki et al., 2004). Also, many signalling proteins that regulate actin organization including the Rho GTPase proteins, Pak, Rho kinase (Rock) and Cofilin have been found to be required for inducing LTP (Wang et al., 2005; Asrar et al., 2009; Rex et al., 2009; Gu et al., 2010; McNair et al., 2010). Thus, unlike LTD, from which spine shrinkage can be dissociated (Zhou et al., 2004), LTP seems to be more tightly coupled with spine enlargement (Kasai et al., 2010).
Membrane and vesicular trafficking
One of the critical output steps of synaptic plasticity is the fusion of recycling endosomes with the plasma membrane, and the exocytosis of membrane proteins including AMPARs. Blockade of exocytosis by tetanus toxin or Botox almost completely blocks LTP and structural plasticity (Lu et al., 2001; Yang et al., 2008). Membrane trafficking between the plasma membrane and recycling endosomes is regulated by a variety of SNAREs (soluble N-ethylmaleimide sensitive fusion protein attachment protein receptors), GTPases and other proteins that confer target specificity and regulate membrane fusion. Of the SNAREs, two have been identified as important for plasticity: syntaxin 13, which directs traffic from early endosomes to the recycling endosome, and syntaxin 4, which is involved in exocytosis at the plasma membrane. Soluble forms of either syntaxin 4 or 13 [produced by the removal of their transmembrane (TM) domains; Syn13ΔTM and Syn4ΔTM], which block membrane fusion, impair AMPAR exocytosis, structural plasticity and LTP (Table 1) (Park et al., 2004; Park et al., 2006; Kennedy et al., 2010). Dominant-negative mutants of proteins required for endosome trafficking, Rab11a (S25N) and the Eps15 homology domain/receptor-mediated endocytosis-1 [Rme1 (G249R)], also block AMPAR exocytosis, structural plasticity, and LTP (Park et al., 2004; 2006;).
In addition to moving proteins to the plasma membrane, it has been hypothesized that exocytosis in the spine could provide additional membrane to aid spine expansion. The total membrane of endosomes in the spine, as measured by electron micrograph, is roughly half that of the spine itself (Park et al., 2006). Simultaneous measurement of spine size and AMPAR exocytosis had been roughly measured on the time scale of minutes with inconclusive results (Kopec et al., 2006; Park et al., 2006). More recently, we measured individual exocytosis events in spines and correlated this with changes in spine size within 10 s of the exocytosis event and found that spine size increases simultaneously with exocytosis (Patterson et al., 2010). This lends credence to the idea that endosomal fusion can provide membrane to the spine.
Protein synthesis
LTP is often delineated into two types: early LTP (E-LTP), which lasts for 1–2 h and is independent of protein synthesis; and late LTP (L-LTP), which persists longer, requires repeated stimuli and is protein synthesis dependent. The role of protein synthesis has been reviewed extensively elsewhere (Kelleher et al., 2004; Sutton and Schuman, 2006), but the gist of the research is that application of protein synthesis inhibitors (typically anisomycin or cyclohexamide) during induction can block LTP.
This work has been recently ported to imaging of structural plasticity. One group used a theta-burst stimulation protocol to induce plasticity and measured structural plasticity at many spines (Yang et al., 2008). They found that normally there is a persistent increase in spine size, but this increase was blocked by application of either anisomycin, or cyclohexamide. Tanaka et al. developed a modified pairing protocol, wherein they patched onto a cell and injected current pulses through the pipette to elicit back-propagating action potentials ∼20 ms after each uncaging pulse, in the presence of Mg2+ (Tanaka et al., 2008). This ‘uncaging-with-spikes’ protocol yielded a transient increase in spine size and uncaging EPSC that increased over the next hour (Table 1). In contrast, unpaired uncaging yielded a more typical time course with a peak followed by a plateau (e.g. Figure 2B). Tanaka et al. tested the protein synthesis dependence of structural plasticity and found that applying anisomycin blocked the gradual plasticity found following ‘uncaging with spikes’ but did not affect the unpaired uncaging results. They further showed that the ‘uncaging with spikes’ protocol was dependent on brain-derived neurotrophic factor (BDNF)-tyrosine kinase receptor B (TrkB) signalling (Table 1). Finally, it has been reported that bath application of BDNF or forskolin during glutamate uncaging is sufficient to induce protein synthesis–dependent spine enlargement (Tanaka et al., 2008; Govindarajan et al., 2011). These results show that protein synthesis is essential for some forms of structural plasticity.
Monitoring signal transduction in single spines
Moving beyond simply measuring structural plasticity, fluorescent sensors – specifically FRET sensors – enable one to measure the activity of signalling molecules directly. These sensors have been optimized for imaging single spines by using two-photon fluorescence lifetime imaging microscopy (2pFLIM) (Yasuda, 2006; Yasuda et al., 2006). Using 2pFLIM, the activities of CaMKII, Ras, Cdc42 and RhoA have been imaged.
2pFLIM
Intracellular signal transduction has been visualized using FRET-based signalling sensors. FRET is the process of energy transfer from an excited donor fluorophore to an acceptor fluorophore via dipole–dipole interaction (Lakowicz, 2006). Because FRET strongly depends on the distance between donor and acceptor and occurs only on the nanometer scale, FRET can be used to monitor protein–protein interactions for proteins fused to fluorophores or conformation changes of a protein tagged with two fluorophores. The fluorescence lifetime of the donor, which is the time between the excitation of the fluorophore and emission of a photon, shortens as FRET increases and thus can be used to measure FRET with high sensitivity independent of the relative concentration of donor and acceptor (Lakowicz, 2006). 2pFLIM, which combines two-photon microscopy with fluorescence lifetime measurement, allows one to quantitatively image FRET signal from the tiny volume of spines in light-scattering brain slices (Svoboda and Yasuda, 2006; Yasuda, 2006). Several sensors designed specifically for 2pFLIM have been developed and used for imaging signal transduction in single dendritic spines (Yasuda et al., 2006; Harvey et al., 2008; Lee et al., 2009; Murakoshi et al., 2011).
While FRET imaging is the only method to access intracellular signalling in individual spines, because FRET imaging relies on overexpressed sensor, one must evaluate the effects of overexpression on the spatiotemporal dynamics of signalling by measuring the relationship between the concentration of the sensor (measured from the brightness) and the spatiotemporal parameters of signalling (e.g. decay time constant, length constant) (Harvey et al., 2008; Lee et al., 2009). Also, the degree of signal perturbation needs to be evaluated (Harvey et al., 2008; Lee et al., 2009).
CaMKII
The dynamics of CaMKII signalling have been measured using biochemical methods, and it was proposed that CaMKII signals last for hours, due to Ca2+-independent, ‘autonomous’ activity produced by the autophoshorylation at T286, to maintain synaptic plasticity (Fukunaga et al., 1993; 1995; Barria et al., 1997; Lengyel et al., 2004). However, inhibition of CaMKII after establishing LTP using various types of inhibitors does not affect the maintenance of LTP (Malinow et al., 1989; Otmakhov et al., 1997; Chen et al., 2001; Buard et al., 2010) (but see Sanhueza et al., 2007). Furthermore, Lengyel et al. (2004) reported that T286 phosphorylation persists long term, while autonomous activity decays within ∼2 min during LTP.
The dynamics of CaMKII activity in neurons have been measured by a FRET sensor, Camui-α (Takao et al., 2005; Lee et al., 2009). Camui-α is a single CaMKIIα molecule tagged with a donor–acceptor fluorescent pair such as ECFP–Venus at each end. Inactive CaMKII subunits rest in a closed configuration (Rosenberg et al., 2005; Chao et al., 2010), causing FRET between the donor and acceptor at its ends; when activated, CaMKII subunits open (Rosenberg et al., 2005; Chao et al., 2010), separating the fluorophores and decreasing FRET.
In order to image CaMKII activity in single dendritic spines in cultured hippocampal slices, Lee et al. (2009) applied 2pFLIM and optimized Camui-α for 2pFLIM. The resulting sensor, named Green Camui-α, in which the monomeric EGFP–resonance energy transfer acceptor chromophore (REACh) FRET pair (Ganesan et al., 2006; Murakoshi et al., 2008) is used instead of ECFP–Venus pair, showed high sensitivity sufficient for single spine imaging under 2pFLIM (Lee et al., 2009).
In response to two-photon glutamate uncaging, CaMKII activity increased in the stimulated spines rapidly, and decayed within 1 min. The detailed analyses showed that the decay time constant of CaMKII is ∼6 s. The role of T286 phosphorylation was also demonstrated using a Green Camui-α mutant deficient in autophosphorylation at T286 [Green Camui-α with CaMKIIα(T286A)]. The mutant Green Camui-α displayed fast inactivation (<2 s), and because of the fast inactivation, the repetitive uncaging activation did not accumulate. This study suggests that CaMKII autophosphorylation is a biochemical memory on the time scale of seconds, but not hours, and helps integrate short Ca2+ signals.
Ras
Pharmacology of Ras activation in single spine
Like CaMKII, there is a fluorescent sensor for Ras activity optimized for 2pFLIM and single spine imaging. The intermolecular FRET sensor FRas consists of two molecules: mEGFP tagged H-Ras (GFP-Ras) and the Ras binding domain (RBD) of Raf1 tagged with mRFP (RFP-RBD) (Yasuda et al., 2006). When Ras is inactive, these two molecules do not interact. However, when GFP-Ras binds GTP, it binds to RFP-RBD, causing FRET. To test Ras's role in synaptic plasticity, this sensor was transfected into CA1 pyramidal neurons, and glutamate uncaging was performed.
Uncaging on spines activated Ras in the stimulated spine within 1 min, and this activation decayed with a time constant of ∼4–5 min (Harvey et al., 2008). Unlike CaMKII, active Ras was not restricted to the stimulated spine and diffused into the dendrite over ∼10 µm and even into adjacent spines.
Harvey et al. investigated the signalling pathways underlying Ras activation by combining Ras imaging with pharmacology. Ras activation was sensitive to inhibitors of CaMKII (KN62), PI3K (LY294002) and PKC (Gö6976), which caused a ∼ 30%, 60% and 40% reduction in Ras activation respectively (Figure 3). Of these, the PI3K inhibitor's effect is most interesting, as PI3K is a known effector of Ras, which implies that there may be a functional Ras-PI3K feedback loop in neurons (Carracedo and Pandolfi, 2008) (Figure 1).
AMPAR exocytosis is regulated by Ras
One of the goals of using fluorescent sensors for signalling activity is to be able to connect specific cellular outcomes with particular signalling pathways. Recently, an assay for imaging AMPAR exocytosis using pHluorin-tagged GluA1 (SEP-GluA1) has been developed (Lin and Huganir, 2007; Yudowski et al., 2007). pHluorins are pH-sensitive fluorophores that are only fluorescent at high pHs (>7), like the pH of ACSF (Miesenbock et al., 1998). Given that the pH of endosomes is typically 5–6, SEP-GluA1 selectively labels surface AMPAR (Kopec et al., 2006). Following the bleaching of all surface receptors, it is possible to image changes in fluorescence due to AMPAR exocytosis (Lin and Huganir, 2007; Yudowski et al., 2007).
Combining SEP-GluA1 with glutamate uncaging, it is possible to determine the spatial profile of AMPAR exocytosis during LTP and structural plasticity induced in single spines (Makino and Malinow, 2009; Patterson et al., 2010). Using this method, it has been found that AMPAR are exocytosed in the stimulated spine (Patterson et al., 2010) and in the parent dendrite within ∼3 µm (Makino and Malinow, 2009; Patterson et al., 2010), just as Ras activity spreads into the dendrite (Harvey et al., 2008). Consistent with this spatial profile, activity-dependent AMPAR exocytosis was inhibited by inhibition of the Ras–ERK pathway by applying ERK inhibitor U0126 or expressing dominant-negative Ras mutant, but not by inhibition of CaMK with KN62 (Patterson et al., 2010). Thus, these studies linked a specific sub-step of LTP (and potentially structural plasticity), AMPAR exocytosis, to Ras signalling (Figure 1).
Rho-GTPases
Rho GTPases are a subfamily of the Ras superfamily of proteins. Because they share structural and biochemical properties with Ras, it is possible to use similar sensors for these molecules. Murakoshi et al. (2011) recently developed sensitive sensors for two Rho proteins, RhoA and Cdc42, and measured their activity in spines during structural plasticity. Induction of spine growth caused rapid Cdc42 and RhoA activation that persisted more than ∼30 min in the stimulated spine. Notably, RhoA and Cdc42 showed contrasting activity patterns: RhoA activity spread over several microns along the dendrite, while Cdc42 activity was restricted to the stimulated spine. Inhibition of CaMKII using KN62 or autocamtide CaMKII inhibitor peptide 2 (AIP2) inhibited the activity of Cdc42 and RhoA partially, suggesting these molecules are downstream of CaMKII (Figure 1) (Murakoshi et al., 2011).
Conclusion
While the use of imaging technology to measure LTP/LTD and associated spine structural plasticity is barely a decade old, it has provided new insights into the signalling mechanisms coupling Ca2+ to the structure and function of dendritic spines. These studies have revealed a complicated signalling network triggering the induction of LTP and spine structural plasticity (Figure 1). Both structural and functional plasticity require similar signalling networks and signal via the mechanisms of actin polymerization as well as the supply of receptors and membrane from the exocytosis of endosomes (Figure 1). Although these pathways are found in experiments performed in slices or primary dissociated neurons, similar pathways may also be used in in vivo spine structural plasticity induced by experience or drug abuse (Holtmaat and Svoboda, 2009; Russo et al., 2010).
Besides using imaging to measure plasticity, the development of fluorescent sensors for signalling molecules has allowed scientists to directly measure signalling activity. Using these sensors, we have found that different signalling molecules have strikingly different spatiotemporal profiles. Inactivation time constants can range from 6 s (CaMKII) to 5 min (Ras) to ∼30 min (RhoA, Cdc42), and perhaps even longer. Some molecules are activated exclusively in the stimulated spine (CaMKII, Cdc42), while others are activated in the spine before diffusing into the dendrite and neighbouring spines (Ras, RhoA). The use of antagonists in combination with these sensors will allow more direct testing of signalling interactions; one example are PI3K antagonists effects on Ras activity (Figure 1).
Besides creating opportunities to monitor signalling pathways, in the future optical techniques will provide new opportunities to manipulate them. Photoactivatable proteins for an adrenergic receptor (Airan et al., 2009), Rho-family GTPases (Levskaya et al., 2009; Wu et al., 2009; Yazawa et al., 2009) and WASP (Leung et al., 2008) have all been recently developed, each of which work by different methods. These tools will allow researchers to precisely manipulate the function of molecules in real time. As more imaging tools are developed, we will hopefully be able to disentangle the complicated signalling responsible for LTP and ultimately memory.
Acknowledgments
We thank members of the laboratory of RY for helpful comments during the preparation of this manuscript, especially Nathan Hedrick for critical reading. MP was supported by NRSA postdoctoral fellowship from the National Institute of Health (NIH). Work in the laboratory of RY is supported by grants from the NIH. RY is an early career scientist of the Howard Hughes Medical Institute.
Glossary
Abbreviations
- 2pFLIM
two-photon fluorescence lifetime imaging microscopy
- ACSF
artificial cerebrospinal fluid
- AMPA
2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid
- AMPAR
AMPA-type glutamate receptor
- BDNF
brain-derived neurotrophic factor
- cLTP
chemical LTP
- CaMKI
Ca2+/calmodulin-dependent kinase I
- CaMKII
Ca2+/calmodulin-dependent kinase II
- CaMKK
Ca2+/calmodulin-dependent kinase kinase
- CaMK
Ca2+/calmodulin-dependent kinase
- Cdc42
Cell division control protein 42 homolog
- dn-Ras
dominant-negative Ras mutant
- DG
dentate gyrus
- E-LTP
early LTP
- FLIM
fluorescence lifetime imaging microscopy
- EPSC
excitatory postsynaptic current
- ERK
extracellular signal-regulated kinase
- FRET
fluorescence resonance energy transfer
- GAP
GTPase activation protein
- GEF
guanosine nucleotide exchange factor
- GRP
general receptor for phosphoinositides
- GTP
guanosine triphosphate
- L-LTP
late LTP
- LatA
latrunculin A
- LTD
long-term depression
- LTP
long-term potentiation
- MNI
4-methoxy-7-nitroindolinyl
- NMDA
N-Methyl-d-asparatic acid
- NMDAR
NMDA-type glutamate receptor
- phophotydilinositol-3
kinase, PI3K
- PH
pleckstrin homology
- PIP2
phosphatidylinositol (4,5)-bisphosphate
- PIP3
phosphatidylinositol (3,4,5)-triphosphate
- PKA
protein kinase A
- PKC
protein kinase C
- PSD
postsynaptic density
- Rho
Ras homolog
- SEP
superecliptic pHluorin
- TEA
tetraethylammonium
- TM
transmembrane
- TrkB
tyrosine kinase receptor B
Conflict of interest
There is no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–3 as PowerPoint slide.
References
- Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC gamma-mutant mice. Cell. 1993;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- Arendt KL, Royo M, Fernandez-Monreal M, Knafo S, Petrok CN, Martens JR, et al. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2009;13:36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW, Jia Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1) Neuropharmacology. 2009;56:73–80. doi: 10.1016/j.neuropharm.2008.06.055. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha -amino-3-hydroxyl-5-methyl4-isoxazole-propionate-type glutamate receptor. Science. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Bjornsti M-A, Houghton PJ. The tor pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Neuronal activity regulates diffusion across the neck of dendritic spines. Science. 2005;310:866–869. doi: 10.1126/science.1114816. [DOI] [PubMed] [Google Scholar]
- Buard I, Coultrap SJ, Freund RK, Lee YS, Dell'Acqua ML, Silva AJ, et al. CaMKII ‘autonomy’ is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci. 2010;30:8214–8220. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, Kuriyan J. Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat Struct Mol Biol. 2010;17:264–272. doi: 10.1038/nsmb.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Strack S, Colbran RJ, Lisman JE. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J Neurophysiol. 2001;85:1368–1376. doi: 10.1152/jn.2001.85.4.1368. [DOI] [PubMed] [Google Scholar]
- Chen H-J, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM Kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. JBC. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Enoki R, Hu Y, Hamilton D, Fine A. Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: optical quantal analysis. Neuron. 2009;62:242–253. doi: 10.1016/j.neuron.2009.02.026. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, et al. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30:11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Muller D, Miyamoto E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long-term potentiation. J Biol Chem. 1995;270:6119–6124. doi: 10.1074/jbc.270.11.6119. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- Ganesan S, Ameer-Beg SM, Ng TT, Vojnovic B, Wouters FS. A dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc Natl Acad Sci U S A. 2006;103:4089–4094. doi: 10.1073/pnas.0509922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the {alpha}?Calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunditz A, Holbro N, Tian L, Zuo Y, Oertner TG. Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J Neurosci. 2008;28:13457–13466. doi: 10.1523/JNEUROSCI.2702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi S-H, Esteban JA, Piccini A, Poncer J-C, Malinow R. Driving AMPA Receptors into Synapses by LTP and CaMKII: requirement for GluR1 and PDZ Domain Interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Holbro N, Grunditz A, Oertner TG. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:15055–15060. doi: 10.1073/pnas.0905110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GCR, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Rusakov DA, Bliss TV, Fine A. Repeated confocal imaging of individual dendritic spines in the living hippocampal slice: evidence for changes in length and orientation associated with chemically induced LTP. J Neurosci. 1995;15:5560–5573. doi: 10.1523/JNEUROSCI.15-08-05560.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu GY, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, et al. Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- Hvalby O, Hemmings HC, Jr, Paulsen O, Czernik AJ, Nairn AC, Godfraind JM, et al. Specificity of protein kinase inhibitor peptides and induction of long-term potentiation. Proc Natl Acad Sci U S A. 1994;91:4761–4765. doi: 10.1073/pnas.91.11.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I, Hidaka H, Sugiyama H. Effects of KN-62, a specific inhibitor of calcium/calmodulin-dependent protein kinase II, on long-term potentiation in the rat hippocampus. Neurosci Lett. 1991;121:119–121. doi: 10.1016/0304-3940(91)90663-e. [DOI] [PubMed] [Google Scholar]
- Karpova A, Sanna PP, Behnisch T. Involvement of multiple phosphatidylinositol 3-kinase-dependent pathways in the persistence of late-phase long term potentiation expression. Neuroscience. 2006;137:833–841. doi: 10.1016/j.neuroscience.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kelly Á, Lynch MA. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology. 2000;39:643–651. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau L-F, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, et al. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Plenum; 2006. [Google Scholar]
- Lee S-JR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel I, Voss K, Cammarota M, Bradshaw K, Brent V, Murphy KP, et al. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur J Neurosci. 2004;20:3063–3072. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci U S A. 2008;105:12797–12802. doi: 10.1073/pnas.0801232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D-T, Huganir RL. PICK1 and phosphorylation of the Glutamate Receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, et al. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 Synapses. Sci STKE. 2006;2006:re11. doi: 10.1126/stke.3562006re11. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A. 1995;92:11175–11179. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Wong KL, Murakami K, Routtenberg A. Protein kinase C inhibitors eliminate hippocampal long-term potentiation. Brain Res. 1987;436:177–183. doi: 10.1016/0006-8993(87)91573-3. [DOI] [PubMed] [Google Scholar]
- Lu W-Y, Man H-Y, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair K, Spike R, Guilding C, Prendergast GC, Stone TW, Cobb SR, et al. A role for RhoB in synaptic plasticity and the regulation of neuronal morphology. J Neurosci. 2010;30:3508–3517. doi: 10.1523/JNEUROSCI.5386-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Lee S-J, Yasuda R. Highly sensitive and quantitative FRET–FLIM imaging in single dendritic spines using improved non-radiative YFP. Brain Cell Biol. 2008;36:31–42. doi: 10.1007/s11068-008-9024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SGN, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, et al. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, et al. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK dependent manner during long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, et al. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 2005;19:2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, et al. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymann KG, Brödemann R, Kase H, Matthies H. Inhibitors of calmodulin and protein kinase C block different phases of hippocampal long-term potentiation. Brain Res. 1988;461:388–392. doi: 10.1016/0006-8993(88)90274-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends in Neurosciences. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–5199. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, et al. Phosphatidylinositol 3-Kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J Neurosci. 2005;25:1281–1290. doi: 10.1523/JNEUROSCI.4086-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM{zeta} Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–719. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Takao K, Okamoto K-I, Nakagawa T, Neve RL, Nagai T, Miyawaki A, et al. Visualization of synaptic Ca2+ /calmodulin-dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GCR, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. Mapk cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Wang JH, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci U S A. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J-H, Kelly PT. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, et al. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, et al. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29:7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang X-b, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Curr Opin Neurobiol. 2006;16:551–561. doi: 10.1016/j.conb.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, Svoboda K. Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci. 2006;9:283–291. doi: 10.1038/nn1635. [DOI] [PubMed] [Google Scholar]
- Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nat Biotech. 2009;27:941–945. doi: 10.1038/nbt.1569. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, et al. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo M-m. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.