

Abstract
Cyclin-dependent kinases (CDKs) promote the initiation of DNA replication and prevent reinitiation before mitosis, presumably through phosphorylation of key substrates at origins of replication. In fission yeast, the p65cdc18 protein is required to initiate DNA replication and interacts with the origin recognition complex (ORC) and the p34cdc2 CDK. Here we report that p65cdc18 becomes highly phosphorylated as cells undergo the G1 → S phase transition. This modification is dependent on p34cdc2 protein kinase activity, as well as six consensus CDK phosphorylation sites within the p65cdc18 polypeptide. Genetic interactions between cdc18+ and the S-phase cyclin cig2+ suggest that CDK-dependent phosphorylation antagonizes cdc18+ function in vivo. Using site-directed mutagenesis, we show that phosphorylation at CDK consensus sites directly targets p65cdc18 for rapid degradation and inhibits its replication activity, as strong expression of a constitutively hypophosphorylated mutant form of p65cdc18 results in large amounts of DNA over-replication in vivo. Furthermore, the over-replication phenotype produced by this mutant p65cdc18 is resistant to increased mitotic cyclin/CDK activity, a known inhibitor of over-replication. Therefore, p65cdc18 is the first example of a cellular initiation factor directly regulated in vivo by CDK-dependent phosphorylation and proteolysis. Regulation of p65cdc18 by CDK phosphorylation is likely to contribute to the CDK-driven “replication switch” that restricts initiation at eukaryotic origins to once per cell cycle.
Keywords: Fission yeast, cell cycle, S phase, cyclin-dependent kinase, over-replication
In all eukaryotes, a conserved group of cyclin-dependent kinases (CDKs) drives the major cell cycle events of DNA replication and mitosis (Forsburg and Nurse 1991; Reed 1992). These CDKs are also involved in quality-control mechanisms (commonly referred to as “checkpoints”), which ensure that, for example, mitosis and cell division occur only after DNA replication has been completed (Enoch and Nurse 1990). Recent work has demonstrated a third major role for CDKs in cell cycle control. Loss of CDK activity in a number of eukaryotic organisms results in ectopic rounds of DNA replication in the absence of mitosis, producing large increases in genome ploidy (Hayles et al. 1994; Dahmann et al. 1995; Sauer et al. 1995). Taken together, these results imply that CDKs actively inhibit the re-firing of replication origins during the G2 and M phases of the cell cycle.
These observations can be explained by a two-state model for origin function (Muzi-Falconi et al. 1996b; Nasmyth 1996; Stillman 1996; Jallepalli and Kelly 1997). In this model, a latent pre-initiation state is established at replication origins during the G1 period, when CDK activity is at a minimum. A subsequent increase in CDK activity at the G1 → S transition leads to phosphorylation of critical (but unidentified) initiation factors at origins of replication. This CDK-catalyzed phosphorylation switches origins into an active initiation state that can support DNA replication. Simultaneously, the phosphorylated proteins at fired origins are rendered incapable of re-entering the pre-initiation state. Because formation of the pre-initation state precedes and is required for initiation, CDK phosphorylation at the origin effectively prevents re-initiation until cells have completed mitosis. This CDK-driven “replication switch” can explain why DNA replication occurs precisely once during each cell cycle (Muzi-Falconi et al. 1996b; Nasmyth 1996; Stillman 1996; Jallepalli and Kelly 1997).
Validation of this model depends on identifying the major in vivo CDK substrates at eukaryotic replication origins. Despite the large number of replication proteins that are potential or actual CDK substrates in vitro, no data are available on their in vivo regulation by CDK phosphorylation. In this study we have examined the phosphorylation of an important replication initiator, the p65cdc18 protein of fission yeast. p65cdc18 normally accumulates only during the G1 phase of the cell cycle, a consequence of periodic transcription coupled with rapid protein turnover (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a). p65cdc18 is a member of a family of replication initiator proteins that are evolutionarily conserved from yeast to man (Bell et al. 1995; Gavin et al. 1995; Muzi-Falconi and Kelly 1995; Piatti et al. 1995; Coleman et al. 1996; Rowles et al. 1996; Williams et al. 1997). p65cdc18 interacts physically with components of the fission yeast origin recognition complex (ORC) and the p34cdc2 kinase, the major CDK in fission yeast (Grallert and Nurse 1996; Leatherwood et al. 1996; Brown et al. 1997). Regulation of p65cdc18 abundance and/or activity could be important in the cell cycle control of DNA replication, as constitutive overproduction of cdc18+ induces over-replication in the absence of mitosis (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a).
Our previous genetic studies suggested that p65cdc18 might be an important target for negative regulation by CDKs, as expression of the fission yeast CDK inhibitor p25rum1 (Correa-Bordes and Nurse 1995; Jallepalli and Kelly 1996; Martin-Castellanos et al. 1996) suppressed the cell cycle defect of a temperature-sensitive cdc18 mutant (Jallepalli and Kelly 1996). Our present analysis has revealed that p65cdc18 becomes highly phosphorylated at the G1 → S transition. p65cdc18 phosphorylation depends on the activity of p34cdc2 kinase, the CDK with which it associates. Mutation of the CDK consensus phosphorylation sites within p65cdc18 demonstrates that phosphorylation directly targets p65cdc18 for rapid proteolysis and limits its replication activity in vivo. We propose therefore that phosphorylation of p65cdc18 by p34cdc2 kinase couples CDK activation at the G1 → S boundary to the inactivation and degradation of the p65cdc18 polypeptide. Down-regulation of p65cdc18 by CDK phosphorylation is likely to contribute to the CDK-driven replication switch that limits initiation at chromosomal origins to once per cell cycle (Muzi-Falconi et al. 1996b; Nasmyth 1996; Stillman 1996; Jallepalli and Kelly 1997).
Results
p65cdc18 is phosphorylated in vivo in a CDK-dependent manner
To determine whether p65cdc18 is phosphorylated in vivo, wild-type fission yeast cells expressing a hemagglutinin (HA)-epitope tagged form of cdc18+ were grown in the presence of [32P]orthophosphate (see Materials and Methods). p65cdc18, recovered by immunoprecipitation with the anti-HA monoclonal antibody 12CA5, was found to have incorporated the radioactive label (Fig. 1A). Acid hydrolysis of the 32P-labeled p65cdc18 immunoprecipitate yielded primarily phosphothreonine and a small amount of phosphoserine (Fig. 1B). To identify the kinase responsible for this modification, we examined p65cdc18 phosphorylation in cells containing a thermolabile form of p34cdc2 kinase. In temperature-sensitive (ts) cdc2-33 mutant cells shifted to the restrictive temperature (36°C), incorporation of [32P]orthophosphate into p65cdc18 was reduced greatly relative to wild-type cells (Fig. 1A). Therefore, we conclude that phosphorylation of p65cdc18 depends on a functional p34cdc2 protein kinase in vivo.
Figure 1.

In vivo phosphorylation of p65cdc18 depends on the p34cdc2 protein kinase. (A) Wild-type fission yeast cells (WT, lanes 1 and 2) or temperature-sensitive cdc2-33 mutant cells (cdc2ts, lanes 3 and 4) expressing hemagglutinin (HA) epitope-tagged cdc18+ from the full-strength nmt1+ promoter (REP3X) were labeled metabolically with [32P]orthophosphate at 36°C (restrictive temperature for cdc2-33). After immunoprecipitation with monoclonal anti-HA antibody 12CA5 (lanes 2,4) or non-immune (NI) antibody (lanes 1,3), samples were resolved by SDS-PAGE. (Top) 32P-labeled p65cdc18 was detected using a PhosphorImager. (Bottom) Total p65cdc18 in each immunoprecipitate was visualized by immunoblotting with HA-specific antibodies. (B) Phosphoamino acid analysis of p65cdc18 phosphorylated in vivo was performed essentially as described (Lin and Desiderio 1993). (P-Ser) Phosphoserine; (P-Thr) phosphothreonine; (P-Tyr) phosphotyrosine. (C) Extracts of cells expressing HA-tagged cdc18+ were immunoprecipitated with anti-HA antibodies. p65cdc18 immunoprecipitates were then incubated without additions (lane 1), with λ protein phosphatase (PPase) (lane 2), or with phosphatase plus the phosphatase inhibitor vanadate (lane 3). Samples were resolved by 6% SDS-PAGE and immunoblotted with antibodies to HA. (D) Dependence of p65cdc18 modification on p34cdc2 kinase activity. Wild-type cells (lane 1) and temperature-sensitive cdc2-33 mutant cells (cdc2ts, lane 2) expressing HA-tagged cdc18+ from the weak nmt1 promoter (REP42X) were shifted to 36°C for 4 hr. Total cell extracts were subjected to modified SDS-PAGE followed by immunoblotting with anti-HA antibodies. The position of the fastest migrating p65cdc18 species is marked by an asterisk.
Modification of p65cdc18 by phosphorylation reduces its electrophoretic mobility during SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1A, bottom). Treatment of p65cdc18 immunoprecipitates with bacteriophage λ protein phosphatase in the absence (but not in the presence) of the phosphatase inhibitor vanadate resulted in increased mobility (Fig. 1C). Individual species of p65cdc18 were best resolved when cell extracts were fractionated directly using a modified SDS-PAGE protocol (see Materials and Methods) and then immunoblotted. Under these conditions, a large fraction of p65cdc18 expressed in cdc2ts cells grown at the restrictive temperature migrated more rapidly than p65cdc18 from wild-type cells, consistent with the reduced phosphorylation of the former (Fig. 1D).
p65cdc18 is phosphorylated at the G1 → S phase transition
We exploited this phosphorylation-induced mobility shift to examine the phosphorylation state of p65cdc18 during a synchronous round of DNA replication. Fission yeast cells constitutively expressing HA-tagged cdc18+ from a heterologous promoter (REP42X) were synchronized in the G1 phase of the cell cycle using a cdc10–V50 mutant block. p65cdc18 produced in G1-arrested cells was largely unmodified, as shown by the absence of slower migrating species (Fig. 2A, lane 2). On release from G1 arrest, slower migrating forms of p65cdc18 were observed just before cells carried out a single round of DNA replication (Fig. 2A, lanes 3–5, and B, left panel).
Figure 2.


Phosphorylation of p65cdc18 occurs at the beginning of S phase and coincides with activation of p34cdc2 kinase. HA-tagged cdc18+ was expressed from the weak nmt1 promoter (REP42X) in temperature-sensitive cdc10-V50 mutant cells. Logarithmically growing cells were synchronized in late G1 by incubation at the restrictive temperature (36°C) for 4 hr, then released by rapid downshift to the permissive temperature (25°C). (A) Extracts were prepared from cells grown asynchronously (lane 1), synchronized in G1 (lane 2), or released from G1 into S phase (lanes 3–5). Additional extracts were prepared from cells released into medium containing the DNA synthesis inhibitor hydroxyurea (lanes 6–8). p65cdc18 modification was resolved using a modified SDS-PAGE protocol; the highest mobility form of p65cdc18 is marked as a reference (*). (B) Flow cytometry analysis of DNA content. Because DNA replication coincides with cytokinesis during normal vegetative growth, asynchronously growing fission yeast cells display a single peak equal to fully replicated (2C) DNA, whereas cells arrested in G1 yield a single peak corresponding to unreplicated (1C) DNA. In untreated cells, this 1C peak was rapidly converted to 2C (left). In contrast, the 1C peak was maintained in cells arrested in early S phase with hydroxyurea (right). (C) Activation of p34cdc2 kinase at the G1 → S transition. p34cdc2 kinase complexes were isolated using p13suc1-agarose from cells synchronized as in A, and kinase reactions performed as described (see Materials and Methods). Histone H1 phosphorylation was quantitated using a PhosphorImager and is presented relative to the activity in G1-arrested cells.
We also measured the activity of p34cdc2 kinase in these synchronized cell populations. As predicted, modification of p65cdc18 closely paralleled the increase in p34cdc2 kinase activity at the G1 → S phase boundary (Fig. 2C). Given that S phase occupies ∼10% of the cell cycle (∼25–30 min under these conditions), the point at which p65cdc18 first becomes modified coincides roughly with the beginning of S phase. This was confirmed by a similar synchronization experiment performed in the presence of the DNA synthesis inhibitor hydroxyurea (HU). This compound leads to depletion of nucleotide precursors and blocks cells in early S phase but probably does not affect the initial firing of replication origins. Slower migrating forms of p65cdc18 appeared with identical kinetics in HU-treated cells, even in the absence of bulk DNA synthesis (Fig. 2A, lanes 6–8, and B, right panel). We conclude that p65cdc18 phosphorylation occurs at or near the initiation of DNA replication, and coincides with the rise in p34cdc2 kinase activity at the G1 → S transition.
Genetic interactions between cdc18+ and the S phase cyclin cig2+ suggest that CDK-dependent phosphorylation of p65cdc18 antagonizes its function in vivo
The fission yeast cig2+ gene encodes a B-type cyclin important for rapid activation of p34cdc2 kinase activity at the G1 → S phase transition (Martin-Castellanos et al. 1996; Mondesert et al. 1996). Consistent with this, we found that deletion of cig2+ delayed the onset of p65cdc18 modification by ∼30 min (Fig. 3A, cf. with Fig. 2A). From this result, we conclude that the S-phase cyclin cig2+ has a role in promoting rapid phosphorylation of p65cdc18 at the G1 → S phase transition.
Figure 3.

S-phase CDK activity promotes phosphorylation of p65cdc18 and antagonizes its function in vivo. (A) Deletion of the S-phase cyclin cig2+ delays the onset of p65cdc18 modification. The G1 synchronization protocol used in Fig. 2 was performed with a similar cdc10–V50 strain also harboring a deletion of the S-phase cyclin cig2+. Note that the appearance of slower migrating forms of p65cdc18 is delayed in Δcig2 cells by ∼30 min (cf. Fig. 2A). (B) Deletion of cig2+ suppresses the cell cycle defect of a temperature-sensitive cdc18 mutation. Fission yeast strains with the indicated genotypes were constructed by standard genetic methods and tested for viability at 25°C (permissive temperature for cdc18–K46) or 36°C (restrictive temperature).
In principle, p45cig2-dependent phosphorylation could either activate or inhibit p65cdc18. If the effect of this phosphorylation is inhibitory, then one would expect that eliminating cig2+ should increase cdc18 function in vivo. To test this genetic prediction, we constructed a double mutant strain harboring a temperature-sensitive allele of cdc18 and a deletion of the S-phase cyclin cig2+ (cdc18–K46 Δcig2) and evaluated its ability to grow at 36°C (the restrictive temperature for the parental cdc18–K46 mutant). Remarkably, deletion of the cig2+ cyclin reversed the lethal cell cycle defect caused by the cdc18 mutation, as the cdc18–K46 Δcig2 strain was able to growth at 36°C (Fig. 3B). This suppression was specific to cig2+, as deletion of cig1+ (a minor G2/M phase cyclin) had no effect (Fig. 3B).
Given that both S phase and mitotic forms of p34cdc2 kinase have been shown to associate with p65cdc18 and phosphorylate it in vitro (Brown et al. 1997), the simplest interpretation of these genetic data is that CDK phosphorylation antagonizes the essential function of p65cdc18, and that elimination of p45cig2-associated CDK activity partially alleviates this antagonism. This conclusion agrees with our earlier finding that increased expression of rum1+ (which encodes a potent inhibitor of both S-phase and mitotic cyclin/CDK complexes in fission yeast; Correa-Bordes and Nurse 1995; Jallepalli and Kelly 1996; Martin-Castellanos et al. 1996) also reverses the replication and cell cycle defects of the cdc18–K46 mutant (Jallepalli and Kelly 1996).
Elimination of CDK phosphorylation sites creates a constitutively hypophosphorylated form of p65cdc18
p65cdc18 contains six sites corresponding to the full consensus for CDK phosphorylation, with five of these sites clustered in the amino-terminal region of the polypeptide (Fig. 4A). The frequency of these sites is consistent with the number of slower-migrating species of p65cdc18 that can be resolved during gel electrophoresis. We changed the putative threonine phosphoacceptor at each of these sites to alanine (both singly and in various combinations), thereby generating a series of mutant cdc18ΔCDK alleles (see Materials and Methods).
Figure 4.

CDK consensus sites are required for phosphorylation of p65cdc18. (A) Primary structure of the p65cdc18 polypeptide. The threonine residue at each of the six CDK consensus sites was replaced with alanine. (B) Dependence of p65cdc18 modification on CDK consensus sites. Extracts from wild-type cells expressing either HA-tagged cdc18+ (WT, lane 1) or a mutant gene lacking all six consensus CDK sites (ΔCDK1–6, lane 2) from the weak nmt1 promoter (REP42X) were analyzed as in Fig. 1D. (C) In vivo phosphorylation of p65cdc18. Cells expressing HA-tagged cdc18+ (lanes 1–3) or cdc18ΔCDK1-6 (lanes 4 and 5) from the full-strength, thiamine-repressible nmt1+ promoter (REP3X) were grown in the absence (−) or presence (+) of thiamine and labeled with [32P]orthophosphate. Extracts were immunoprecipitated and analyzed as in Fig. 1A. (D) In vivo 32P incorporation is shown as a percent of that in the wild type (solid bars). Wild-type and ΔCDK1-5 mutant forms of p65cdc18 purified from yeast as GST fusion proteins (Brown et al. 1997) were subjected to in vitro phosphorylation by exogenous p34cdc2 kinases (p45cig2 CDK, open bars; p56cdc13 CDK, hatched bars). 32P incorporation was quantitated using a PhosphorImager.
Several lines of evidence demonstrated that mutation of these sites greatly reduces phosphorylation of p65cdc18 in vivo. Very little [32P]orthophosphate was incorporated into a mutant protein lacking all six CDK consensus sites (p65cdc18ΔCDK1-6) in vivo (Fig. 4C). Consistent with its lack of modification, this mutant polypeptide migrated as a single species of increased mobility relative to wild-type p65cdc18 (Fig. 4B). In addition, a purified glutathione-S-transferase (GST) fusion protein lacking the first five CDK consensus sites (GST–Cdc18ΔCDK1-5) was a relatively poor substrate for p34cdc2 kinases in vitro (Fig. 4D), although it bound p34cdc2 kinase as tightly as wild-type GST–Cdc18 (Brown et al. 1997). We conclude that CDK consensus sites account for the majority of p65cdc18 phosphorylation.
Characterization of cdc18ΔCDK mutants in vivo
Taken together, the experiments presented above demonstrate that the CDK consensus sites are critical for phosphorylation of p65cdc18 both in vitro and in vivo and are probably the actual phosphoacceptor sites used by p34cdc2 kinase. The functional significance of this phosphorylation was tested in fission yeast. Based on our previous genetic data (Fig. 3; Jallepalli and Kelly 1996), hypophosphorylated p65cdc18 is expected to be an active form of the replication initiator protein. Plasmids expressing either wild-type or various ΔCDK forms of cdc18 from the native cdc18+ promoter were transformed into a fission yeast strain in which the endogenous copy of cdc18+ is under the control of the thiamine-repressible nmt1 promoter (YMF15; Muzi-Falconi et al. 1996a). Endogenous cdc18+ expression was then shut off by adding thiamine, allowing us to test whether the plasmid-borne sequences were capable of complementing the replication and cell-cycle defects conferred by loss of cdc18+. Cells containing either wild-type cdc18+ or any of the cdc18ΔCDK alleles [including those lacking the first five (ΔCDK1-5) or all six (ΔCDK1-6) consensus phosphorylation sites] were capable of growth in the presence of thiamine (data not shown). Therefore, each of the cdc18ΔCDK alleles is functional in fission yeast.
However, the cdc18ΔCDK mutants exhibited several phenotypes (described below) consistent with hyperactivation or hyperaccumulation of cdc18+ in vivo. These phenotypes were particularly apparent when the cdc18ΔCDK plasmids were introduced into a wild-type fission yeast strain and are therefore genetically dominant (Fig. 5). Following transformation, we observed that the cdc18ΔCDK1-5 mutant gave rise to colonies with decreased efficiency compared with wild-type cdc18+. Those cdc18ΔCDK1-5 transformants that formed colonies displayed a reduced colony size, suggesting severe retardation of cell growth (Fig. 5). Microscopic observation of these colonies revealed many highly elongated cells, a phenotype indicative of cell division cycle (cdc) arrest or delay (P.V. Jallepalli and T.J. Kelly, unpubl.; Nurse and Bisset 1976). These dominant slow-growth and cdc phenotypes are consistent with the idea that phosphorylation at these sites negatively regulates cdc18+, as similar phenotypes have been observed in cells overproducing wild-type cdc18+ (Kelly et al. 1993a).
Figure 5.

Characterization of cdc18ΔCDK mutants in fission yeast. Wild-type cells (TK2) were transformed with plasmids expressing wild-type cdc18+ or cdc18ΔCDK1-5 from the cdc18+ promoter, or with vector alone (pIRTL) as a control. Following transformation, cells were plated on EMM plates and incubated at 30°C for 3 days. Examples of small, slow-growing cdc18ΔCDK1-5 transformant colonies are marked by the white arrows. Further growth was observed after several more days at 30°C, yielding colonies large enough for subsequent manipulation and analysis.
ΔCDK mutant forms of p65cdc18 are resistant to rapid degradation
To explore the biochemical basis of these phenotypes, we examined the abundance of wild-type and mutant forms of p65cdc18 by immunoblotting (Fig. 6A). We found that the mutant p65cdc18ΔCDK polypeptides accumulated to higher than normal steady-state levels when expressed from the cdc18+ promoter (Fig. 6A). To test whether the mutant polypeptides were resistant to proteolysis in vivo, wild-type and mutant cdc18 genes were expressed from a weak form of the thiamine-repressible nmt1 promoter (REP42X). We monitored the degradation of wild-type and mutant p65cdc18 after adding thiamine to repress transcription and prevent de novo synthesis. Wild-type p65cdc18 was degraded very rapidly, consistent with its short half-life (t1/2 8 min; Muzi-Falconi et al. 1996a). In contrast, a mutant polypeptide lacking all six CDK sites was significantly more stable, persisting for more than 60 min after shutoff (t1/2 ≅ 35 min). Intermediate degrees of stabilization were observed for mutant p65cdc18ΔCDK polypeptides in which fewer sites were eliminated (Fig. 6B). We obtained similar results when new protein synthesis was inhibited simultaneously with cycloheximide, indicating that the observed differences were independent of translation (P.V. Jallepalli and T.J. Kelly, unpubl.). We conclude that CDK-dependent phosphorylation at these sites normally targets p65cdc18 for rapid degradation. This conclusion is consistent with our earlier demonstration that p65cdc18 accumulates to very high levels in fission yeast cells with low p34cdc2 kinase activity, such as those overproducing the CDK inhibitor p25rum1 (Jallepalli and Kelly 1996).
Figure 6.
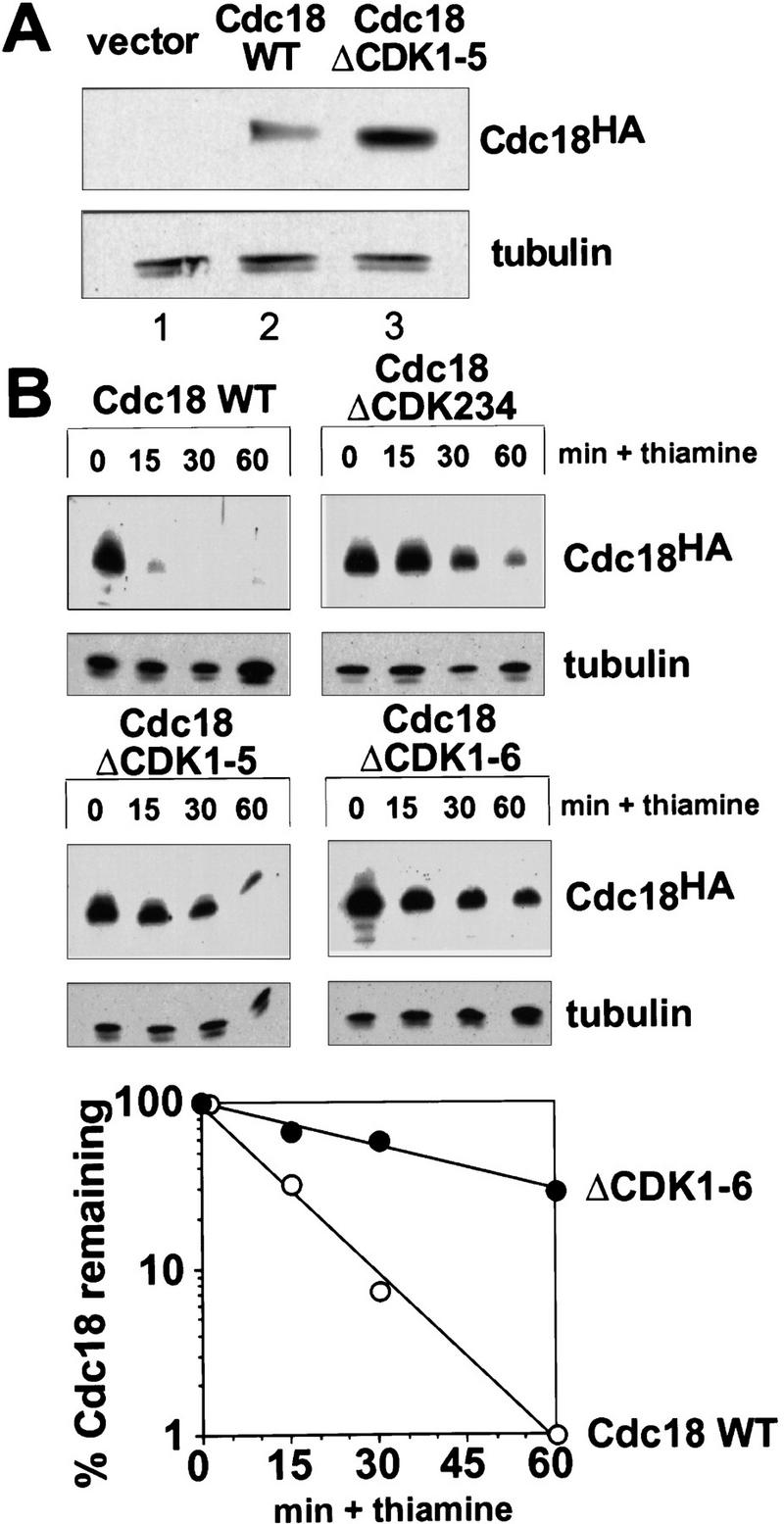
Phosphorylation down-regulates the stability of p65cdc18 in vivo. (A) Increased abundance of p65cdc18ΔCDK1-5. Fission yeast cells were transformed with plasmids expressing HA-tagged cdc18+ or cdc18ΔCDK1-5 under the control of the cdc18+ promoter, or with vector alone (pIRTL) as a control. Extracts were resolved by standard SDS-PAGE and immunoblotted with monoclonal antibodies to HA (to detect p65cdc18) and tubulin (to provide a loading control). (B) Increased stability of mutant p65cdc18 proteins. Wild-type cdc18+ or various cdc18ΔCDK mutants, each under the control of the weak nmt1 promoter (REP42X) and containing the HA epitope tag, were expressed in wild-type cells in the absence of thiamine. At time 0, cdc18 expression was repressed by addition of thiamine (Muzi-Falconi et al. 1996a). Samples harvested at the indicated times were analyzed by SDS-PAGE and immunoblotted with monoclonal antibodies to HA or tubulin. (Bottom) The abundance of wild-type p65cdc18 (○) and p65cdc18ΔCDK1-6 (•) was quantitated using 35S-labeled secondary antibodies and a PhosphorImager. These data were used to calculate the apparent half-life (t1/2) of each protein.
Elimination of CDK phosphorylation sites strongly enhances cdc18-induced over-replication of the genome
As a further test of p65cdc18 function in vivo, we compared the ability of wild-type and mutant p65cdc18 polypeptides to cause over-replication when expressed from the full-strength nmt1+ promoter (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a). We found that strong expression of cdc18+ appeared to saturate the proteolytic machinery, so that comparable levels of the wild-type and mutant polypeptides accumulated (Fig. 7A). Under these conditions, cells expressing wild-type p65cdc18 became elongated, and a modest percentage over-replicated their DNA (Fig. 7B; Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a). In contrast, the mutant p65cdc18ΔCDK1-5 protein caused a much larger fraction of cells to over-replicate their DNA and increased the extent of over-replication to between 8 and 16 genome equivalents per cell (Fig. 7B). A significant proportion of these cells displayed grossly swollen nuclei that distended the fission yeast cell wall (Fig. 7C). As expected, this strong over-replication phenotype was incompatible with further cell proliferation (Fig. 7D). We conclude that the mutant p65cdc18ΔCDK1-5 polypeptide is a more potent inducer of over-replication than wild-type p65cdc18, even when both are expressed at similar levels. These results suggest that CDK phosphorylation directly inhibits the activity of p65cdc18 by an additional mechanism that is independent of protein turnover.
Figure 7.
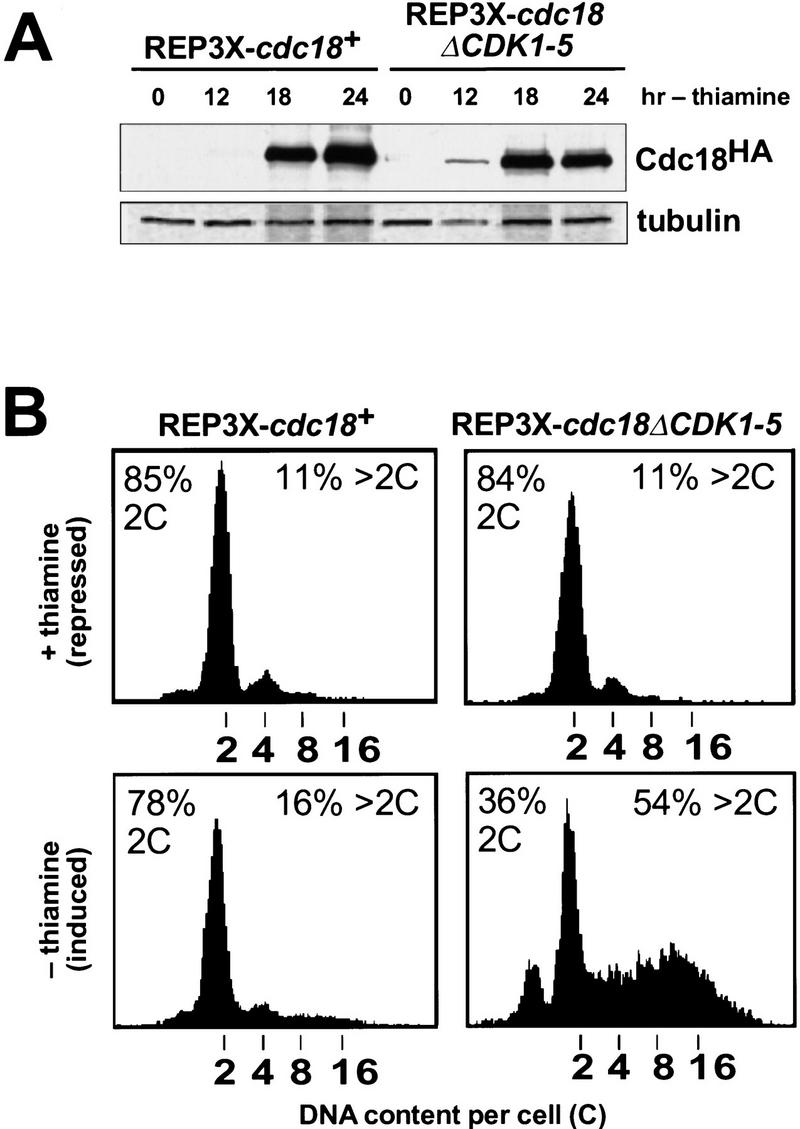


Increased replication activity associated with p65cdc18ΔCDK1-5. (A) Expression of HA-tagged cdc18+ (lanes 1–4) or cdc18ΔCDK1-5 (lanes 5–8) from the full-strength nmt1+ promoter (REP3X) was induced by growth in thiamine-free medium for the indicated times. Cell extracts were separated by SDS-PAGE and immunoblotted with monoclonal antibodies to HA and tubulin. Quantitation of p65cdc18 abundance as in Fig. 5B demonstrated that the wild-type protein accumulated to the same or slightly higher level as the ΔCDK mutant polypeptide by 18 hr. (B) The same strains were grown in the presence (upper panels) or absence of thiamine (lower panels) for 30 hr. DNA content per cell was measured by flow cytometry and is presented on a logarithmic scale. The percent of cells with normal (2C) and over-replicated (>2C) DNA content is indicated. (C) Cells in B were examined by DAPI staining of nuclei and fluorescence microscopy. The fields shown illustrate qualitatively the range of phenotypes observed in several independent experiments. When grown in the presence of thiamine, cells containing REP3X–cdc18ΔCDK1-5 were indistinguishable from those containing REP3X–cdc18+ (data not shown). (D) Wild-type cells transformed with the indicated plasmids were streaked onto EMM plates either with (+) or without (−) thiamine and incubated for 4 days at 30°C.
Over-replication induced by constitutively hypophosphorylated p65cdc18 is resistant to increased mitotic cyclin/CDK activity
The high level of over-replication induced by the p65cdc18ΔCDK1-5 mutant allowed us to investigate the mechanism of over-replication and in particular the role of the mitotic cyclin p56cdc13 in this process. To date, two classes of over-replication mutants have been described in fission yeast. The first class comprises mutants in which mitotic (i.e., p56cdc13-associated) CDK activity has been eliminated quantitatively, for example, through deletion of the cdc13+ gene or overproduction of CDK inhibitors (Broek et al. 1991; Hayles et al. 1994; Moreno and Nurse 1994). The second class consists of mutants overproducing the essential replication initiator protein p65cdc18 (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a; this paper). In the latter class, mitotic cyclin/CDK activity seems to be reduced slightly (about two- to threefold) as cells begin to over-replicate (Nishitani and Nurse 1995). Moreover, it is not known whether the small decline in mitotic cyclin/CDK activity contributes to over-replication, or whether active DNA synthesis itself elicits a cell cycle checkpoint that delays entry into mitosis and activation of mitotic cyclin/CDK complexes (Enoch and Nurse 1990; Enoch et al. 1992; Kelly et al. 1993a,b; Navas et al. 1995).
To address these issues, we examined mitotic cyclin/CDK activity during p65cdc18ΔCDK1-5-induced over-replication. Cells harboring the REP3X–cdc18ΔCDK1-5 construct were grown in the presence or absence of thiamine and mitotic cyclin/CDK complexes immunoprecipitated using polyclonal antibodies directed against p56cdc13 (Fig. 8A, lanes 1 and 2). Interestingly, we found that overexpression of p65cdc18ΔCDK1-5 produced little or no decline in p56cdc13-associated kinase activity, even though the majority of cells had undergone multiple cycles of DNA replication (Fig. 8B, left panel). These results suggest that inhibition of mitotic cyclin/CDK activity is not required for p65cdc18ΔCDK1-5-induced over-replication.
Figure 8.
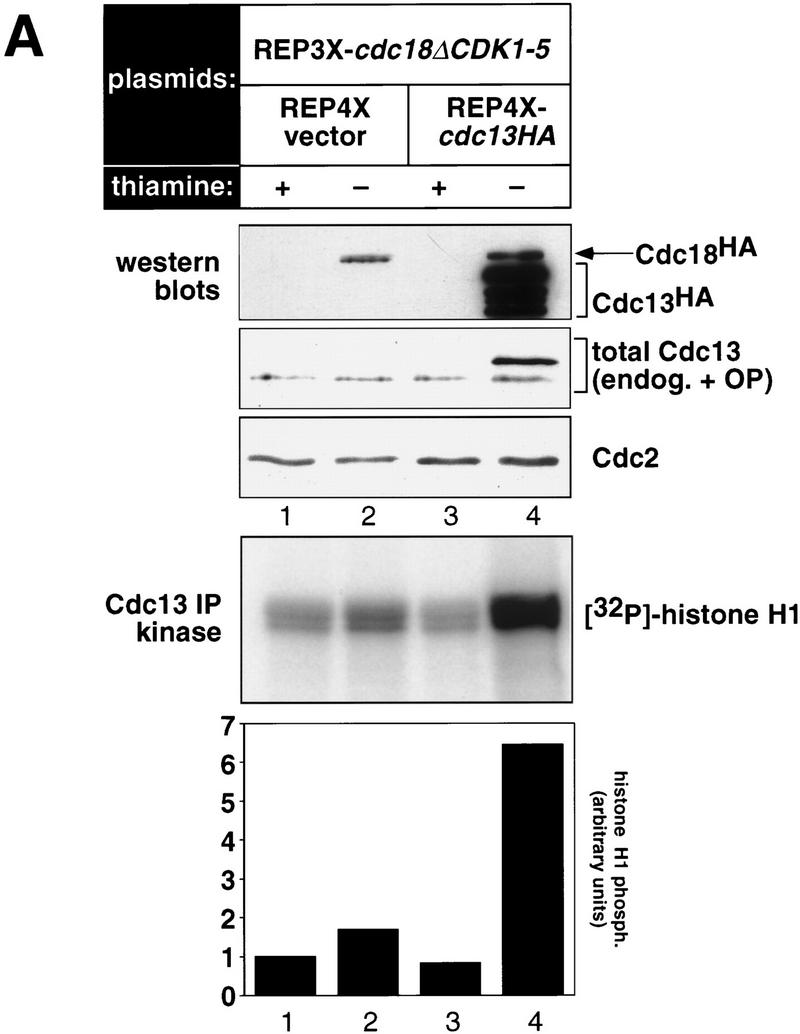

Over-replication induced by p65cdc18ΔCDK1-5 does not involve inhibition of mitotic CDKs and is resistant to increased mitotic CDK activity. (A) Strains expressing HA-tagged cdc18ΔCDK1-5 as in Fig. 7 were transformed with a plasmid expressing the mitotic cyclin cdc13+ (also HA-tagged) from the full-strength, thiamine-repressible nmt1+ promoter (REP4X–cdc13HA), or with empty vector (REP4X) as a control. Strains were subsequently grown in the presence (+) or absence (−) of thiamine for 24 hr. (Upper panels) Cell extracts were resolved by SDS-PAGE and immunoblotted with either monoclonal antibodies to HA (12CA5) or polyclonal antiserum to p56cdc13 (min56). Immunoblots were subsequently probed with monoclonal antibodies to p34cdc2 (Y100) as a loading control. (Bottom panel) Cell extracts were immunoprecipitated with antiserum specific for p56cdc13, and immobilized mitotic cyclin/CDK complexes were incubated with histone H1 and [γ-32P]ATP as described (Jallepalli and Kelly 1996). Histone H1 phosphorylation was quantitated using a PhosphorImager and is presented relative to the activity in normal diploid cells (lane 1). (B) The DNA content of cells in A was measured by flow cytometry. The percent of cells with normal (2C) and over-replicated (>2C) DNA content is indicated.
To demonstrate conclusively that over-replication is independent of effects on mitotic CDK activity, we examined the effect of high mitotic CDK levels on this process. Cells expressing p65cdc18ΔCDK1-5 were cotransformed with a plasmid expressing HA-tagged p56cdc13 from the same promoter (REP4X–cdc13HA). As shown in Figure 8, growth of this strain in thiamine-free medium induced high levels of p56cdc13, which was detected readily by immunoblotting with HA-specific monoclonal antibodies (Fig. 8A, top). Overexpressed p56cdc13 was also detected with polyclonal antibodies directed against p56cdc13 (Fig. 8A, middle), therefore demonstrating that total p56cdc13 levels were increased significantly. p56cdc13-associated kinase activity also increased dramatically, reaching a level about six or seven times higher than in normal diploid cells (Fig. 8A, bottom; cf. lanes 1 and 4). However, this increase in mitotic cyclin/CDK activity was unable to block re-initation caused by p65cdc18ΔCDK1-5, as most cells had accumulated higher than normal DNA contents, averaging about eight genome equivalents per cell (Fig. 8B, right panels). We conclude that over-replication caused by the p65cdc18ΔCDK1-5 polypeptide is resistant to increased mitotic CDK activity, which has been been proposed to block re-activation of chromosomal origins during the G2 and M phases of the cell cycle (Hayles et al. 1994).
Discussion
Our results demonstrate that p65cdc18 is an important substrate for CDK-dependent phosphorylation at the G1 → S transition. This phosphorylation is not needed for p65cdc18 to promote DNA replication; rather, CDK phosphorylation regulates p65cdc18 in a negative manner, in part by providing a signal for its rapid degradation. Phosphorylation of p65cdc18 depends on the activity of p34cdc2 kinase, the CDK with which it interacts physically (Brown et al. 1997). CDK-dependent phosphorylation of p65cdc18 (both in vivo and in vitro by p34cdc2 kinase) also depends on six consensus CDK phosphorylation sites, five of which are clustered in the amino-terminal region of the polypeptide. We have shown recently that, although this amino-terminal region is also important for the association of p65cdc18 with p34cdc2 kinase, the CDK consensus phosphorylation sites within this region are dispensable for this physical interaction (Brown et al. 1997). This observation suggests that the mutations described in this paper specifically affect p65cdc18 phosphorylation while sparing other structural and functional domains within the protein.
Phosphorylation-dependent proteolysis can account for the fact that p65cdc18 must be synthesized de novo during the G1 phase of each cell cycle (Muzi-Falconi et al. 1996a). p65cdc18 is the first example of a cellular replication initiator protein regulated in vivo by CDK-dependent phosphorylation and proteolysis. Similar mechanisms also control the abundance of the mammalian recombination activator protein RAG-2 (Lin and Desiderio 1993; Li et al. 1996) and the yeast G1 cyclin CLN2 (Lanker et al. 1996) during the cell cycle. We have found recently that degradation of p65cdc18 also requires the activity of the 26S proteasome and appears to proceed via a covalent, multiubiquitinated intermediate (P.V. Jallepalli and T.J. Kelly, unpubl.; Kominami and Toda 1997). Phosphorylation of p65cdc18 may promote its recognition by specific E2 (ubiquitin-conjugating enzyme) and/or E3 (ubiquitin-protein ligase) proteins, although the identity of these factors remains unknown. In the case of CLN2, the CDC53 protein has been shown to interact with the phosphorylated G1 cyclin and promote its ubiquitin-mediated degradation (Willems et al. 1996). Recently, the fission yeast pop1 mutant was reported to up-regulate expression of the cdc18+ gene product (Kominami and Toda 1997), but whether this reflects reduced degradation of p65cdc18, or increased transcription or translation of cdc18+ mRNAs, is not clear. Similarly, degradation of the budding yeast homolog of p65cdc18, CDC6, depends on the activity of CDC4, a gene closely related to pop1+ (Piatti et al. 1996). Given the early divergence of fission and budding yeasts during evolution (Sipiczki 1989), these results suggest that rapid, phosphorylation-coupled proteolysis of these initiator proteins is an ancient and conserved feature of DNA replication, at least among simple eukaryotes.
Our results also suggest that, in addition to promoting its degradation, CDK phosphorylation of p65cdc18 directly inhibits its replication activity in vivo. This conclusion comes from the fact that a hypophosphorylated form of p65cdc18 (p65cdc18ΔCDK1-5) induced much more over-replication than wild-type p65cdc18, even though both proteins were expressed at the same level (Fig. 7). Exactly how CDK phosphorylation inhibits the replication activity of p65cdc18 remains unknown, but several potential mechanisms exist that could be tested in future experiments. For example, CDK phosphorylation could disrupt the association of p65cdc18 with ORC or minichromosome maintenance (MCM) factors bound to chromatin (Coleman et al. 1996; Grallert and Nurse 1996; Leatherwood et al. 1996), or inhibit an enzymatic activity of p65cdc18 itself. Importantly, p65cdc18 contains consensus ATP-binding motifs and shares homology with the largest ORC subunit, ORC1, which has been shown recently to possess origin-regulated ATPase activity (Klemm et al. 1997). It will be of interest to determine whether p65cdc18 has a similar activity in vitro and if it is subject to regulation by CDK phosphorylation.
We have begun to address the mechanism by which the p65cdc18ΔCDK1-5 protein promotes over-replication in fission yeast by examining the role of the mitotic cyclin p56cdc13 in this process. We find little or no decline in mitotic cyclin/CDK activity in cells undergoing over-replication caused by p65cdc18ΔCDK1-5 (Fig. 8A). Moreover, overexpression of p56cdc13, which produced mitotic CDK levels about six to seven times higher than in normal (i.e., non-over-replicating) cells, did not reduce the extent of over-replication (Fig. 8B). Therefore, p65cdc18ΔCDK1-5-induced over-replication does not require inhibition of mitotic cyclin/CDK activity and in fact is resistant to increased mitotic cyclin/CDK activity in vivo. These results suggest that p65cdc18ΔCDK1-5 promotes reinitiation at replication origins by acting at a point downstream of mitotic CDK regulation (Nishitani and Nurse 1995; see also Jallepalli and Kelly 1996).
Whatever the precise mechanism, our findings demonstrate that CDK phosphorylation normally limits the stability and in vivo replication activity of p65cdc18 as cells traverse the G1 → S boundary, therefore contributing to the CDK-driven replication switch that restricts origin firing to once per cell cycle (Fig. 9; Muzi-Falconi et al. 1996b; Nasmyth 1996; Stillman 1996; Jallepalli and Kelly 1997). An important (but until now hypothetical) feature of this switch is that CDK phosphorylation triggers the inactivation and/or destruction of one or more critical initiator proteins, thereby preventing re-initiation at previously fired origins. The results presented in this paper demonstrate that p65cdc18—a protein essential for the initiation of DNA replication—is subject to precisely this kind of negative regulation. Inhibition of p65cdc18 provides a general paradigm for how CDK phosphorylation of multiple origin-bound replication factors could limit S phase to once per cell cycle.
Figure 9.

Regulation of p65cdc18 by CDK phosphorylation as an element of the CDK-driven replication switch (Jallepalli and Kelly 1997). Under this model, CDK phosphorylation of origin-bound replication proteins alters their activities in two important ways. First, CDK phosphorylation converts one or more of these factors from a latent pre-initiation state to an active initiation state, which promotes origin unwinding and subsequent events required for DNA replication. Second, CDK phosphorylation of several key replication proteins (including p65cdc18) prevents them from regenerating the pre-initiation state by inhibiting their enzymatic activity and/or promoting their degradation. Because establishment of the pre-initiation state precedes and is required for initiation, origins fired previously are incompetent for re-initiation during the same cell cycle. However, in cells in which hypophosphorylated forms of p65cdc18 are in abundance, fired origins fail to remain incompetent, and high levels of over-replication are observed (Fig. 7). Under normal circumstances, CDK-dependent inhibition of origin competence persists until anaphase, when mitotic cyclins are degraded and CDK activity falls to very low levels. This event allows unphosphorylated replication factors (including p65cdc18) to accumulate during the subsequent G1 interval and re-establish the pre-initiation state at each chromosomal origin, which therefore becomes competent for a new round of DNA replication. This CDK-driven replication switch can explain why DNA replication occurs once and only once per cell cycle.
We believe that there may be components of the CDK-driven replication switch in addition to p65cdc18, as cells expressing low levels of mutant p65cdc18ΔCDK polypeptides exhibit very slow growth and cell cycle delay, but do not have grossly increased DNA contents as measured by flow cytometry (Fig. 5; P.V. Jallepalli and T.K. Kelly, unpubl.). Activation of p34cdc2 kinase at the G1 → S transition is likely to bring about the phosphorylation of additional CDK substrates at origins of replication, including potentially several subunits of ORC and the MCM family of proteins (Fig. 9; Coue et al. 1996; Hendrickson et al. 1996; Leatherwood et al. 1996). Phosphorylation of these replication factors could act in parallel with p65cdc18 phosphorylation to block re-activation of replication origins under some circumstances. When p65cdc18 is in abundance, however, these controls are apparently insufficient to prevent over-replication (Nishitani and Nurse 1995; Muzi-Falconi et al. 1996a). Under such conditions, we find that the phosphorylation state of p65cdc18 determines the frequency and degree of over-replication (Fig. 7). If p65cdc18 is present in a constitutively hypophosphorylated state (through mutation of CDK phosphorylation sites), the resulting cycles of re-initiation are resistant to increased mitotic cyclin/CDK activity, a known inhibitor of over-replication (Fig. 8; Hayles et al. 1994).
The fact that CDK phosphorylation inhibits p65cdc18 function through both degradative and nondegradative pathways may explain how the Xenopus and human homologs of p65cdc18 are regulated during chromosomal DNA replication, even though their abundance appears to be relatively constant throughout the cell cycle (Coleman et al. 1996; Williams et al. 1997). One possibility is that these metazoan proteins have evolved a simpler control mechanism in which CDK phosphorylation still regulates their biochemical activity and/or intranuclear distribution, but no longer promotes their rapid destruction. Consistent with this idea, consensus CDK phosphorylation sites have been conserved among all known homologs of p65cdc18 (Piatti et al. 1995,1996; Coleman et al. 1996; Williams et al. 1997), suggesting that the negative regulation of these initiator proteins by CDK phosphorylation is a general feature of DNA replication in all eukaryotes.
Materials and Methods
Growth and genetic manipulation of fission yeast strains
Strains used in this study are listed in Table 1 or have been described previously (Nasmyth and Nurse 1981; Connolly and Beach 1993; Jallepalli and Kelly 1996; Muzi-Falconi et al 1996a). Schizosaccharomyces pombe was grown in rich yeast-extract based medium (YE5S) or minimal synthetic medium (EMM) containing the required supplements. Genetic crosses and tetrad analysis were performed using standard techniques (Moreno et al. 1991).
Table 1.
Fission yeast strains used in this study
| TK2 | h− leu1-32 |
| TK13 | h+ cdc2-33 leu1-32 ade6–M210 ura4–D18 |
| TK117 | h+ leu1-32 ade6–M210 ura4–D18 his3–D1 |
| PJ252 | h+ cdc10–V50 leu1-32 ade6–M210 ura4–D18 |
| PJ273 | h+ cdc18–K46 leu1-32 ura4–D18 his3–D1 |
| PJ274 | h+ cdc18–K46 Δcig1::ura4+ leu1-32 ade6-704 ura4–D18 his3–D1 |
| PJ275 | h+ cdc18–K46 Δcig2::sup3-5 leu1-32 ade6-704 ura4–D18 his3–D1 |
| PJ300 | h+ cdc10–V50 Δcig2::sup3-5 leu1-32 ade6-704 ura4–D18 |
Protein preparation and immunoblotting
Total S. pombe protein was prepared by glass bead lysis of 108 cells in the presence of SDS-PAGE sample buffer, followed by boiling. Protein samples were subjected to SDS-PAGE (see below) and then transferred to nitrocellulose membranes for immunoblotting. Mouse monoclonal antibodies to HA (12CA5), tubulin (B512), and p34cdc2 (Y100) were used at 1:5000, in a solution of 5% nonfat dry milk in phosphate-buffered saline (PBS) plus 0.2% Tween 20. Rabbit polyclonal antibodies to p56cdc13 (min56) were affinity-purified and used at 1:500. For routine immunoblots, anti-mouse and anti-rabbit secondary antibody-horseradish peroxidase conjugates were used at 1:5000, and signals developed using an enhanced chemiluminescent reagent system (SuperSignal, Pierce). Alternatively, for quantitative immunoblots, a 35S-labeled anti-mouse secondary antibody (Amersham) was used at a concentration of 0.1 μCi/ml, and the radioactive signals quantified using a PhosphorImager (Molecular Dynamics).
Resolution of differentially phosphorylated p65cdc18 species by modified SDS-PAGE
A modified SDS-PAGE protocol was used to detect phosphorylation-induced changes in p65cdc18 mobility. Briefly, a 6% gel was prepared in a 24-cm Hoefer apparatus using solutions prepared from ultrapure reagents and filtered before use. Electrophoresis was performed at 200 V for 1 hr, 300 V for 1 hr, then 400 V for 3–4 hr; high voltage was critical in maximizing resolution. For other experiments, SDS-PAGE was performed using standard 18-cm gels (Hoefer) or 7-cm minigels (Bio-Rad Mini-PROTEAN system). Under these conditions, modified species of p65cdc18 are not resolved from one another but migrate as a single band.
Labeling of fission yeast cells with [32P]orthophosphate, immunoprecipitation, and phosphoamino acid analysis
[32P]Orthophosphate labeling was performed essentially as described (Moreno et al. 1991). Briefly, cells were grown overnight in phosphate-free EMM supplemented with 1 mm sodium phosphate, plus 5 μg/ml of thiamine (where indicated) to repress transcription from the nmt1+ promoter. Cells were then resuspended in fresh phosphate-free EMM supplemented with 50 μm sodium phosphate and labeled with 2.5 mCi [32P]orthophosphate for 90 min (in the experiment in Fig. 1, cells were shifted to 36°C for 150 min before labeling). Cells were washed twice with cold H2O, then spheroplasted and lysed with 1% Triton X-100 on ice. After adding SDS to 1%, extracts were boiled and diluted 20-fold with RIPA buffer lacking SDS. Equal cpm of each 32P-labeled extract were immunoprecipitated with anti-HA antibody or non-immune antibody, followed by RNase A digestion on ice for 15 min. Samples were resolved by 6% SDS-PAGE and subjected to autoradiography and/or PhosphorImager analysis (to detect 32P-labeled p65cdc18) or transferred to nitrocellulose membranes and immunoblotted with anti-HA antibodies (to detect total p65cdc18).
Phosphoamino acid analysis of the 32P-labeled p65cdc18 immunoprecipitate was performed essentially as described (Lin and Desiderio 1993). Briefly, the protein sample was precipitated with ice-cold trichloroacetic acid (with 30 μg of RNase A as a carrier) and washed four times with ice-cold ethanol. The air-dried pellet was then resuspended in 100 μl of 6N hydrochloric acid and boiled at 110°C for 1 hr, resulting in partial hydrolysis and release of 32P-labeled phosphoamino acids. After lyophilization, the pellet was resuspended in 10 μl of pH 1.9 buffer (Boyle et al. 1991) containing cold phosphoserine, phosphothreonine, and phosphotyrosine (100 μg/ml each) and 1% phenol red. Samples were applied to a thin-layer chromatography (TLC) plate and electrophoresed in pH 1.9 buffer at 2000 V for 2 cm, followed by pH 3.5 buffer (Boyle et al. 1991) at 2000 V for 9 cm. The position of the phosphoamino acid standards was determined by ninhydrin staining of the TLC plate, which was then exposed to film with an intensifying screen at −70°C for 4–7 days.
G1 synchronization by cdc10–V50 mutant block and p34cdc2 kinase assays
To synchronize cells in the G1 phase of the cell cycle, a cdc10–V50 mutant strain expressing HA-tagged cdc18+ from a weak heterologous promoter (REP42X) was grown to mid-log phase at 25°C and then shifted to 36°C for 4 hr. The G1-arrested culture was then chilled quickly on ice and incubated at 25°C to allow synchronous entry into S phase. [In some experiments, hydroxyurea (25 mm) was added at this point to arrest cells in early S phase.] Samples were taken at various timepoints throughout the experiment to monitor p65cdc18 phosphorylation, cellular DNA content, and p34cdc2 kinase activity.
p34cdc2 kinase activity was measured as follows. About 5 × 108 cells from each timepoint were lysed with glass beads in HB buffer (Moreno et al. 1991). After clarification at 15,000g for 30 min, 400 μg of each extract was incubated with 20 μl of p13suc1–agarose beads (Upstate Biotechnology) for 1 hr at 4°C. The immobilized p34cdc2 kinase complexes were washed three times in HB buffer and then incubated with 40 μl of kinase buffer [HB buffer supplemented with 200 μm ATP (containing 40 μCi/ml [γ-32P]ATP) and 1 mg/ml of histone H1] for 20 min at 30°C. Reactions were terminated by adding SDS sample buffer and boiling for 3 min. Phosphorylation of histone H1 was analyzed by 12% SDS-PAGE and quantitated using a PhosphorImager.
Measurement of mitotic cyclin/CDK activity using antibodies specific for p56cdc13 (min56) and protein A–Sepharose beads has been described already (Jallepalli and Kelly 1996).
Site-directed mutagenesis of CDK phosphorylation sites in cdc18+
The threonine residues at each of six CDK consensus phosphorylation sites in cdc18+ were mutated to alanine using the QuikChange site-directed mutagenesis method (Stratagene). In the case of CDK sites 1 and 4, an adjacent minimal phosphorylation site (T-P) was also changed to alanine. Mutagenesis reactions were performed on a plasmid carrying cdc18+ under the control of its own promoter and tagged at the amino terminus with three copies of the HA epitope (pIRTL–3HAcdc18+). Correct substitutions were assessed first by introduction of novel restriction endonuclease sites and confirmed by sequencing of the mutated region. The protein-coding regions of the cdc18ΔCDK1-5 and cdc18ΔCDK1-6 genes were also sequenced in their entirety. Fragments encompassing these regions were subcloned into plasmids containing either the full-strength thiamine-repressible nmt1+ promoter (REP3X) or a weak nmt1 derivative (REP42X), which is transcribed at 10- to 50-fold lower levels (Basi et al. 1993; Forsburg 1993).
To measure the half-life of mutant and wild-type forms of p65cdc18, transcription from the weak nmt1 promoter (REP42X) was repressed by addition of thiamine (10 μg/ml). Cells (108) were withdrawn from each culture just before thiamine addition (time 0) and at the indicated timepoints, pelleted, and quick-frozen in a dry ice/ethanol bath. Cells were subsequently lysed with glass beads in SDS-PAGE sample buffer and analyzed by immunoblotting. In other experiments, new protein synthesis was also inhibited by adding cycloheximide (100 μg/ml final) together with thiamine. This concentration of cycloheximide inhibited 95% of protein synthesis in fission yeast, as measured by incorporation of [35S]methionine into TCA-precipitable protein (P.V. Jallepalli and T.J. Kelly, unpubl.; Ayscough and Warren 1994).
Purification of GST–Cdc18 fusion proteins and in vitro phosphorylation by fission yeast CDKs
The purification of wild-type and ΔCDK1-5 mutant forms of p65cdc18 from fission yeast as GST fusion proteins has been described already (Brown et al. 1997). For the in vitro phosphorylation experiments in Figure 4, wild-type fission yeast strains were made to express HA-tagged versions of the S-phase cyclin cig2+ or the mitotic cyclin cdc13+. Extracts derived from these strains were then immunoprecipitated with the anti-HA monoclonal antibody 12CA5 and protein A–Sepharose beads. The immobilized S-phase and mitotic cyclin/CDK complexes were then incubated with purified GST–Cdc18 or GST–Cdc18ΔCDK1-5 proteins and [γ-32P]ATP as described (Brown et al. 1997). Phosphorylation of the input proteins was detected by SDS-PAGE and quantified using a PhosphorImager.
Analysis of cdc18-induced over-replication
For the cdc18-induced over-replication experiments in Figure 7, a wild-type fission yeast strain was transformed with plasmids expressing wild type or ΔCDK mutant forms of cdc18+ under the control of the full-strength, thiamine-repressible nmt1+ promoter (REP3X). Transformants were selected and propagated in minimal medium containing 5 μg/ml thiamine. At the beginning of each experiment, cells were washed five times and cultured in thiamine-free medium to induce transcription from the nmt1+ promoter, which becomes fully active ∼14–16 hr after removal of thiamine. Aliquots of each culture were taken for protein analysis and immunoblotting, for measurement of DNA content by propidium iodide staining and flow cytometry, and for direct examination of cell morphology by DAPI staining and fluorescence microscopy (Jallepalli and Kelly 1996). In some experiments, cells were also transformed with plasmids expressing an HA-tagged form of the mitotic cyclin cdc13+ under the control of the nmt1+ promoter (REP4X–cdc13HA) or with the empty vector as a control (REP4X). Growth and induction of these strains was performed exactly as described above.
We find that expression of cdc18+ from the full-strength nmt1+ promoter (REP3X) consistently induces over-replication to a lesser extent than that reported by Nishitani and Nurse (1995). The basis for this difference is unclear, but could relate to several differences in the conditions used to express cdc18+. Our constructs transcribe cdc18+ sequences starting with the first codon (ATG) of the open reading frame (ORF) and contain 3′ elements required for transcription termination. Moreover, our constructs are maintained extrachromosomally as autonomously replicating plasmids, rather than being integrated in multiple copies within the genome. The more modest over-replication phenotype produced by wild-type cdc18+ under these conditions is consistent with our previous observations (Muzi-Falconi et al. 1996a) and has enabled us to detect the increased over-replication potential of the mutant cdc18ΔCDK alleles.
Acknowledgments
We thank Stephen Desiderio, Susan Forsburg, Daniel Nathans, and our colleagues in the Kelly laboratory for helpful discussions and comments on the manuscript. We are grateful to Ann Marie Egloff and Stephen Desiderio for their assistance with phosphoamino acid analysis. We also thank Kathy Gould and Hiroyuki Yamano for generous gifts of antibodies to p56cdc13 and p34cdc2. This work was made possible by grants from the National Institutes of Health to T. J. K. and from the American Cancer Society to G.W.B., and a Medical Scientist Training Program Award to P.V.J.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tom_kelly@qmail.bs.jhu.edu; FAX (410) 955-0831.
References
- Ayscough K, Warren G. Inhibition of protein synthesis disrupts the Golgi apparatus in the fission yeast, Schizosaccharomyces pombe. Yeast. 1994;10:1–11. doi: 10.1002/yea.320100102. [DOI] [PubMed] [Google Scholar]
- Basi G, Schmid E, Maundrell K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene. 1993;123:131–136. doi: 10.1016/0378-1119(93)90552-e. [DOI] [PubMed] [Google Scholar]
- Bell SP, Mitchell J, Leber J, Kobayashi R, Stillman B. The multidomain structure of Orc1p reveals similarity to regulators of DNA replication and transcriptional silencing. Cell. 1995;83:563–568. doi: 10.1016/0092-8674(95)90096-9. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–148. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- Broek D, Bartlett R, Crawford K, Nurse P. Involvement of p34cdc2 in establishing the dependency of S phase on mitosis. Nature. 1991;349:388–393. doi: 10.1038/349388a0. [DOI] [PubMed] [Google Scholar]
- Brown GW, Jallepalli PV, Huneycutt BJ, Kelly TJ. Interaction of the S phase regulator Cdc18 with cyclin-dependent kinase in fission yeast. Proc Natl Acad Sci. 1997;94:6142–6147. doi: 10.1073/pnas.94.12.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TR, Carpenter PB, Dunphy WG. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell. 1996;87:53–63. doi: 10.1016/s0092-8674(00)81322-7. [DOI] [PubMed] [Google Scholar]
- Connolly T, Beach D. Interaction between the Cig1 and Cig2 B-type cyclins in the fission yeast cell cycle. Mol Cell Biol. 1993;14:768–776. doi: 10.1128/mcb.14.1.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Bordes J, Nurse P. p25rum1 orders S phase and mitosis by acting as an inhibitor of the p34cdc2 mitotic kinase. Cell. 1995;83:1001–1009. doi: 10.1016/0092-8674(95)90215-5. [DOI] [PubMed] [Google Scholar]
- Coue M, Kearsey SE, Mechali M. Chromatin binding, nuclear localization and phosphorylation of Xenopus cdc21 are cell-cycle dependent and associated with the control of initiation of DNA replication. EMBO J. 1996;15:1085–1097. [PMC free article] [PubMed] [Google Scholar]
- Dahmann C, Diffley JFX, Nasmyth K. S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transition of replication origins to a pre-replicative state. Curr Biol. 1995;5:1257–1269. doi: 10.1016/s0960-9822(95)00252-1. [DOI] [PubMed] [Google Scholar]
- Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–673. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- Enoch T, Carr AM, Nurse P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes & Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- Forsburg SL. Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 1993;21:2955–2956. doi: 10.1093/nar/21.12.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL, Nurse P. Cell cycle regulation in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe. Annu Rev Cell Biol. 1991;7:227–256. doi: 10.1146/annurev.cb.07.110191.001303. [DOI] [PubMed] [Google Scholar]
- Gavin KA, Hidaka M, Stillman B. Conserved initiator proteins in eukaryotes. Science. 1995;270:1667–1671. doi: 10.1126/science.270.5242.1667. [DOI] [PubMed] [Google Scholar]
- Grallert B, Nurse P. The ORC1 homolog orp1 in fission yeast plays a key role in regulating onset of S phase. Genes & Dev. 1996;10:2644–2654. doi: 10.1101/gad.10.20.2644. [DOI] [PubMed] [Google Scholar]
- Hayles J, Fisher D, Woollard A, Nurse P. Temporal order of S phase and mitosis in fission yeast is determined by the state of the p34cdc2-mitotic B cyclin complex. Cell. 1994;78:813–822. doi: 10.1016/s0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- Hendrickson M, Madine M, Dalton S, Gautier J. Phosphorylation of MCM4 by cdc2 protein kinase inhibits the activity of the minichromosome maintenance complex. Proc Natl Acad Sci. 1996;93:12223–12228. doi: 10.1073/pnas.93.22.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Kelly TJ. Rum1 and Cdc18 link inhibition of cyclin dependent kinase to the initiation of DNA replication in S. pombe. Genes & Dev. 1996;10:541–552. doi: 10.1101/gad.10.5.541. [DOI] [PubMed] [Google Scholar]
- ————— Cyclin-dependent kinase and initiation at eukaryotic origins: A replication switch? Curr Opin Cell Biol. 1997;9:358–363. doi: 10.1016/s0955-0674(97)80008-7. [DOI] [PubMed] [Google Scholar]
- Kelly TJ, Martin GS, Forsburg SL, Stephen RJ, Russo A, Nurse P. The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell. 1993a;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- Kelly TJ, Nurse P, Forsburg SL. Coupling DNA replication to the cell cycle. Cold Spring Harbor Symp Quant Biol. 1993b;58:637–644. doi: 10.1101/sqb.1993.058.01.071. [DOI] [PubMed] [Google Scholar]
- Klemm RD, Austin RJ, Bell SP. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell. 1997;88:493–502. doi: 10.1016/s0092-8674(00)81889-9. [DOI] [PubMed] [Google Scholar]
- Kominami K, Toda T. Fission yeast WD-repeat protein pop1 regulates genome ploidy through ubiquitin-proteasome-mediated degradation of the CDK inhibitor Rum1 and the S-phase initiator Cdc18. Genes & Dev. 1997;11:1548–1560. doi: 10.1101/gad.11.12.1548. [DOI] [PubMed] [Google Scholar]
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science. 1996;271:1597–1601. doi: 10.1126/science.271.5255.1597. [DOI] [PubMed] [Google Scholar]
- Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature. 1996;379:360–363. doi: 10.1038/379360a0. [DOI] [PubMed] [Google Scholar]
- Li Z, Dordai DI, Lee J, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- Lin W-C, Desiderio S. Regulation of V(D)J recombination activator protein RAG-2 by phosphorylation. Science. 1993;260:953–959. doi: 10.1126/science.8493533. [DOI] [PubMed] [Google Scholar]
- Martin-Castellanos C, Labib K, Moreno S. B-type cyclins regulate G1 progression in fission yeast in opposition to the p25rum1 cdk inhibitor. EMBO J. 1996;15:839–849. [PMC free article] [PubMed] [Google Scholar]
- Mondesert O, McGowan CH, Russell P. Cig2, a B-type cyclin, promotes the onset of S in Schizosaccharomyces pombe. Mol Cell Biol. 1996;16:1527–1533. doi: 10.1128/mcb.16.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Moreno S, Nurse P. Regulation of progression through the G1 phase of the cell cycle by the rum1+ gene. Nature. 1994;367:236–242. doi: 10.1038/367236a0. [DOI] [PubMed] [Google Scholar]
- Muzi-Falconi M, Kelly T J. Orp1, a member of the Cdc18/Cdc6 family of S-phase regulators, is homologous to a component of the origin recognition complex. Proc Natl Acad Sci. 1995;92:12475–12479. doi: 10.1073/pnas.92.26.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzi-Falconi M, Brown GW, Kelly TJ. cdc18+ regulates initiation of DNA replication in Schizosaccharomyces pombe. Proc Natl Acad Sci. 1996a;93:1566–1570. doi: 10.1073/pnas.93.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— DNA replication: Controlling initiation during the cell cycle. Curr Biol. 1996b;6:229–233. doi: 10.1016/s0960-9822(02)00464-5. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Viewpoint: Putting the cell cycle in order. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Nurse P. Cell division cycle mutants altered in DNA replication and mitosis in the fission yeast Schizosaccharomyces pombe. Mol & Gen Genet. 1981;182:119–124. doi: 10.1007/BF00422777. [DOI] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Nurse P. p65cdc18 plays a major role controlling the initiation of DNA replication in fission yeast. Cell. 1995;83:397–495. doi: 10.1016/0092-8674(95)90117-5. [DOI] [PubMed] [Google Scholar]
- Nurse P, Bisset Y. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol & Gen Genet. 1976;146:167–178. doi: 10.1007/BF00268085. [DOI] [PubMed] [Google Scholar]
- Piatti S, Bohm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Piatti S, Lengauer C, Nasmyth K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a “reductional” anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J. 1995;14:3788–3799. doi: 10.1002/j.1460-2075.1995.tb00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SI. The role of p34 kinases in the G1 to S-phase transition. Annu Rev Cell Biol. 1992;8:529–561. doi: 10.1146/annurev.cb.08.110192.002525. [DOI] [PubMed] [Google Scholar]
- Rowles A, Chong JP, Brown L, Howell M, Evan GI, Blow JJ. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell. 1996;87:287–296. doi: 10.1016/s0092-8674(00)81346-x. [DOI] [PubMed] [Google Scholar]
- Sauer K, Knoblich JA, Richardson H, Lehner CF. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila melanogaster. Genes & Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- Sipiczki M. Taxonomy and phylogenesis. In: Nasim A, Young P, Johnson B, editors. Molecular biology of the fission yeast. New York, NY: Academic Press; 1989. pp. 431–452. [Google Scholar]
- Stillman B. Cell cycle control of DNA replication. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Williams RS, Shohet RV, Stillman B. A human protein related to yeast Cdc6p. Proc Natl Acad Sci. 1997;94:142–147. doi: 10.1073/pnas.94.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]